Type I Neurofibromatosis: Case Report and Review of the Literature Focused on Oral and Cutaneous Lesions


Abstract
:1. Introduction
2. Background
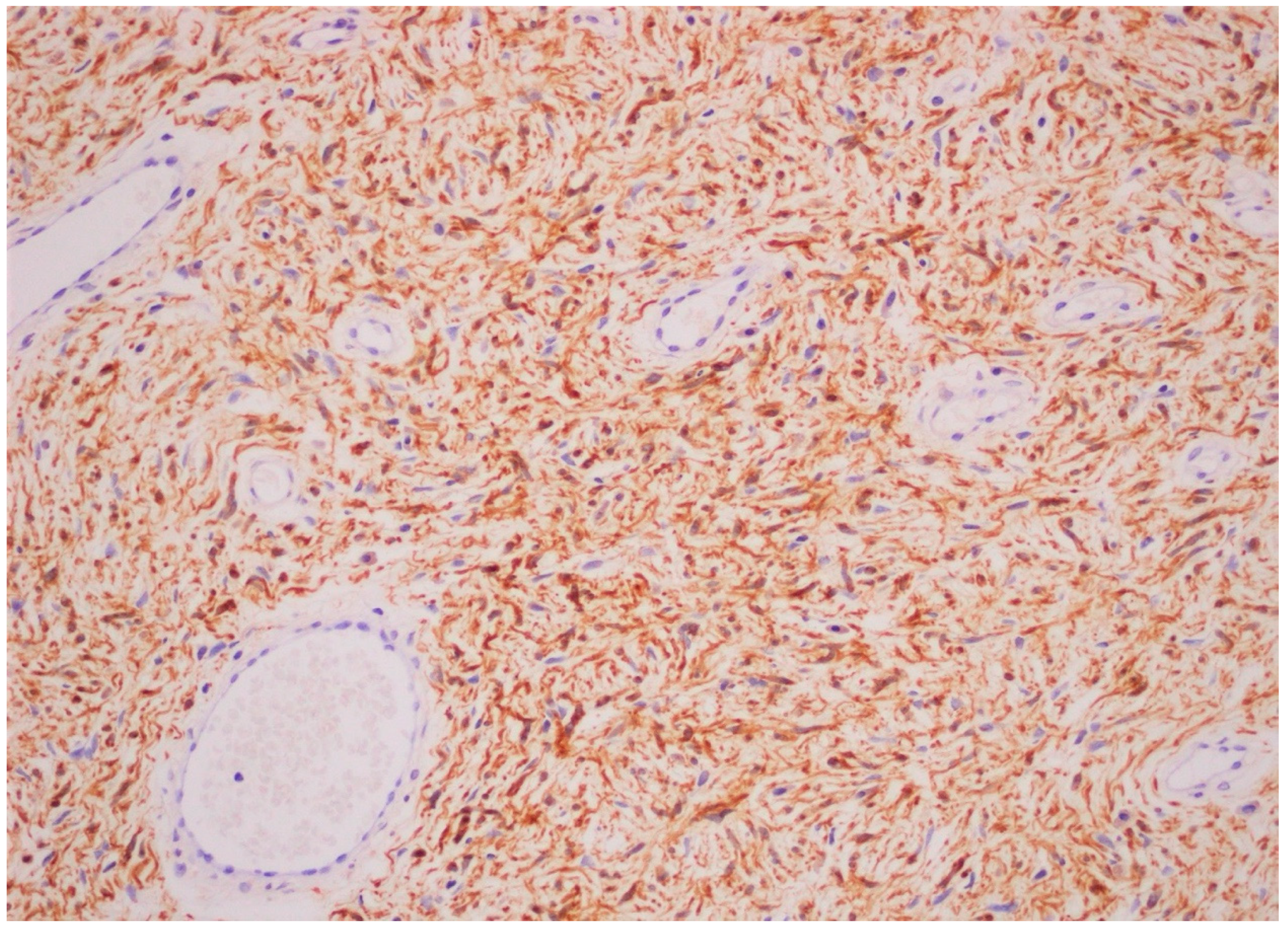
3. Case Report
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bachelet, J.T.; Combemale, P.; Devic, C.; Foray, N.; Jouanneau, E.; Breton, P. Prise en charge des atteintes craniofaciales de la neurofibromatose de type 1 [Management of craniofacial type 1 neurofibromatosis]. Rev. Stomatol. Chir. Maxillofac. Chir. Orale 2015, 116, 209–214. [Google Scholar] [PubMed]
- Shetty, B.; Umesh, Y.; Kranti, K.; Seshan, H. Periodontal manifestations of von Recklinghausen neuro fibromatosis. J. Indian Soc. Periodontol. 2013, 17, 253–256. [Google Scholar] [CrossRef]
- Visnapuu, V.; Peltonen, S.; Alivuotila, L.; Happonen, R.-P.; Peltonen, J. Craniofacial and oral alterations in patients with Neurofibromatosis 1. Orphanet J. Rare Dis. 2018, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Javed, F.; Ramalingam, S.; Ahmed, H.B.; Gupta, B.; Sundar, C.; Qadri, T.; Al-Hezaimi, K.; Romanos, G.E. Oral manifestations in patients with neurofibromatosis type-1: A comprehensive literature review. Crit. Rev. Oncol. 2014, 91, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Bongiorno, M.; Pistone, G.; Arico, M. Manifestations of the tongue in Neurofibromatosis type 1. Oral Dis. 2006, 12, 125–129. [Google Scholar] [CrossRef]
- Santoro, R.; Santoro, C.; Loffredo, F.; Romano, A.; Perrotta, S.; Serpico, R.; Lauritano, D.; Lucchese, A. Oral Clinical Manifestations of Neurofibromatosis Type 1 in Children and Adolescents. Appl. Sci. 2020, 10, 4687. [Google Scholar] [CrossRef]
- Visnapuu, V.; Peltonen, S.; Tammisalo, T.; Peltonen, J.; Happonen, R.-P. Radiographic Findings in the Jaws of Patients with Neurofibromatosis 1. J. Oral Maxillofac. Surg. 2012, 70, 1351–1357. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, F.; Masurel, A.; Olivier-Faivre, L.; Vabres, P. Juvenile Xanthogranuloma and Nevus Anemicus in the Diagnosis of Neurofibromatosis Type 1. JAMA Dermatol. 2014, 150, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Cunha, K.S.; Barboza, E.P.; Dias, E.P.; Oliveira, F.M. Neurofibromatosis type I with periodontal manifestation. A case report and literature review. Br. Dent. J. 2004, 196, 457–460. [Google Scholar] [CrossRef] [Green Version]
- Bergqvist, C.; Network, N.F.; Servy, A.; Valeyrie-Allanore, L.; Ferkal, S.; Combemale, P.; Wolkenstein, P. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J. Rare Dis. 2020, 15, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Jouhilahti, E.-M.; Visnapuu, V.; Soukka, T.; Aho, H.; Peltonen, S.; Happonen, R.-P.; Peltonen, J. Oral soft tissue alterations in patients with neurofibromatosis. Clin. Oral Investig. 2011, 16, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Christen-Zaech, S.; Vernez, M. Syndromes néoplasiques héréditaires avec atteinte cutanée. Rev. Med. Suisse. 2008, 4, 1095–1102. [Google Scholar] [PubMed]
- García de Marcos, J.A.; Dean Ferrer, A.; Alamillos Granados, F.; Ruiz Masera, J.J.; García de Marcos, M.J.; Vidal Jiménez, A.; Valenzuela Salas, B.; García Lainez, A. Gingival neurofibroma in a neurofibromatosis type 1 patient. Med. Oral Patol. Oral Cir. Bucal. 2007, 12, E287–E291. [Google Scholar] [PubMed]
- Thompson, L.D.R.; Koh, S.S.; Lau, S.K. Sporadic Neurofibroma of the Tongue Unassociated with Neurofibromatosis Type I: A Clinicopathologic Study of Ten Cases. Head Neck Pathol. 2020, 14, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Bouimetarhan, L.; Bellamlih, H.; En-Nafaa, I.; Fenni, J.E.; Amil, T.; Radouane, B. Neurofibfrome plexiforme cervical: À propos d’un cas [Plexiform cervical neurofibroma: About a case]. Pan Afr. Med. J. 2018, 30, 41. [Google Scholar] [CrossRef]
- Prud’Homme, T.; Dajean-Trutaud, S.; Badran, Z.; Hyon, I.; Wotjiuk, F. Dental Management of Neurofibromatosis Type 1: A Case Report and Literature Review. Int. J. Clin. Pediatr. Dent. 2019, 12, 577–581. [Google Scholar] [CrossRef]
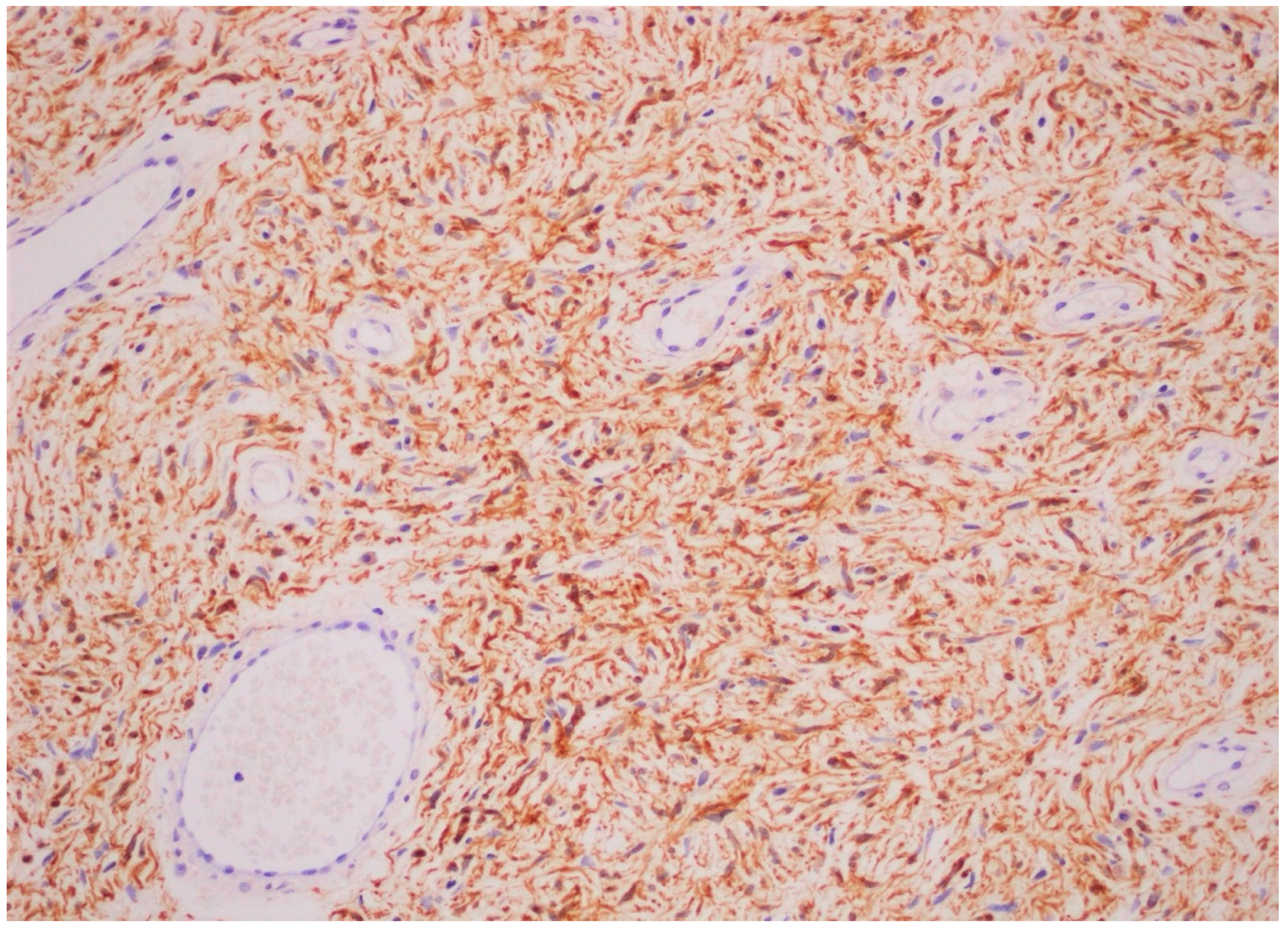
- Campos, M.S.; Fontes, A.; Marocchio, L.S.; Nunes, F.D.; De Sousa, S.C.O.M. Clinicopathologic and immunohistochemical features of oral neurofibroma. Acta Odontol. Scand. 2011, 70, 577–582. [Google Scholar] [CrossRef]
- Amaral, F.R.; Ferreira, M.V.L.; Costa, L.A.P.; De Oliveira, P.A.D.; Soares, B.M.; Souza, P.E.A.; De Sousa, G.R. Use of Surgical Laser for Excision of a Neurofibroma Associated With Neurofibromatosis Type-1. J. Lasers Med. Sci. 2018, 9, 219–222. [Google Scholar] [CrossRef]
- Kubota, S.; Imai, T.; Iwai, S.; Nakazawa, M.; Uzawa, N. Gingival Neurofibroma with Teardrop-Shaped Defects of the Interdental Alveolar Bone: An Unusual Oral Manifestation of Neurofibromatosis Type 1. J. Craniofac. Surg. 2019, 30, e205–e207. [Google Scholar] [CrossRef]
- Stumpf, D.; Alksne, J.; Annegers, J.; Brown, S.S.; Conneally, P.M.; Housman, D.; Leppert, M.F.; Miller, J.P.; Moss, M.L.; Pileggi, A.J.; et al. Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch. Neurol. 1988, 45, 575–578. [Google Scholar]
- Shofty, B.; Constantini, S.; Ben-Shachar, S. Advances in Molecular Diagnosis of Neurofibromatosis Type 1. Semin. Pediatr. Neurol. 2015, 22, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Uusitalo, E.; Rantanen, M.; Kallionpää, R.A.; Pöyhönen, M.; Leppävirta, J.; Ylä-Outinen, H.; Riccardi, V.M.; Pukkala, E.; Pitkäniemi, J.; Peltonen, S.; et al. Distinctive Cancer Associations in Patients with Neurofibromatosis Type 1. J. Clin. Oncol. 2016, 34, 1978–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janardhanan, M.; Rakesh, S.; Kumar, R.V. Intraoral presentation of multiple malignant peripheral nerve sheath tumors associated with neurofibromatosis-1. J. Oral Maxillofac. Pathol. 2011, 15, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Pramanick, D.; Ghose, S.; Mazumdar, A. First case report of tongue squamous cell carcinoma in a neurofibromatosis type 1 patient and review of pathogenesis of carcinoma in neurofibromatosis type 1. Indian J. Pathol. Microbiol. 2020, 63, 112–115. [Google Scholar] [CrossRef]
- Gross, A.M.; Wolters, P.L.; Dombi, E.; Baldwin, A.; Whitcomb, P.; Fisher, M.J.; Weiss, B.; Kim, A.; Bornhorst, M.; Shah, A.C.; et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N. Engl. J. Med. 2020, 382, 1430–1442. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
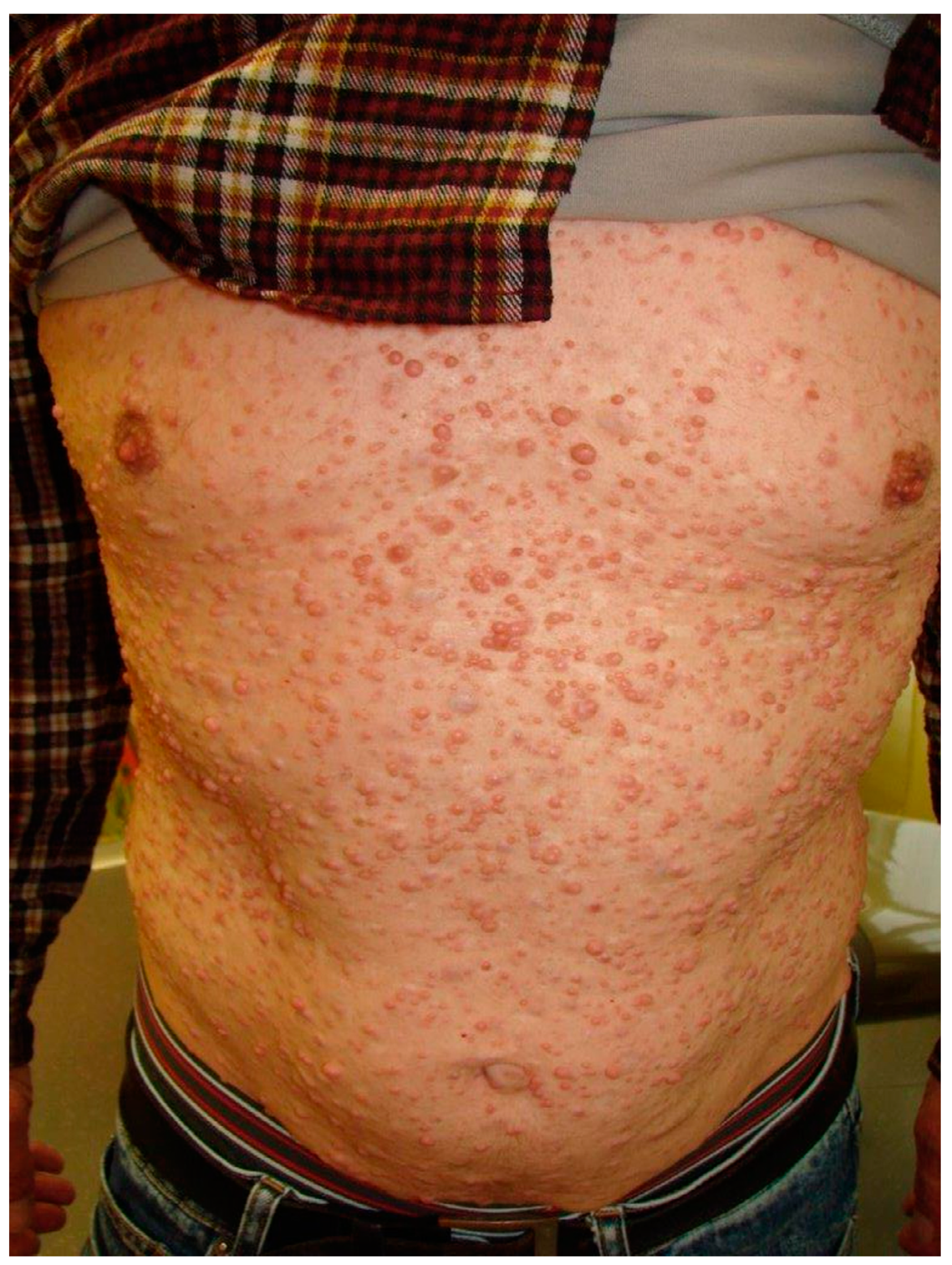
| Cutaneous | Café au lait spots *, axillary or inguinal freckling * (Crowe’s sign), cutaneous neurofibromas * (localized or plexiform). |
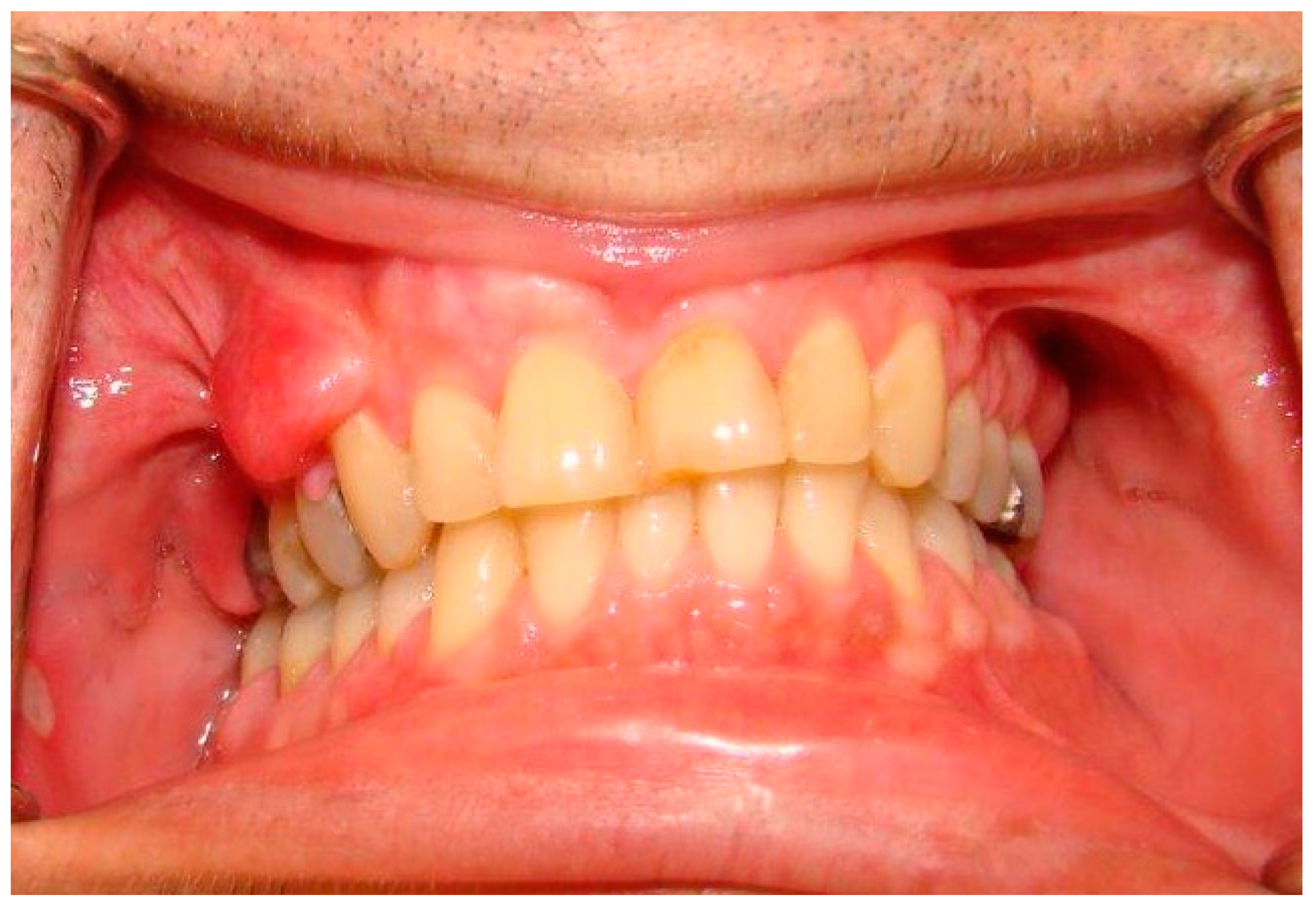
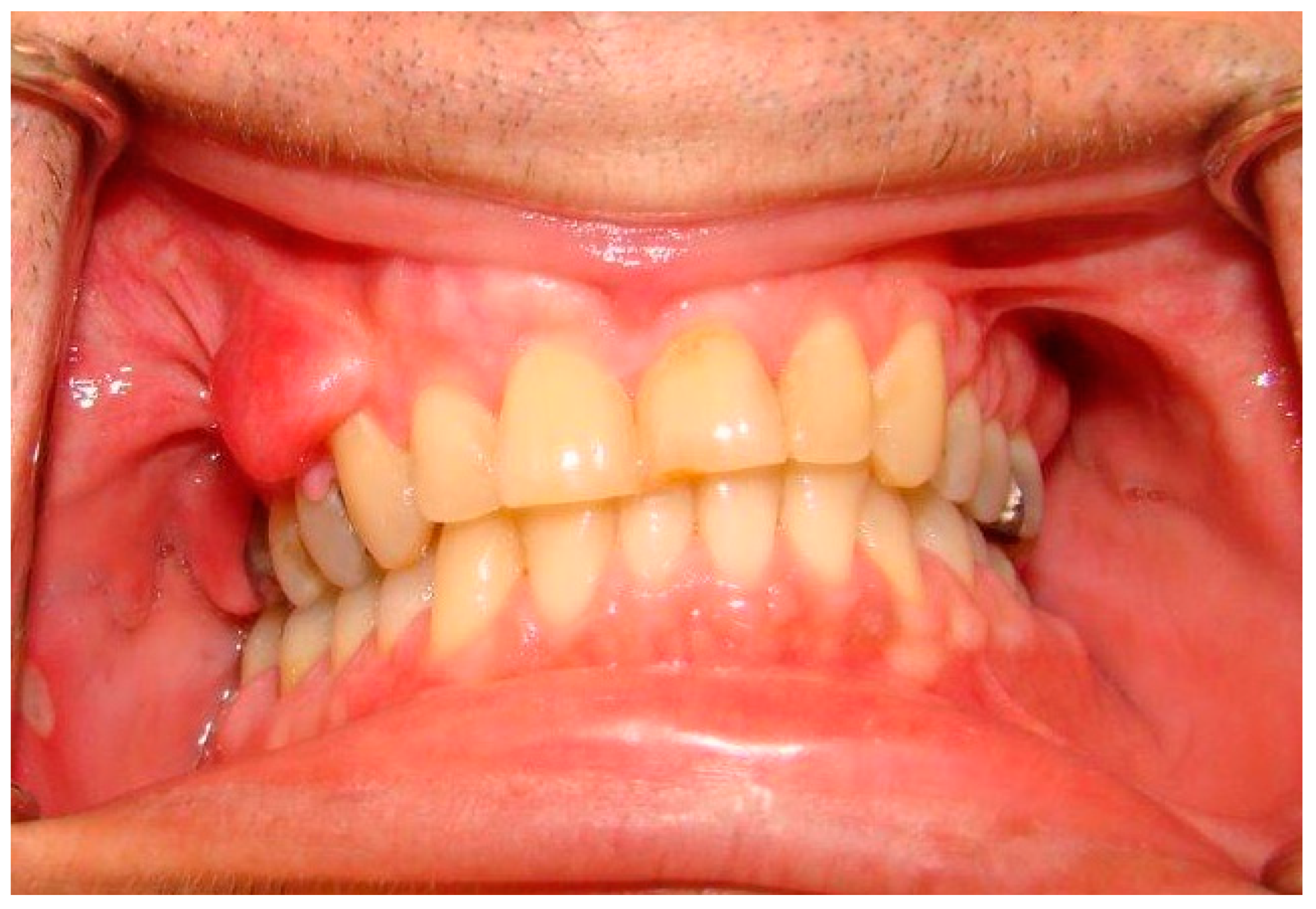
| Oral soft tissue | Prominent lingual papillae (50% cases); Mucosal and gingival neurofibromas * (25% of cases): mostly the tongue, followed by buccal mucosa, lips, and gingiva, and less commonly the palate, the floor of the mouth, the major salivary glands and the pharynx; Macroglossia in relation to plexiform neurofibromas arising inside the tongue; Melanin pigmentation of the gingiva (rare); Gingivitis or periodontitis in relation to oral neurofibromas prohibiting a proper oral hygiene. |
| Cranio-facial | Orbital dysplasia (may lead to exophthalmia), sphenoidal wings dysplasia *; Widening of the mandibular canal without relation with any tumor mass +/− irregular border of the canal and enlarged mandibular foramina; Short mandibular body, ramus, and condyle, undergrowth maxilla with hypoplasia of the maxillary tuberosity and short cranial base (inducing retrognathia); Intra-osseus neurofibromas of the maxilla/mandible and the temporo-mandibular joint (well-defined unilocular and occasionally multilocular radiolucent lesions); Notching of the posterior border of the mandibular ramus, elongated coronoid process with a deep sigmoid notch, hypoplasia of the condyle and zygomatic processes; Periapical cement dysplasia (only NF1 females affected), central giant cell granuloma, and osteolytic bone lesions linked to cherubism. |
| Dental | Retained or displaced teeth, agenesia, or hyperdontia, impaired growth of alveolar bone in association with gingival or bone neurofibromas and especially plexiform neurofibromas arising from the trigeminal nerve; Enamel defects such as molar-incisor hypomineralization, enamel hypoplasia, or opacities; Predisposition to caries is controversial. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buchholzer, S.; Verdeja, R.; Lombardi, T. Type I Neurofibromatosis: Case Report and Review of the Literature Focused on Oral and Cutaneous Lesions. Dermatopathology 2021, 8, 17-24. https://0-doi-org.brum.beds.ac.uk/10.3390/dermatopathology8010003
Buchholzer S, Verdeja R, Lombardi T. Type I Neurofibromatosis: Case Report and Review of the Literature Focused on Oral and Cutaneous Lesions. Dermatopathology. 2021; 8(1):17-24. https://0-doi-org.brum.beds.ac.uk/10.3390/dermatopathology8010003
Chicago/Turabian StyleBuchholzer, Samanta, Raùl Verdeja, and Tommaso Lombardi. 2021. "Type I Neurofibromatosis: Case Report and Review of the Literature Focused on Oral and Cutaneous Lesions" Dermatopathology 8, no. 1: 17-24. https://0-doi-org.brum.beds.ac.uk/10.3390/dermatopathology8010003