
Multi-Enzyme Systems in Flow Chemistry


1
Faculty of Engineering, Universidade Lusófona, 1749-024 Lisboa, Portugal
2
DREAMS, Universidade Lusófona, 1749-024 Lisboa, Portugal
3
Department of Bioengineering, Instituto Superior Técnico, Universidade de Lisboa, 1049-001 Lisbon, Portugal
4
iBB—Institute for Bioengineering and Biosciences, Instituto Superior Técnico, Universidade de Lisboa, 1049-001 Lisbon, Portugal
*
Authors to whom correspondence should be addressed.
Processes 2021, 9(2), 225; https://0-doi-org.brum.beds.ac.uk/10.3390/pr9020225
Submission received: 31 December 2020
/
Revised: 19 January 2021
/
Accepted: 22 January 2021
/
Published: 25 January 2021
(This article belongs to the Special Issue Biocatalysis, Enzyme and Process Engineering)
Abstract
:Recent years have witnessed a growing interest in the use of biocatalysts in flow reactors. This merging combines the high selectivity and mild operation conditions typical of biocatalysis with enhanced mass transfer and resource efficiency associated to flow chemistry. Additionally, it provides a sound environment to emulate Nature by mimicking metabolic pathways in living cells and to produce goods through the systematic organization of enzymes towards efficient cascade reactions. Moreover, by enabling the combination of enzymes from different hosts, this approach paves the way for novel pathways. The present review aims to present recent developments within the scope of flow chemistry involving multi-enzymatic cascade reactions. The types of reactors used are briefly addressed. Immobilization methodologies and strategies for the application of the immobilized biocatalysts are presented and discussed. Key aspects related to the use of whole cells in flow chemistry are presented. The combination of chemocatalysis and biocatalysis is also addressed and relevant aspects are highlighted. Challenges faced in the transition from microscale to industrial scale are presented and discussed.
1. Introduction
Flow bioreactors present large surface-to-volume ratio that is advantageous in enzymatic reactions, but parameters such as fluid velocity which affect residence time, influence significantly enzyme kinetics. Nevertheless, they provide the ability to use cascades of bioreactors, each containing a different enzyme operating at its best condition, for the synthesis of compounds requiring multi-steps [1,2,3]. Immobilization of the enzymes on the walls of the reactors or in carriers allows their reuse or continuous use, and simplifies downstream processing as the enzymes will be retained inside the reactor [4].
The application of whole cells in flow reactors allows multi-enzyme reactions to be carry out with natural cofactor regeneration, usually with high region- and stereo-selectivity, at lower costs than if pure enzymes were used since no purification steps are required [5,6,7]. The use of the natural ability of the cells to form biofilms on surfaces and to live in extreme environments has been used to naturally immobilize the cells on the walls of the reactors and to perform the reactions in the presence of e.g., organic solvents [8,9]. Synthetic biology and metabolic engineering tools have resulted in the production of compounds by new synthetic routes [10,11].
When compared to batch processes, flow systems have putatively the potential to accelerate biotransformations by favoring mass transfer and to facilitate large-scale production, as smaller bioreactors and shorter reaction times are possible [12]. Both bioprocess intensification and simple numbering up of bioreactors are possible, which facilitates scale-up [13].
In this review, we discuss the achievements and bottlenecks of multi-enzyme systems in flow chemistry.
2. Flow Bioreactors
The concept of flow chemistry revolves around continuously pumping fluid(s) containing the starting material(s) through a reactor in a continuous manner to produce a stream of product. The reactors can be fabricated from glass, polymeric materials (e.g., polydimethylsiloxane (PDMS), polymethyl methacrylate (PMMA), polystyrene (PS), perfluoroalkoxy (PFA)) and metal (e.g., stainless steel) or ceramics, the two later chosen when the reaction is performed under high temperature/pressure [14,15]. Depending on the volumetry and one characteristic dimension, reactors can be classified as microreactors (or microfluidic) reactors, mesoreactors and macroreactors (Table 1).
Given these respective features, microreactors display high mass and heat transfer and allow operation in laminar flow, where all the particles in the fluid move in parallel layers, with no mixing between layers, opposite to turbulent flow, where the particles in the fluid move in a random and chaotic manner. However, they have poor throughput, processing solids is challenging and they are prone to channel blockage and high pressure drops; on the other hand, mesoreactors have higher throughput and are less sensitive to pressure drops, although mass and heat transfer is less effective than in microreactors and operation in laminar flow may not be feasible. Macroreactors abridge those reactors that display volumes above a few mL [12,16,17].
Different types of reactors are used, but they can be grouped in four different types: tubular reactors, packed-bed reactors, monolith reactors and chip-based reactors [14,18].
Tubular reactors are the simplest of all: fluid flows through the channel with negligible backpressure, and hydrodynamics and heat transfer are easy to control. In tubular reactors the inner wall of the vessel is used as the carrier for enzyme immobilization (wall-coated reactors). Even for capillary-sized vessels, the surface area to volume ratio is smaller than for other microreactors; hence enzyme loading is poorer with a negative impact on the reaction efficiency. Several strategies have been suggested to improve enzyme loading, mostly involving the deposition of nanomaterials on the inner cell wall of the reactor, as recently reviewed [19]. An inner diameter of capillary as small as possible is also advised to minimize diffusion path [18,19]. Wall-coated reactors were used in cascade reactions, such as the two-step conversion of bis(p-nitrophenol)phosphate monosodium salt into p-nitrophenoxide and inorganic phosphate [20,21], and three-step production of resorufin from lactose [21]. In the later case, the coated-wall microreactor was outperformed by an equivalent packed-bed reactor based on the calculated specific substrate conversion efficiency [21].
Packed-bed reactors contain the immobilizing support and are designed in order to maximize enzyme loading. Hence, enzymes are immobilized on, e.g., hydrogels or inorganic particles that are then packed in a channel. Unlike conventional, macroscale packed-bed reactors, where mm size particles are used with negative impact in heat and mass transfer, in microreactors the particle size should be under 50 nm. The high surface area to volume ratio also results in shorter diffusion paths when compared with coated-wall reactors. Still, relatively high backpressure is required to achieve the intended flow rate [14,19]. The production of (1S,2S)-1-phenylpropane-1,2-diol through an enzymatic cascade involving fusion enzymes benzoylformate decarboxylase and alcohol dehydrogenase separately immobilize on HaloLinkTM resin. The sequential packed beds were operated for several weeks with high conversion, stereoselectivity and space–time yields up to 1850 g L−1 day−1 [22]. Pyrroline-5-carboxylate reductase and lysine-6-dehydrogenase were co-immobilized in agarose microbeads and applied in a packed-bed reactor for the continuous synthesis of L-pipecolic acid from L-lysine. Almost quantitative molar conversion was achieved in 30 min residence time with a space–time yield up to 2.5 g L−1 h−1 [23]. Ethyl esters were produced using combi-CLEAS of lipases from different sources. The optimized formulation was applied in a packed-bed reactor and operated for 30 days with constant conversion yields of ~50% and an average productivity of 1.94 gethyl esters gsubstrate−1 h−1 [24]. Tyrosinase and DOPA-decarboxylase were immobilized in functionalized silica beads. The formulations were applied in sequential packed-bed reactors for the production of dopamine from tyrosine to achieve an overall yield of 30% [25].
Despite of their high enzyme loading capacity, packed-bed reactors still display some drawbacks, e.g., high pressure drops, limited heat transfer and risk of leakage at high flow rates. To overcome these, monolith reactors were developed that display a network of meso- or microporous structures, hence high void volume easing fluid flow and lower pressure drop. Moreover, the synthesis of monoliths is performed within the channel, thus packing procedures are avoided, although the preparation can be time-consuming and reproducibility is questionable. Monoliths can be silica- or polymeric-based; the former endure organic solvents, but are sensitive to extreme pH whereas the later are not affected by pH, but their pore structure may be affected by organic solvents [14,18,19]. Invertase, glucose oxidase and horseradish peroxidase were immobilized in a polymeric monolith to produce resorufin from sucrose. The authors established that high product yields were only observed if the enzymes were immobilized in the correct sequence. Moreover, the preparation displayed high operational stability [26]. Deoxyribonuclease I, snake venom phosphodiesterase and alkaline phosphatase were immobilized in a silica-based capillary monolith for the digestion of genomic DNA. Enzymatic activity was significantly enhanced upon immobilization and the monolith displayed high operational and storage stability [27]. Chip reactors typically display a bankcard or microscope slide footprint. The internal structural designs of microfluidic channels inside the chip have a wide variety, such as a straight, serpentine or zig-zag microchannel to enhance the effective volume for immobilization, multiple channels, inclusion of single or multi-chambers and/or wells, among others. They are made of glass, poly(dimethylsiloxane), poly(ethylene terephthalate) or poly(methyl methacrylate), among other materials. Immobilization can be performed either directly on the channel, as for tubular wall-coated reactors, or particles or monolith can be packed in the channel/chambers [12,14,16,18]. As example, β-galactosidade, glucose oxidase and horseradish peroxidase were individually immobilized in three compartments in a chip-type reactor for the quantification of lactose [28]. The different configurations of microreactors are illustrated in Figure 1.
2.1. Immobilization
Enzyme immobilization consists in the confinement of those specialized proteins in a defined region of space while retaining their catalytic activity. This approach enables either enzyme reuse or continuous use while simplifying the downstream processing, since reactor effluents are enzyme-free [4]. Besides the wide versatility in the biocatalytic set-up and high volume specific biocatalyst loading enabled by enzyme immobilization [4,29,30], when properly implemented, this technology conveys improved activity, resilience, selectivity and stability to the biocatalyst [29,30,31,32], enhanced space–time yield [33] and can increase enzyme purity [34]. Immobilization is not without some drawbacks, namely: the cost of enzyme carriers and of immobilization procedures, plus complexity of the later; disposal of exhausted immobilized enzyme formulations; risk of fouling; and decrease in observed reaction rates as compared to free enzymes, either due to mass transfer limitations or enzyme inactivation during immobilization [4,35]. Immobilization of enzymes is paramount within the scope of flow chemistry as to retain the biocatalyst in tubular reactors. Additionally, when cascade enzymes are used, the possibility to contain the individual enzymes through immobilization in specific compartments can enhance the overall kinetics by providing reduced, more efficient paths for the intermediates to move between them as enzymes are close to one another, preventing unwanted cross-reactions and easing the regeneration of cofactors [1,30,36,37]. There is no universal method for enzyme immobilization, but an ideal approach requires favorable interaction between enzyme and the carrier (if used), which should provide a high surface area and display chemical, mechanical and thermal stability, high rigidity and endurance to microbial degradation, alongside with ease of regeneration. Moreover, the carrier should be nontoxic and have a low cost and be easy to produce. Simultaneous compliance with all these requirements is hard if not impossible to achieve, hence a compromise is often needed [15,30]. Roughly, enzyme immobilization can be carried through either chemical or physical methods, most of which involve interaction with or containment inside a solid carrier. Immobilization of an enzyme onto a carrier can be achieved through (a) covalent binding, where noncatalytic enzyme residues, e.g., lysine, cysteine and aspartic and glutamic acids, form covalent bonds with active groups of the carrier; (b) adsorption, where the enzyme attaches to the surface of the carrier through weak forces; (c) ionic binding, which involves ionic interaction between charged enzyme residues and oppositely charged carriers; (d) affinity binding, where either the enzyme is conjugated to a molecule that displays affinity towards the carrier or the carrier is previously coupled to an affinity ligand for the envisaged enzyme; (e) alternatively to carrier binding, carrier-free immobilized enzyme formulations can be produced by chemically crosslinking enzyme aggregates to deliver micro- to millimeter-sized crosslinked enzyme aggregates (CLEAs or combi-CLEAS, if multiple enzymes are immobilized), using a bifunctional reagent; (f) entrapment, where the enzyme is physically contained within a polymeric network, rather than attached to the surface of the carrier; and (g) encapsulation, where the enzyme is physically contained within a membrane [30,38,39] (Figure 2). Several reviews have been published recently providing comprehensive overviews on methodologies for enzyme immobilization and characterization [30,40,41,42,43], including some where focus is given to the particular requirements for multi-enzyme immobilization [1,37,44,45,46,47,48,49] and to the integration of immobilized enzyme systems and continuous flow reactors [14,18,50].
Briefly: (a) In covalent binding, the enzyme molecules are linked to the carrier typically through either amide, carbamate, ether or thioether bonds. This method offers the strongest bond between enzyme and carrier, hence high reusability and resilience under extreme condition and often enhances stability, yet the random orientation of the enzyme molecule during immobilization can hamper activity [14,30,51,52]. It is mostly irreversible, thus the whole enzyme formulation has to be discarded once activity is exhausted, safe for a few exceptions involving disulfide bonds [41,42]. Conformational changes as an outcome of immobilization and risk of amino acid residues involved in catalytic activity being compromised in the covalent bonding are two reported drawbacks, but these can be overcome by careful selection/design of the carrier [53,54,55], enzyme engineering [56] and blockage of the active site of the enzyme through the use of the substrate or of a competitive inhibitor during immobilization [14]. (b) Adsorption involves interaction between enzyme and carrier through weak, nonspecific forces, e.g., van der Waals forces, hydrogen bonds and hydrophobic interactions. The risks of conformational changes upon immobilization are low, due to the mild nature of the method, although the random orientation of the enzyme in the carrier may again cause concern, and the process is typically reversible. This can prove useful, as the carrier can be washed and reloaded with fresh enzyme, but unwanted enzyme leakage is most prone to occur [14,52,57,58,59]. (c) Ionic binding involves the interaction between oppositely charged functional groups of the carrier and of the enzyme. The immobilization process can be controlled by a suitable adjustment of the pH of the medium according to the isoelectric points of enzyme and carrier [60,61]. Binding strength is superior to adsorption and reversibility can be achieved through proper manipulation of pH, temperature and ionic strength, but again unwanted enzyme leakage may occur, as well as conformation changes upon immobilization [14,41]. (d) Affinity binding evolves around the interaction between enzyme and carrier through specific ligands. To achieve this, the enzyme is typically decorated, either chemically [62] or genetically [44], with a suitable tag (e.g., a peptide tag, a given protein domain) that attaches to the complementary ligand in the carrier, through either covalent or noncovalent interactions, which convey high enzyme load, stability and proper enzyme orientation, albeit at increased cost and complexity when compared to other methods [14,30]. Typical examples are His-tag/metal binding [63,64], avidin/ biotin [65], ssDNA/ssDNA [66], Spytag/Spycatcher formation of isopeptide bond [67] or Zbasic2 module/silica [68]; comprehensive information can be found elsewhere [34,44,69]. Affinity binding can be reversed under adequate conditions (pH, temperature, chemical processing), thus allowing the reuse of the carrier once enzyme activity is depleted [14,15]. (e) CLEAs and combi-CLEAs are typically formed using glutaraldehyde as crosslinking agent, due to low cost and wide availability, but other reagents such as dextran polyaldehyde [70], genipin [71] and polyethyleneimine [72] have been used. By avoiding the use of a carrier, process cost decreases and productivity (kgproduct/kgenzyme formulation) increases as carriers account for about 90% to 99% of the total mass of the enzyme formulation (CLEAs, combi-CLEAs). This irreversible immobilization method is easily implemented and has low cost, but poor mechanical stability has been often associated to CLEA [73,74], as well as a trend towards size increase and cluster formation, when these enzyme formulations are recovered from the reaction medium by centrifugation or filtration, resulting in mass transfer limitations [74,75]. To tackle these issues, magnetic combi-CLEA were developed that allow for easy recovery from the reaction media [39,76]. The use of magnetic materials within the scope of multi-enzyme immobilization has been advantageously used when methods other than combi-CLEAs were used [77,78,79,80,81], as recently reviewed [82]. (f) Entrapment is a typically mild, easy to implement, physical irreversible immobilization process where the enzyme is physically contained within a matrix, e.g., hydrogels [71,72], metal–organic frameworks [83], a type of highly porous and thermally stable material, with tunable functionalities, composed of metal ions/clusters bound by organic ligands [54,84] or membranes [58,85]. The method is highly dependent on enzyme size to pore size of the carrier and prone to mass transfer limitations and enzyme leakage [14]. (g) Encapsulation can be considered a variation of entrapment whereby the enzyme is contained within a semipermeable membrane while maintaining the native form of the enzyme structure [86,87], thus sharing mostly the advantages and drawbacks of entrapment [14]. It should be pointed out that often in the literature, entrapment and encapsulation are used indiscriminately. Recent concise comparative evaluations of the advantages and limitation of the different immobilization methods can be found elsewhere [14,88]. Some further recent representative examples of the application of the different methods for the immobilization of enzyme cascades, including some where different immobilization methods are combined, are given in Table 2.
When cascade reactions catalyzed by multiple immobilized enzymes are considered, three different strategies can be considered: stepwise immobilization of enzymes, mixed immobilization of enzymes and co-immobilization of enzymes. In the former two cases, enzymes are separately immobilized on the carrier, whereas in the latter case, which is possibly the most disseminated in multi-enzyme systems, the enzymes are immobilized on the same carrier (Figure 3) [1,46,49]. Briefly, stepwise immobilization is based on the use of several units, organized in a sequential manner, each unit consisting of an individual immobilized enzyme, thus catalyzing one reaction [28,58]. Proper enzyme sequence in the flow sense is mandatory to achieve high product yields [21,26]. The approach is quite flexible, allows optimization and detection of activity and stability of each formulation, as well as the adjustment of the reaction conditions for each step, the overall catalytic efficiency is relatively low and is more energy-consuming when compared with the remaining methods [50]. Still, co-immobilization failed to improve methanol production from CO2 in a three-step enzyme reaction when compared with sequential immobilization, due to the unfavorable trade-off between product inhibition and low substrate concentration for the adjacent enzymes [58]. Mixed immobilization is achieved by mingling individual immobilized enzymes. The relative proportion of the different enzymes is easy to control and when used for the transesterification of soybean oil with methanol this approach allowed to minimize methanol inhibition as compared to the use of co-immobilized enzymes. However, higher initial reaction rate was observed in the latter case [101]. The synthesis of tauroursodeoxycholic acid from taurochenodeoxycholic acid by 7α- and 7β-hydroxysteroid dehydrogenases immobilized in activated chitosan microspheres through covalent binding was evaluated using the three different immobilization strategies. Substrate conversion of 73%, 80% and 90% and product yields of 22%, 41% and 62% were observed for stepwise immobilization, mixed immobilization and co-immobilization, respectively. This trend was partially ascribed to the close proximity of the enzymes in the latter case and concomitant reduction of diffusional limitations [102]. Mixed immobilization is seldom used, for when compared with co-immobilization it mostly displays lower activity and poorer kinetics. Again, this trend is ascribed to the closer proximity between enzymes in the latter methodology, hence reducing diffusional restrictions related to the transfer of intermediates in the cascade [103,104,105]. In co-immobilization the different enzymes of the cascade are simultaneously immobilized in the same carrier. This approach has been clearly privileged in recent publications as it has been shown that it typically enhances the reaction rate. Thus, due to the close proximity of the enzymes, high initial concentration of intermediate products can be obtained, hence enabling the remaining enzymes to express all the activity from the onset of the reaction [46,49]. Still, effective immobilization of multiple enzymes in a single carrier is particularly challenging, since the properties of the carrier, e.g., hydrophilicity, acidity, porosity, will most likely affect differently the conformation of the different enzymes, hence carrier loading and activity/stability of the biocatalyst [1,37,49]. A suitable compromise involving multiple enzymes and a single carrier may not be achieved; hence, more than one carrier may have to be used [37]. Detailed insight on the advantages and drawbacks of co-immobilization of enzymes can be found elsewhere [49]. Co-immobilization can be implemented through random co-immobilization, which is the simplest approach to assemble an immobilization system, where enzyme solutions are mixed with either carriers or a crosslinking agent and the classical immobilization methods take place [75,76,106,107]. Nevertheless, this approach hardly complies with the attempt to control the immobilization pattern and the ratio of immobilized enzyme [32,35,71], an approach that attempts to mimic the structure of the cellular environment where multi-enzymatic reactions take place [1,46,50,100,108,109], and positional co-immobilization that allows for setting the enzymes sequentially according to the pathway/reaction rate and enzyme loading, which is often used for direct immobilization to surfaces [46,59,65], e.g., the inner walls of flow reactors [46,50].
2.2. Engineering Aspects
From an engineering perspective, flow bioreactors are simple reactors through which a fluid containing the substrate(s) and/or the biocatalyst is pumped to yield a stream containing the product(s). The large surface-to-volume ratio in microfluidic reactors is advantageous for enzyme loading. However, several parameters such as fluid velocity, which directly influence residence time and mass transfer, may be responsible for the behavior of the biocatalyst(s) and for the success of the proposed bioprocess. Several parameters and structures that may be engineered in a flow bioreactor are summarized in Figure 4.
Flow patterns inside the reactors may be generated by modifications of the internal geometry to increase mixing. Holvey et al. showed that by changing the mixing geometry of the mixing zone, chaotic advection may be induced as well as changes in the mixing time scale [110]. The authors used a mixing zone with a tangential, SZ-shaped or caterpillar mixer, and found that this zone was the main contributor to the overall pressure drop in the reactor due to significant chaotic secondary flow patterns. These caused a quadratic relation between pressure drop and velocity, whilst in the serpentine-shaped main channel, pressure drop decreased linearly and laminar flow with negligible formation of vortices was maintained.
Nakagawa et al. demonstrated that enzyme activity is affected by the backpressure caused by the channel shape [111]. The authors immobilized a protease from Bacillus licheniformis in a freeze-dried polyvinyl alcohol (PVA) micromonolith directly prepared in the microchannels of five microreactors, which differed from each other by having microchannels consisting of interconnected straight and elbow segments or a plain straight segment. Microchannel patterning and flow rates were found to significantly influence protease activity during the nine days of the assay, the results suggesting that there is an optimal combination of straight and elbow sections that maximizes reaction rate. When the mean diameter of the fluid path decreased, the Reynolds number increased, and the access of substrate to the enzyme via diffusion was thus affected by fluid flow.
The small size of the reactors also enhances wall–fluid heat transfer rates as result of the surface-to-volume ratio [112]. Several devices have been proposed and tested to maintain temperature or to create a temperature gradient in microfluidic chips, including in-chip, out-of-chip and noncontact heating/cooling systems [45,46,47]. For economical reasons, in-chip heating/cooling systems should be avoided in disposable bioreactor chips and a multi-use chip holder containing the required electronics, sensors and actuators, should be used. Furthermore, the small size of channels hampers the placement of classic sensors and temperature control relies often on the measurement of outlet temperatures. Since heat transfer coefficients up to 10 kW/m2.K have been reported, energy balances are usually neglected and operators assume isothermal conditions or microreactors may be designed for isothermal operation [48,49]. Calorimetric measurements allow the assessment of both heat transfer characteristics of the bioreactor and that caused by a biochemical reaction [50,51].
Calorimeters can measure enthalpy using temperature variation, power compensation and heat conduction, and can be used to study protein–ligand interactions, enzyme activity and protein folding [50,52]. Wei et al. developed a multi-channel calorimetric simultaneous assay (MCSA) platform containing the measurement circuit and calorimeter array in a single block [113]. The MCSA platform could detect 38 mV/K during temperature change and detect heating in the 7.865 V/J range. The temperature change range correlated linearly with catalase activity. Recently, van Schie et al. showed that a microfluidic calorimeter could be successfully used to determine enzyme activity by developing a new calibration method [114]. Deprotonation of phosphates provided a significant amount of heat during sufficient time for calibration purposes and to determine the sensitivity of each thermopile used. The authors used the hydrolysis of 5 mM para-nitrophenyl phosphate by 10 nM of alkaline phosphatase to para-nitrophenyl and phosphate. The latter inhibited enzyme activity. The enthalpy of the reaction was found to be −43.7 kJ/mol by isothermal titration calorimetry and the system was sensible enough to assess the inhibitory effect of phosphate. The system could be used to screen for substrates targeting drug discovery or to identify enzyme mutants, which can also be further characterized by more sensitive methods such as isothermal titration calorimetry.
A recent study showed how isothermal titration calorimetry may be used for the systematic characterization of the catalytic efficiency of human soluble epoxide hydrolase (hsEH) towards its epoxy-fatty acids substrates [115]. Using a single-injection method, the intrinsic heat of hsEH-mediated hydrolysis of the native substrates could be measured continuously in real time allowing the determination of the reaction rate without the need for synthetic substrates, or for detectable changes in the physicochemical properties of substrates or products over time to monitor their concentration.
All the previous techniques described to improve and control engineering aspects, such as heat and mass transfer inside microreactors, may be used in multi-enzymatic systems. Theoretically, cascades of microreactors operating at different temperatures and flow rates could be placed in series to produce compounds requiring multi-steps to be synthesized. The spatial organization of the enzymes, to simulate natural compartmentalization inside living organisms, may be achieved by co-immobilization of multi-enzyme systems on solid materials, by using microreactors with separate areas for each enzyme, or through cascades of microreactors (for reviews see [1,2,3]).
Luckarift et al. used individual microfluidic chips containing the following catalysts: (i) metallic zinc to catalyze the reduction of nitrobenzene to hydroxylaminobenzene, (ii) silica-immobilized hydroxylaminobenzene mutase for the bioconversion of hydroxylaminobenzene to 2-aminophenol and (iii) silica-immobilized soybean peroxidase for the polymerization of the latter to 2-aminophenoxazin-3-one [116]. This product is an intermediate in the synthesis of actinomycins. The silica-immobilized enzymes were packed into the channel of the chip with an equal volume of agarose beads to prevent silica particles from packing and to reduce void volume. Relatively low overall yields were observed, but the system may be used to assess the chemoenzymatic conversion of nitroarene substrates into the corresponding phenoxazinones.
Heinzler et al. developed a cascade of immobilized microfluidic enzyme reactors where intermediate products are transported for further conversion in subsequent reactor modules under optimal conditions for the production of glycan [117]. Six different enzymes, namely galactokinase, UDP-sugar pyrophosphorylase, UDP-glucose-dehydrogenase, NADH oxidase, β1,4-galactosyltransferase and glucuronosyltransferase, were immobilized on magnetic particles and loaded to each microreactor compartment at the best loading yield (g immobilized enzyme per L of settled beads). The reactor system was connected to an ESI–Q–ToF MS for in-line reaction control and product analysis. A yield of 96% of the nonsulfated human natural killer cell-1 glycan epitope was achieved, which was ca. 40% higher than that achieved with soluble enzymes. This was mainly the result of using the best reaction conditions for each enzyme in the different compartments.
A different approach was tested by Logan et al.: multiple enzymes were placed on porous polymer monoliths within microfluidic devices by photopatterning [26]. The system was tested with (i) glucose oxidase and horseradish peroxidase, and with (ii) invertase, glucose oxidase and horseradish peroxidase. Both systems were tested at different flow directions, but significant product formation was only observed in the correct sequential order. This demonstrated that control of the sequence of multi-enzymatic reactions is possible by patterning enzymes in microfluidic reactors.
Vong et al. used a DNA-directed immobilization strategy to immobilize enzymes in a closed fused silica microchannel for the production of gluconolactone from glucose mono-acetate [66]. The first enzyme of the system, Candida antarctica lipase B, and the third, horseradish peroxidase, were immobilized by ssDNA-ssDNA in discrete zones of the capillary wall, and the two microchannels were connected to each other by a piece of unmodified fused silica tubing of variable length. The second enzyme necessary in the system was added in the mobile phase. Product formation increased with increasing distance between the two microchannels containing the immobilized enzymes. The reaction time of the glucose oxidase being transported in the reaction medium could be varied independently of the other two enzymes, which allowed the study of engineering aspects in this complex reaction system.
3. Reactions with Whole Cells
Whole cells allow multi-enzymatic reactions, with cofactor regeneration, with high region- and stereo-selectivity, while maintaining the enzymes under their theoretically best optimal conditions (for reviews see [5,6,7,10]). They also allow lower bioprocess costs than pure enzymes, which require expensive isolation and recovery procedures. Since undesired reactions may take place in the cell, and some interesting biocatalysts such as extremophiles may require harsh reaction conditions, systems biology and metabolic engineering tools have been used to develop industrially relevant bioprocesses. This improved the production of traditional metabolites such as ethanol and lysine, but most importantly, the production of nearly any desired molecule such as artemisinic acid [11] and spider silk [118].
One of the major advantages of using whole cells is their ability to regenerate cofactors naturally. Due to the high cost of NADH and NADPH, cofactor recycling may hamper the implementation of a bioprocess at industrial scale. However, side reactions may occur. In a recent review evaluating NAD(P)H regeneration, the authors compared six approaches: enzymatic, chemical, homogenous catalytic, electrochemical photocatalytic and heterogeneous catalytic methods [119]. They suggested that since whole-cell immobilization can cause side reactions, it might not be appropriate for chiral drug and fine chemical synthesis. Nevertheless, there are several successful examples of application of microbial cells in the literature (Table 3). Immobilized cells have been reported to synthesize several classes of chiral compounds, including alcohols, amines, amides, sulfides, carboxylic acids and lactones (for a review, see [120]).
Immobilization of whole cells may increase their lifetime in organic solvents and non-natural environments, specific biocatalyst loading, and simplify recycling and downstream processing [6,60,61]. The natural ability of whole cells to form biofilms on surfaces, which protect the cells from toxic compounds and harsh environments, may also be used in biocatalysis as an improved form of immobilization [8,62,63].
Rhamnolipids, which are a group of industrially relevant biosurfactants, could be produced in biofilms of P. putida containing the rhlAB operon from the opportunist pathogen P. aeruginosa, in flow cells [125]. Clostridium beijerinckii BA101 adsorbed onto clay brick was able to produce acetone, butanol and ethanol from glucose in a continuous packed-bed reactor [126]. The reactor worked for 25 days and a maximum solvent yield of 0.45 g/g was achieved at a dilution rate of 0.3 h−1.
Whole cells of Lactobacillus kefiri were used for the reduction of (2,5)-hexanedione to yield (5R)-hydroxyhexane-2-one at an enantiomeric excess >99% [121]. When the cells were used immobilized in sodium cellulose sulfate in a plug flow reactor, they reached high selectivity (95%) and space–time yield (87 g/L day) for six days, maintaining 68% of residual activity at the end of the assay. Additionally, the productivity in the plug-flow reactor was 14 and 23 times larger than that observed in batch reactors with immobilized and free cells, respectively.
Continuous ethanol production was attained in packed-bed reactors with polypropylene or plastic composite supports [127]. The latter contained 75% polypropylene with 20% ground soybean hulls and 5% zein for Zymomonas mobilis, or 5% soybean flour for S. cerevisiae. An ethanol productivity of 536 g/h.L, corresponding to 39% yield, was attained with Z. mobilis, whilst S. cerevisiae produced 499 g/h.L, corresponding to a yield of 37%. In the biofilm reactors, ethanol productivities were 15- and 100-fold higher than in suspension cultures for S. cerevisiae and Z. mobilis, respectively. Z. mobilis in biofilm can produce ethanol from lignocellulosic hydrolysates, which usually inhibit the growth and enzymatic activity of microorganisms. Rice straw hydrolysate could be converted into ethanol by two strains of Z. mobilis in a laboratory-scale packed-bed biofilm reactor with plastic composite corn silk as biofilm support operated continuously or in batch mode [9].
A bacterial–yeast consortium containing Brevibacillus laterosporus and Galactomyces geotrichum was used for the decolorization of two effluents from the textile industry [128]. In a triple-layer fixed-bed bioreactor, made with three layers of noncorrosive wire mesh 1 cm apart from each other, the cells could maintain over 80% decolorization for a period of 7 days and ca. 78% reduction in chemical oxygen demand (COD).
Co-immobilization was also used for the multi-step synthesis of enantiopure chiral compounds: whole cells of E. coli with Chromobacterium violaceum ω-transaminase activity were co-immobilized with Lodderomyces elongisporus with ketoreductase activity [122]. Even in continuous flow mode, the whole-cell system could carry out the cascade of reactions necessary for the conversion of the racemic 4-phenylbutan-2-amine or heptan-2-amine into the corresponding enantiomerically pure R amine and S alcohol. The co-immobilized whole cells presented higher activity up to 24 h of continuous flow operation and final conversion (46.7%) than when the cells were used in a single-cell cascade (41.6%).
To improve L-malic acid by S. cerevisiae, the cells were immobilized on a microchannel bioreactor by covalent immobilization and their membranes were permeabilized with hexadecyltrimethylammonium bromide to improve mass transfer across them [129]. Both the biocatalyst and volumetric productivities observed in the microreactor were nearly four times higher than reported for a membrane reactor, although the substrate concentrations tested were more than 10 times lower, indicating process intensification.
A different application of whole cells in microreactors may be used for drug testing and development. One example is the “human gut-on-a-chip” microdevice where two microfluidic channels are separated by a porous membrane coated with an extracellular matrix and lined by human intestinal epithelial (Caco-2) cells [130]. The device mimics the complex structure and physiology of the human intestine by flowing fluid at low rate and by exerting cyclic strain as peristaltic motions. This type of flow microdevice could be applied to mimic other human organs and allow the study of the multi-enzymatic reactions of the human metabolism while assessing the transport, conversion and toxicity of substrates and/or therapeutic drugs.
4. Chemoenzymatic Reactions
Enzymatic one pot cascades have been shown to provide a sound approach for the production of a plethora of chemicals [46,131]. Still, there is a growing demand for new molecules, namely of chiral nature, that can be used as building blocks or end-products, which cannot be entirely fulfilled solely through a purely enzyme-based approach, given the relatively limited substrate scope and reactivity of enzymes. This limitation can be overcome by integrating chemocatalysis and biocatalysis (Table 3). The resulting chemoenzymatic cascades combine the high selectivity of enzymes with the high productivity of chemocatalysts. Still, the implementation of this strategy is quite challenging, namely because the typical operational conditions are quite diverse in chemocatalysis and biocatalysis, the former involving harsh thermal, pH and pressure environments and nonaqueous media, the latter involving mild pH and temperature environments, atmospheric pressure and mostly aqueous media, and such incompatibilities must be overcome [132,133]. Continuous flow operation where the catalysts are contained in tubular reactors allows for compartmentalization, e.g., chemocatalysis and biocatalysis being performed in separate, sequential reactors, a strategy that allows for overcoming incompatibility issues. In cases where kinetics, operational conditions and stability of the chemical and enzymatic steps are compatible, catalysts formulations can be packed in a single reactor, which simplifies the experimental set-up and reduces costs [134]. To favor this approach, efforts are made to develop biocatalyst with improved thermostability [135]. The integration of chemocatalysis and biocatalysis towards the implementation of multi-step production systems is relatively recent, but it has gained growing attention and the concomitant developments have been recently published in comprehensive reviews [136,137]. Still, some representative examples are provided.
Compartmentalization in alternating sequential flow reactors was used for the production of the (S)-N-Boc-phenylalanine benzylamide enantiomer through dynamic kinetic resolution of racemic N-Boc-phenylalanine ethyl thioester by the selective amidation with benzylamine of the (S) enantiomer using Alcalase immobilized by adsorption on ethyl-grafted macroporous silica gel. Racemization of the residual (R) ethyl thioester enantiomer was carried out using the non-nucleophilic base 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU) attached also to ethyl-grafted macroporous silica gel. Each of the catalytic active gels was packed in columns that were intercalated in series to alternate kinetic resolution (Alcalase driven) with racemization (DBU driven), in a total of 11 columns, with tert-amyl alcohol as common solvent. The compartmentalization allowed the use of optimal temperature for each step, 50 °C for the kinetic resolution and 150 °C for racemization. Continuous operation throughout 120 h was undertaken with unaltered catalyst performance allowing high conversion (79%), purity (98% enantiomeric excess) and volumetric productivity of 8.17 g L−1 h−1 [138].
The integration of chemo- and biocatalyst in the same reactor was implemented for the dynamic kinetic resolution of benzylic amines. This goal was achieved by packing in a single reactor sol−gel entrapped lipase B from Candida antarctica for the kinetic resolution with isopropyl 2-ethoxyacetate for selective amidation and palladium on 3-aminopropyl-functionalized silica for the racemization of the nonreacting enantiomer. Continuous operation was performed with 2-methyl-2-butanol as solvent at temperatures within 60 to 70 °C. Both catalysts were active and stable under these operation conditions. To achieve high conversion yields, columns packed only with the immobilized enzyme were also used for kinetic resolution solely. Continuous flow operation was performed to produce (R) enantiomers of the corresponding amines in high yields (57−96%) and purity (enantiomeric excess over 99.8%). This outcome was obtained using none or only one or two columns for the kinetic resolution, prior to that used for dynamic kinetic resolution. One of these systems was monitored for 48 h at 60 °C, displayed high operational stability and produced highly pure (R) enantiomer (enantiomeric excess over 99.8%) with a volumetric productivity of 4.3 g L−1 h−1 [139].
Compartmentalization in two flow reactors was used for the production of (E)-4-hydroxystilbene and of the pharmacologically active resveratrol and pterostilbene from coumaric acid. This was performed through the sequential use of phenolic acid decarboxylase from Bacillus subtilis entrapped in calcium alginate beads and a heterogenous Pd catalyst. The catalysts were individually packed in column reactors. The enzymatic decarboxylation to vinylphenol was performed at 30 °C and the ensuing cross coupling step (Heck coupling) was performed at 145 °C with a suitable aryl iodide. A choline chloride/glycerol deep eutectic solvent and phosphate buffer was used as solvent. Under continuous operation, quantitative conversion of coumaric acid to vinylphenol in the enzymatic step was observed but final yields of 54%, 32% and 50% were obtained for (E)-4-hydroxystilbene, resveratrol and pterostilbene, respectively, suggesting the need for improvement in the Heck coupling step. This can be partially overcome by minimizing the leakage of Pd typically observed when heterogeneous Pd catalysts are involved, through the use of a reverse flow reactor [140]. Under optimized condition, continuous operation for the production of (E)-4-hydroxystilbene was monitored for ~60 h, with constant conversion (close to 100%) and overall yield ~25%, with space–time yield of 4.8 g L−1 h−1 for decarboxylation and 0.52 g L−1 h−1 for Heck coupling [123].
Compartmentalization in two catalytic flow reactors, intercalated with an in-line purification of the key intermediates was used for chemoenzymatic synthesis of given active pharmaceutical ingredients (APIs), namely procainamide, procaine and butacaine, from vinyl 4-nitrobenzoate. Different nucleophiles were used, depending on the final product intended, with toluene as solvent in the enzymatic step, which was performed at 28 °C. The process was implemented through the sequential use of an acyltransferase immobilized on glyoxyl–agarose to produce amide and ester intermediates and a Pd-based catalyst for their hydrogenation, each formulation packed in column reactors. The in-line purification step consisted of sulphonyl chloride that was packed into a column connected with the enzymatic reactor, to remove excess of the nucleophiles. Upon solvent evaporation, the intermediates were redissolved in methanol (amide intermediate) or ethyl acetate (ester intermediate) and hydrogenated (60 °C, 1 × 106 Pa). Yields in excess of 99% were reported [124].
Chemoenzymatic Baeyer−Villiger oxidation with C. antarctica lipase B immobilized by adsorption on multi-walled carbon nanotubes packed in a column reactor operating at 40 °C has been recently reported. The biocatalyst enabled the generation of peracids in situ, hence avoiding the need to handle those chemicals. The peracids formed oxidize cyclic ketones to lactones. Ethyl acetate and n-octanoic acid were evaluated as peracid precursors and aqueous hydrogen peroxide as the primary oxidant. Ethyl acetate was ultimately preferred as it could also be used as solvent. To establish the scope of the adding to the substrate, several ketones were evaluated. In all cases, the corresponding lactones were obtained in high yields (83% to 99%) and selectivities (100%). In the particular case of the oxidation of 2-methylcyclohexanone to 6-methyl-ε-caprolactone, high product yield (87%) and selectivity (over 99%) were observed in 5 min reaction time. Moreover, high operational stability was observed, as ketone conversion remained in excess of 90% after 8 h of continuous operation [141].
5. Implementation Challenges at Processing Scale
A method to transpose biocatalytic systems and flow reactors from micro- to industrial scale could be a cornerstone for the real application of several interesting research processes. One key point is how many times a biocatalyst may be used to reduce the product–catalyst costs. It has been estimated that the total turnover number (the moles of product formed per mole of catalyst consumed) of a bioconversion should be higher than 1000 for expensive products being produced at small scale, and higher than 50,000 for large-scale or cheaper products [142]. When catalyst-to-substrate ratios are greater than 1:10,000, possible efforts to recycle the catalyst are superfluous [143].
When compared to batch processes, flow systems have the potential to accelerate bioconversions by enhancing mass transfer and to make large-scale production economically feasible in smaller bioreactors, at shorter reaction times and with up to 650-fold increase in space–time yield [12]. The small dimensions and the allowed process control of reaction parameters helps yields and productivities while minimizing interruptions in production and waste generation [144]. Additionally, the module nature of flow reactors allows the scale-up of bioprocesses by simple numbering up where the original-sized bioreactors are simply added in large number in series and/or parallel to increase production. This is probably the major feature of these reactors due to the simplification in process development.
In general, large-scale flow reactors either use static mixing, which rely on turbulent flow and/or baffles, or dynamic mixing, which is achieved by mechanical stirrers [145]. The latter has advantages over statically mixed systems when long reaction times, demanding mixing conditions or solids are required. An example of such a system is the Coflore Agitated Tube Reactor, which is agitated by loose agitator elements and mechanical shaking of the reactor body [75,76]. In biocatalytic systems requiring oxygen, such as the oxidation of glucose to glucono-1,5-lactone by glucose oxidase, this reactor allows a doubling of the overall reaction rate by increasing oxygen transfer rate [146].
Reaction systems with whole cells pose additional problems since cell growth and physiological behavior depend highly on local environmental conditions, which may vary along the length of a flow reactor. To overcome the complexity of reaction systems, excessive biomass growth and mass transfer limitations usually observed during scale-up of biofilm systems, Gross et al. proposed a scale-up approach by parallelization and numbering-up of membrane tubes in a membrane fiber module to keep the conditions close to the small-scale bioreactor [8]. The authors used a Pseudomonas sp. strain containing a native styrene monooxygenase StyAB and the introduced alkane monooxygenase system alkBGT/alkST for the production of (S)-styrene and 1-octanol. Two approaches were tested for process intensification: (1) by enhancement of biological activity, which indicated that an increase in specific activity reduced the reactor module number (and thus reactor area) and the costs related to glucose input and water consumption, and (2) by increasing the membrane surface, which would increase the amount of product achievable, but carbon source cost would increase to produce the necessary biomass to cover the entire membrane.
6. Final Remarks
Flow bioreactors using multi-enzymatic assemblies or whole cells present a credible technology for the production of compounds from screening systems to industrial-scale production. Although some technological aspects still need to be developed, their simplicity, ease of operation and the possibility to scaling-up by numbering-up, make flow reactors good candidates in bioprocess application.
Author Contributions
Conceptualization, C.C.C.R.d.C. and P.F.; writing—original draft preparation, C.C.C.R.d.C. and P.F.; writing—review and editing, C.C.C.R.d.C. and P.F. All authors have read and agreed to the published version of the manuscript.
Funding
The iBB—Institute for Bioengineering and Biosciences—acknowledges funding from FCT—Portuguese Foundation for Science and Technology (UID/BIO/04565/2020).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Velasco-Lozano, S.; López-Gallego, F. Wiring step-wise reactions with immobilized multi-enzyme systems. Biocatal. Biotransform. 2018, 36, 184–194. [Google Scholar] [CrossRef]
- Schoffelen, S.; van Hest, J.C.M. Multi-enzyme systems: Bringing enzymes together in vitro. Soft Matter 2012, 8, 1736–1746. [Google Scholar] [CrossRef]
- Gruber, P.; Marques, M.P.C.; O’Sullivan, B.; Baganz, F.; Wohlgemuth, R.; Szita, N. Conscious coupling: The challenges and opportunities of cascading enzymatic microreactors. Biotechnol. J. 2017, 12, 1700030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, A.; Serban, S. Industrial applications of immobilized enzymes—A review. Mol. Catal. 2019, 479, 110607. [Google Scholar] [CrossRef]
- de Carvalho, C.C.C.R. Whole cell biocatalysts: Essential workers from Nature to the industry. Microb. Biotechnol. 2017, 10, 250–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polakovič, M.; Švitel, J.; Bučko, M.; Filip, J.; Neděla, V.; Ansorge-Schumacher, M.B.; Gemeiner, P. Progress in biocatalysis with immobilized viable whole cells: Systems development, reaction engineering and applications. Biotechnol. Lett. 2017, 39, 667–683. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, C.C.C.R.; da Fonseca, M.M.R. Biotransformations. In Comprehensive Biotechnology, 3rd ed.; Moo-Young, M., Ed.; Pergamon: Oxford, UK, 2017; pp. 574–585. [Google Scholar]
- Gross, R.; Buehler, K.; Schmid, A. Engineered catalytic biofilms for continuous large scale production of n-octanol and (S)-styrene oxide. Biotechnol. Bioeng. 2013, 110, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Todhanakasem, T.; Salangsing, O.-L.; Koomphongse, P.; Kaewket, S.; Kanokratana, P.; Champreda, V. Zymomonas mobilis Biofilm Reactor for Ethanol Production Using Rice Straw Hydrolysate Under Continuous and Repeated Batch Processes. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.; Tao, Y. Whole-cell biocatalysts by design. Microb. Cell Factories 2017, 16, 106. [Google Scholar] [CrossRef] [Green Version]
- Ro, D.-K.; Paradise, E.M.; Ouellet, M.; Fisher, K.J.; Newman, K.L.; Ndungu, J.M.; Ho, K.A.; Eachus, R.A.; Ham, T.S.; Kirby, J.; et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 2006, 440, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Tamborini, L.; Fernandes, P.; Paradisi, F.; Molinari, F. Flow Bioreactors as Complementary Tools for Biocatalytic Process Intensification. Trends Biotechnol. 2018, 36, 73–88. [Google Scholar] [CrossRef] [PubMed]
- De Vitis, V.; Dall’Oglio, F.; Tentori, F.; Contente, M.L.; Romano, D.; Brenna, E.; Tamborini, L.; Molinari, F. Bioprocess Intensification Using Flow Reactors: Stereoselective Oxidation of Achiral 1,3-diols with Immobilized Acetobacter aceti. Catalysts 2019, 9, 208. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Chen, Q.; Shao, L.; Jia, Y.; Zhang, X. Microfluidic immobilized enzyme reactors for continuous biocatalysis. React. Chem. Eng. 2020, 5, 9–32. [Google Scholar] [CrossRef]
- Britton, J.; Majumdar, S.; Weiss, G.A. Continuous flow biocatalysis. Chem. Soc. Rev. 2018, 47, 5891–5918. [Google Scholar] [CrossRef]
- De Santis, P.; Meyer, L.-E.; Kara, S. The rise of continuous flow biocatalysis—Fundamentals, very recent developments and future perspectives. React. Chem. Eng. 2020, 5, 2155–2184. [Google Scholar] [CrossRef]
- Wohlgemuth, R.; Plazl, I.; Žnidaršič-Plazl, P.; Gernaey, K.V.; Woodley, J.M. Microscale technology and biocatalytic processes: Opportunities and challenges for synthesis. Trends Biotechnol. 2015, 33, 302–314. [Google Scholar] [CrossRef]
- Meller, K.; Szumski, M.; Buszewski, B. Microfluidic reactors with immobilized enzymes—Characterization, dividing, perspectives. Sens. Actuators B Chem. 2017, 244, 84–106. [Google Scholar] [CrossRef]
- Arshi, S.; Nozari-Asbemarz, M.; Magner, E. Enzymatic Bioreactors: An Electrochemical Perspective. Catalysts 2020, 10, 1232. [Google Scholar] [CrossRef]
- Britton, J.; Dyer, R.P.; Majumdar, S.; Raston, C.L.; Weiss, G.A. Ten-Minute Protein Purification and Surface Tethering for Continuous-Flow Biocatalysis. Angew. Chem. Int. Ed. 2017, 56, 2296–2301. [Google Scholar] [CrossRef]
- Boehm, C.R.; Freemont, P.S.; Ces, O. Design of a prototype flow microreactor for synthetic biology in vitro. Lab A Chip 2013, 13, 3426–3432. [Google Scholar] [CrossRef] [PubMed]
- Döbber, J.; Gerlach, T.; Offermann, H.; Rother, D.; Pohl, M. Closing the gap for efficient immobilization of biocatalysts in continuous processes: HaloTag™ fusion enzymes for a continuous enzymatic cascade towards a vicinal chiral diol. Green Chem. 2018, 20, 544–552. [Google Scholar] [CrossRef]
- Roura Padrosa, D.; Benítez-Mateos, A.I.; Calvey, L.; Paradisi, F. Cell-free biocatalytic syntheses of L-pipecolic acid: A dual strategy approach and process intensification in flow. Green Chem. 2020, 22, 5310–5316. [Google Scholar] [CrossRef]
- Poppe, J.K.; Matte, C.R.; de Freitas, V.O.; Fernandez-Lafuente, R.; Rodrigues, R.C.; Záchia Ayub, M.A. Enzymatic synthesis of ethyl esters from waste oil using mixtures of lipases in a plug-flow packed-bed continuous reactor. Biotechnol. Prog. 2018, 34, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Brás, E.J.S.; Domingues, C.; Chu, V.; Fernandes, P.; Conde, J.P. Microfluidic bioreactors for enzymatic synthesis in packed-bed reactors—Multi-step reactions and upscaling. J. Biotechnol. 2020, 323, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Logan, T.C.; Clark, D.S.; Stachowiak, T.B.; Svec, F.; Fréchet, J.M.J. Photopatterning Enzymes on Polymer Monoliths in Microfluidic Devices for Steady-State Kinetic Analysis and Spatially Separated Multi-Enzyme Reactions. Anal. Chem. 2007, 79, 6592–6598. [Google Scholar] [CrossRef]
- Yin, J.; Xu, T.; Zhang, N.; Wang, H. Three-Enzyme Cascade Bioreactor for Rapid Digestion of Genomic DNA into Single Nucleosides. Anal. Chem. 2016, 88, 7730–7737. [Google Scholar] [CrossRef]
- Fornera, S.; Kuhn, P.; Lombardi, D.; Schlüter, A.D.; Dittrich, P.S.; Walde, P. Sequential Immobilization of Enzymes in Microfluidic Channels for Cascade Reactions. ChemPlusChem 2012, 77, 98–101. [Google Scholar] [CrossRef]
- Rodrigues, R.C.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Fernández-Lafuente, R. Modifying enzyme activity and selectivity by immobilization. Chem. Soc. Rev. 2013, 42, 6290–6307. [Google Scholar] [CrossRef]
- Romero-Fernández, M.; Paradisi, F. Protein immobilization technology for flow biocatalysis. Curr. Opin. Chem. Biol. 2020, 55, 1–8. [Google Scholar] [CrossRef]
- Bommarius, A.S.; Paye, M.F. Stabilizing biocatalysts. Chem. Soc. Rev. 2013, 42, 6534–6565. [Google Scholar] [CrossRef]
- Chen, R.; Wei, Q.; Wei, X.; Liu, Y.; Zhang, X.; Chen, X.; Yin, X.; Xie, T. Stable and efficient immobilization of bi-enzymatic NADPH cofactor recycling system under consecutive microwave irradiation. PLoS ONE 2020, 15, e0242564. [Google Scholar] [CrossRef] [PubMed]
- Peschke, T.; Bitterwolf, P.; Hansen, S.; Gasmi, J.; Rabe, K.S.; Niemeyer, C.M. Self-Immobilizing Biocatalysts Maximize Space–Time Yields in Flow Reactors. Catalysts 2019, 9, 164. [Google Scholar] [CrossRef] [Green Version]
- Sánta-Bell, E.; Molnár, Z.; Varga, A.; Nagy, F.; Hornyánszky, G.; Paizs, C.; Balogh-Weiser, D.; Poppe, L. “Fishing and Hunting”—Selective Immobilization of a Recombinant Phenylalanine Ammonia-Lyase from Fermentation Media. Molecules 2019, 24, 4146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamad, N.R.; Marzuki, N.H.C.; Buang, N.A.; Huyop, F.; Wahab, R.A. An overview of technologies for immobilization of enzymes and surface analysis techniques for immobilized enzymes. Biotechnol. Biotechnol. Equip. 2015, 29, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Küchler, A.; Yoshimoto, M.; Luginbühl, S.; Mavelli, F.; Walde, P. Enzymatic reactions in confined environments. Nat. Nanotechnol. 2016, 11, 409–420. [Google Scholar] [CrossRef]
- Xu, K.; Chen, X.; Zheng, R.; Zheng, Y. Immobilization of Multi-Enzymes on Support Materials for Efficient Biocatalysis. Front. Bioeng. Biotechnol. 2020, 8, 660. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.-Q.; Wang, S.-S.; Li, L.-N.; Gao, J.; Zhang, Y.-W. Combined Cross-Linked Enzyme Aggregates as Biocatalysts. Catalysts 2018, 8, 460. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, R.A. CLEAs, Combi-CLEAs and ‘Smart’ Magnetic CLEAs: Biocatalysis in a Bio-Based Economy. Catalysts 2019, 9, 261. [Google Scholar] [CrossRef] [Green Version]
- Homaei, A.A.; Sariri, R.; Vianello, F.; Stevanato, R. Enzyme immobilization: An update. J. Chem. Biol. 2013, 6, 185–205. [Google Scholar] [CrossRef] [Green Version]
- Sirisha, V.L.; Jain, A.; Jain, A. Chapter Nine—Enzyme Immobilization: An Overview on Methods, Support Material, and Applications of Immobilized Enzymes. In Advances in Food and Nutrition Research; Kim, S.-K., Toldrá, F., Eds.; Academic Press: Cambridge, MA, USA, 2016; Volume 79, pp. 179–211. [Google Scholar]
- Zdarta, J.; Meyer, A.S.; Jesionowski, T.; Pinelo, M. A General Overview of Support Materials for Enzyme Immobilization: Characteristics, Properties, Practical Utility. Catalysts 2018, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Taheri-Kafrani, A.; Kharazmi, S.; Nasrollahzadeh, M.; Soozanipour, A.; Ejeian, F.; Etedali, P.; Mansouri-Tehrani, H.-A.; Razmjou, A.; Yek, S.M.-G.; Varma, R.S. Recent developments in enzyme immobilization technology for high-throughput processing in food industries. Crit. Rev. Food Sci. Nutr. 2020, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Kazenwadel, F.; Franzreb, M.; Rapp, B.E. Synthetic enzyme supercomplexes: Co-immobilization of enzyme cascades. Anal. Methods 2015, 7, 4030–4037. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Wu, Y.; Zhang, S.; Tian, Y.; Yang, D.; Jiang, Z. Bioinspired construction of multi-enzyme catalytic systems. Chem. Soc. Rev. 2018, 47, 4295–4313. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.T.; Lee, S. Multienzymatic Cascade Reactions via Enzyme Complex by Immobilization. ACS Catal. 2019, 9, 4402–4425. [Google Scholar] [CrossRef]
- Ren, S.; Li, C.; Jiao, X.; Jia, S.; Jiang, Y.; Bilal, M.; Cui, J. Recent progress in multienzymes co-immobilization and multienzyme system applications. Chem. Eng. J. 2019, 373, 1254–1278. [Google Scholar] [CrossRef]
- Giannakopoulou, A.; Gkantzou, E.; Polydera, A.; Stamatis, H. Multienzymatic Nanoassemblies: Recent Progress and Applications. Trends Biotechnol. 2020, 38, 202–216. [Google Scholar] [CrossRef]
- Arana-Peña, S.; Carballares, D.; Morellon-Sterlling, R.; Berenguer-Murcia, Á.; Alcántara, A.R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Enzyme co-immobilization: Always the biocatalyst designers’ choice…or not? Biotechnol. Adv. 2020, 107584. [Google Scholar] [CrossRef]
- Ji, Q.; Wang, B.; Tan, J.; Zhu, L.; Li, L. Immobilized multienzymatic systems for catalysis of cascade reactions. Process Biochem. 2016, 51, 1193–1203. [Google Scholar] [CrossRef]
- Brena, B.; González-Pombo, P.; Batista-Viera, F. Immobilization of Enzymes: A Literature Survey. In Immobilization of Enzymes and Cells, 3rd ed.; Guisan, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 15–31. [Google Scholar]
- Wahab, R.A.; Elias, N.; Abdullah, F.; Ghoshal, S.K. On the taught new tricks of enzymes immobilization: An all-inclusive overview. React. Funct. Polym. 2020, 152, 104613. [Google Scholar] [CrossRef]
- Mateo, C.; Fernandez-Lorente, G.; Rocha-Martin, J.; Bolivar, J.M.; Guisan, J.M. Oriented Covalent Immobilization of Enzymes on Heterofunctional-Glyoxyl Supports. In Immobilization of Enzymes and Cells, 3rd ed.; Guisan, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 73–88. [Google Scholar]
- Rodriguez-Abetxuko, A.; Sánchez-deAlcázar, D.; Muñumer, P.; Beloqui, A. Tunable Polymeric Scaffolds for Enzyme Immobilization. Front. Bioeng. Biotechnol. 2020, 8, 830. [Google Scholar] [CrossRef]
- Guisan, J.M.; López-Gallego, F.; Bolivar, J.M.; Rocha-Martín, J.; Fernandez-Lorente, G. The Science of Enzyme Immobilization. In Immobilization of Enzymes and Cells: Methods and Protocols; Guisan, J.M., Bolivar, J.M., López-Gallego, F., Rocha-Martín, J., Eds.; Springer: New York, NY, USA, 2020; pp. 1–26. [Google Scholar]
- Wu, J.C.Y.; Hutchings, C.H.; Lindsay, M.J.; Werner, C.J.; Bundy, B.C. Enhanced Enzyme Stability through Site-Directed Covalent Immobilization. J. Biotechnol. 2015, 193, 83–90. [Google Scholar] [CrossRef]
- Li, Y.; Ogorzalek, T.L.; Wei, S.; Zhang, X.; Yang, P.; Jasensky, J.; Brooks, C.L.; Marsh, E.N.G.; Chen, Z. Effect of immobilization site on the orientation and activity of surface-tethered enzymes. Phys. Chem. Chem. Phys. 2018, 20, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Meyer, A.S.; Mateiu, R.V.; Pinelo, M. Cascade catalysis in membranes with enzyme immobilization for multi-enzymatic conversion of CO2 to methanol. New Biotechnol. 2015, 32, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Küchler, A.; Adamcik, J.; Mezzenga, R.; Schlüter, A.D.; Walde, P. Enzyme immobilization on silicate glass through simple adsorption of dendronized polymer–enzyme conjugates for localized enzymatic cascade reactions. RSC Adv. 2015, 5, 44530–44544. [Google Scholar] [CrossRef]
- Torres, R.; Pessela, B.C.C.; Mateo, C.; Ortiz, C.; Fuentes, M.; Guisan, J.M.; Fernandez-Lafuente, R. Reversible Immobilization of Glucoamylase by Ionic Adsorption on Sepabeads Coated with Polyethyleneimine. Biotechnol. Prog. 2004, 20, 1297–1300. [Google Scholar] [CrossRef] [PubMed]
- Hanefeld, U.; Gardossi, L.; Magner, E. Understanding enzyme immobilisation. Chem. Soc. Rev. 2009, 38, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Ngo, T.T.; Lenhoff, H.M.; Ivy, J. Biotinyl-Glucose-6-Phosphate Dehydrogenase Preparation, Kinetics, and Modulation by Avidin. Appl. Biochem. Biotechnol. 1982, 7, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Peschke, T.; Bitterwolf, P.; Rabe, K.S.; Niemeyer, C.M. Self-Immobilizing Oxidoreductases for Flow Biocatalysis in Miniaturized Packed-Bed Reactors. Chem. Eng. Technol. 2019, 42, 2009–2017. [Google Scholar] [CrossRef]
- Plž, M.; Petrovičová, T.; Rebroš, M. Semi-Continuous Flow Biocatalysis with Affinity Co-Immobilized Ketoreductase and Glucose Dehydrogenase. Molecules 2020, 25, 4278. [Google Scholar] [CrossRef]
- Fornera, S.; Bauer, T.; Schlüter, A.D.; Walde, P. Simple enzyme immobilization inside glass tubes for enzymatic cascade reactions. J. Mater. Chem. 2012, 22, 502–511. [Google Scholar] [CrossRef]
- Vong, T.; Schoffelen, S.; van Dongen, S.F.M.; van Beek, T.A.; Zuilhof, H.; van Hest, J.C.M. A DNA-based strategy for dynamic positional enzyme immobilization inside fused silica microchannels. Chem. Sci. 2011, 2, 1278–1285. [Google Scholar] [CrossRef]
- Zhang, G.; Johnston, T.; Quin, M.B.; Schmidt-Dannert, C. Developing a Protein Scaffolding System for Rapid Enzyme Immobilization and Optimization of Enzyme Functions for Biocatalysis. ACS Synth. Biol. 2019, 8, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Valikhani, D.; Bolivar, J.M.; Viefhues, M.; McIlroy, D.N.; Vrouwe, E.X.; Nidetzky, B. A Spring in Performance: Silica Nanosprings Boost Enzyme Immobilization in Microfluidic Channels. ACS Appl. Mater. Interfaces 2017, 9, 34641–34649. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Han, P.; You, C. Facilitation of cascade biocatalysis by artificial multi-enzyme complexes—A review. Chin. J. Chem. Eng. 2020, 28, 2799–2809. [Google Scholar] [CrossRef]
- Sahutoglu, A.S.; Akgul, C. Immobilisation of Aspergillus oryzae α-amylase and Aspergillus niger glucoamylase enzymes as cross-linked enzyme aggregates. Chem. Pap. 2015, 69, 433–439. [Google Scholar] [CrossRef]
- De Martino, M.T.; Tonin, F.; Yewdall, N.A.; Abdelghani, M.; Williams, D.S.; Hanefeld, U.; Rutjes, F.P.J.T.; Abdelmohsen, L.K.E.A.; van Hest, J.C.M. Compartmentalized cross-linked enzymatic nano-aggregates (c-CLEnA) for efficient in-flow biocatalysis. Chem. Sci. 2020, 11, 2765–2769. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Gui, X.; Wang, G.; Yan, Y. Improving Stability and Activity of Cross-linked Enzyme Aggregates Based on Polyethylenimine in Hydrolysis of Fish Oil for Enrichment of Polyunsaturated Fatty Acids. Appl. Biochem. Biotechnol. 2012, 166, 925–932. [Google Scholar] [CrossRef]
- Wilson, L.; Illanes, A.; Pessela, B.C.C.; Abian, O.; Fernández-Lafuente, R.; Guisán, J.M. Encapsulation of crosslinked penicillin G acylase aggregates in lentikats: Evaluation of a novel biocatalyst in organic media. Biotechnol. Bioeng. 2004, 86, 558–562. [Google Scholar] [CrossRef]
- Noori, R.; Perwez, M.; Sardar, M. Cross-linked Enzyme Aggregates: Current Developments and Applications. In Biocatalysis: Enzymatic Basics and Applications; Husain, Q., Ullah, M.F., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 83–112. [Google Scholar]
- Talekar, S.; Pandharbale, A.; Ladole, M.; Nadar, S.; Mulla, M.; Japhalekar, K.; Pattankude, K.; Arage, D. Carrier free co-immobilization of alpha amylase, glucoamylase and pullulanase as combined cross-linked enzyme aggregates (combi-CLEAs): A tri-enzyme biocatalyst with one pot starch hydrolytic activity. Bioresour. Technol. 2013, 147, 269–275. [Google Scholar] [CrossRef]
- Perwez, M.; Ahmed Mazumder, J.; Sardar, M. Preparation and characterization of reusable magnetic combi-CLEA of cellulase and hemicellulase. Enzym. Microb. Technol. 2019, 131, 109389. [Google Scholar] [CrossRef]
- Chen, S.; Wen, L.; Svec, F.; Tan, T.; Lv, Y. Magnetic metal–organic frameworks as scaffolds for spatial co-location and positional assembly of multi-enzyme systems enabling enhanced cascade biocatalysis. RSC Adv. 2017, 7, 21205–21213. [Google Scholar] [CrossRef] [Green Version]
- Talekar, S.; Joshi, A.; Kambale, S.; Jadhav, S.; Nadar, S.; Ladole, M. A tri-enzyme magnetic nanobiocatalyst with one pot starch hydrolytic activity. Chem. Eng. J. 2017, 325, 80–90. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, D.; Wu, C.; Yao, K.; Li, Z.; Shi, N.; Wen, F.; Gates, I.D. Co-immobilization of cellulase and lysozyme on amino-functionalized magnetic nanoparticles: An activity-tunable biocatalyst for extraction of lipids from microalgae. Bioresour. Technol. 2018, 263, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulou, A.; Patila, M.; Spyrou, K.; Chalmpes, N.; Zarafeta, D.; Skretas, G.; Gournis, D.; Stamatis, H. Development of a Four-Enzyme Magnetic Nanobiocatalyst for Multi-Step Cascade Reactions. Catalysts 2019, 9, 995. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; He, Z.; Wang, X.; Zhang, Q.; Wei, Q.; Ma, S.; Ma, C.; Li, J.; Wang, Q. Cascade enzymes within self-assembled hybrid nanogel mimicked neutrophil lysosomes for singlet oxygen elevated cancer therapy. Nat. Commun. 2019, 10, 240. [Google Scholar] [CrossRef]
- Del Arco, J.; Alcántara, A.R.; Fernández-Lafuente, R.; Fernández-Lucas, J. Magnetic micro-macro biocatalysts applied to industrial bioprocesses. Bioresour. Technol. 2021, 322, 124547. [Google Scholar] [CrossRef]
- Zhong, X.; Xia, H.; Huang, W.; Li, Z.; Jiang, Y. Biomimetic metal-organic frameworks mediated hybrid multi-enzyme mimic for tandem catalysis. Chem. Eng. J. 2020, 381, 122758. [Google Scholar] [CrossRef]
- Ye, N.; Kou, X.; Shen, J.; Huang, S.; Chen, G.; Ouyang, G. Metal-Organic Frameworks: A New Platform for Enzyme Immobilization. Chembiochem 2020, 21, 2585–2590. [Google Scholar] [CrossRef]
- Cen, Y.-K.; Liu, Y.-X.; Xue, Y.-P.; Zheng, Y.-G. Immobilization of Enzymes in/on Membranes and their Applications. Adv. Synth. Catal. 2019, 361, 5500–5515. [Google Scholar] [CrossRef]
- Zhao, Z.; Fu, J.; Dhakal, S.; Johnson-Buck, A.; Liu, M.; Zhang, T.; Woodbury, N.W.; Liu, Y.; Walter, N.G.; Yan, H. Nanocaged enzymes with enhanced catalytic activity and increased stability against protease digestion. Nat. Commun. 2016, 7, 10619. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.-L.; Zhu, J.; Wheeldon, I. Synthetic Protein Scaffolds for Biosynthetic Pathway Colocalization on Lipid Droplet Membranes. ACS Synth. Biol. 2017, 6, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Eş, I.; Vieira, J.D.G.; Amaral, A.C. Principles, techniques, and applications of biocatalyst immobilization for industrial application. Appl. Microbiol. Biotechnol. 2015, 99, 2065–2082. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, F.; Fernández-Lucas, J.; de la Fuente, D.; Zheng, C.; Bavaro, T.; Peters, B.; Massolini, G.; Annunziata, F.; Conti, P.; de la Mata, I.; et al. Immobilized enzyme reactors based on nucleoside phosphorylases and 2′-deoxyribosyltransferase for the in-flow synthesis of pharmaceutically relevant nucleoside analogues. Bioresour. Technol. 2020, 307, 123258. [Google Scholar] [CrossRef]
- Robescu, M.S.; Serra, I.; Terreni, M.; Ubiali, D.; Bavaro, T. A Multi-Enzymatic Cascade Reaction for the Synthesis of Vidarabine 5′-Monophosphate. Catalysts 2020, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Tamborini, L.; Previtali, C.; Annunziata, F.; Bavaro, T.; Terreni, M.; Calleri, E.; Rinaldi, F.; Pinto, A.; Speranza, G.; Ubiali, D.; et al. An Enzymatic Flow-Based Preparative Route to Vidarabine. Molecules 2020, 25, 1223. [Google Scholar] [CrossRef] [Green Version]
- Hartley, C.J.; Williams, C.C.; Scoble, J.A.; Churches, Q.I.; North, A.; French, N.G.; Nebl, T.; Coia, G.; Warden, A.C.; Simpson, G.; et al. Engineered enzymes that retain and regenerate their cofactors enable continuous-flow biocatalysis. Nat. Catal. 2019, 2, 1006–1015. [Google Scholar] [CrossRef]
- Peschke, T.; Skoupi, M.; Burgahn, T.; Gallus, S.; Ahmed, I.; Rabe, K.S.; Niemeyer, C.M. Self-Immobilizing Fusion Enzymes for Compartmentalized Biocatalysis. ACS Catal. 2017, 7, 7866–7872. [Google Scholar] [CrossRef]
- Zhong, C.; Duić, B.; Bolivar, J.M.; Nidetzky, B. Three-Enzyme Phosphorylase Cascade Immobilized on Solid Support for Biocatalytic Synthesis of Cello−oligosaccharides. ChemCatChem 2020, 12, 1350–1358. [Google Scholar] [CrossRef]
- Bitterwolf, P.; Ott, F.; Rabe, K.S.; Niemeyer, C.M. Imine Reductase Based All-Enzyme Hydrogel with Intrinsic Cofactor Regeneration for Flow Biocatalysis. Micromachines 2019, 10, 783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peschke, T.; Bitterwolf, P.; Gallus, S.; Hu, Y.; Oelschlaeger, C.; Willenbacher, N.; Rabe, K.S.; Niemeyer, C.M. Self-Assembling All-Enzyme Hydrogels for Flow Biocatalysis. Angew. Chem. Int. Ed. 2018, 57, 17028–17032. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Obst, F.; Haefner, S.; Heroldt, T.; Peiter, M.; Simon, F.; Richter, A.; Voit, B.; Appelhans, D. Hydrogel/enzyme dots as adaptable tool for non-compartmentalized multi-enzymatic reactions in microfluidic devices. React. Chem. Eng. 2019, 4, 67–77. [Google Scholar] [CrossRef]
- Lefrançois, P.; Goudeau, B.; Arbault, S. Dynamic monitoring of a bi-enzymatic reaction at a single biomimetic giant vesicle. Analyst 2020. [Google Scholar] [CrossRef] [PubMed]
- Hakala, T.A.; Bialas, F.; Toprakcioglu, Z.; Bräuer, B.; Baumann, K.N.; Levin, A.; Bernardes, G.J.L.; Becker, C.F.W.; Knowles, T.P.J. Continuous Flow Reactors from Microfluidic Compartmentalization of Enzymes within Inorganic Microparticles. ACS Appl. Mater. Interfaces 2020, 12, 32951–32960. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, Z.; Liu, Y.; Shao, C.; Bian, F.; Zhao, Y. Biomimetic enzyme cascade reaction system in microfluidic electrospray microcapsules. Sci. Adv. 2018, 4, eaat2816. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Kim, S.B.; Yoo, H.Y.; Lee, J.H.; Park, C.; Han, S.O.; Kim, S.W. Kinetic modeling of biodiesel production by mixed immobilized and co-immobilized lipase systems under two pressure conditions. Korean J. Chem. Eng. 2013, 30, 1272–1276. [Google Scholar] [CrossRef]
- Ji, Q.; Tan, J.; Zhu, L.; Lou, D.; Wang, B. Preparing tauroursodeoxycholic acid (TUDCA) using a double-enzyme-coupled system. Biochem. Eng. J. 2016, 105, 1–9. [Google Scholar] [CrossRef]
- Huang, X.; Li, M.; Mann, S. Membrane-mediated cascade reactions by enzyme–polymer proteinosomes. Chem. Commun. 2014, 50, 6278–6280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubey, N.C.; Tripathi, B.P.; Müller, M.; Stamm, M.; Ionov, L. Bienzymatic Sequential Reaction on Microgel Particles and Their Cofactor Dependent Applications. Biomacromolecules 2016, 17, 1610–1620. [Google Scholar] [CrossRef]
- Sun, J.; Ge, J.; Liu, W.; Lan, M.; Zhang, H.; Wang, P.; Wang, Y.; Niu, Z. Multi-enzyme co-embedded organic–inorganic hybrid nanoflowers: Synthesis and application as a colorimetric sensor. Nanoscale 2014, 6, 255–262. [Google Scholar] [CrossRef]
- Cui, C.; Chen, H.; Chen, B.; Tan, T. Genipin Cross-Linked Glucose Oxidase and Catalase Multi-enzyme for Gluconic Acid Synthesis. Appl. Biochem. Biotechnol. 2017, 181, 526–535. [Google Scholar] [CrossRef]
- Mafra, A.C.; Ulrich, L.G.; Kornecki, J.F.; Fernandez-Lafuente, R.; Tardioli, P.W.; Ribeiro, M.P. Combi-CLEAs of Glucose Oxidase and Catalase for Conversion of Glucose to Gluconic Acid Eliminating the Hydrogen Peroxide to Maintain Enzyme Activity in a Bubble Column Reactor. Catalysts 2019, 9, 657. [Google Scholar] [CrossRef] [Green Version]
- Hwang, E.T.; Seo, B.-K.; Gu, M.B.; Zeng, A.-P. Successful bi-enzyme stabilization for the biomimetic cascade transformation of carbon dioxide. Catal. Sci. Technol. 2016, 6, 7267–7272. [Google Scholar] [CrossRef]
- Rabe, K.S.; Müller, J.; Skoupi, M.; Niemeyer, C.M. Cascades in Compartments: En Route to Machine-Assisted Biotechnology. Angew. Chem. Int. Ed. 2017, 56, 13574–13589. [Google Scholar] [CrossRef] [PubMed]
- Holvey, C.P.; Roberge, D.M.; Gottsponer, M.; Kockmann, N.; Macchi, A. Pressure drop and mixing in single phase microreactors: Simplified designs of micromixers. Chem. Eng. Process. Process Intensif. 2011, 50, 1069–1075. [Google Scholar] [CrossRef]
- Nakagawa, K.; Tamura, A.; Chaiya, C. Preparation of proteolytic microreactors by freeze-drying immobilization. Chem. Eng. Sci. 2014, 119, 22–29. [Google Scholar] [CrossRef]
- Illg, T.; Hessel, V.; Löb, P.; Schouten, J.C. Novel process window for the safe and continuous synthesis of tert.-butyl peroxy pivalate in a micro-reactor. Chem. Eng. J. 2011, 167, 504–509. [Google Scholar] [CrossRef]
- Wei, H.-C.; Huang, S.-H.; Jiang, J.-A.; Lee, Y.-C. A Multichannel Calorimetric Simultaneous Assay Platform Using a Microampere Constant-Current Looped Enthalpy Sensor Array. Sensors 2017, 17, 292. [Google Scholar] [CrossRef] [Green Version]
- Van Schie, M.M.C.H.; Ebrahimi, K.H.; Hagen, W.R.; Hagedoorn, P.-L. Fast and accurate enzyme activity measurements using a chip-based microfluidic calorimeter. Anal. Biochem. 2018, 544, 57–63. [Google Scholar] [CrossRef]
- Abis, G.; Pacheco-Gómez, R.; Bui, T.T.T.; Conte, M.R. Isothermal Titration Calorimetry Enables Rapid Characterization of Enzyme Kinetics and Inhibition for the Human Soluble Epoxide Hydrolase. Anal. Chem. 2019, 91, 14865–14872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luckarift, H.R.; Ku, B.S.; Dordick, J.S.; Spain, J.C. Silica-immobilized enzymes for multi-step synthesis in microfluidic devices. Biotechnol. Bioeng. 2007, 98, 701–705. [Google Scholar] [CrossRef]
- Heinzler, R.; Fischöder, T.; Elling, L.; Franzreb, M. Toward Automated Enzymatic Glycan Synthesis in a Compartmented Flow Microreactor System. Adv. Synth. Catal. 2019, 361, 4506–4516. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.-X.; Qian, Z.-G.; Ki, C.S.; Park, Y.H.; Kaplan, D.L.; Lee, S.Y. Native-sized recombinant spider silk protein produced in metabolically engineered Escherichia coli results in a strong fiber. Proc. Natl. Acad. Sci. USA 2010, 107, 14059–14063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Saba, T.; Yiu, H.H.P.; Howe, R.F.; Anderson, J.A.; Shi, J. Cofactor NAD(P)H Regeneration Inspired by Heterogeneous Pathways. Chem 2017, 2, 621–654. [Google Scholar] [CrossRef] [Green Version]
- Kisukuri, C.M.; Andrade, L.H. Production of chiral compounds using immobilized cells as a source of biocatalysts. Org. Biomol. Chem. 2015, 13, 10086–10107. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.W.I.; Fischbach, M.; Huebner, H.; Buchholz, R.; Hummel, W.; Daussmann, T.; Wandrey, C.; Liese, A. Synthesis of enantiopure (5R)-hydroxyhexane-2-one with immobilised whole cells of Lactobacillus kefiri. Appl. Microbiol. Biotechnol. 2006, 71, 289–293. [Google Scholar] [CrossRef]
- Nagy-Gyor, L.; Abahazi, E.; Bodai, V.; Satorhelyi, P.; Erdelyi, B.; Balogh-Weiser, D.; Paizs, C.; Hornyanszky, G.; Poppe, L. Co-immobilized Whole Cells with omega-Transaminase and Ketoreductase Activities for Continuous-Flow Cascade Reactions. Chembiochem 2018, 19, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- Grabner, B.; Schweiger, A.K.; Gavric, K.; Kourist, R.; Gruber-Woelfler, H. A chemo-enzymatic tandem reaction in a mixture of deep eutectic solvent and water in continuous flow. React. Chem. Eng. 2020, 5, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Annunziata, F.; Letizia Contente, M.; Betti, D.; Pinna, C.; Molinari, F.; Tamborini, L.; Pinto, A. Efficient Chemo-Enzymatic Flow Synthesis of High Value Amides and Esters. Catalysts 2020, 10, 939. [Google Scholar] [CrossRef]
- Wigneswaran, V.; Nielsen, K.F.; Sternberg, C.; Jensen, P.R.; Folkesson, A.; Jelsbak, L. Biofilm as a production platform for heterologous production of rhamnolipids by the non-pathogenic strain Pseudomonas putida KT2440. Microb. Cell Factories 2016, 15, 181. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, N.; Schripsema, J.; Lienhardt, J.; Blaschek, H.P. Continuous solvent production by Clostridium beijerinckii BA101 immobilized by adsorption onto brick. World J. Microbiol. Biotechnol. 2000, 16, 377–382. [Google Scholar] [CrossRef]
- Kunduru, M.R.; Pometto, A.L. Continuous ethanol production by Zymomonas mobilis and Saccharomyces cerevisiae in biofilm reactors. J. Ind. Microbiol. 1996, 16, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Kurade, M.B.; Waghmode, T.R.; Patil, S.M.; Jeon, B.-H.; Govindwar, S.P. Monitoring the gradual biodegradation of dyes in a simulated textile effluent and development of a novel triple layered fixed bed reactor using a bacterium-yeast consortium. Chem. Eng. J. 2017, 307, 1026–1036. [Google Scholar] [CrossRef]
- Stojkovic, G.; Znidarsic-Plazl, P. Continuous synthesis of L-malic acid using whole-cell microreactor. Process Biochem. 2012, 47, 1102–1107. [Google Scholar] [CrossRef]
- Kim, H.J.; Huh, D.; Hamilton, G.; Ingber, D.E. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab A Chip 2012, 12, 2165–2174. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Moore, B.S. Enzymatic Cascade Reactions in Biosynthesis. Angew. Chem. Int. Ed. 2019, 58, 6846–6879. [Google Scholar] [CrossRef] [PubMed]
- Rudroff, F.; Mihovilovic, M.D.; Gröger, H.; Snajdrova, R.; Iding, H.; Bornscheuer, U.T. Opportunities and challenges for combining chemo- and biocatalysis. Nat. Catal. 2018, 1, 12–22. [Google Scholar] [CrossRef]
- Schmidt, S.; Castiglione, K.; Kourist, R. Overcoming the Incompatibility Challenge in Chemoenzymatic and Multi-Catalytic Cascade Reactions. Chem. A Eur. J. 2018, 24, 1755–1768. [Google Scholar] [CrossRef]
- Clayton, A.D.; Labes, R.; Blacker, A.J. Combination of chemocatalysis and biocatalysis in flow. Curr. Opin. Green Sustain. Chem. 2020, 26, 100378. [Google Scholar] [CrossRef]
- Suresh, A.; Shravan Ramgopal, D.; Panchamoorthy Gopinath, K.; Arun, J.; SundarRajan, P.; Bhatnagar, A. Recent advancements in the synthesis of novel thermostable biocatalysts and their applications in commercially important chemoenzymatic conversion processes. Bioresour. Technol. 2021, 323, 124558. [Google Scholar] [CrossRef]
- Losada-Garcia, N.; Cabrera, Z.; Urrutia, P.; Garcia-Sanz, C.; Andreu, A.; Palomo, J.M. Recent Advances in Enzymatic and Chemoenzymatic Cascade Processes. Catalysts 2020, 10, 1258. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, P.; Gao, S.; Wang, Z.; Luan, P.; González-Sabín, J.; Jiang, Y. Construction of chemoenzymatic cascade reactions for bridging chemocatalysis and Biocatalysis: Principles, strategies and prospective. Chem. Eng. J. 2020, 127659. [Google Scholar] [CrossRef]
- Falus, P.; Cerioli, L.; Bajnóczi, G.; Boros, Z.; Weiser, D.; Nagy, J.; Tessaro, D.; Servi, S.; Poppe, L. A Continuous-Flow Cascade Reactor System for Subtilisin A- Catalyzed Dynamic Kinetic Resolution of N-tert-Butyloxycarbonylphenylalanine Ethyl Thioester with Benzylamine. Adv. Synth. Catal. 2016, 358, 1608–1617. [Google Scholar] [CrossRef]
- Farkas, E.; Oláh, M.; Földi, A.; Kóti, J.; Éles, J.; Nagy, J.; Gal, C.A.; Paizs, C.; Hornyánszky, G.; Poppe, L. Chemoenzymatic Dynamic Kinetic Resolution of Amines in Fully Continuous-Flow Mode. Org. Lett. 2018, 20, 8052–8056. [Google Scholar] [CrossRef] [PubMed]
- Lackner, F.; Hiebler, K.; Grabner, B.; Gruber-Woelfler, H. Optimization of a Catalytic Chemoenzymatic Tandem Reaction for the Synthesis of Natural Stilbenes in Continuous Flow. Catalysts 2020, 10, 1404. [Google Scholar] [CrossRef]
- Szelwicka, A.; Zawadzki, P.; Sitko, M.; Boncel, S.; Czardybon, W.; Chrobok, A. Continuous Flow Chemo-Enzymatic Baeyer–Villiger Oxidation with Superactive and Extra-Stable Enzyme/Carbon Nanotube Catalyst: An Efficient Upgrade from Batch to Flow. Org. Process Res. Dev. 2019, 23, 1386–1395. [Google Scholar] [CrossRef]
- Blaser, H.-U.; Pugin, B.; Studer, M. Enantioselective Heterogeneous Catalysis: Academic and Industrial Challenges. In Chiral Catalyst Immobilization and Recycling; Vos, D.E.D., Vankelecom, I.F.J., Jacobs, P.A., Eds.; Wiley: Weinheim, Germany, 2000; pp. 1–17. [Google Scholar]
- Kragl, U.; Dwars, T. The development of new methods for the recycling of chiral catalysts. Trends Biotechnol. 2001, 19, 442–449. [Google Scholar] [CrossRef]
- Jones, E.; McClean, K.; Housden, S.; Gasparini, G.; Archer, I. Biocatalytic oxidase: Batch to continuous. Chem. Eng. Res. Des. 2012, 90, 726–731. [Google Scholar] [CrossRef]
- Gasparini, G.; Archer, I.; Jones, E.; Ashe, R. Scaling Up Biocatalysis Reactions in Flow Reactors. Org. Process Res. Dev. 2012, 16, 1013–1016. [Google Scholar] [CrossRef]
- Toftgaard Pedersen, A.; de Carvalho, T.M.; Sutherland, E.; Rehn, G.; Ashe, R.; Woodley, J.M. Characterization of a continuous agitated cell reactor for oxygen dependent biocatalysis. Biotechnol. Bioeng. 2017, 114, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Different types of reactors that are used for biocatalytic reactions carried out under continuous operation in flow chemistry with immobilized biocatalysts. (a) Basic configuration, where reagents are continuously pumped into a reactor containing the immobilized biocatalyst. The reactor can be a tubular type of reactor, such as (b1) open tubular, capillary wall-coated vessel, either as straight channel or coiled; (b2) packed-bed reactor; (b3) monolith reactor; or chip-type reactor with (c1) straight channel; (c2) serpentine channel; (c3) multiple channels; (c4) inclusion of single chambers; (c5) inclusion of multiple chambers. In c1–c5, gray areas correspond to reaction zones where biocatalysts are immobilized.
Figure 1.
Different types of reactors that are used for biocatalytic reactions carried out under continuous operation in flow chemistry with immobilized biocatalysts. (a) Basic configuration, where reagents are continuously pumped into a reactor containing the immobilized biocatalyst. The reactor can be a tubular type of reactor, such as (b1) open tubular, capillary wall-coated vessel, either as straight channel or coiled; (b2) packed-bed reactor; (b3) monolith reactor; or chip-type reactor with (c1) straight channel; (c2) serpentine channel; (c3) multiple channels; (c4) inclusion of single chambers; (c5) inclusion of multiple chambers. In c1–c5, gray areas correspond to reaction zones where biocatalysts are immobilized.

Figure 2.
Different immobilization techniques involving chemical methods (a–e) and physical methods (f,g): (a) covalent binding, (b) adsorption, (c) ionic binding, (d) affinity binding, (e) CLEAs, (f) entrapment and (g) encapsulation [14,30].

Figure 3.
Different methodologies for multi-enzyme immobilization: (a) stepwise immobilization, (b) mixed immobilization, (c) co-immobilization, (c1) random co-immobilization, (c2) compartmentalization and (c3) positional immobilization.
Figure 3.
Different methodologies for multi-enzyme immobilization: (a) stepwise immobilization, (b) mixed immobilization, (c) co-immobilization, (c1) random co-immobilization, (c2) compartmentalization and (c3) positional immobilization.

Figure 4.
Critical aspects in microreactors, such as the mixing zone, the shape of the channel and fluid velocity, may be engineered to improve mass and heat transfer, and thus biocatalyst activity.
Figure 4.
Critical aspects in microreactors, such as the mixing zone, the shape of the channel and fluid velocity, may be engineered to improve mass and heat transfer, and thus biocatalyst activity.


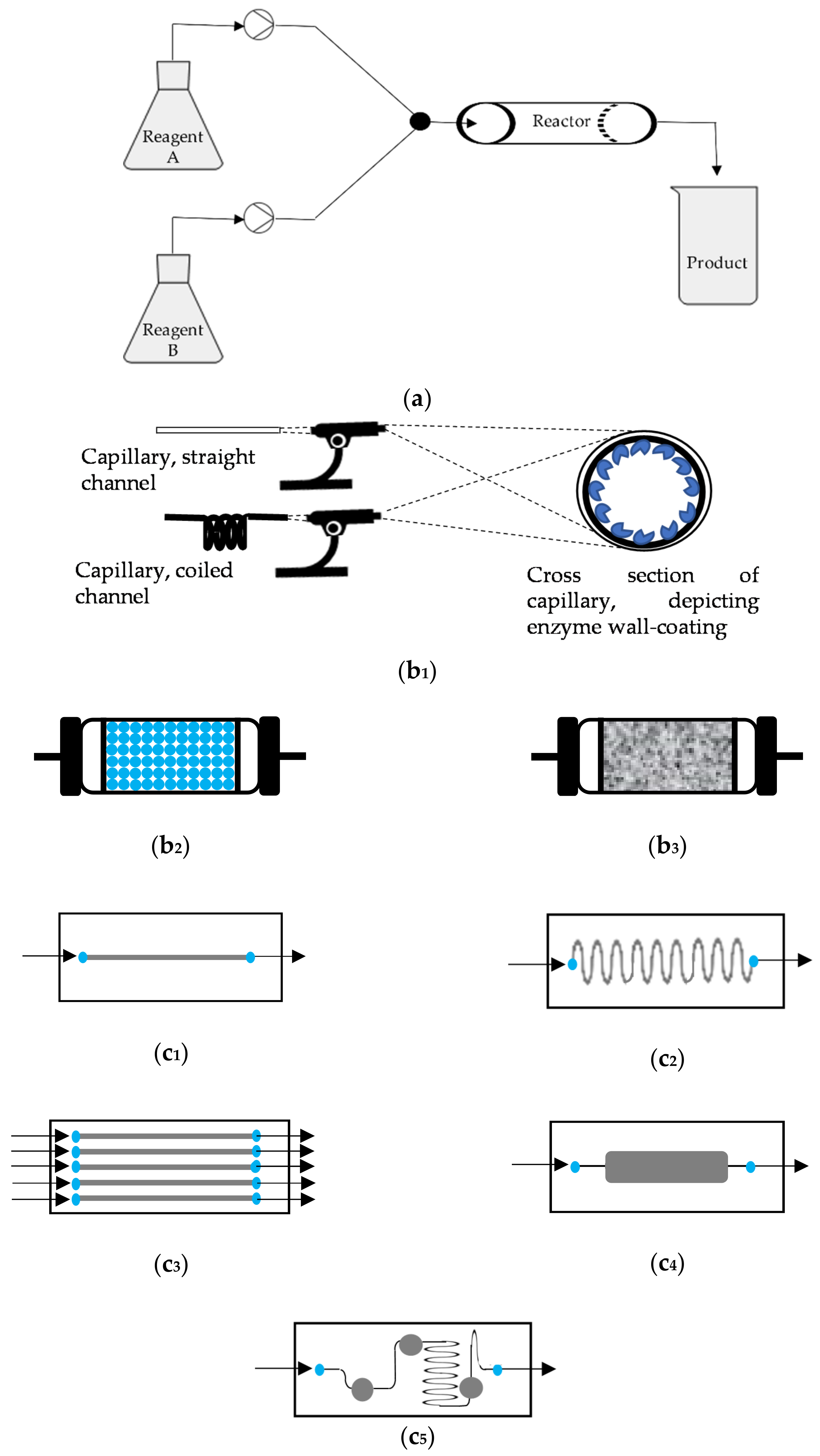{kind=link}
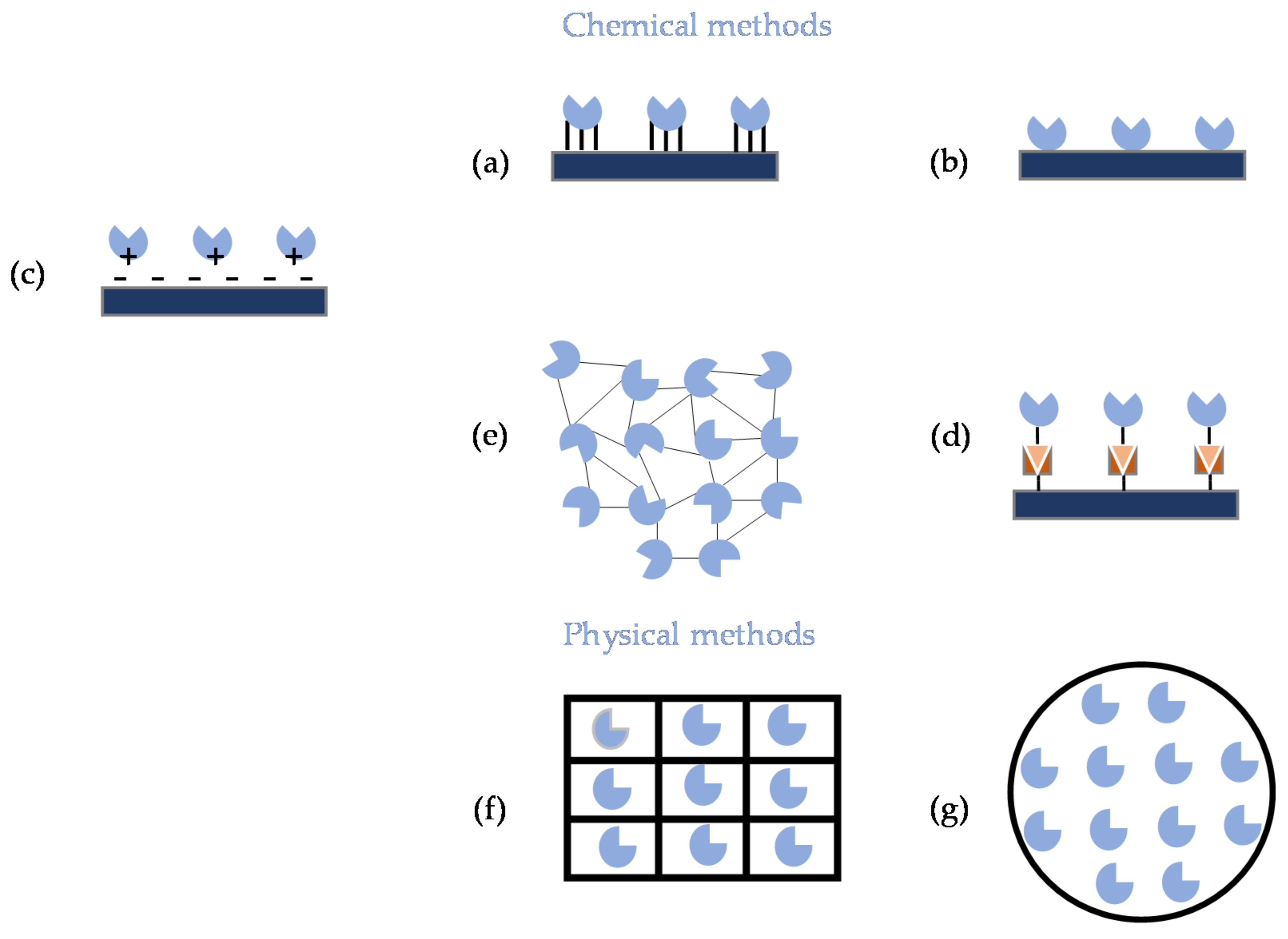{kind=link}
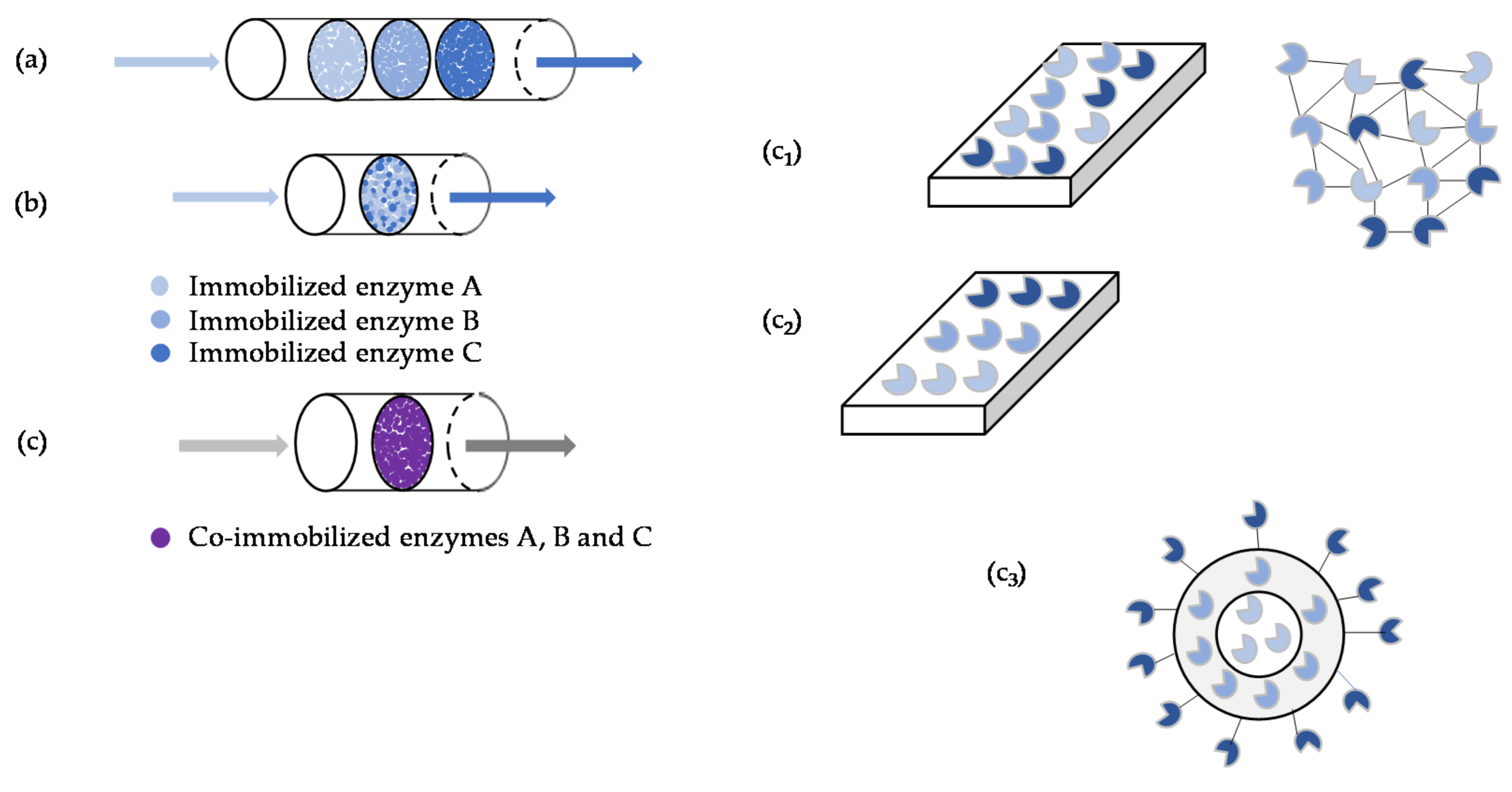{kind=link}
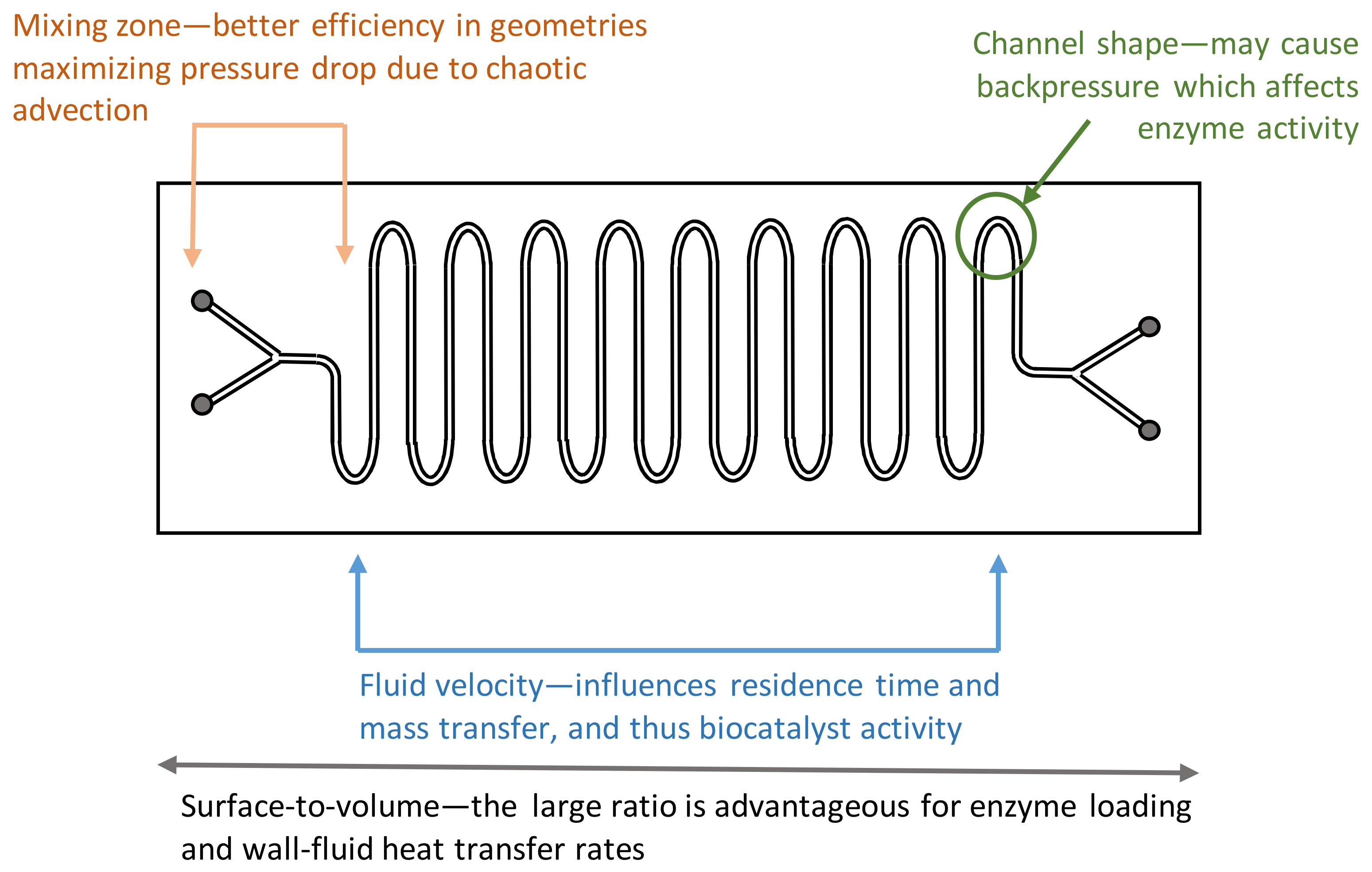{kind=link}
Table 1.
Typical metrics of micro-, meso- and macroreactors.
| Microreactors | Mesoreactors | Macroreactors |
|---|---|---|
| At least one dimension between 10 µm and 500 µm | At least one dimension between 500 µm and a few mm | Dimensions above mm scale |
| Specific area 1 up to 50,000 m2/m3 | Specific area up to 50,000 m2/m3 | Specific area around 100 and 1000 m2/m3 |
| µL range | mL range | L to kL range |
| mg to g scale | multi-g to kg scale | kg to ton scale |
1 Specific area: surface to volume ratio of the reactor.
Table 2.
Recent examples illustrative of the use of different methods for the immobilization of enzyme cascades.
Table 2.
Recent examples illustrative of the use of different methods for the immobilization of enzyme cascades.
| Immobilization Method | System | Comments | Reference |
|---|---|---|---|
| Covalent | Two-step: keto reductase (KRE) and glucose dehydrogenase (GDH) | Production of (R)-4-chloro-3-hydroxybutanoate. The enzymes were co-immobilized in mesocellular siliceous foams through microwave irradiation with p-benzoquinone as crosslinking agent. The formulation retained more than 90% residual activity upon 30 days of storage at 4 °C. The formulation retained over 50% residual activity after 6 repeated batch conversion cycles, whereas upon co-immobilization by entrapment in calcium alginate the residual activity dropped to ~20%. | [32] |
| Two-step: uridine phosphorylase (UP) and purine nucleoside phosphorylase (pNP) phosphorylase | Synthesis of arabinosyladenine, an antiviral nucleoside. Enzymes were co-immobilized on glutaraldehyde activated monolithic aminopropyl silica carrier. Recirculation through the column at a flow rate of 0.5 mL.min−1 of adenine (nucleobase, 1 mM) and arabinosyluracil (sugar donor, 2 mM) resulted in 60% conversion after 24 h. | [89] | |
| Covalent and ionic | Three-step: UP, pNP and deoxyadenosine kinase (dAK) | Synthesis of vidarabine 5′-monophosphate (antiviral drug). UP and pNP were covalently bound individually to glyoxyl-agarose and dAK was bound by ionic interaction ionic interaction to functionalized Sepabeads® EC-EP. Close to full conversion of the substrate (adenine, 25 mM) was reported. | [90] |
| Covalent and affinity | Two-step: UP and pNP, each fused with His-tag binding peptide | Synthesis of vidarabine The enzymes were co-immobilized either covalently on glyoxyl-agarose or by metal-ion affinity on a hydrophylic polymer-coated controlled porosity glass beads, EziG1 (Opal). Each one of the resulting formulations was packed in a glass column that was fed with arabinofuranosyluracil (16 mM) as sugar donor and adenine (8 mM) as sugar acceptor. Eighty percent conversion was reached after either 4 h of residence time (covalent-based formulation) or 80 min (affinity-based formulation). The later displayed poor operational stability, hence the former was used for continuous production of vidarabine. Under a residence time 2 h (67% conversion) operation proceeded for 8 days, after which the product was recovered (55% yield, over 99% purity). | [91] |
| Three-step: glycerol kinase (GK)/acetate kinase (AK) + glycerol-3-phosphate dehydrogenase (GPD)/NADH oxidase (NOX) + fructose aldolase (FA), each harboring a harboring a maleimide–thiol conjugation module | Conversion of glycerol to a chiral d-fagomine precursor. GK/AK and GPD/NOX were produced as modular biocatalysts that retain and recycle their cofactors as fusion proteins, to which cofactors were covalently tethered. GK/AK, GPD/NOX and FA were covalently bound through the conjugation module to chemically trifluoroketone activated agarose beads and each of the three formulations added in packed-bed reactors disposed sequentially. Space–time yields of 70 g L−1 h−1 g−1 and total turnover numbers above 10,000 were reported | [92] | |
| Affinity | Two-step: (R)-selective alcohol dehydrogenase (RADH), (S)-selective methylglyoxal reductase and GDH, each fused with streptavidin binding peptide | Selective reduction of 5-nitrononane-2,8-dione. Each enzyme was bound to magnetic microbeads coated with streptavidin, which were introduced in a compartmentalized microfluidic packed-bed reactor. Under selected flow conditions and ratio of immobilized enzymes load, an initial conversion of 73.6% stereoselectivity exceeding 99:1 and product space–time yield of 106 g L−1day−1 were reported. | [93] |
| Two-step: RADH, and GDH, each fused with His-tag binding peptide | Selective reduction of 5-nitrononane-2,8-dione. Each enzyme was bound to Co2+ functionalized magnetic microbeads, which were introduced in a compartmentalized microfluidic packed-bed reactor. Despite noticeable decrease inactivity upon immobilization, namely for GDH, under selected immobilized enzyme load and flow conditions, 98% substrate conversion of 98% and product space–time yield of 131 g L−1day−1 was reported. The outcome compared favorably with that of a previous work [93], albeit at the cost of a decrease (~60%) in specific productivity. | [63] | |
| Two-step: KRE and GDH, each fused with His-tag binding peptide | Reduction of keto-ester (ethyl-2-methylacetoacetate) and bulky ketones (4-phenyl-2-butanone; 3′-hydroxyacetophenone) to secondary alcohols. The enzymes from crude extracts were co-immobilized under optimized ratios in a column Ni2+ functionalized crosslinked agarose, which was packed in a flow reactor. Co-immobilization reduced cofactor requirements and immobilization enhanced tolerance to high substrate concentrations (130 mM and above) as compared to the free enzymes. The immobilized enzymes were used in 20 (keto-ester) and 13 (bulky ketones) repeated batch conversion cycles with 95% substrate conversion with substrate concentration of 130 mM. | [64] | |
| Affinity (cont.) | Three-step: β-galactosidase (bGAL), glucose oxidase GOx and horseradish peroxidase (HRP) | Conversion of lactose to resorufin. Streptavidin-coated microbeads functionalized with either individual or a mixture of chemically biotinylated enzymes. The formulations were introduced in microfluidic channel, where the former immobilization approach outperformed the later. Operational parameters, e.g., flow rate, relative amount of enzymes, initial substrate concentration and total amount of biocatalyst, were optimized. | [21] |
| Three-step: sucrose phosphorylase, cellobiose phosphorylase and cellodextrin phosphorylase, each fused with Zbasic2 binding module | Synthesis of soluble cello-oligosaccharides with degree of polymerization ≤6. The enzymes were co-immobilized according to previously established ratio of activities on macroporous polymethacrylate particles coated with sulfonate groups harboring the negative charges to interact with the Zbasic2 module. The formulation was used through five repeated batch conversion cycles allowing the synthesis of 10 to 12.5 g L−1 of the intended cello-oligosaccharides from ~68 g L−1 sucrose and 12 g L−1 glucose, and retaining ~85% of the overall initial activity. Some leakage of cellobiose phosphorylase was observed, the trend ascribed to the excess of negative surface charges of the fused enzyme. | [94] | |
| Two-step: Imine reductase and GDH fused with SpyTag and SpyCatcher domain, respectively, to generate two complementary building blocks | Conversion of cyclic imines to the corresponding secondary amines. Upon incubation in magnesium-supplemented potassium phosphate buffer the two fused enzymes self-assembled to a porous hydrogel through the formation of a covalent isopeptide bond between the activated lysin residue of the SpyCatcher and the aspartic acid residue on the SpyTag domains. The catalytic hydrogel exhibited a stereoselectivity over 99%. The gel was packed in a microfluidic (150 µL volume) channel. After 40 h days of operation (10 µL min−1 feeding rate, 5 mM 3,4-dihydroisoquinoline solution) ~90% conversion was observed, whereas after 5 h of operation with unbound GDH roughly no conversion was observed, due to GDH leakage. Space–time yield of 150 g L−1.day−1 was observed at a flow rate of 100 µL.min−1. | [95] | |
| Entrapment (cont.) | Two-step: ADH and GDH fused with SpyCatcher and SpyTag domain, respectively, to generate two complementary homo-tetrameric building blocks | Selective reduction of 5-nitrononane-2,8-dione, acetophenone, 4′-chloroacetophenone and trans-4-phenyl-3-buten-2-one to the corresponding R alcohols. The two fused enzymes self-assembled to a porous hydrogel containing 77% of enzyme. The gel was packed in a microfluidic (150 µL volume) channel. After 7 days of operation (10 µL min−1 feeding rate, 5 mM substrate solution) ~70% conversion was observed with no enzyme leakage, whereas after 2 h of operation with the free enzymes roughly full leakage was observed. Spacetime yield 4.5-fold higher than previously reported [93] was observed. Mass transfer limitations were advantageously used: co-entrapment of NADP+ (cofactor) allowed for 30 h of continuous conversion with no cofactor in the feed. Stereoselectivity over 99% was observed in all reactions after 10 h of continuous operation. The gel could be stored for 30 days at 4 °C with no loss in activity. | [96] |
| Entrapment (cont.) | Two cascades, each three-step: bGAL GOx and HRP (cascade 1); and phospholipase D, choline oxidase and HRP (cascade 2) | Detection of lactose and/or glucose (cascade 1) and of phosphatidylcholine (cascade 2). The enzymes of each cascade were co-immobilized in a noncompartmentalized manner in a hydrogel matrix composed of poly(ethylene glycol) diacrylate, 2-(dimethylamino)ethyl methacrylate, and 2-hydroxyethyl methacrylate, either as bulky hydrogels or as dots (350 μm diameter) integrated into polydimethylsiloxane (PDMS)-on-glass microfluidic reactors to perform the reaction under continuous flow. Overall, immobilization increased the catalytic activity of the cascades as compared to the free form. | [97] |
| Encapsulation | Two-step: GOx and HRP | Conversion of glucose to resorufin. A mixture of GOx and HRP was encapsulated in giant unilamellar vesicles (GUV) sized from 10 to 200 μm, produced out of a liposome suspension prepared from phospholipids present in the soybean polar extract. | [98] |
| Two-step: GOx and HRP | Conversion of glucose to resorufin. The two enzymes were encapsulated inside silica microparticles. The formulation was packed in a microfluidic chamber and assessed for monitoring glucose concentration. The device operated within the range of glucose concentration found in saliva and sweat. | [99] | |
| Encapsulation and entrapment | Two-step: Alcohol oxidase and catalase | Alcohol oxidase and catalase were individually entrapped in inverse opal particles and the whole embedded in calcium alginate microcapsules to mimic hepatocytes for elementary alcohol detoxification. | [100] |
| Combi-CLEAs and covalent binding | Multi-step: cellulase, pectinase and xylanase | Saccharification of cellulose and hemicellulose. The enzymes were individually immobilized in amino-functionalized magnetic particles which were afterwards crosslinked with glutaraldehyde to yield magnetic combi-CLEAs. Immobilization improved the thermal stability of the enzymes and the formulation was used through 12 repeated batch conversion cycles with minor loss of activity. Moreover, when integrated in simultaneous saccharification and fermentation of wheat straw the formulation allowed a 1.82-fold increase in bioethanol concentration as compared to use of free enzymes. | [76] |
| Combi-CLEAs and encapsulation | Two-step: GOx and HRP | Conversion of glucose to resorufin. A mixture of GOx and HRP was engulfed inside the bowl-shaped polymersomes and the enzyme molecules were crosslinked with either genipin or glutaraldehyde to produce crosslinked enzymatic nanoaggregates inside the submicron-sized vesicles (c-CLEnA). | [71] |
Table 3.
Examples of multi-step bioconversions using whole cells, chemo- and enzymatic biocatalysts.
Table 3.
Examples of multi-step bioconversions using whole cells, chemo- and enzymatic biocatalysts.
| Biocatalyst | Bioreactor | Reaction | Reference |
|---|---|---|---|
| Lactobacillus kefiri | Plug flow reactor |  | [121] |
| Escherichia coli with Chromobacterium violaceum ωws-transaminase activity and Lodderomyces elongisporus with ketoreductase activity | Continuous flow reactor |  | [122] |
| Immobilized 7α- and 7β-hydroxysteroid dehydrogenases | Two column bioreactors with each enzyme or single column with both immobilized enzymes |  | [102] |
| Phenolic acid decarboxylase and a chemical Pd-catalyst | Packed-bed reactor |  | [123] |
| Acyltransferase from Mycobacterium smegmatis, in-line purification with SO2Cl, and hydrogenation step | Continuous flow reactor |  | [124] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fernandes, P.; de Carvalho, C.C.C.R. Multi-Enzyme Systems in Flow Chemistry. Processes 2021, 9, 225. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9020225
AMA Style
Fernandes P, de Carvalho CCCR. Multi-Enzyme Systems in Flow Chemistry. Processes. 2021; 9(2):225. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9020225
Chicago/Turabian StyleFernandes, Pedro, and Carla C. C. R. de Carvalho. 2021. "Multi-Enzyme Systems in Flow Chemistry" Processes 9, no. 2: 225. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9020225
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.