
TP53 Abnormalities and MMR Preservation in 5 Cases of Proliferating Trichilemmal Tumours

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
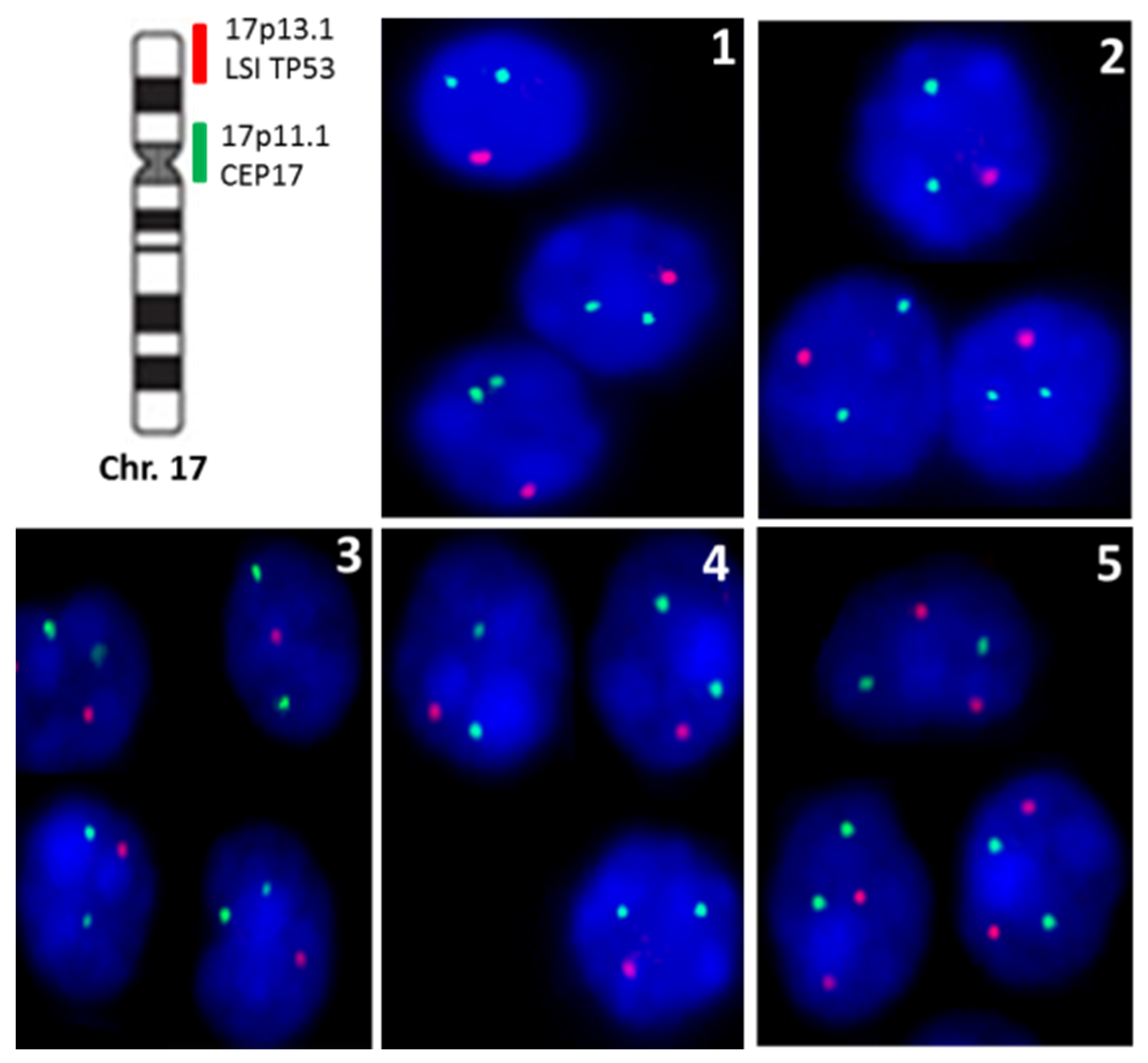
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Pinkus, H. “Sebaceous cysts” are trichilemmal cysts. Arch. Dermatol. 1969, 99, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Kamyab, K.; Kianfar, N.; Dasdar, S.; Salehpour, Z.; Nasimi, M. Cutaneous cysts: A clinicopathologic analysis of 2438 cases. Int. J. Dermatol. 2020, 59, 457–462. [Google Scholar] [CrossRef]
- Leppard, B.J.; Sanderson, K.; Wells, R. Hereditary trichilemmal cysts. Hereditary pilar cysts. Clin. Exp. Dermatol. 1977, 2, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Satyaprakash, A.K.; Sheehan, D.J.; Sangueza, O.P. Proliferating trichilemmal tumors: A review of the literature. Dermatol. Surg. 2007, 33, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.W. Proliferating epidermoid cysts. Arch. Dermatol. 1966, 94, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Noto, G. ‘Benign’ proliferating trichilemmal tumour: Does it really exist? Histopathology 1999, 35, 386–387. [Google Scholar] [CrossRef]
- Ye, J.; Nappi, O.; Swanson, P.E.; Patterson, J.W.; Wick, M.R. Proliferating pilar tumors: A clinicopathologic study of 76 cases with a proposal for definition of benign and malignant variants. Am. J. Clin. Pathol. 2004, 122, 566–574. [Google Scholar] [CrossRef]
- Baptista, A.P.; e Silva, L.G.; Born, M.C. Proliferating trichilemmal cyst. J. Cutan. Pathol. 1983, 10, 178–187. [Google Scholar] [CrossRef]
- Bhavya, P.M.; Letha, V.; Anilkumar, V. Malignant Proliferating Trichilemmal Tumor: A Rare Adnexal Neoplasm. Indian J. Dermatopathol. Diagn. Dermatol. 2020, 7, 40–41. [Google Scholar] [CrossRef]
- Park, B.S.; Yang, S.G.; Cho, K.H. Malignant Proliferating Trichilemmal Tumor Showing Distant Metastases. Am. J. Dermatopathol. 1997, 19, 536–539. [Google Scholar] [CrossRef]
- Sau, P.; Graham, J.H.; Helwig, E.B. Proliferating epithelial cysts. Clinicopathological analysis of 96 cases. J. Cutan. Pathol. 1995, 22, 394–406. [Google Scholar] [CrossRef]
- Takata, M.; Rehman, I.; Rees, J.L. A trichilemmal carcinoma arising from a proliferating trichilemmal cyst: The loss of the wild-type p53 is a critical event in malignant transformation. Hum. Pathol. 1998, 29, 193–195. [Google Scholar] [CrossRef]
- Singh, P.; Usman, A.; Motta, L.; Khan, I. Malignant proliferating trichilemmal tumour. BMJ Case Rep. 2018, 2018, 224460. [Google Scholar] [CrossRef] [PubMed]
- Chaichamnan, K.; Satayasoontorn, K.; Puttanupaab, S.; Attainsee, A. Malignant proliferating trichilemmal tumors with CD34 expression. J. Med. Assoc. Thail. 2010, 93, S28–S34. [Google Scholar]
- Vargas-Mora, P.; Orlandi, D.; Morales, C.; Araya, I. Proliferating Trichilemmal Cysts: A Clinicopathological Study of 14 Cases. Int. J. Trichol. 2019, 11, 258–259. [Google Scholar] [CrossRef]
- Haas, N.; Audring, H.; Sterry, W. Carcinoma arising in a proliferating trichilemmal cyst expresses fetal and trichilemmal hair phenotype. Am. J. Dermatopathol. 2002, 24, 340–344. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Matsuo, S.; Iizuka, H. A DNA-flow cytometric analysis of trichilemmal carcinoma, proliferating trichilemmal cyst and trichilemmal cyst. Acta Dermato-Venereol. 1994, 74, 358–360. [Google Scholar]
- Takata, M.; Quinn, A.G.; Hashimoto, K.; Rees, J.L. Low Frequency of loss and heterozygosity at the nevoid basal cell carcinoma locus and other selected loci in appendageal tumors. J. Investig. Dermatol. 1996, 106, 1141–1144. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Figueras, M.T.; Casalots, A.; Puig, L.; Llatjós, R.; Ferrándiz, C.; Ariza, A. Proliferating trichilemmal tumor: p53 immunoreactivity in association with p27Kip1 over-expression indicates a low-grade carcinoma profile. Histopathology 2001, 38, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Hörer, S.; Marrakchi, S.; Radner, F.P.; Zolles, G.; Heinz, L.; Eichmann, T.O.; Has, C.; Salavei, P.; Mahfoudh, N.; Turki, H.; et al. A Monoallelic Two-Hit Mechanism in PLCD1 Explains the Genetic Pathogenesis of Hereditary Trichilemmal Cyst Formation. J. Investig. Dermatol. 2019, 139, 2154–2163.e5. [Google Scholar] [CrossRef]
- Kolodney, M.S.; Coman, G.C.; Smolkin, M.B.; Hagen, R.; Katzman, J.A.; Katzman, S.N.; Holliday, A.C.; Kolodney, J.A. Hereditary Trichilemmal Cysts are Caused by Two Hits to the Same Copy of the Phospholipase C Delta 1 Gene (PLCD1). Sci. Rep. 2020, 10, 6035. [Google Scholar] [CrossRef] [Green Version]
- Rutty, G.; Richman, P.; Laing, J. Malignant change in trichilemmal cysts: A study of cell proliferation and DNA content. Histopathology 1992, 21, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Herrero, J.; Monteagudo, C.; Ruiz, A.; Llombart-Bosch, A. Malignant proliferating trichilemmal tumours: An histopathological and immunohistochemical study of three cases with DNA ploidy and morphometric evaluation. Histopathology 1998, 33, 542–546. [Google Scholar] [CrossRef]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar]
- Moreira, L.; Balaguer, F.; Lindor, N.; De La Chapelle, A.; Hampel, H.; Aaltonen, L.A.; Hopper, J.L.; Le Marchand, L.; Gallinger, S.; Newcomb, P.A.; et al. Identification of lynch syndrome among patients with colorectal cancer. JAMA 2012, 308, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Le, S.; Ansari, U.; Mumtaz, A.; Malik, K.; Patel, P.; Doyle, A.; Khachemoune, A. Lynch Syndrome and Muir-Torre Syndrome: An update and review on the genetics, epidemiology, and management of two related disorders. Dermatol. Online J. 2017, 23, 13030. [Google Scholar] [PubMed]
- Kruse, R.; Ruzicka, T. DNA mismatch repair and the significance of a sebaceous skin tumor for visceral cancer prevention. Trends Mol. Med. 2004, 10, 136–141. [Google Scholar] [CrossRef]
- Hall, M.J.; Neumann, C.C.; Lamont, J.T.; Grover, S. Lynch syndrome (hereditary nonpolyposis colorectal cancer): Clinical manifestations and diagnosis. In UpToDate; Post, T.W., Ed.; UpToDate: Waltham, MA, USA, 2020; Available online: https://www.uptodate.com/contents/lynch-syndrome-hereditary-nonpolyposis-colorectal-cancer-clinical-manifestations-and-diagnosis (accessed on 24 May 2021).
- Everett, J.N.; Raymond, V.M.; Dandapani, M.; Marvin, M.; Kohlmann, W.; Chittenden, A.; Koeppe, E.; Gustafson, S.L.; Else, T.; Fullen, D.R.; et al. Screening for germline mismatch repair mutations following diagnosis of sebaceous neoplasm. JAMA Dermatol. 2014, 150, 1315–1321. [Google Scholar] [CrossRef] [Green Version]
- Boennelycke, M.; Thomsen, B.M.; Holck, S. Sebaceous neoplasms and the immunoprofile of mismatch-repair proteins as a screening target for syndromic cases. Pathol. Res. Pract. 2015, 211, 78–82. [Google Scholar] [CrossRef]
- Lamba, A.R.; Moore, A.Y.; Moore, T.; Rhees, J.; Arnold, M.A.; Boland, C.R. Defective DNA mismatch repair activity is common in sebaceous neoplasms, and may be an ineffective approach to screen for Lynch syndrome. Fam. Cancer 2015, 14, 259–264. [Google Scholar] [CrossRef]
- Kuwabara, K.; Suzuki, O.; Chika, N.; Kumamoto, K.; Minabe, T.; Fukuda, T.; Arai, E.; Tamaru, J.-I.; Akagi, K.; Eguchi, H.; et al. Prevalence and molecular characteristics of DNA mismatch repair protein-deficient sebaceous neoplasms and keratoacanthomas in a Japanese hospital-based population. Jpn. J. Clin. Oncol. 2018, 48, 514–521. [Google Scholar] [CrossRef]
- Hatta, N.; Takata, A.; Ishizawa, S.; Niida, Y. Family with MSH2 mutation presenting with keratoacanthoma and precancerous skin lesions. J. Dermatol. 2015, 42, 1087–1090. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.R.; Roggero, E.; Sudilovsky, E.C.; Tuthill, R.J.; Wood, G.S.; Sudilovsky, O. Alterations of mismatch repair protein expression in benign melanocytic nevi, melanocytic dysplastic nevi, and cutaneous malignant melanomas. Am. J. Dermatopathol. 2001, 23, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Perrett, C.; Harwood, C.; McGregor, J.; Warwick, J.; Cerio, R.; Karran, P. Expression of DNA mismatch repair proteins and MSH2 polymorphisms in nonmelanoma skin cancers of organ transplant recipients. Br. J. Dermatol. 2009, 162, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Eiberg, H.; Hansen, L.; Hansen, C.; Mohr, J.; Teglbjaerg, P.S.; Kjaer, K.W.; Teglbjærg, P.S. Mapping of hereditary trichilemmal cyst(TRICY1)to chromosome 3p24-p21.2 and exclusion of β-CATENIN and MLH1. Am. J. Med. Genet. Part A 2005, 133A, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Cilona, M.; Locatello, L.G.; Novelli, L.; Gallo, O. The mismatch repair system (MMR) in head and neck carcinogenesis and its role in modulating the response to immunotherapy: A critical review. Cancers 2020, 12, 3006. [Google Scholar] [CrossRef] [PubMed]
- Reichrath, J.; Rass, K. Ultraviolet Damage, DNA Repair and Vitamin D in Nonmelanoma Skin Cancer and in Malignant Melanoma. Adv. Exp. Med. Biol. 2014, 810, 208–233. [Google Scholar] [CrossRef]
- Loureiro, J.B.; Abrantes, M.; Oliveira, P.; Saraiva, L. P53 in skin cancer: From a master player to a privileged target for prevention and therapy. Biochim. Biophys. Acta BBA Rev. Cancer 2020, 1874, 188438. [Google Scholar] [CrossRef]
- Vasan, K.; Anand, S.; Satgunaseelan, L.; Asher, R.; Low, H.; Palme, C.E.; Lee, J.H.; Clark, J.R.; Gupta, R. Mismatch repair protein loss in cutaneous head and neck squamous cell carcinoma. J. Surg. Oncol. 2020, 122, 1755–1760. [Google Scholar] [CrossRef]
- El Hassani, Y.; Beaulieu, J.-Y.; Tschanz, E.; Marcheix, P.-S. Localisation inhabituelle pulpaire d’un kyste trichilemmal proliférant. Chir. Main 2013, 32, 117–119. [Google Scholar] [CrossRef]
- Makiese, O.; Chibbaro, S.; Hamdi, S.; Mirone, G.; George, B. Huge proliferating trichilemmal tumors of the scalp: Report of six cases. Plast. Reconstr. Surg. 2010, 126, 18e–19e. [Google Scholar] [CrossRef] [PubMed]
- López-Ríos, F.; Rodríguez-Peralto, J.L.; Aguilar, A.; Hernández, L.; Gallego, M. Proliferating trichilemmal cyst with focal invasion. Am. J. Dermatopathol. 2000, 22, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Erdem, H.; Uzunlar, A.K.; Ozcelik, D.; Yildirim, U.; Sahiner, C.; Toplu, G. Posttraumatic giant proliferating trichilemmal cysts on the parietal region of the scalp. Indian J. Dermatol. Venereol. Leprol. 2011, 77, 707–709. [Google Scholar] [CrossRef]
- Al-Shanawani, B.; Abdelhamid, M.M.; Al-Shomer, F.M. Giant proliferating trichilemmal tumor. Arch. Plast. Surg. 2013, 40, 461–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, M.; Soua, Y. Giant proliferating trichilemmal cyst. Pan Afr. Med. J. 2014, 18, 195. [Google Scholar] [CrossRef] [PubMed]
- Challita, R.; Halabi, S. Giant aggressive forehead tumor: A 15-year follow-up. Clin. Pract. 2019, 9, 1172. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, E. Scratching the surface of skin development. Nature 2007, 445, 834–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zhao, H.; Qiao, J.; Zhang, S.; Liu, S.; Li, N.; Lei, X.; Ning, L.; Cao, Y.; Duan, E. Expansion of hair follicle stem cells sticking to isolated sebaceous glands to generate in vivo epidermal structures. Cell Transplant. 2016, 25, 2071–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, F.; Ito, M.; Nakamura, A.; Sato, Y. Proliferating trichilemmal cyst with apocrine-acrosyringeal and sebaceous differentiation. J. Cutan. Pathol. 1991, 18, 137–141. [Google Scholar] [CrossRef]
- Dekio, S.; Imaoka, C.; Jidoi, J. Proliferating trichilemmal tumor with apocrine sweat glands. J. Dermatol. 1990, 17, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, S.F.; Aickara, D.; Price, A.; Pavlis, J.; Elgart, G.; Cho-Vega, J.H.; Wei, E.X. Giant proliferating trichilemmal cyst arising from a nevus sebaceus growing for 30 years. J. Cutan. Pathol. 2017, 44, 639–642. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, S.; Huang, X.; Chen, K.; Shen, J.; Wang, Z. Involvement of p53 mutation and mismatch repair proteins dysregulation in NNK-induced malignant transformation of human bronchial epithelial cells. BioMed Res. Int. 2014, 2014, 920275. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Ramamurthi, K.; Mishima, M.; Kondo, A.; Christen, R.D.; Howell, S.B. P53 modulates the effect of loss of DNA mis-match repair on the sensitivity of human colon cancer cells to the cytotoxic and and mutagenic effects of cisplatin. Cancer Res. 2001, 61, 1508–1516. [Google Scholar]
- Liang, S.B.; Furihata, M.; Takeuchi, T.; Sonobe, H.; Ohtsuki, Y. Reduced human mismatch repair protein expression in the development of precancerous skin lesions to squamous cell carcinoma. Virchows Arch. 2001, 439, 622–627. [Google Scholar] [CrossRef]
- Ciavattini, A.; Piccioni, M.; Tranquilli, A.L.; Filosa, A.; Pieramici, T.; Goteri, G. Immunohistochemical expression of DNA mismatch repair (MMR) system proteins (hMLH1, hMSH2) in cervical preinvasive and invasive lesions. Pathol. Res. Pract. 2005, 201, 21–25. [Google Scholar] [CrossRef]
- Young, L.; Listgarten, J.; Trotter, M.; Andrew, S.; Tron, V.A. Evidence that dysregulated DNA mismatch repair characterizes human nonmelanoma skin cancer. Br. J. Dermatol. 2007, 158, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.S.; Grayson, W.; Redston, M.; Diwan, A.H.; Warneke, C.L.; McKee, P.H.; Lev, D.; Lyle, S.; Calonje, E.; Lazar, A.J.F. Site and tumor type predicts DNA mismatch repair status in cutaneous sebaceous neoplasia. Am. J. Surg. Pathol. 2008, 32, 936–942. [Google Scholar] [CrossRef]
- North, J.P.; Golovato, J.; Vaske, C.J.; Sanborn, J.Z.; Nguyen, A.; Wu, W.; Goode, B.; Stevers, M.; McMullen, K.; White, B.E.P.; et al. Cell of origin and mutation pattern define three clinically distinct classes of sebaceous carcinoma. Nat. Commun. 2018, 9, 1894. [Google Scholar] [CrossRef] [PubMed]
- Campos, M.A.; Macedo, S.; Fernandes, M.S.; Pestana, A.; Pardal, J.; Batista, R.; Vinagre, J.; Sanches, A.; Baptista, A.; Lopes, J.M.; et al. Prognostic significance of RAS mutations and P53 expression in cutaneous squamous cell carcinomas. Genes 2020, 11, 751. [Google Scholar] [CrossRef]
- Hwang, L.-A.; Phang, B.H.; Liew, O.W.; Iqbal, J.; Koh, X.H.; Koh, X.Y.; Othman, R.; Xue, Y.; Richards, A.M.; Lane, D.P.; et al. Monoclonal antibodies against specific p53 hotspot mutants as potential tools for precision medicine. Cell Rep. 2018, 22, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Norberg, E.; Vakifahmetoglu-Norberg, H. Mutant p53 as a regulator and target of autophagy. Front. Oncol. 2021, 10, 607149. [Google Scholar] [CrossRef] [PubMed]
- Boeckmann, L.; Martens, M.C.; Emmert, S. Molecular biology of basal and squamous cell carcinomas. Adv. Exp. Med. Biol. 2020, 1268, 171–191. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, A.; Shafi, R.; Nelson, J.; Cantrell, W.; Subhadarshani, S.; Andea, A.; Athar, M.; Elmets, C. The clinical course of actinic keratosis correlates with underlying molecular mechanisms. Br. J. Dermatol. 2019, 182, 995–1002. [Google Scholar] [CrossRef] [Green Version]
- Javor, S.; Gasparini, G.; Biatta, C.M.; Cozzani, E.; Cabiddu, F.; Ravetti, J.L.; Vellone, V.G.; Parodi, A. P53 staining index and zonal staining patterns in actinic keratoses. Arch. Dermatol. Res. 2021, 313, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Reichrath, J.; Reichrath, S.; Vogt, T.; Römer, K. Crosstalk between vitamin D and p53 signaling in cancer: An update. Adv. Exp. Med. Biol. 2020, 1268, 307–318. [Google Scholar] [CrossRef]
- Berman, B.; Cockerell, C.J. Pathobiology of actinic keratosis: Ultraviolet-dependent keratinocyte proliferation. J. Am. Acad. Dermatol. 2013, 68, S10–S19. [Google Scholar] [CrossRef] [PubMed]
- Murnyák, B.; Hortobágyi, T. Immunohistochemical correlates of TP53 somatic mutations in cancer. Oncotarget 2016, 7, 64910–64920. [Google Scholar] [CrossRef] [Green Version]
- Niyaz, M.; Ainiwaer, J.; Abudureheman, A.; Zhang, L.; Sheyhidin, I.; Turhong, A.; Cai, R.; Hou, Z.; Awut, E. Association between TP53 gene deletion and protein expression in esophageal squamous cell carcinoma and its prognostic significance. Oncol. Lett. 2020, 20, 1855–1865. [Google Scholar] [CrossRef]
- McGraw, K.L.; Nguyen, J.; Komrokji, R.S.; Sallman, D.; Al Ali, N.H.; Padron, E.; Lancet, J.E.; Moscinski, L.C.; List, A.F.; Zhang, L. Immunohistochemical pattern of p53 is a measure of TP53 mutation burden and adverse clinical outcome in myelodysplastic syndromes and secondary acute myeloid leukemia. Haematologica 2016, 101, e320–e323. [Google Scholar] [CrossRef]
- Chang, H.; Yeung, J.; Qi, C.; Xu, W. Aberrant nuclear p53 protein expression detected by immunohistochemistry is associated with hemizygous P53 deletion and poor survival for multiple myeloma. Br. J. Haematol. 2007, 138, 324–329. [Google Scholar] [CrossRef]
- Chen, M.-H.; Qi, C.X.; Saha, M.N.; Chang, H. p53 Nuclear Expression Correlates with Hemizygous TP53 Deletion and Predicts an Adverse Outcome for Patients with Relapsed/Refractory Multiple Myeloma Treated with Lenalidomide. Am. J. Clin. Pathol. 2012, 137, 208–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynt, E.; Bisht, K.; Sridharan, V.; Ortiz, M.; Towfic, F.; Thakurta, A. Prognosis, Biology, and Targeting of TP53 Dysregulation in Multiple Myeloma. Cells 2020, 9, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zetner, D.B.; Bisgaard, M.L. Familial Colorectal Cancer Type X. Curr. Genom. 2017, 18, 341–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]



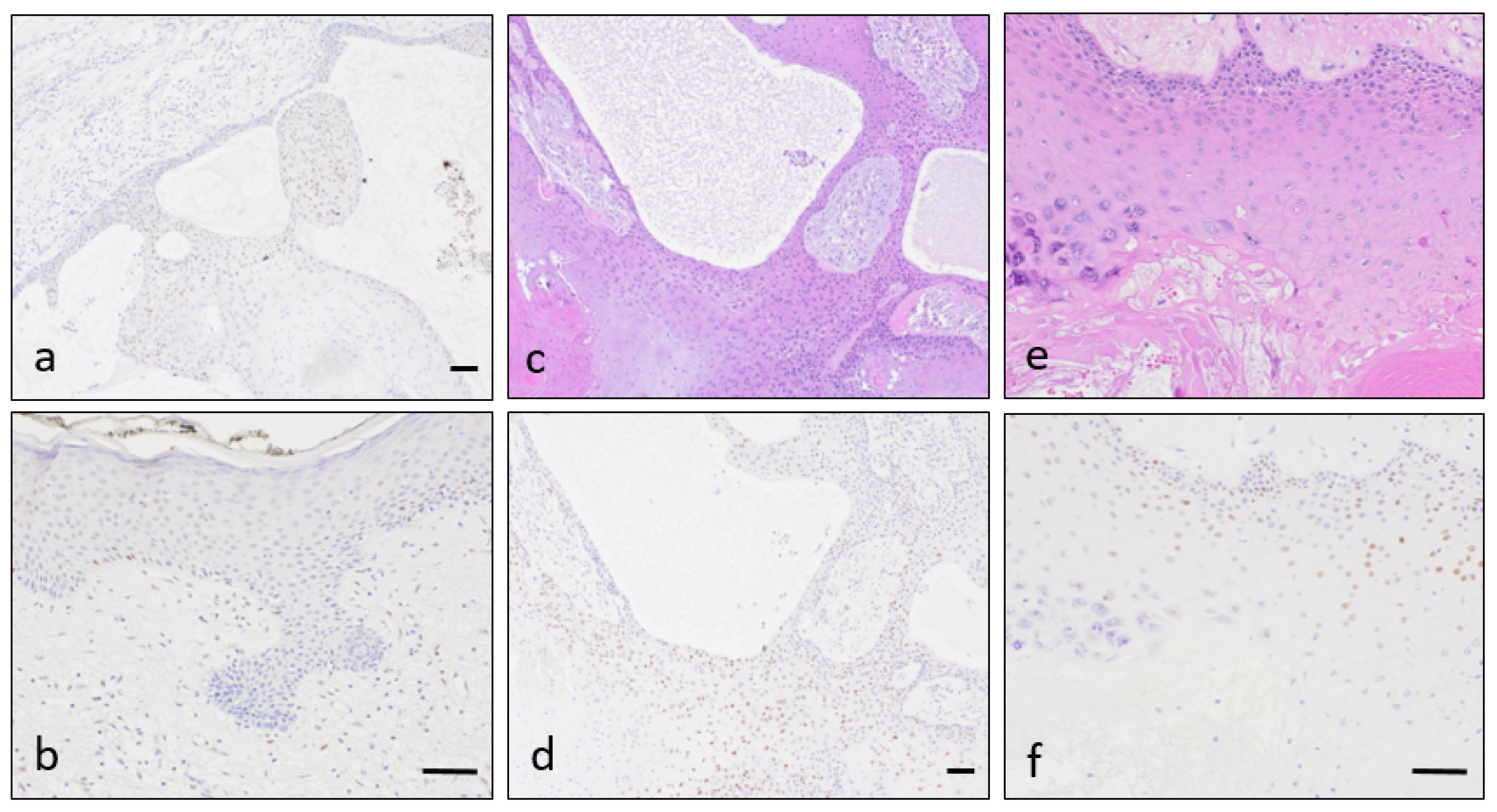


{kind=link}
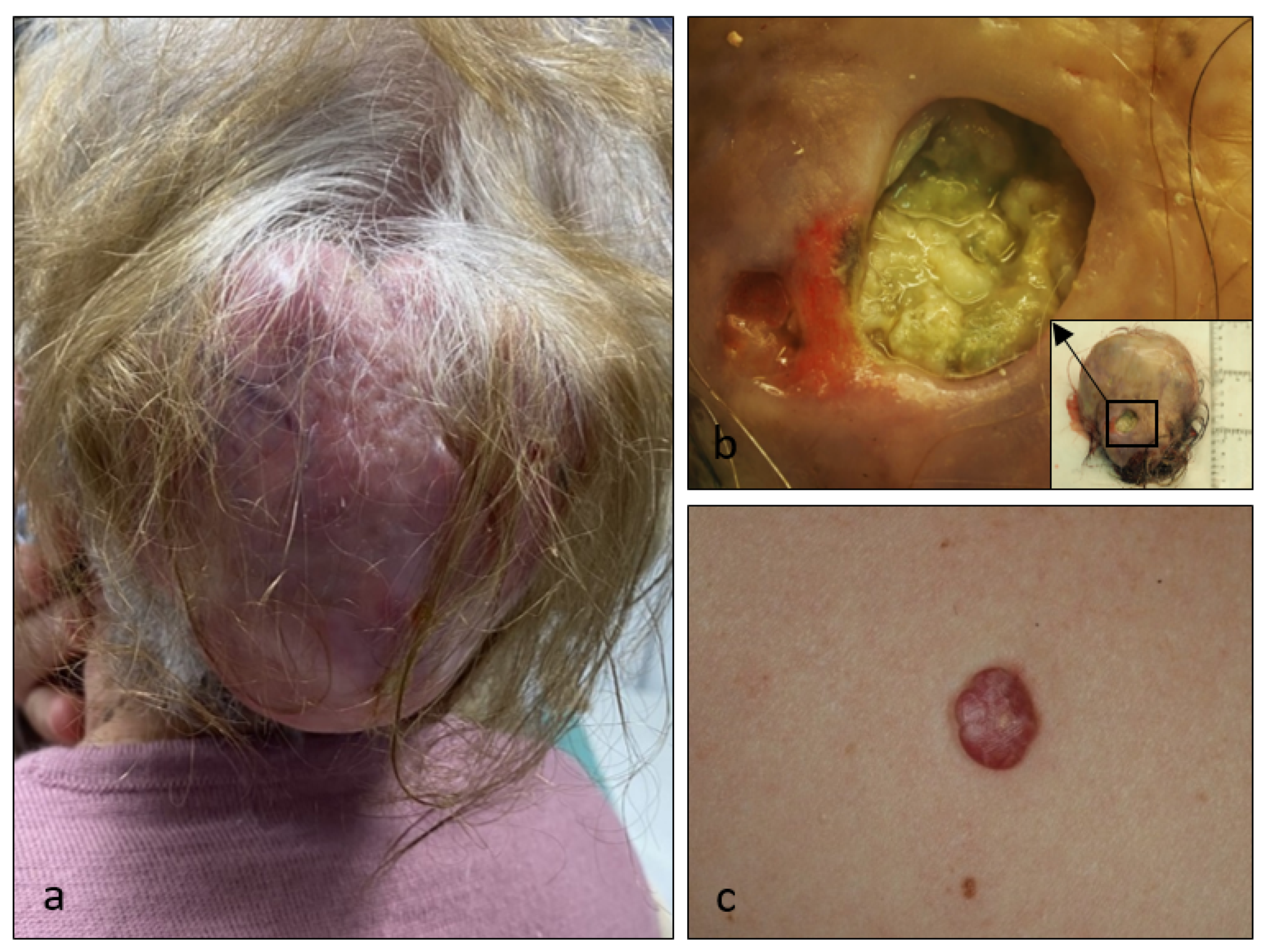
{kind=link}
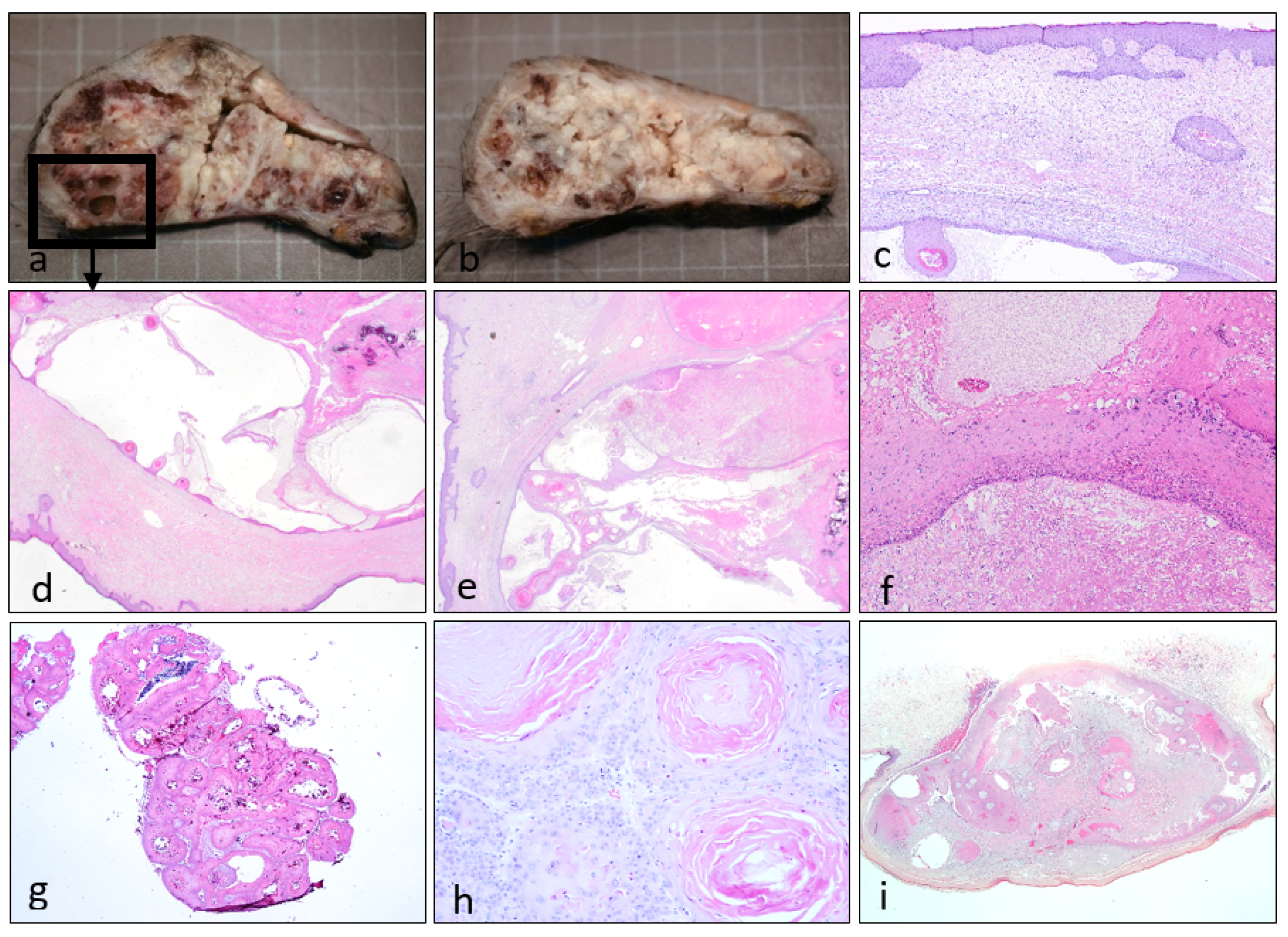
{kind=link}
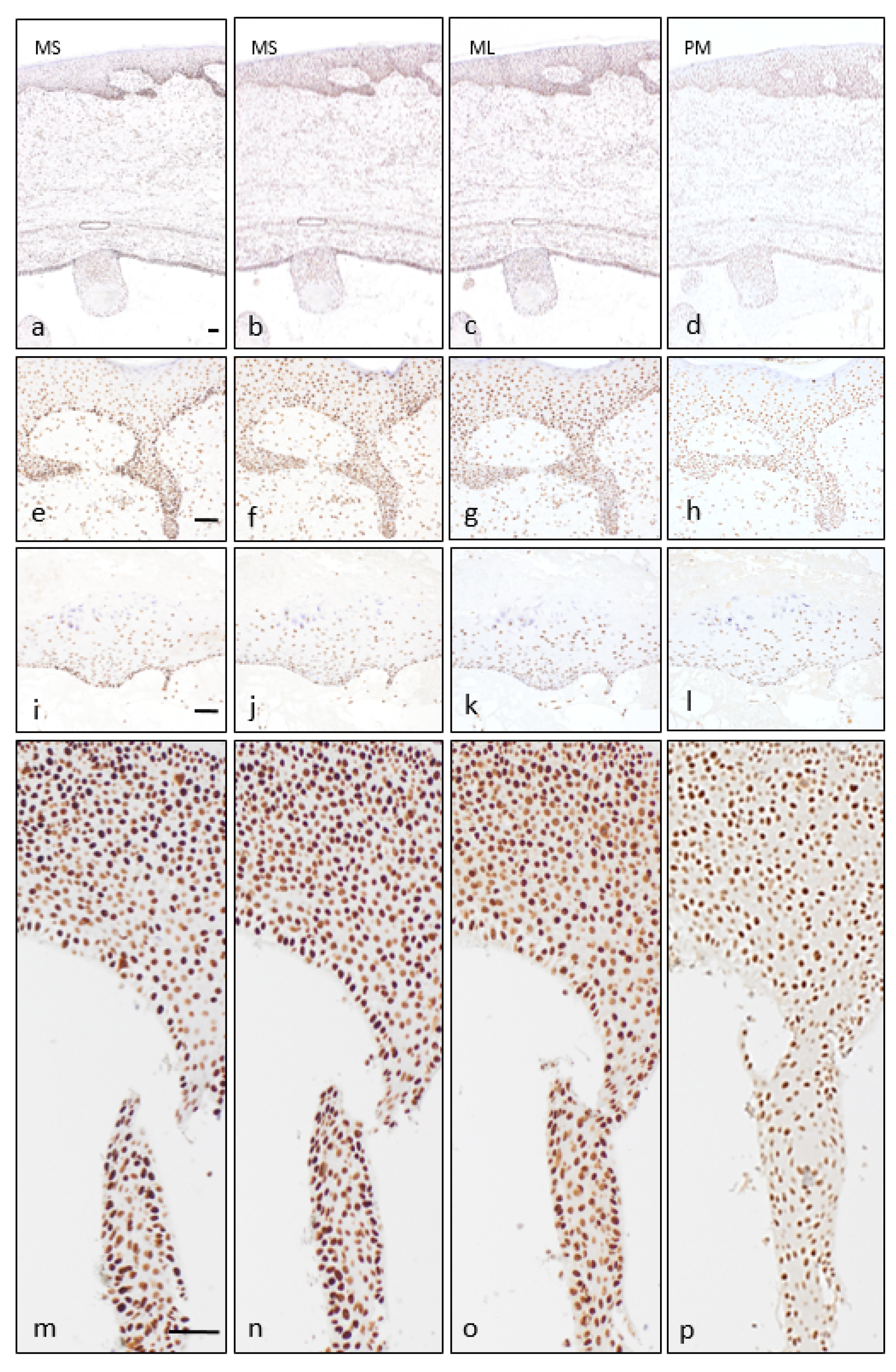
{kind=link}
{kind=link}
| Case | Gender | Age | Topography | Size |
|---|---|---|---|---|
| Case 1 | Female | 88 | Scalp (occipital) | 9 cm |
| Case 2 | Female | 45 | Back | 1 cm |
| Case 3 | Female | 84 | Scalp (frontal) | 3 cm |
| Case 4 | Female | 63 | Scalp (occipital) | 3 cm |
| Case 5 | Female | 69 | Scalp (parietal) | 1.5 cm |
| Case | MSH2 | MSH6 | MLH1 | PMS2 | P53 IHQ | TP53 Loss |
|---|---|---|---|---|---|---|
| Case 1 | 80% | 80% | 60% | 70% | Weak | 63% |
| Case 2 | 60% | 60% | 40% | 30% | Negative | 65% |
| Case 3 | 90% | 80% | 80% | 80% | Negative | 52% |
| Case 4 | 90% | 90% | 90% | 90% | Weak | 80% |
| Case 5 | 90% | 90% | 80% | 70% | Negative | 0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martín-Sanz, R.; Sayagués, J.M.; García-Cano, P.; Azcue-Mayorga, M.; Parra-Pérez, M.d.C.; Pacios-Pacios, M.Á.; Piqué-Durán, E.; Feito, J. TP53 Abnormalities and MMR Preservation in 5 Cases of Proliferating Trichilemmal Tumours. Dermatopathology 2021, 8, 147-158. https://0-doi-org.brum.beds.ac.uk/10.3390/dermatopathology8020021
Martín-Sanz R, Sayagués JM, García-Cano P, Azcue-Mayorga M, Parra-Pérez MdC, Pacios-Pacios MÁ, Piqué-Durán E, Feito J. TP53 Abnormalities and MMR Preservation in 5 Cases of Proliferating Trichilemmal Tumours. Dermatopathology. 2021; 8(2):147-158. https://0-doi-org.brum.beds.ac.uk/10.3390/dermatopathology8020021
Chicago/Turabian StyleMartín-Sanz, Raquel, José María Sayagués, Pilar García-Cano, Mikel Azcue-Mayorga, María del Carmen Parra-Pérez, María Ángeles Pacios-Pacios, Enric Piqué-Durán, and Jorge Feito. 2021. "TP53 Abnormalities and MMR Preservation in 5 Cases of Proliferating Trichilemmal Tumours" Dermatopathology 8, no. 2: 147-158. https://0-doi-org.brum.beds.ac.uk/10.3390/dermatopathology8020021