
Digital Twin of mRNA-Based SARS-COVID-19 Vaccine Manufacturing towards Autonomous Operation for Improvements in Speed, Scale, Robustness, Flexibility and Real-Time Release Testing




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
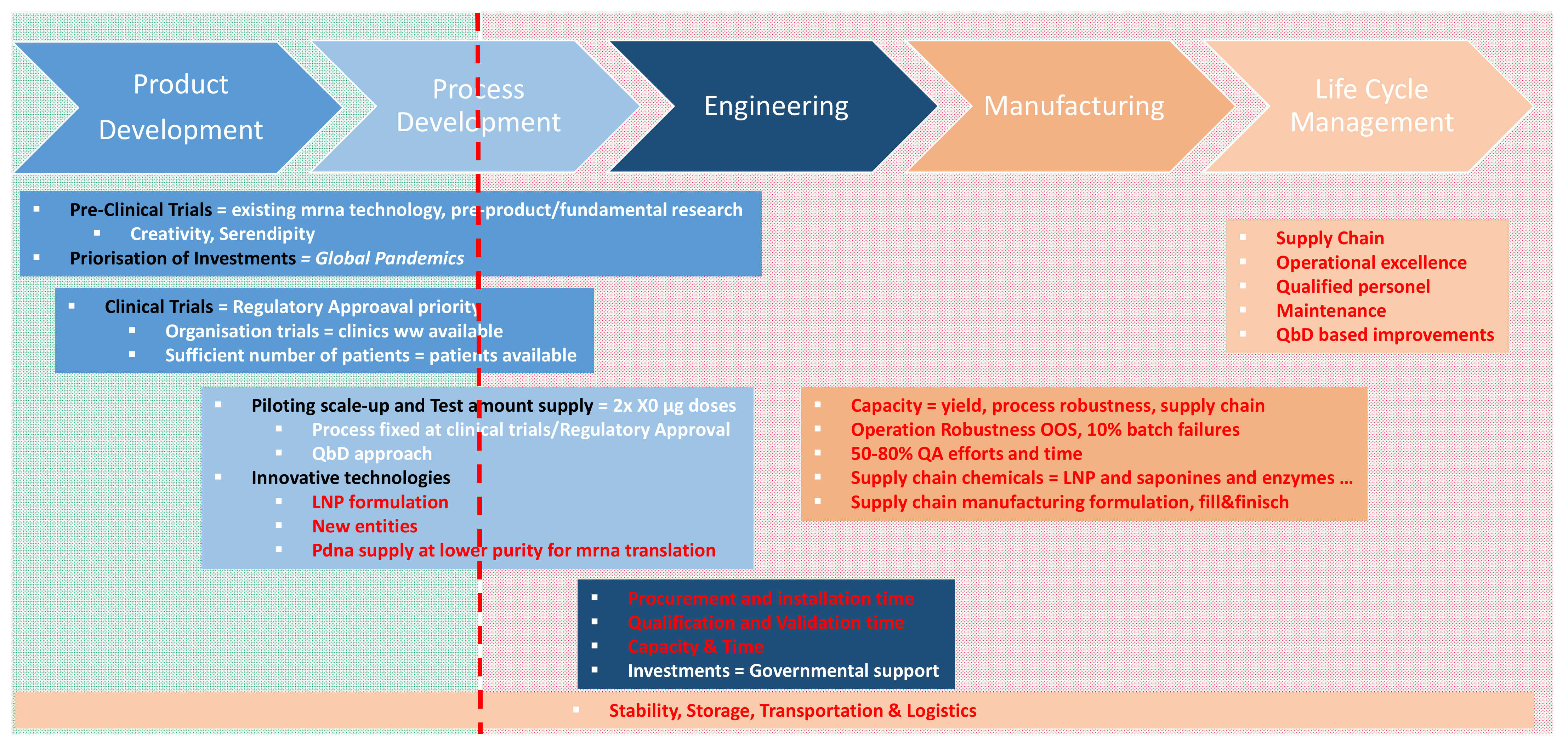
:1. Introduction
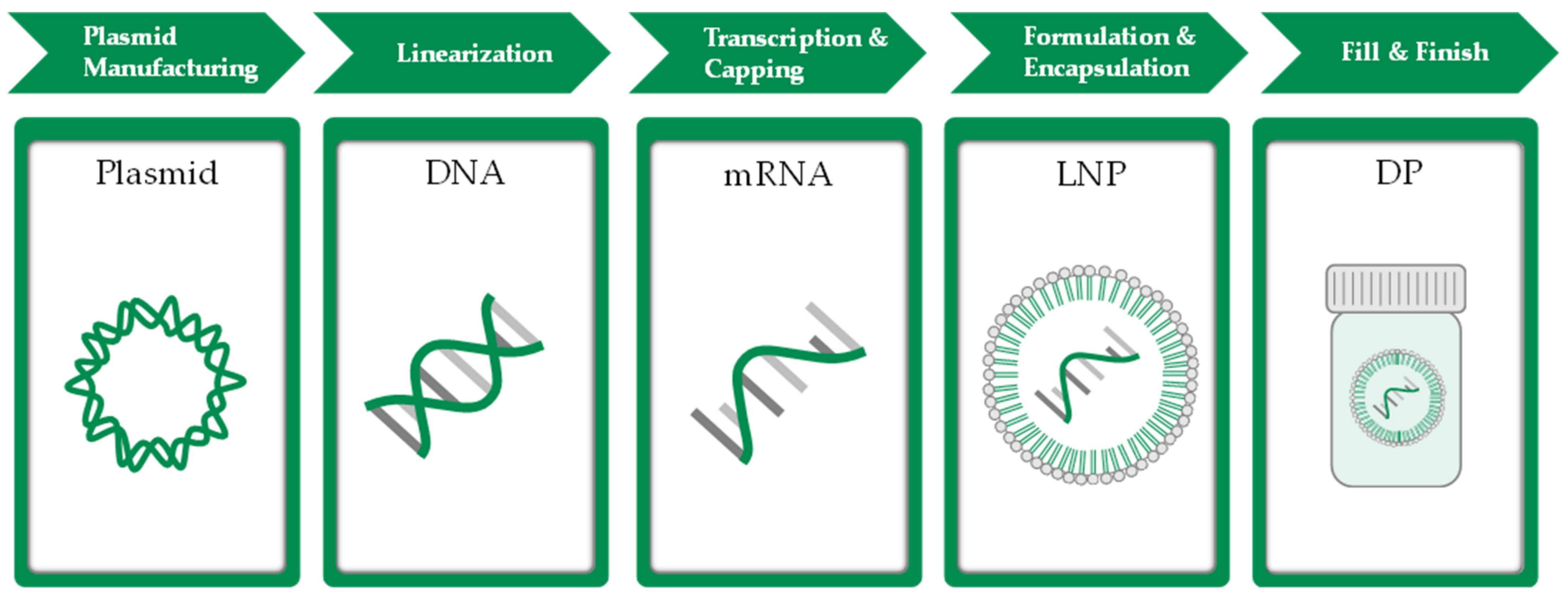
1.1. State-of-The-Art in mRNA Manufacturing
2. Material and Methods
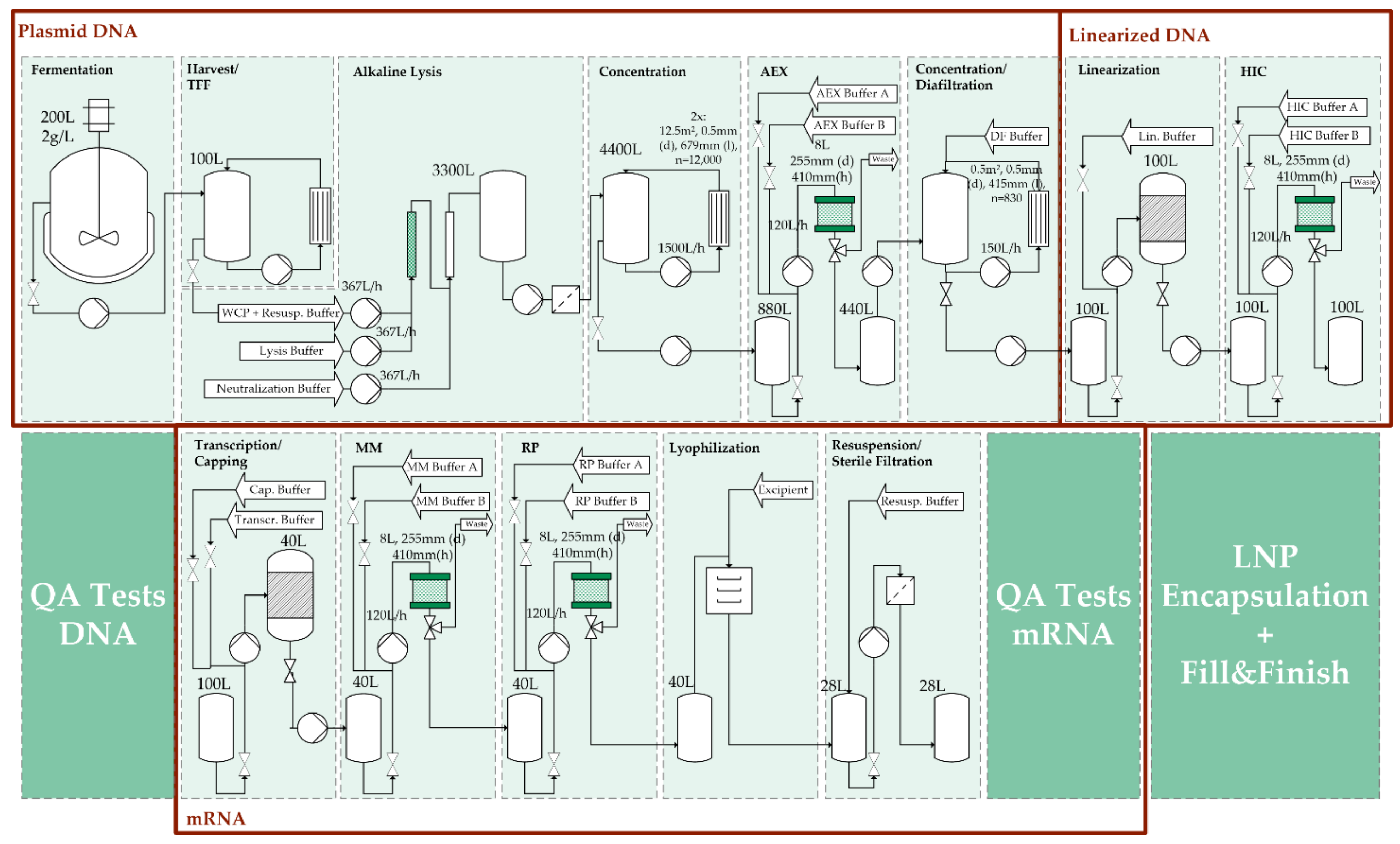
2.1. Model Overview
2.1.1. Fermentation
2.1.2. Alkaline Lysis
2.1.3. Concentration by Ultrafiltration
2.1.4. Purification with Adsorption Monoliths
2.1.5. Linearization
2.1.6. Transcription
2.1.7. Lyophilization of mRNA
3. Results
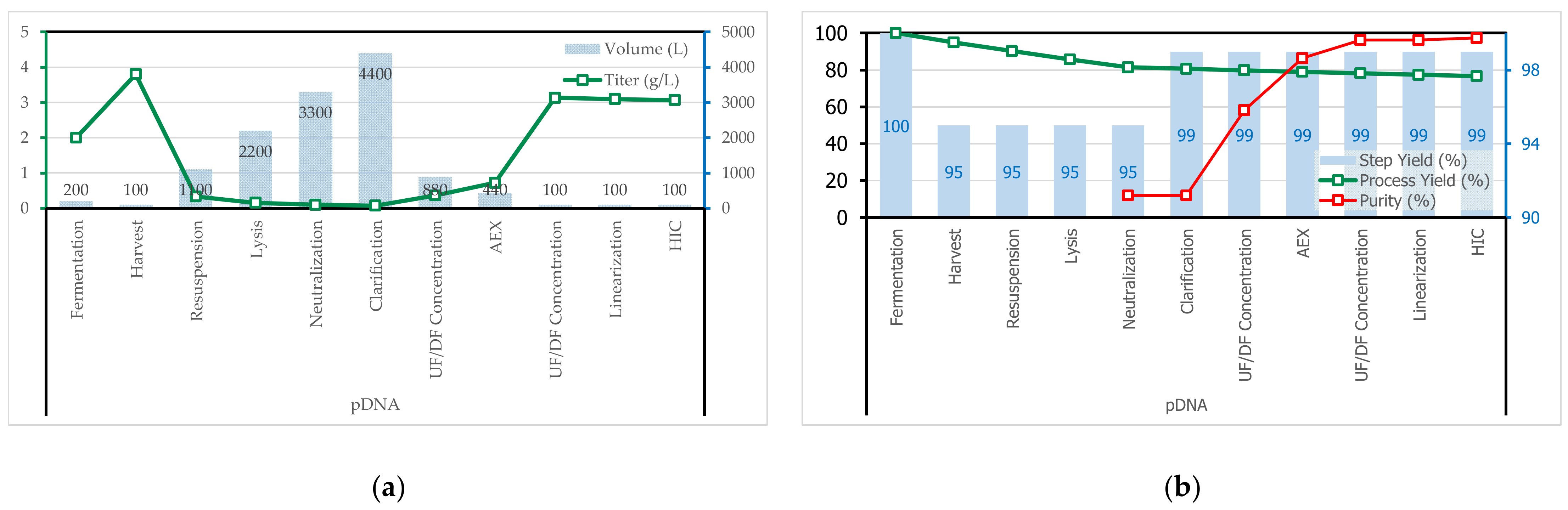
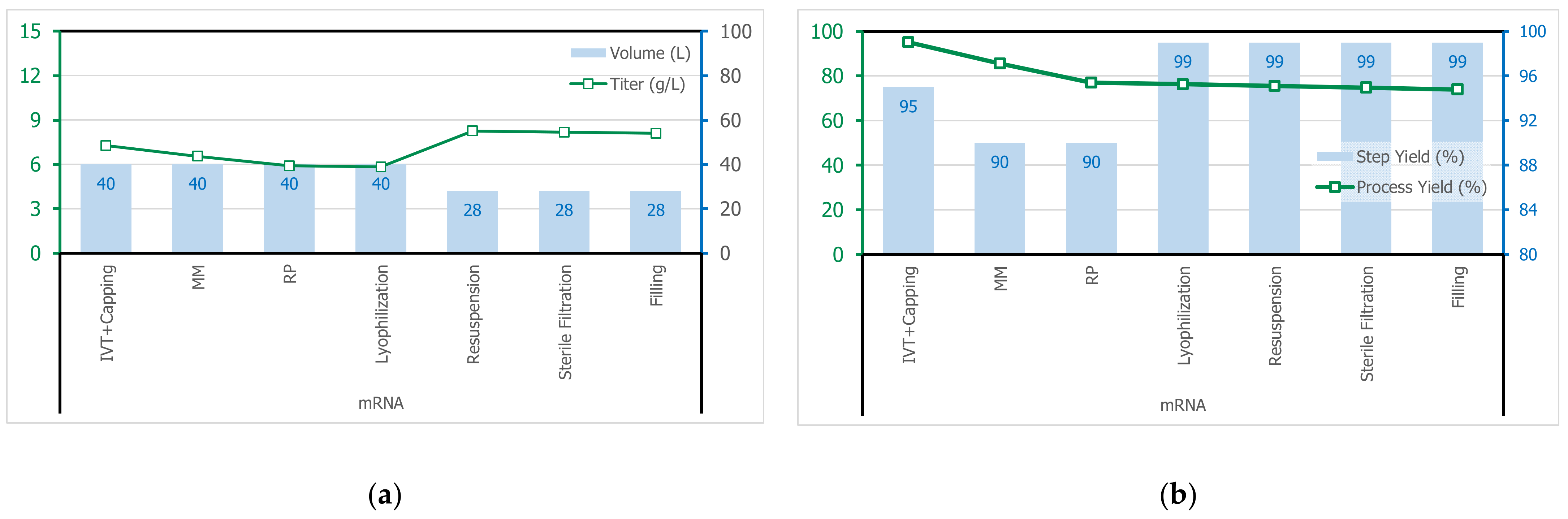
3.1. Process Synthethis
3.2. Process Simulation
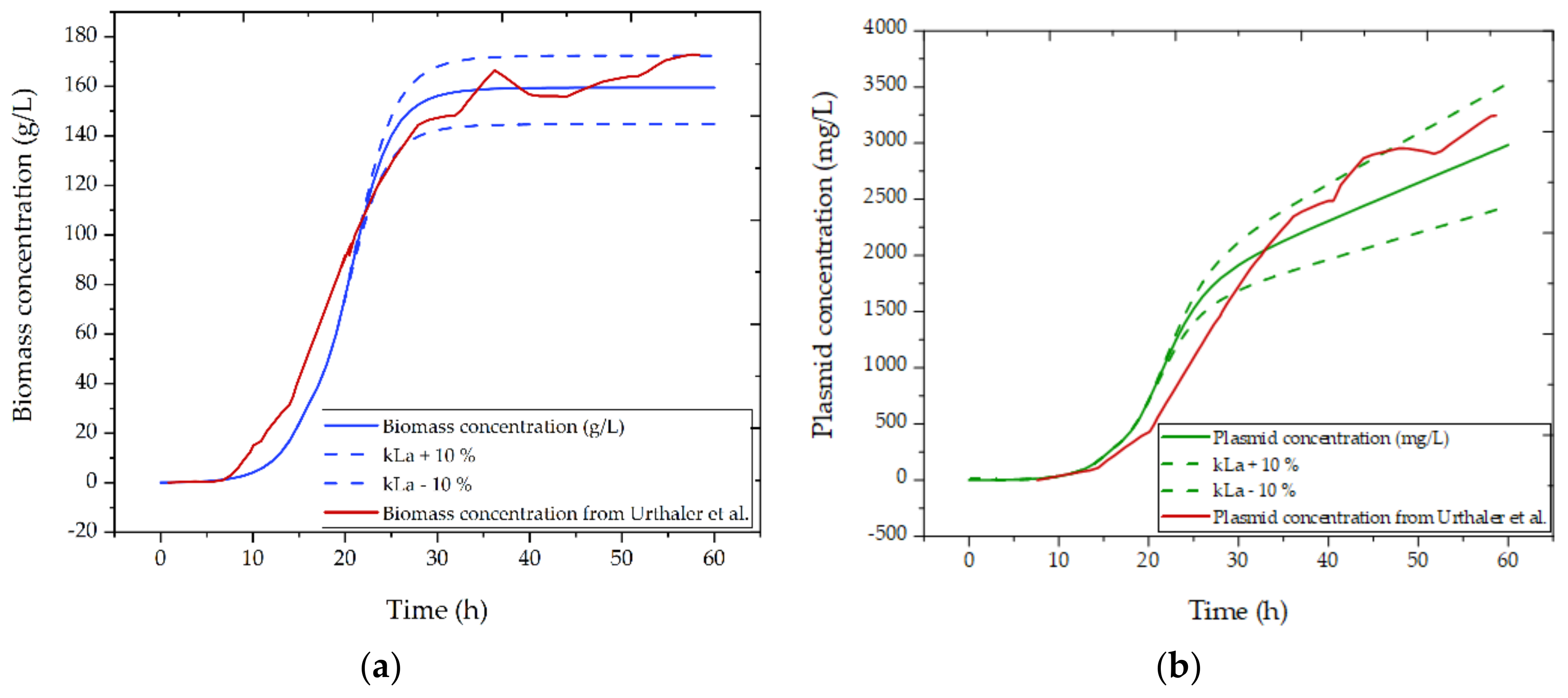
3.2.1. Fermentation
3.2.2. Resuspension and Alkaline Lysis
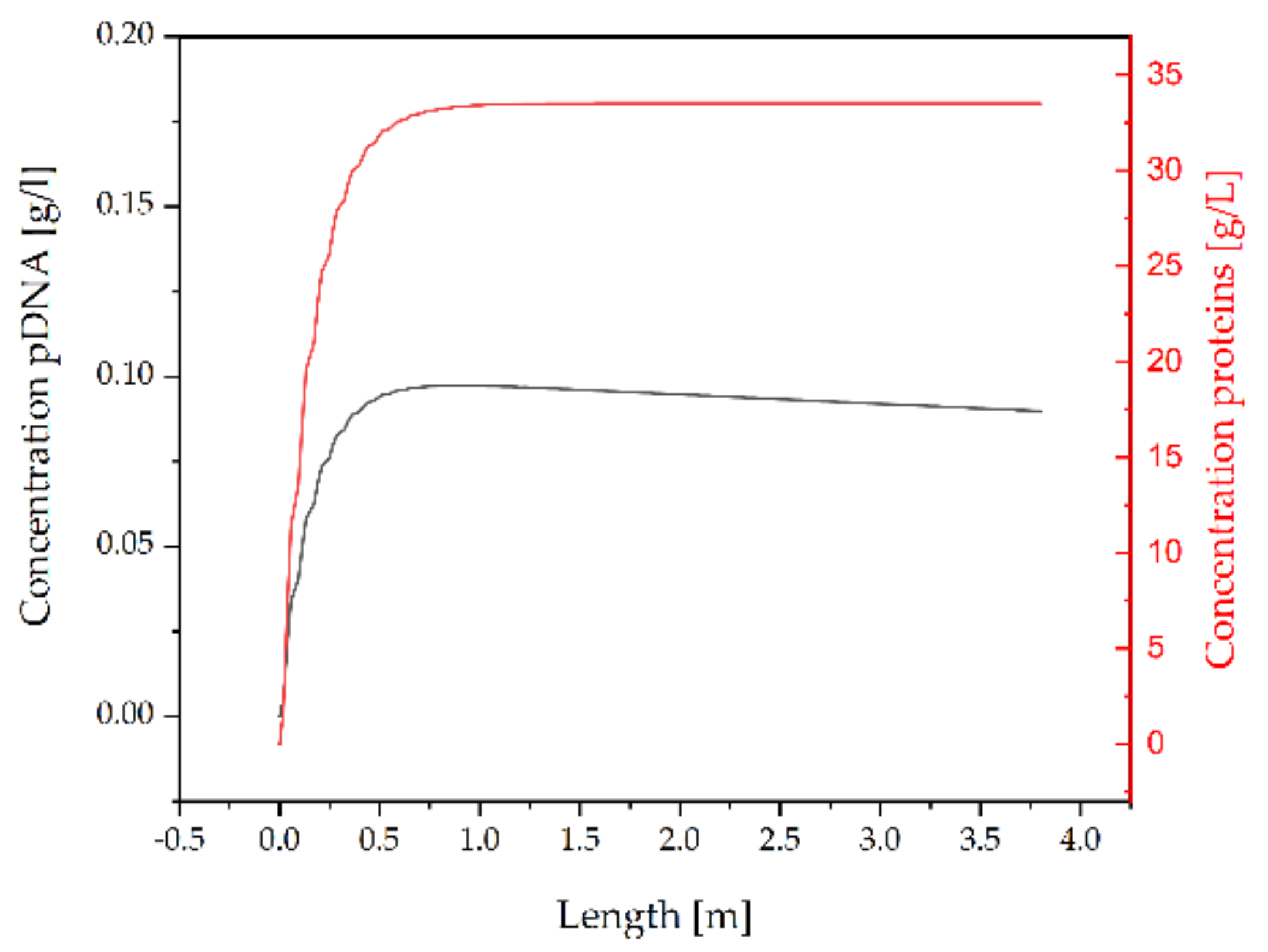
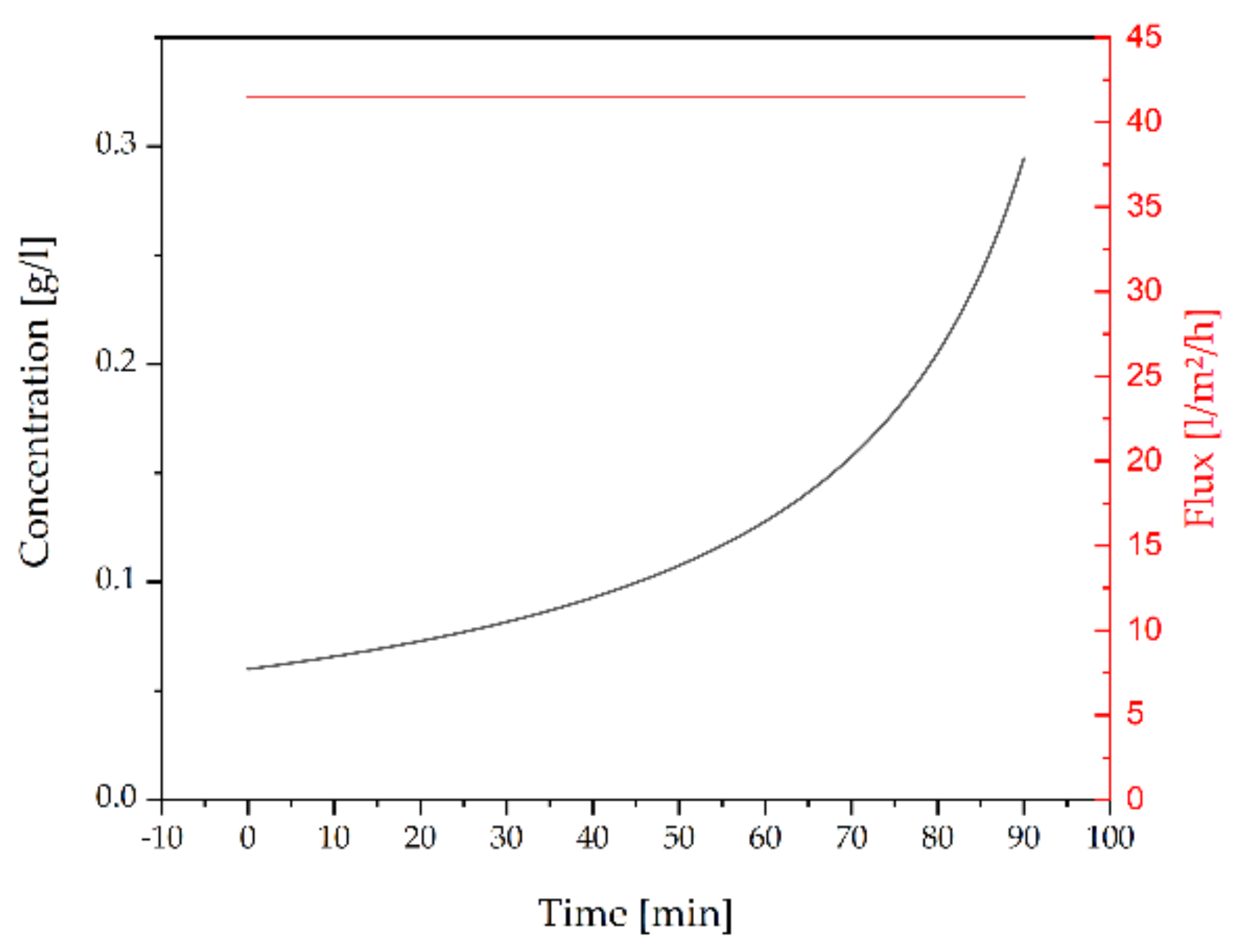
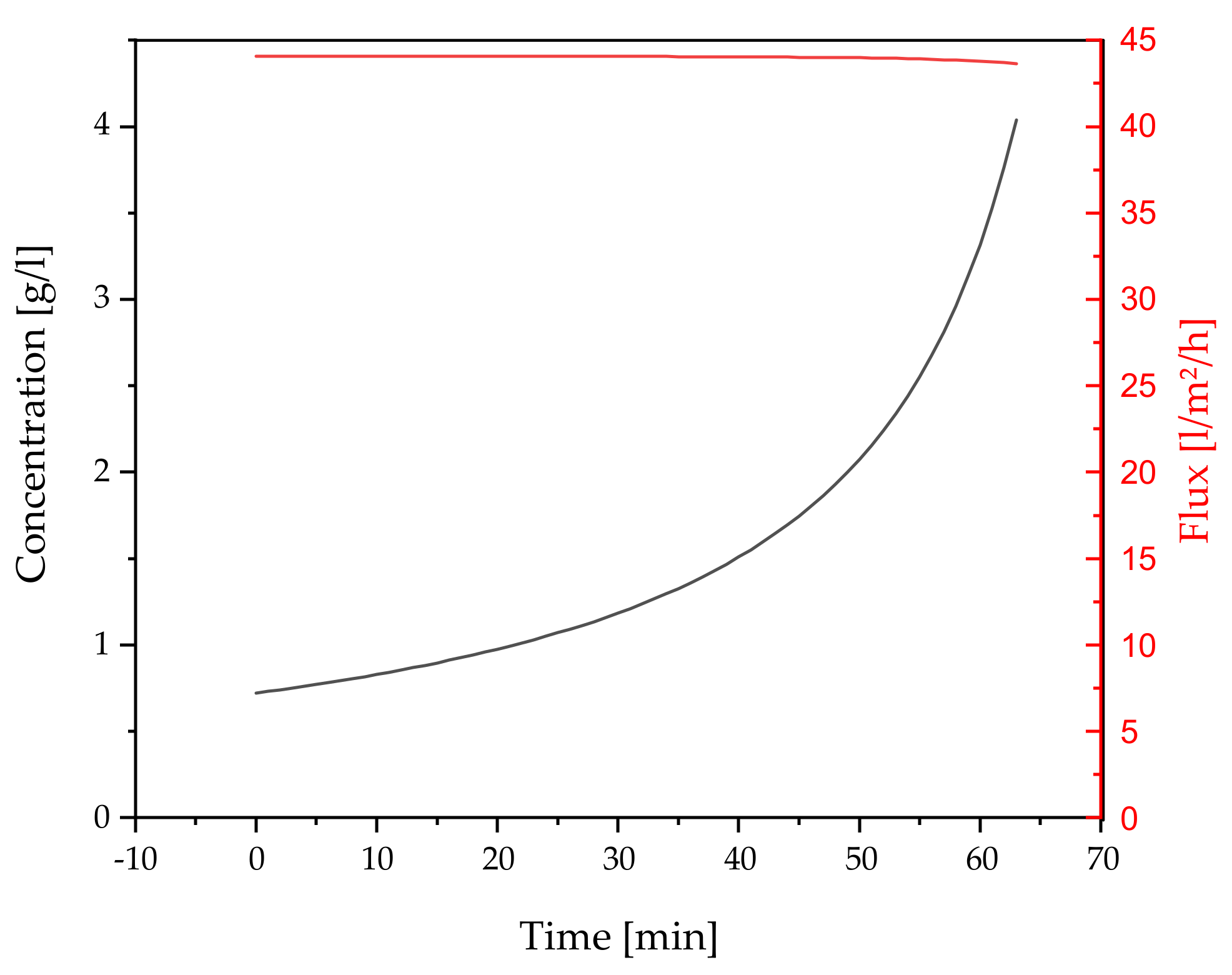
3.2.3. Concentration by Ultrafiltration
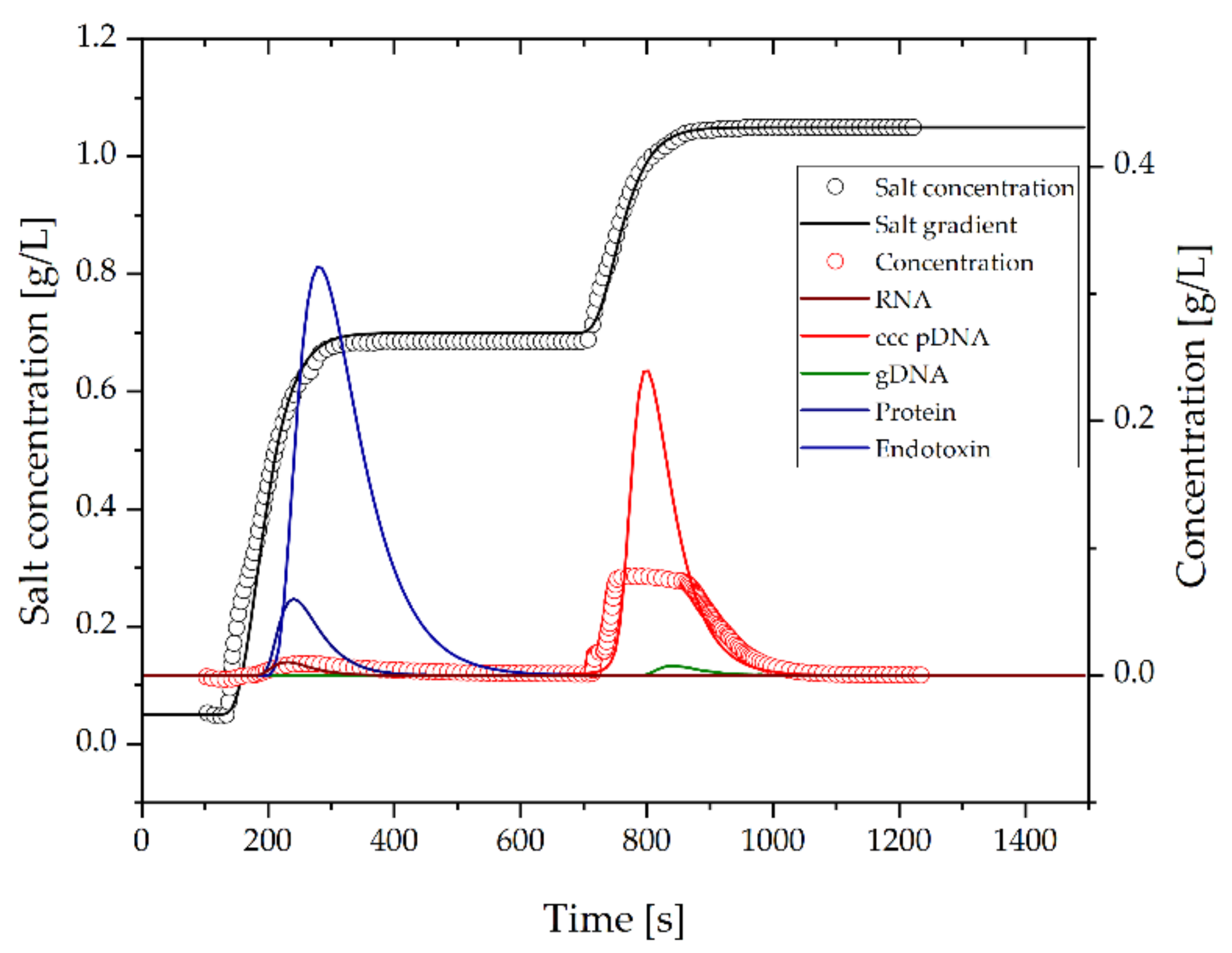
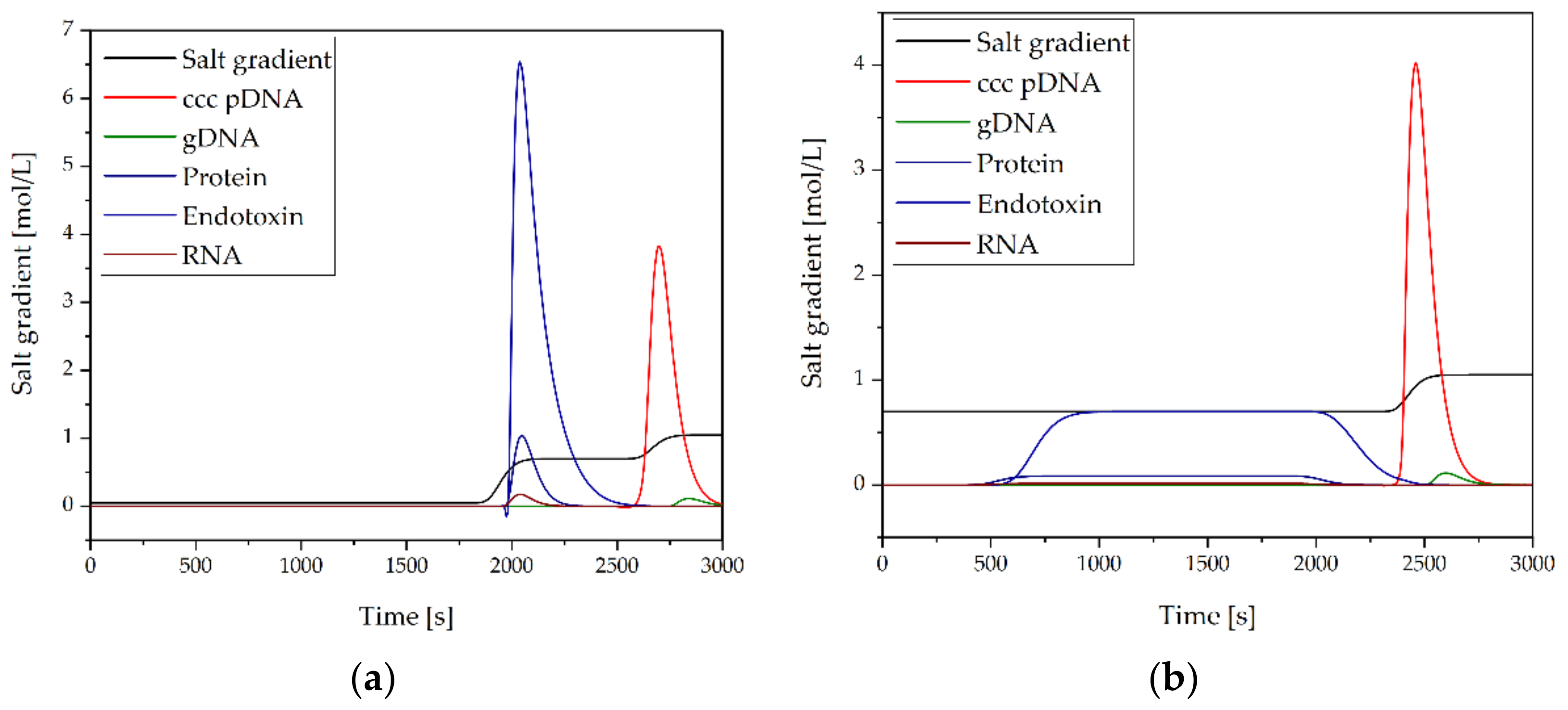
3.2.4. Purification of pDNA by AEX
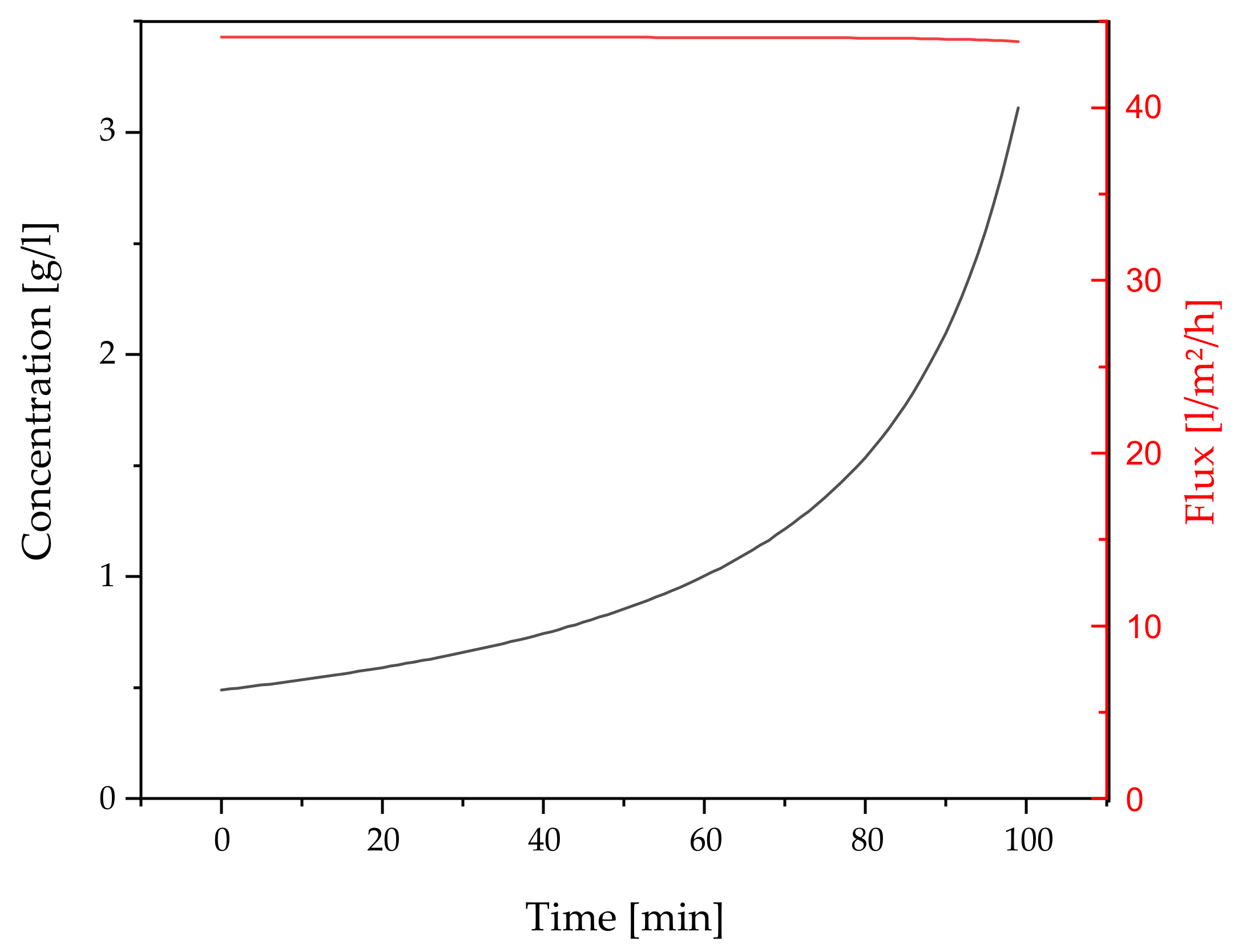
3.2.5. Concentration and Diafiltration by UF/DF
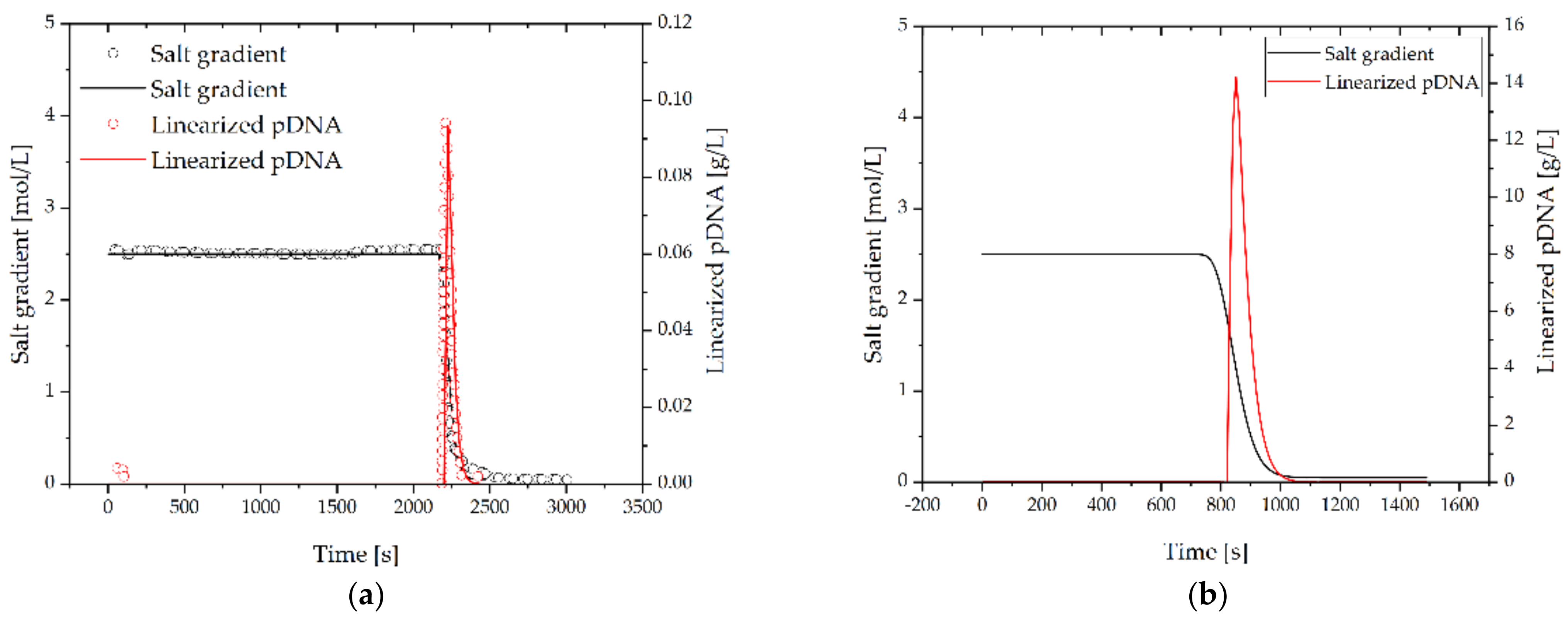
3.2.6. Linearization
3.2.7. Purification of Linearized ds-DNA by HIC
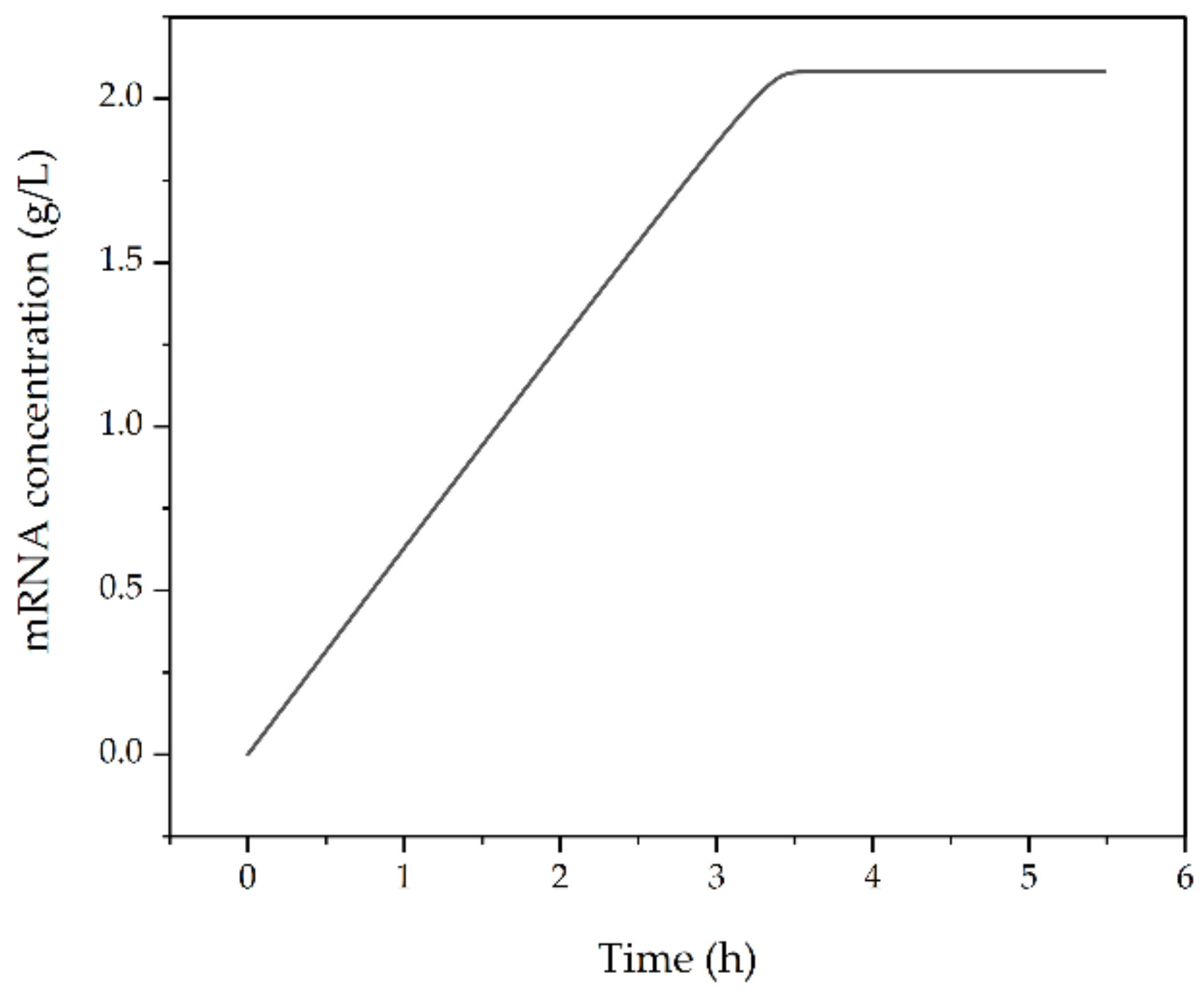
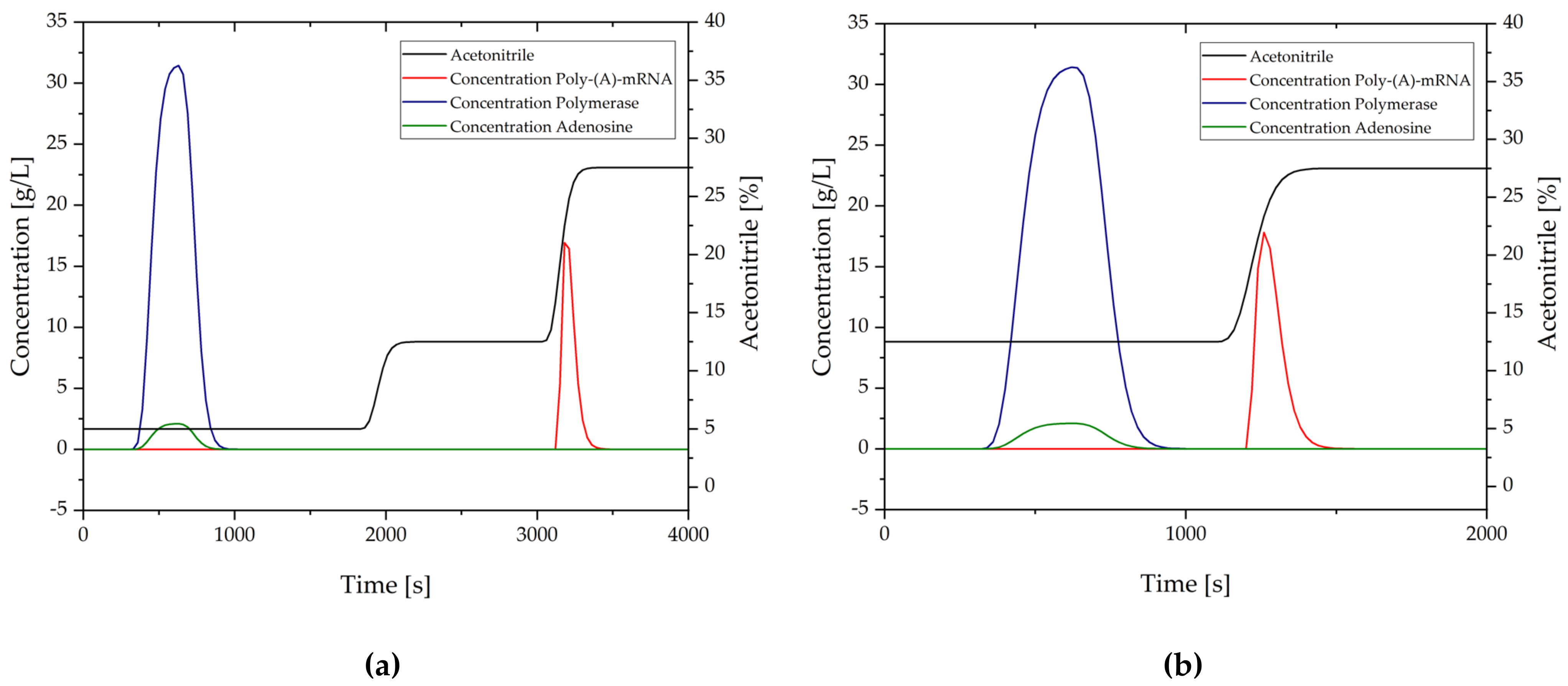
3.2.8. Transcription
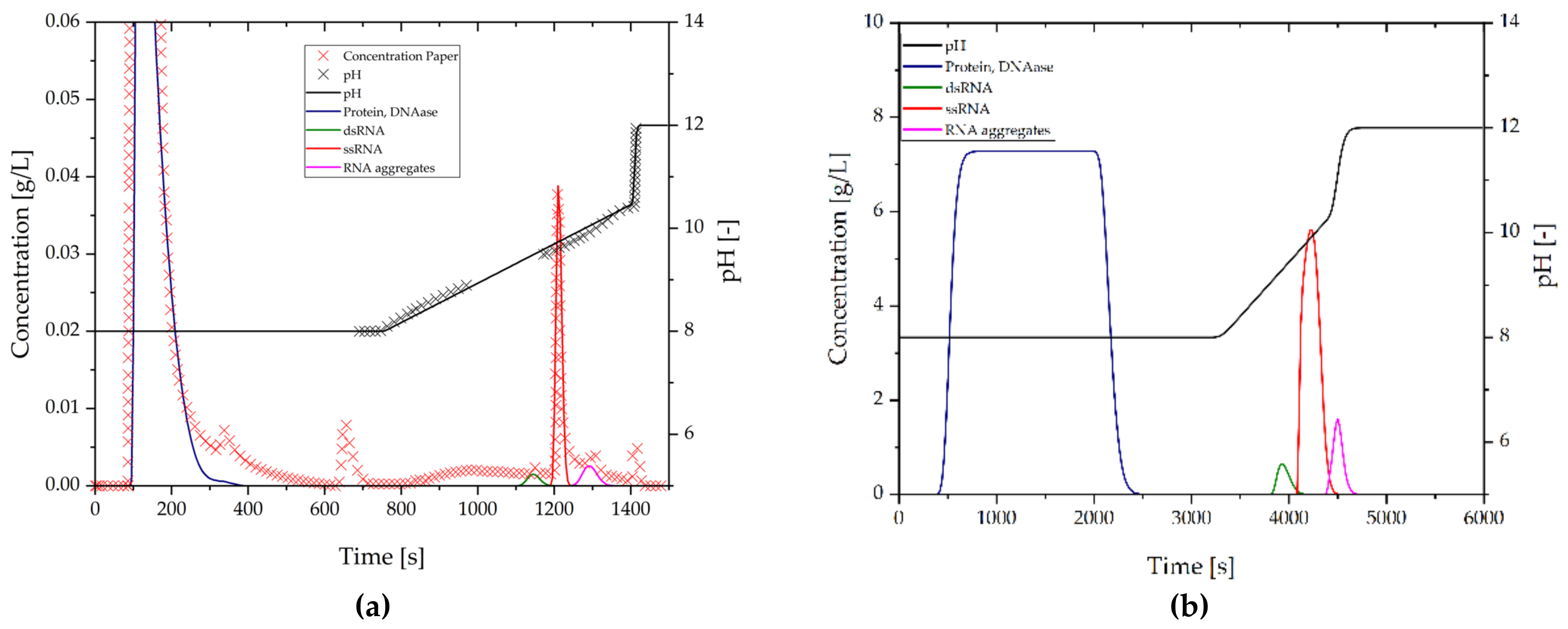
3.2.9. Purification of mRNA by MMC
3.2.10. Purification of mRNA by RPC
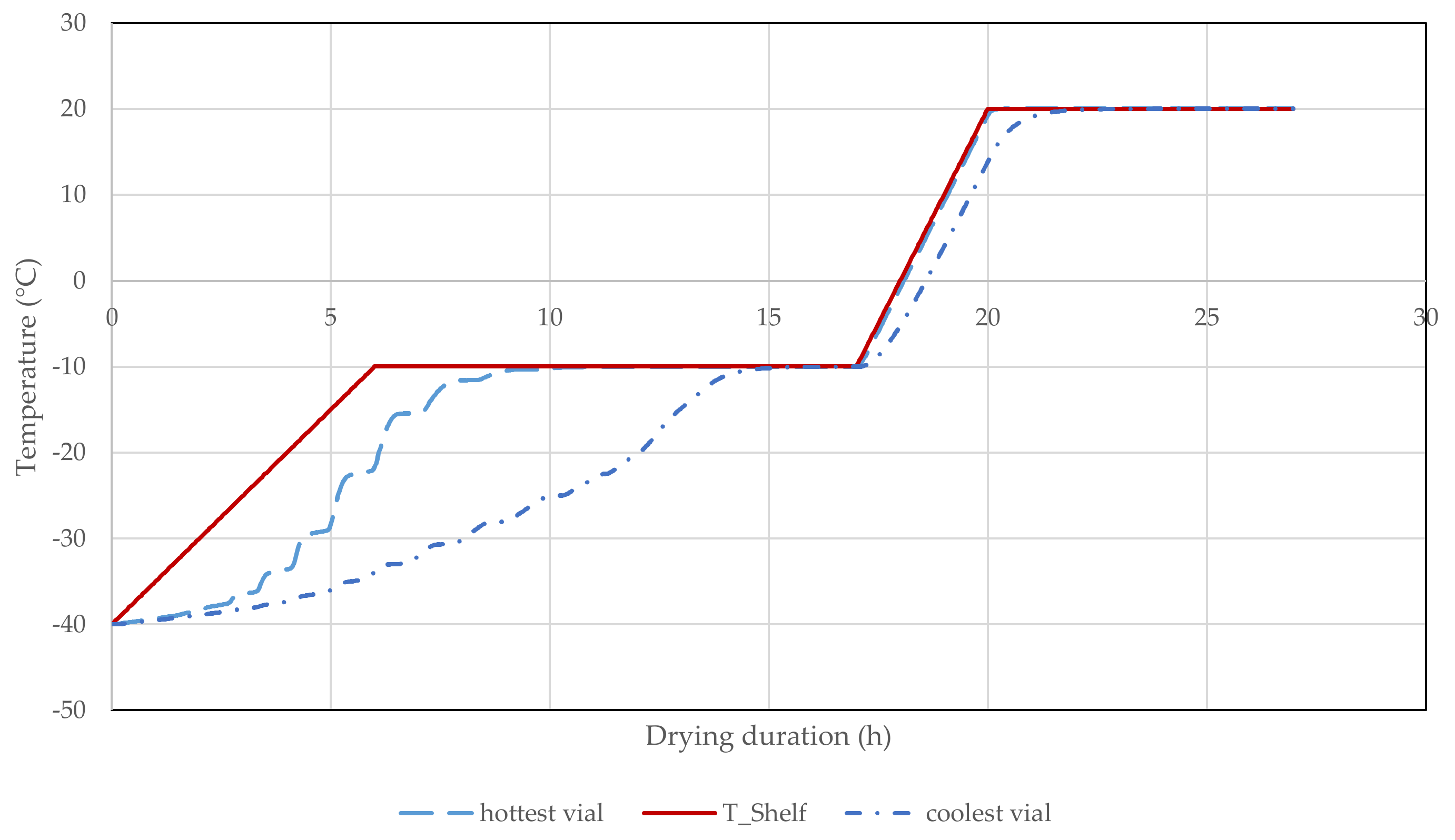
3.2.11. Lyophilization of mRNA
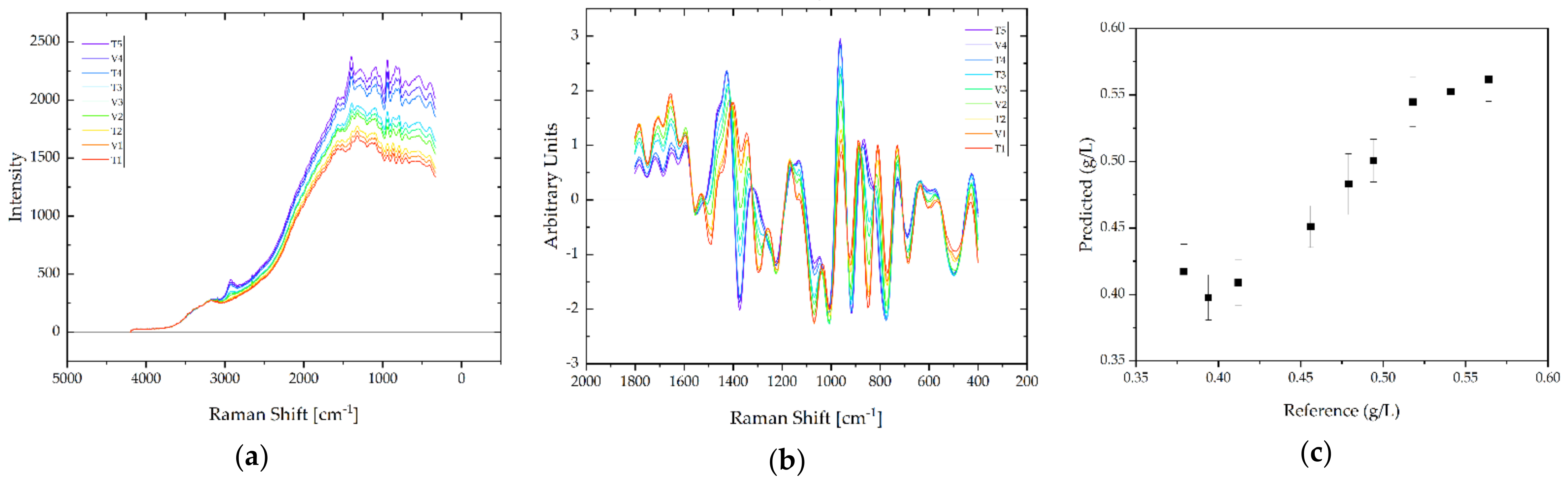
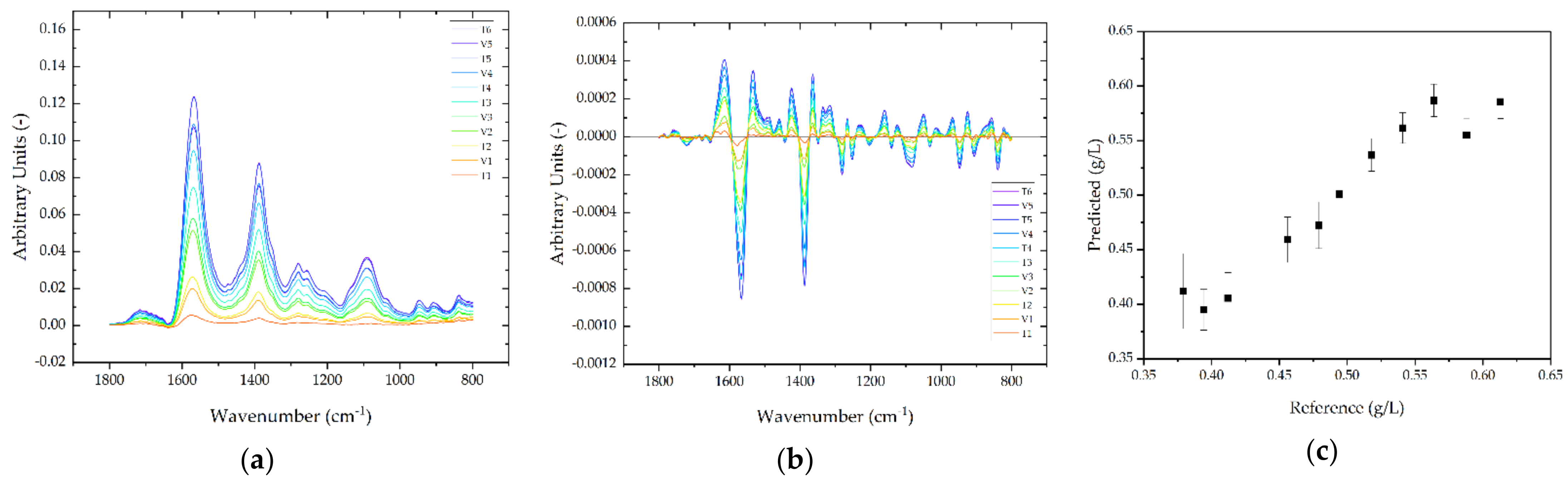
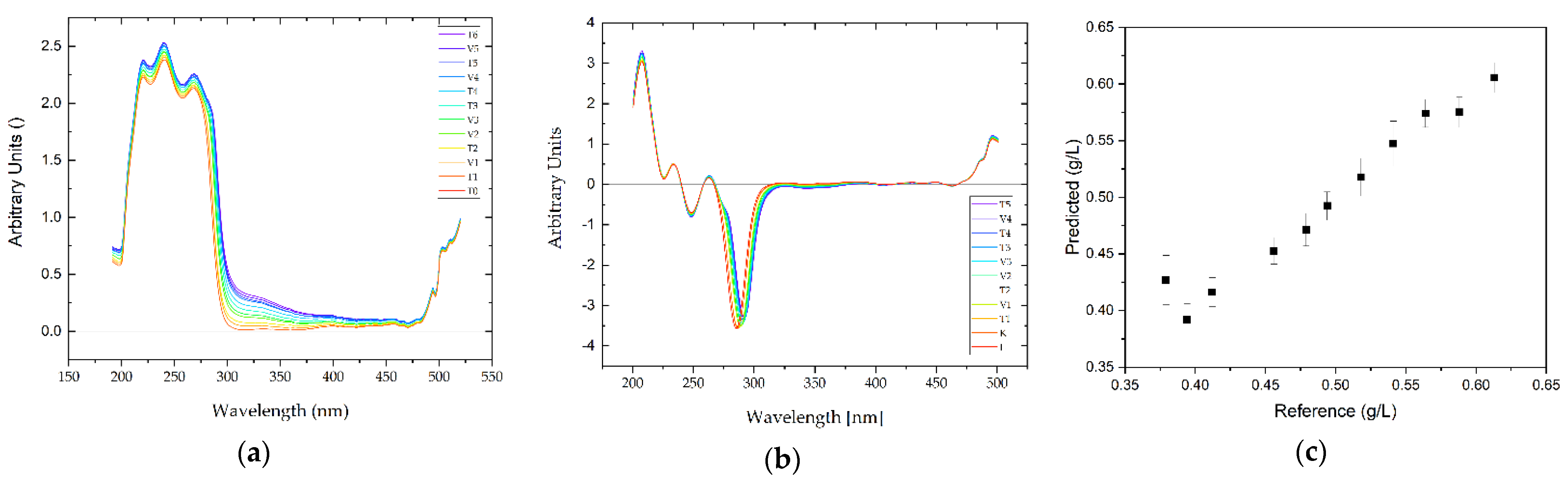
3.3. Feasibility of PAT for APC
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sheila Rocchio. COVID-19 Vacine Development at Full Tilt: Will the Rapod Development of COVID-19 Vaccines Lead to Unsafe Therapies?; The Medicine Make. 2020. Available online: https://themedicinemaker.com/manufacture/covid-19-vaccine-development-at-full-tilt (accessed on 2 March 2021).
- Hu, Y. Graphics: How Soon Can We Get a COVID-19 Vaccine? 2020. Available online: https://news.cgtn.com/news/2020-04-07/Graphics-How-soon-can-we-get-a-COVID-19-vaccine--PuPkfQbGxi/index.html (accessed on 2 March 2021).
- GSK. How We Develop New Vaccines. 2021. Available online: https://www.gsk.com/en-gb/research-and-development/development/how-we-develop-new-vaccines/ (accessed on 2 March 2021).
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef]
- Alia, C. COVID-19 Vaccines Explained in Maps and Charts. Available online: https://www.aljazeera.com/features/2020/11/24/covid-19-vaccines-explained-in-maps-and-charts (accessed on 2 March 2021).
- McCarthy, N. The Cost Per Jab Of Covid-19 Vaccine Candidates. 2020. Available online: https://0-www-statista-com.brum.beds.ac.uk/chart/23658/reported-cost-per-dose-of-covid-19-vaccines/ (accessed on 2 March 2021).
- McCarthy, N. How Effective Are The Covid-19 Vaccines? 2021. Available online: https://0-www-statista-com.brum.beds.ac.uk/chart/23510/estimated-effectiveness-of-covid-19-vaccine-candidates/ (accessed on 2 March 2021).
- Pietromarchi, V. WHO Against Mandatory Coronavirus Vaccines: Latest Updates. 2020. Available online: https://www.aljazeera.com/news/2020/12/7/millions-in-california-face-stay-at-home-order-live-news (accessed on 2 March 2021).
- Sloan, C.; Cappio, K.; Vidulich, A.; George, K.L.; Shaw, M.; Chepigin, C. Recent History Shows Variability in Vaccine Development Timeline. 2020. Available online: https://avalere.com/press-releases/recent-history-shows-variability-in-vaccine-development-timeline (accessed on 2 March 2021).
- Thompson, S.A. How Long Will a Vaccine Really Take? 2020. Available online: https://www.nytimes.com/interactive/2020/04/30/opinion/coronavirus-covid-vaccine.html (accessed on 2 March 2021).
- Plotkin, S.A.; Plotkin, S.L. The development of vaccines: How the past led to the future. Nat. Rev. Microbiol. 2011, 9, 889–893. [Google Scholar] [CrossRef]
- Sprenger, R.K. Magie des Konflikts: Warum ihn jeder braucht und wie er uns weiterbringt, 1. Auflage; Deutsche Verlags-Anstalt: München, Germany, 2020; ISBN 3421048541. [Google Scholar]
- Brors, P.; Hofmann, S. Dietmar Hopp will Mit Curevac “Rennen um Besten Impfstoff Gewinnen”. Available online: https://www.handelsblatt.com/unternehmen/management/der-risikoinvestor-dietmar-hopp-will-mit-curevac-rennen-um-besten-impfstoff-gewinnen/26154156.html?ticket=ST-13985126-bIPozoMDRzkCluynaUe7-ap3 (accessed on 11 March 2021).
- Bill & Melinda Gates Foundation. Bill & Melinda Gates Foundation, Wellcome, and Mastercard Launch Initiative to Speed Development and Access to Therapies for COVID-19: Press Release. Available online: https://www.gatesfoundation.org/Media-Center/Press-Releases/2020/03/COVID-19-Therapeutics-Accelerator (accessed on 11 March 2021).
- Müller, E.; Noe, M. Die Pläne der Biontech-Geldgeber. 2021. Available online: https://www.manager-magazin.de/unternehmen/pharma/biontech-thomas-struengmann-helmut-jeggle-und-michael-motschmann-ueber-ihre-plaene-a-00000000-0002-0001-0000-000174925829 (accessed on 11 March 2021).
- Pfizer Inc. Pfizer and BioNTech Announce Further Details on Collaboration to Accelerate Global COVID-19 Vaccine Development. 2020. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-and-biontech-announce-further-details-collaboration (accessed on 6 March 2021).
- GlaxoSmithKline plc. Sanofi and GSK to Join Forces in Unprecedented Vaccine Collaboration to Fight COVID-19. 2020. Available online: https://www.gsk.com/en-gb/media/press-releases/sanofi-and-gsk-to-join-forces-in-unprecedented-vaccine-collaboration-to-fight-covid-19/ (accessed on 6 March 2021).
- CureVac AG. CureVac and Bayer join forces on COVID-19 vaccine candidate CVnCoV: Joint Press Release. 2021. Available online: https://www.curevac.com/en/2021/01/07/curevac-and-bayer-join-forces-on-covid-19-vaccine-candidate-cvncov/ (accessed on 6 March 2021).
- Lonza Group Ltd. Moderna and Lonza Announce Worldwide Strategic Collaboration to Manufacture Moderna’s Vaccine (mRNA-1273) Against Novel Coronavirus. Available online: http://e3.marco.ch/publish/lonza/551_1203/200501_Press_Release__Moderna_Lonza_COVID_FINAL.pdf (accessed on 6 March 2021).
- Thermo Fisher Scientific. Challenges, Risks and Strategies for Biologic Substance Manufacturing. 2018. Available online: https://patheon.com/resource-library/whitepapers/challenges-risks-and-strategies-for-biologic-substance-manufacturing/ (accessed on 6 March 2021).
- Ward, J. Solutions to Today’s Biomanufacturing Challenges. 2018. Available online: https://patheon.com/resource-library/whitepapers/solutions-to-todays-biomanufacturing-challenges/ (accessed on 6 March 2021).
- ISR Reports. Changes in Manufacturing Pipeline Focus. 2020. Available online: https://www.bioprocessonline.com/doc/changes-in-manufacturing-pipeline-focus-0002 (accessed on 6 March 2021).
- Unger, B. Top 10 Most-Cited MHRA GMP Inspection Deficiencies by Annex/Chapter in 2019. 2021. Available online: https://www.pharmaceuticalonline.com/doc/top-most-cited-mhra-gmp-inspection-deficiencies-by-annex-chapter-in-0001 (accessed on 6 March 2021).
- Unger, B. An Analysis of MHRA’s Annual GMP Inspection Deficiencies Report. 2020. Available online: https://www.pharmaceuticalonline.com/doc/an-analysis-of-mhra-s-annual-gmp-inspection-deficiencies-report-0001 (accessed on 6 March 2021).
- Unger, B. An Analysis of 2018 FDA Warning Letters Citing Data Integrity Failures. 2019. Available online: https://www.pharmaceuticalonline.com/doc/an-analysis-of-fda-warning-letters-citing-data-integrity-failures-0001 (accessed on 6 March 2021).
- Wouters, O.J.; Shadlen, K.C.; Salcher-Konrad, M.; Pollard, A.J.; Larson, H.J.; Teerawattananon, Y.; Jit, M. Challenges in ensuring global access to COVID-19 vaccines: Production, affordability, allocation, and deployment. Lancet 2021, 397, 1023–1034. [Google Scholar] [CrossRef]
- Sommerfeld, S.; Strube, J. Challenges in biotechnology production—generic processes and process optimization for monoclonal antibodies. Chem. Eng. Process. Process Intensif. 2005, 44, 1123–1137. [Google Scholar] [CrossRef]
- Strube, J.; Sommerfeld, S.; Lohrmann, M. Process Development and Optimization for Biotechnology Production—Monoclonal Antibodies. In Bioseparation and Bioprocessing, 2nd ed.; Subramanian, G., Ed.; Wiley-VCH: Weinheim, Germany; New York, NY, USA, 2007. [Google Scholar]
- Verband Forschender Arzneimittelhersteller e.V. Impfstoffe zum Schutz vor der Coronavirus-Infektion Covid-19. 2021. Available online: https://www.vfa.de/de/arzneimittel-forschung/woran-wir-forschen/impfstoffe-zum-schutz-vor-coronavirus-2019-ncov (accessed on 2 March 2021).
- Melinek, B.; Colant, N.; Stamatis, C.; Lennon, C.; Farid, S.S.; Polizzi, K.; Carver, M.; Bracewell, D.G. Toward a Roadmap for Cell-Free Synthesis in Bioprocessing. Available online: https://bioprocessintl.com/upstream-processing/expression-platforms/toward-a-roadmap-for-cell-free-synthesis-in-bioprocessing/ (accessed on 2 March 2021).
- Ipsen Biopharm Ltd. Novel Production Process for a Highly Potent Recombinant Protein Using Doggybone DNA (dbDNA) Vector and Cell Free Expression Technology. 2020. Available online: https://gtr.ukri.org/projects?ref=104201 (accessed on 3 June 2020).
- GenScript ProBio. Accelerating Vaccine development against COVID-19. 2020. Available online: https://www.genscriptprobio.com/gsfiles/techfiles/GPB-COVID19-GMP%20plasmid-GPB_052020.pdf (accessed on 6 March 2021).
- Shin, D.; Azizian, K.T.; Henderson, J.M.; Hogrefe, R.I.; Lebedev, A.; Houston, M.; McCaffrey, A.P. CleanCap® Co-transcriptional Capping Streamlines mRNA Manufacturing. Available online: https://www.trilinkbiotech.com/media/contentmanager/content/mRNA_WVC2_1.pdf (accessed on 6 March 2021).
- Geipel-Kern, A. Covid-19-Impfstoff—warum bei den Rohstoff-Herstellern die Kasse klingelt. 2021. Available online: https://www.process.vogel.de/covid-19-impfstoff-warum-bei-den-rohstoff-herstellern-die-kasse-klingelt-a-1004361/ (accessed on 6 March 2021).
- Kis, Z.; Kontoravdi, C.; Shattock, R.; Shah, N. Resources, Production Scales and Time Required for Producing RNA Vaccines for the Global Pandemic Demand. Vaccines 2020, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Von der Mülbe, F.; Reidel, L.; Ketterer, T.; Gontcharova, L.; Bauer, S.; Pascolo, S.; Probst, J.; Schmid, A. Method for Producing RNA. U.S. Patent No. 10,017,826, 10 July 2018. [Google Scholar]
- Stark, A. Novartis startet Produktion von Curevac-Impfstoff im zweiten Quartal. 2021. Available online: https://www.process.vogel.de/novartis-startet-produktion-von-curevac-impfstoff-im-zweiten-quartal-a-1005737/?cmp=nl-98&uuid=B2EB5C9F-EC8E-64F3-11C6F1C278C9BE12 (accessed on 11 March 2021).
- Nemec, K.S.; Cernigoj, U.; Vidic, J.; Livk, A.G.; Goricar, B.; Bozic, K.; Celjar, A.M.; Skok, J.; Mencin, N.; Kralj, S.; et al. High yield mRNA production process from E. coli to highly pure mRNA. 2020. Available online: https://www.biaseparations.com/en/library/seminars-webinars/1098/high-yield-mrna-production-process-from-ecoli-to-highly-pure-mrna (accessed on 2 March 2021).
- Weise, E.; Weintraub, K. A COVID-19 Vaccine Life Cycle: From DNA to Doses. 2021. Available online: https://eu.usatoday.com/in-depth/news/health/2021/02/07/how-covid-vaccine-made-step-step-journey-pfizer-dose/4371693001/ (accessed on 6 March 2021).
- BioNTech SE. BioNTech to Acquire GMP Manufacturing Site to Expand COVID-19 Vaccine Production Capacity in First Half 2021. 2020. Available online: https://investors.biontech.de/news-releases/news-release-details/biontech-acquire-gmp-manufacturing-site-expand-covid-19-vaccine (accessed on 6 March 2021).
- Zobel-Roos, S.; Schmidt, A.; Mestmäcker, F.; Mouellef, M.; Huter, M.; Uhlenbrock, L.; Kornecki, M.; Lohmann, L.; Ditz, R.; Strube, J. Accelerating Biologics Manufacturing by Modeling or: Is Approval under the QbD and PAT Approaches Demanded by Authorities Acceptable Without a Digital-Twin? Processes 2019, 7, 94. [Google Scholar] [CrossRef] [Green Version]
- Kornecki, M.; Strube, J. Accelerating Biologics Manufacturing by Upstream Process Modelling. Processes 2019, 7, 166. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.; Strube, J. Distinct and Quantitative Validation Method for Predictive Process Modeling with Examples of Liquid-Liquid Extraction Processes of Complex Feed Mixtures. Processes 2019, 7, 298. [Google Scholar] [CrossRef] [Green Version]
- Zobel-Roos, S.; Mouellef, M.; Ditz, R.; Strube, J. Distinct and Quantitative Validation Method for Predictive Process Modelling in Preparative Chromatography of Synthetic and Bio-Based Feed Mixtures Following a Quality-by-Design (QbD) Approach. Processes 2019, 7, 580. [Google Scholar] [CrossRef] [Green Version]
- Huter, M.J.; Strube, J. Model-Based Design and Process Optimization of Continuous Single Pass Tangential Flow Filtration Focusing on Continuous Bioprocessing. Processes 2019, 7, 317. [Google Scholar] [CrossRef] [Green Version]
- Lohmann, L.J.; Strube, J. Accelerating Biologics Manufacturing by Modeling: Process Integration of Precipitation in mAb Downstream Processing. Processes 2020, 8, 58. [Google Scholar] [CrossRef] [Green Version]
- Klepzig, L.S.; Juckers, A.; Knerr, P.; Harms, F.; Strube, J. Digital Twin for Lyophilization by Process Modeling in Manufacturing of Biologics. Processes 2020, 8, 1325. [Google Scholar] [CrossRef]
- Korz, D.J.; Rinas, U.; Hellmuth, K.; Sanders, E.A.; Deckwer, W.-D. Simple fed-batch technique for high cell density cultivation of Escherichia coli. J. Biotechnol. 1995, 39, 59–65. [Google Scholar] [CrossRef]
- Urthaler, J.; Buchinger, W.; Necina, R. Improved downstream process for the production of plasmid DNA for gene therapy. Acta Biochim. Pol. 2005, 52, 703–711. [Google Scholar] [CrossRef]
- Urthaler, J.; Ascher, C.; Wöhrer, H.; Necina, R. Automated alkaline lysis for industrial scale cGMP production of pharmaceutical grade plasmid-DNA. J. Biotechnol. 2007, 128, 132–149. [Google Scholar] [CrossRef]
- Urthaler, J.; Buchinger, W.; Necina, R. Industrial Scale cGMP Purification of Pharmaceutical Grade Plasmid-DNA. Chem. Eng. Technol. 2005, 28, 1408–1420. [Google Scholar] [CrossRef]
- Grote, F.; Fröhlich, H.; Strube, J. Integration of Ultrafiltration Unit Operations in Biotechnology Process Design. Chem. Eng. Technol. 2011, 34, 673–687. [Google Scholar] [CrossRef]
- Gritti, F.; Piatkowski, W.; Guiochon, G. Study of the mass transfer kinetics in a monolithic column. J. Chromatogr. A 2003, 983, 51–71. [Google Scholar] [CrossRef]
- Hahn, R.; Panzer, M.; Hansen, E.; Mollerup, J.; Jungbauer, A. Mass transfer properties of monoliths. Sep. Sci. Technol. 2007, 37, 1545–1565. [Google Scholar] [CrossRef]
- Boi, C.; Dimartino, S.; Sarti, G.C. Modelling and simulation of affinity membrane adsorption. J. Chromatogr. A 2007, 1162, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, H.; Villian, L.; Melzner, D.; Strube, J. Membrane Technology in Bioprocess Science. Chem. Ing. Tech. 2012. [Google Scholar] [CrossRef]
- Schwellenbach, J.; Kosiol, P.; Sölter, B.; Taft, F.; Villain, L.; Strube, J. Controlling the polymer-nanolayer architecture on anion-exchange membrane adsorbers via surface-initiated atom transfer radical polymerization. React. Funct. Polym. 2016, 106, 32–42. [Google Scholar] [CrossRef]
- Zobel-Roos, S.; Stein, D.; Strube, J. Evaluation of Continuous Membrane Chromatography Concepts with an Enhanced Process Simulation Approach. Antibodies 2018, 7, 13. [Google Scholar] [CrossRef] [Green Version]
- Guiochon, G. Fundamentals of Preparative and Nonlinear Chromatography, 2nd ed.; Elsevier Acad. Press: Amsterdam, The Netherlands, 2006; ISBN 9780123705372. [Google Scholar]
- Zobel-Roos, S. Entwicklung, Modellierung und Validierung von integrierten kontinuierlichen Gegenstrom-Chromatographie-Prozessen. Ph. D. Thesis, Shaker Verlag GmbH, Technische Universität, Clausthal-Zellerfeld, Germany, 2018. [Google Scholar]
- Brooks, C.A.; Cramer, S.M. Steric mass-action ion exchange: Displacement profiles and induced salt gradients. AIChE J. 1992, 38, 1969–1978. [Google Scholar] [CrossRef]
- Carta, G.; Jungbauer, A. Protein Chromatography: Process Development and Scale-Up; Wiley-VCH: Weinheim, Germany, 2010; ISBN 978-3-527-31819-3. [Google Scholar]
- Seidel-Morgenstern, A.; Guiochon, G. Modelling of the competitive isotherms and the chromatographic separation of two enantiomers. Chem. Eng. Sci. 1993, 48, 2787–2797. [Google Scholar] [CrossRef]
- Langmuir, I. The adsorption of gases on plane surfaces of glass, mica and platinum. J. Am. Chem. Soc. 1918, 40, 1361–1403. [Google Scholar] [CrossRef] [Green Version]
- Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000; ISBN 3527306730.
- Wiesel, A.; Schmidt-Traub, H.; Lenz, J.; Strube, J. Modelling gradient elution of bioactive multicomponent systems in non-linear ion-exchange chromatography. J. Chromatogr. A 2003, 1006, 101–120. [Google Scholar] [CrossRef]
- Leśko, M.; Åsberg, D.; Enmark, M.; Samuelsson, J.; Fornstedt, T.; Kaczmarski, K. Choice of Model for Estimation of Adsorption Isotherm Parameters in Gradient Elution Preparative Liquid Chromatography. Chromatographia 2015, 78, 1293–1297. [Google Scholar] [CrossRef] [Green Version]
- Zobel, S.; Helling, C.; Ditz, R.; Strube, J. Design and operation of continuous countercurrent chromatography in biotechnological production. Ind. Eng. Chem. Res. 2014, 53, 9169–9185. [Google Scholar] [CrossRef]
- Seidel-Morgenstern, A. Experimental determination of single solute and competitive adsorption isotherms. J. Chromatogr. A 2004, 1037, 255–272. [Google Scholar] [CrossRef] [PubMed]
- BIA Separations. Purification of mRNA with CIMmultus PrimaS™: Technical Note. 2020. Available online: https://www.biaseparations.com/en/library/technical-notes/1088/purification-of-mrna-with-cimmultus-primastm (accessed on 2 March 2021).
- Tarmann, C.; Jungbauer, A. Adsorption of plasmid DNA on anion exchange chromatography media. J. Sep. Sci. 2008, 31, 2605–2618. [Google Scholar] [CrossRef]
- Wright, D.J.; Jack, W.E.; Modrich, P. The kinetic mechanism of EcoRI endonuclease. J. Biol. Chem. 1999, 274, 31896–31902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urthaler, J.; Schuchnigg, H.; Garidel, P.; Huber, H. Industrial Manufacturing of Plasmid-DNA Products for Gene Vaccination and Therapy. In Gene Vaccines; Thalhamer, J., Weiss, R., Scheiblhofer, S., Eds.; Springer Vienna: Vienna, Austria, 2012; pp. 311–330. ISBN 978-3-7091-0438-5. [Google Scholar]
- Latulippe, D.R.; Ager, K.; Zydney, A.L. Flux-dependent transmission of supercoiled plasmid DNA through ultrafiltration membranes. J. Membr. Sci. 2007, 294, 169–177. [Google Scholar] [CrossRef]
- Latulippe, D.R.; Zydney, A.L. Salt-induced changes in plasmid DNA transmission through ultrafiltration membranes. Biotechnol. Bioeng. 2008, 99, 390–398. [Google Scholar] [CrossRef]
- Latulippe, D.R.; Zydney, A.L. Separation of plasmid DNA isoforms by highly converging flow through small membrane pores. J. Colloid Interface Sci. 2011, 357, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J. Allowing the Process to Determine Filtration Technology: Hollow Fiber vs. Flat Sheet. 2020. Available online: https://vimeo.com/478502171 (accessed on 12 March 2021).
- Madigan, J.; Dobbs, C.; Henderson, J.; Lebedev, A.; McCaffrey, A.P.; Powers, J. Considerations for the Development, Scale-up and Manufacturing of mRNA Therapeutics. 2021. Available online: https://www.trilinkbiotech.com/media/contentmanager/content/mRNA_OTS1.pdf (accessed on 2 March 2021).
- Zhu, T.; Moussa, E.M.; Witting, M.; Zhou, D.; Sinha, K.; Hirth, M.; Gastens, M.; Shang, S.; Nere, N.; Somashekar, S.C.; et al. Predictive models of lyophilization process for development, scale-up/tech transfer and manufacturing. Eur. J. Pharm. Biopharm. 2018, 128, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Rambhatla, S.; Pikal, M.J. Heat and mass transfer scale-up issues during freeze-drying, I: Atypical radiation and the edge vial effect. AAPS PharmSciTech 2003, 4, E14. [Google Scholar] [CrossRef] [Green Version]
- Zobel-Roos, S.; Mouellef, M.; Siemers, C.; Strube, J. Process Analytical Approach towards Quality Controlled Process Automation for the Downstream of Protein Mixtures by Inline Concentration Measurements Based on Ultraviolet/Visible Light (UV/VIS) Spectral Analysis. Antibodies 2017, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Kornecki, M.; Schmidt, A.; Strube, J. PAT as key-enabling technology for QbD in pharmaceutical manufacturing A conceptual review on upstream and downstream processing. Chim. Oggi Chem. Today 2018, 36, 44–48. [Google Scholar]
- Helgers, H.; Schmidt, A.; Lohmann, L.J.; Vetter, F.L.; Juckers, A.; Jensch, C.; Mouellef, M.; Zobel-Roos, S.; Strube, J. Towards Autonomous Operation by Advanced Process Control—Process Analytical Technology for Continuous Biologics Antibody Manufacturing. Processes 2021, 9, 172. [Google Scholar] [CrossRef]
- Vetter, F.L.; Zobel-Roos, S.; Strube, J. PAT for Continuous Chromatography Integrated into Continuous Manufacturing of Biologics towards Autonomous Operation. Processes 2021, 9, 472. [Google Scholar] [CrossRef]
- Lohmann, L.J.; Strube, J. Process Analytical Technology for Precipitation Process Integration into Biologics Manufacturing towards Autonomous Operation—mAb Case Study. Processes 2021, 9, 488. [Google Scholar] [CrossRef]
- C Technologies, Inc. SoloVPE. 2021. Available online: https://www.ctechnologiesinc.com/products/solovpe (accessed on 11 March 2021).
- Wikimedia Foundation, I. QS-21. 2021. Available online: https://en.wikipedia.org/wiki/QS-21 (accessed on 18 March 2021).
- Wikimedia Foundation, I. Quillaia. 2021. Available online: https://en.wikipedia.org/wiki/Quillaia (accessed on 18 March 2021).
- Fleck, J.D.; Betti, A.H.; Da Silva, F.P.; Troian, E.A.; Olivaro, C.; Ferreira, F.; Verza, S.G. Saponins from Quillaja saponaria and Quillaja brasiliensis: Particular Chemical Characteristics and Biological Activities. Molecules 2019, 24, 171. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Palanisamy, A.; Dhama, K.; Mal, G.; Singh, B.; Singh, K.P. Exploring the possible use of saponin adjuvants in COVID-19 vaccine. Hum. Vaccin. Immunother. 2020, 16, 2944–2953. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Uhlenbrock, L.; Strube, J. Technical Potential for Energy and GWP Reduction in Chemical–Pharmaceutical Industry in Germany and EU—Focused on Biologics and Botanicals Manufacturing. Processes 2020, 8, 818. [Google Scholar] [CrossRef]
- Uhlenbrock, L.; Sixt, M.; Tegtmeier, M.; Schulz, H.; Hagels, H.; Ditz, R.; Strube, J. Natural Products Extraction of the Future—Sustainable Manufacturing Solutions for Societal Needs. Processes 2018, 6, 177. [Google Scholar] [CrossRef] [Green Version]
- Lenz, C.; Reichen, P. Ganz Europa Schaut Nach Visp—und Jetzt Ruckelts bei Lonza. Available online: https://www.bazonline.ch/im-schweizer-impf-eldorado-knirscht-es-im-system-772070807325 (accessed on 18 March 2021).
- Jiang, M.; Severson, K.A.; Love, J.C.; Madden, H.; Swann, P.; Zang, L.; Braatz, R.D. Opportunities and challenges of real-time release testing in biopharmaceutical manufacturing. Biotechnol. Bioeng. 2017, 114, 2445–2456. [Google Scholar] [CrossRef] [PubMed]

























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, A.; Helgers, H.; Vetter, F.L.; Juckers, A.; Strube, J. Digital Twin of mRNA-Based SARS-COVID-19 Vaccine Manufacturing towards Autonomous Operation for Improvements in Speed, Scale, Robustness, Flexibility and Real-Time Release Testing. Processes 2021, 9, 748. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9050748
Schmidt A, Helgers H, Vetter FL, Juckers A, Strube J. Digital Twin of mRNA-Based SARS-COVID-19 Vaccine Manufacturing towards Autonomous Operation for Improvements in Speed, Scale, Robustness, Flexibility and Real-Time Release Testing. Processes. 2021; 9(5):748. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9050748
Chicago/Turabian StyleSchmidt, Axel, Heribert Helgers, Florian Lukas Vetter, Alex Juckers, and Jochen Strube. 2021. "Digital Twin of mRNA-Based SARS-COVID-19 Vaccine Manufacturing towards Autonomous Operation for Improvements in Speed, Scale, Robustness, Flexibility and Real-Time Release Testing" Processes 9, no. 5: 748. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9050748