
Cutaneous Melanomas Arising during Childhood: An Overview of the Main Entities

 , , and
, , and
Abstract
:1. Introduction
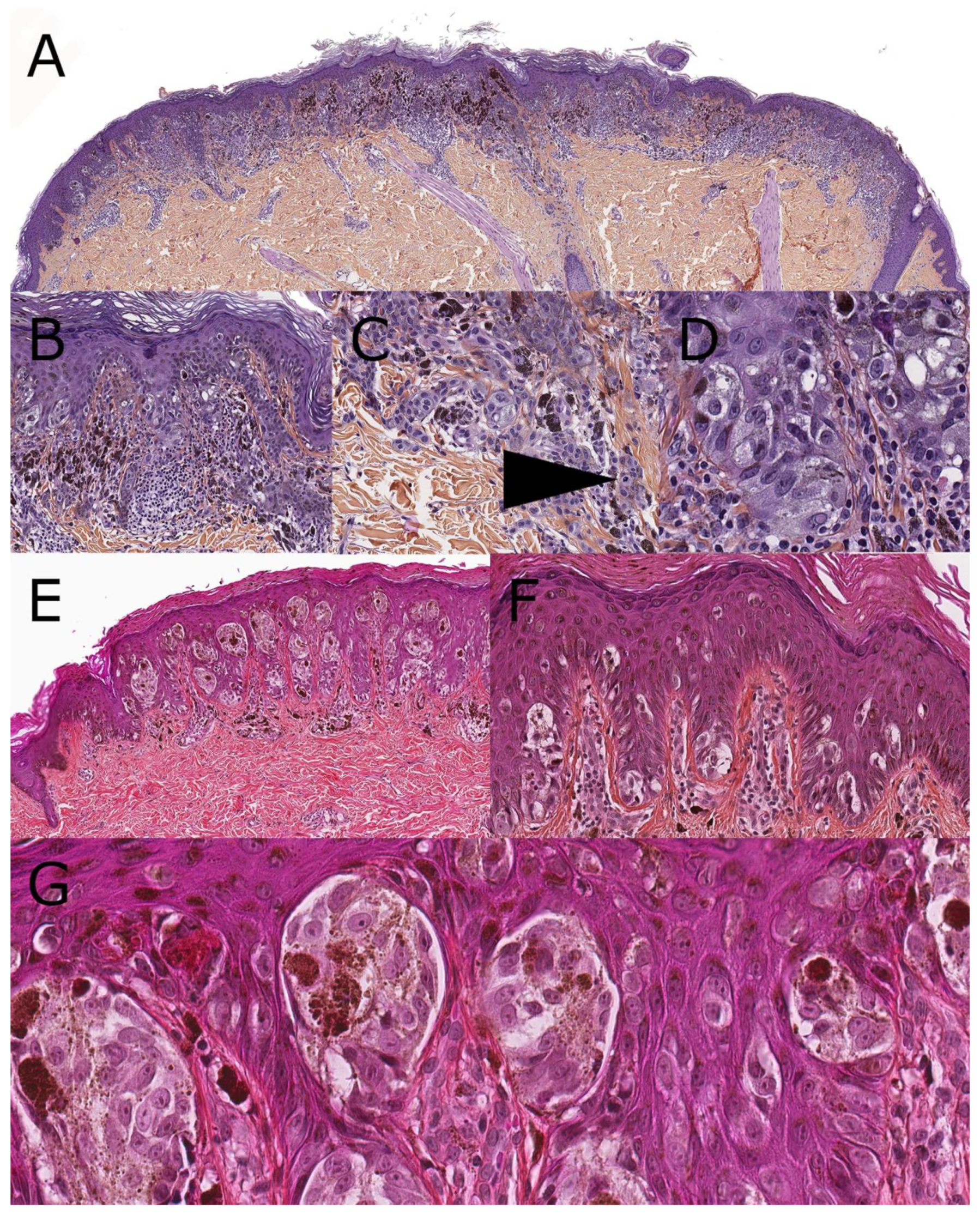
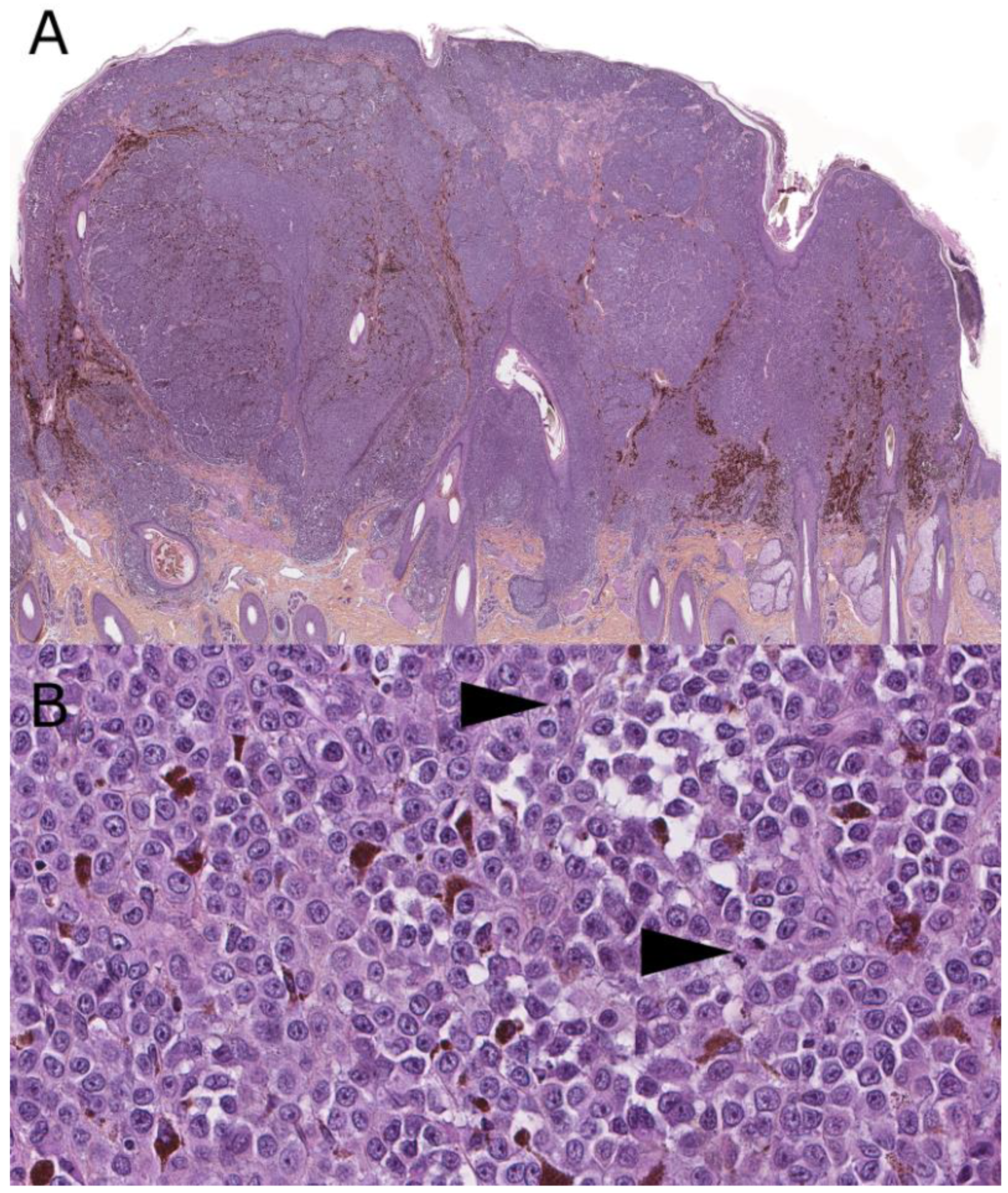
2. Superficial Spreading Melanoma (SSM)
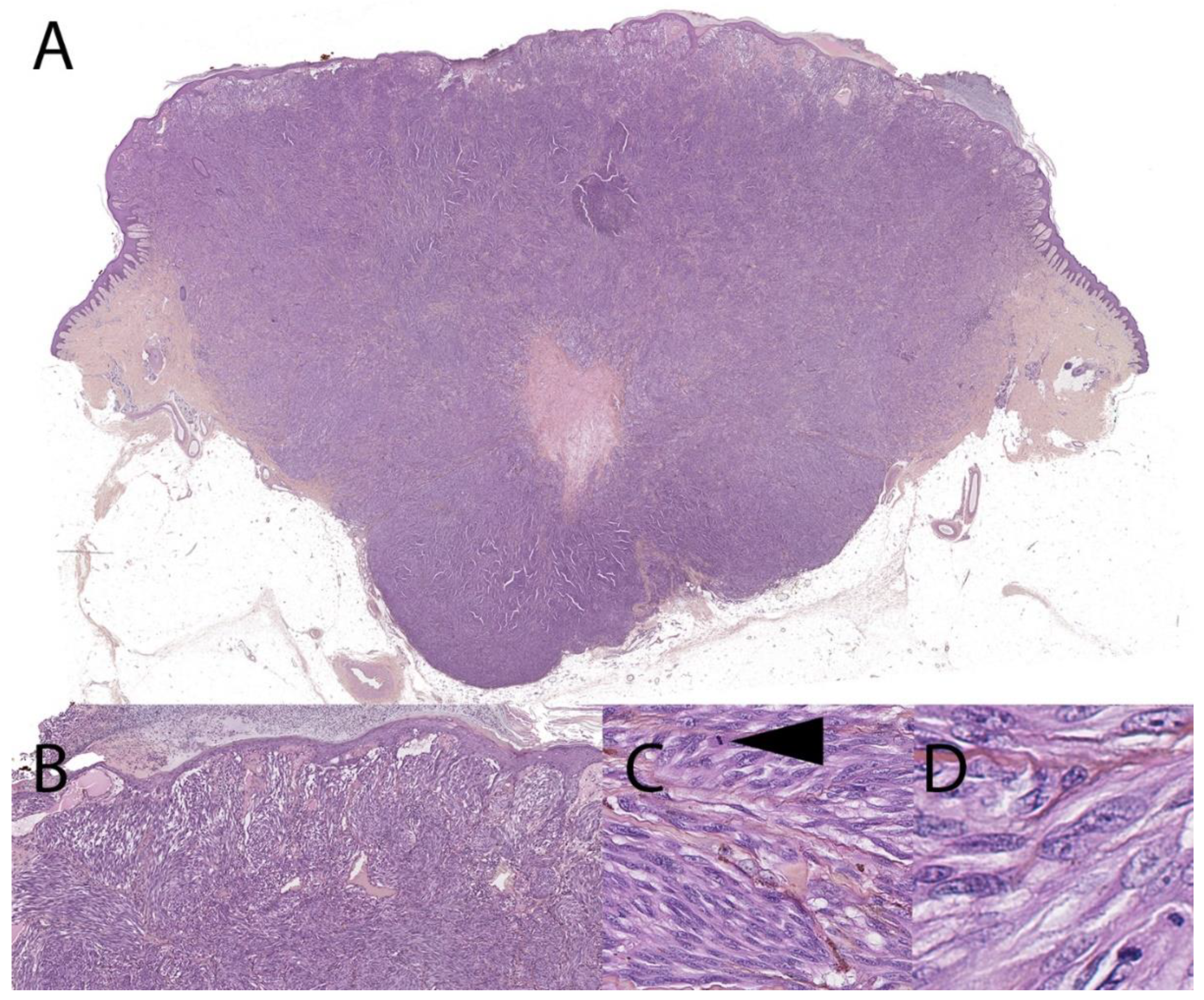
3. Malignant Spitz Tumor (Melanoma with a Spitzoid Morphology and Specific Genetic Features)
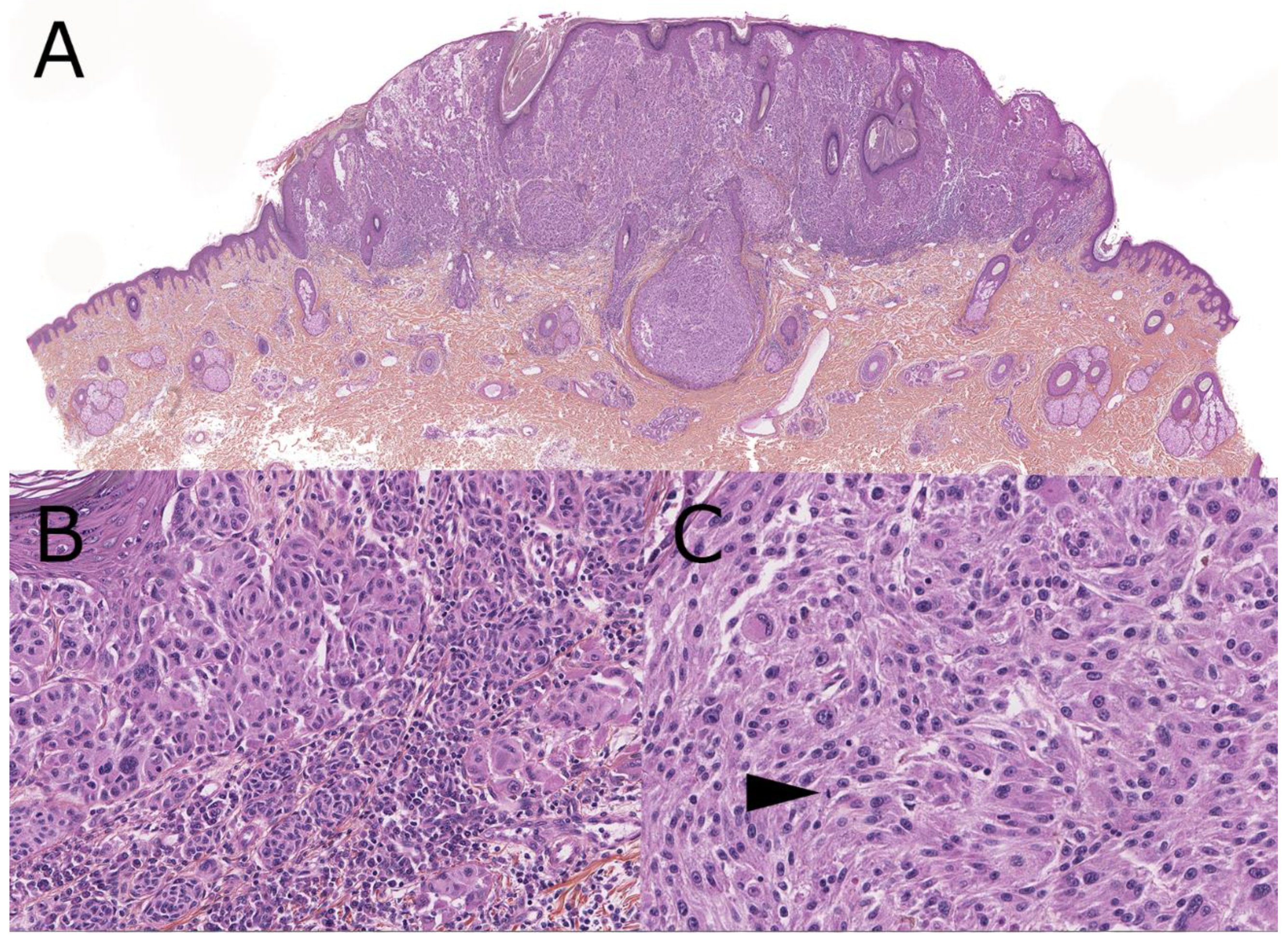
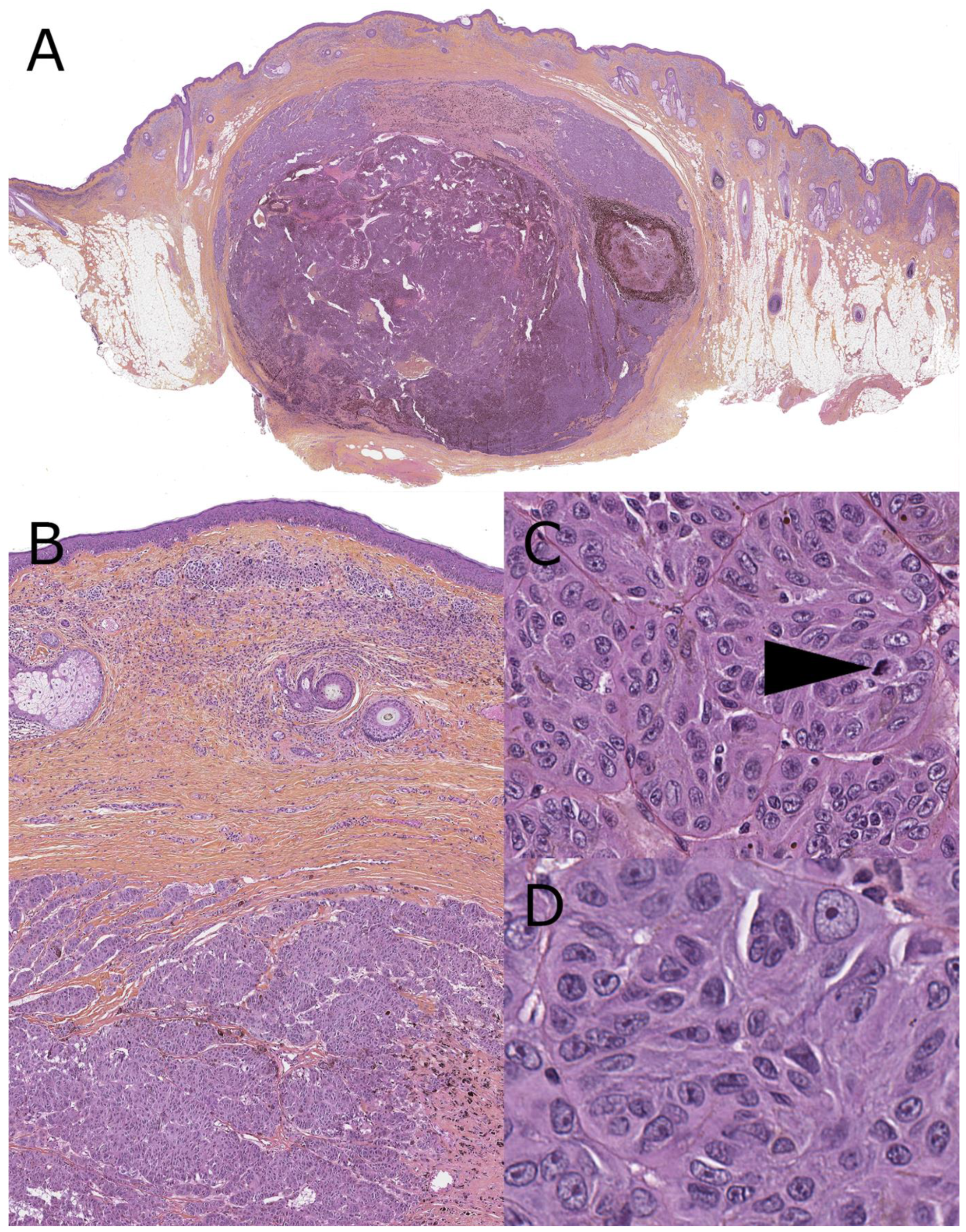
4. Melanoma Arising from a Congenital Nevus
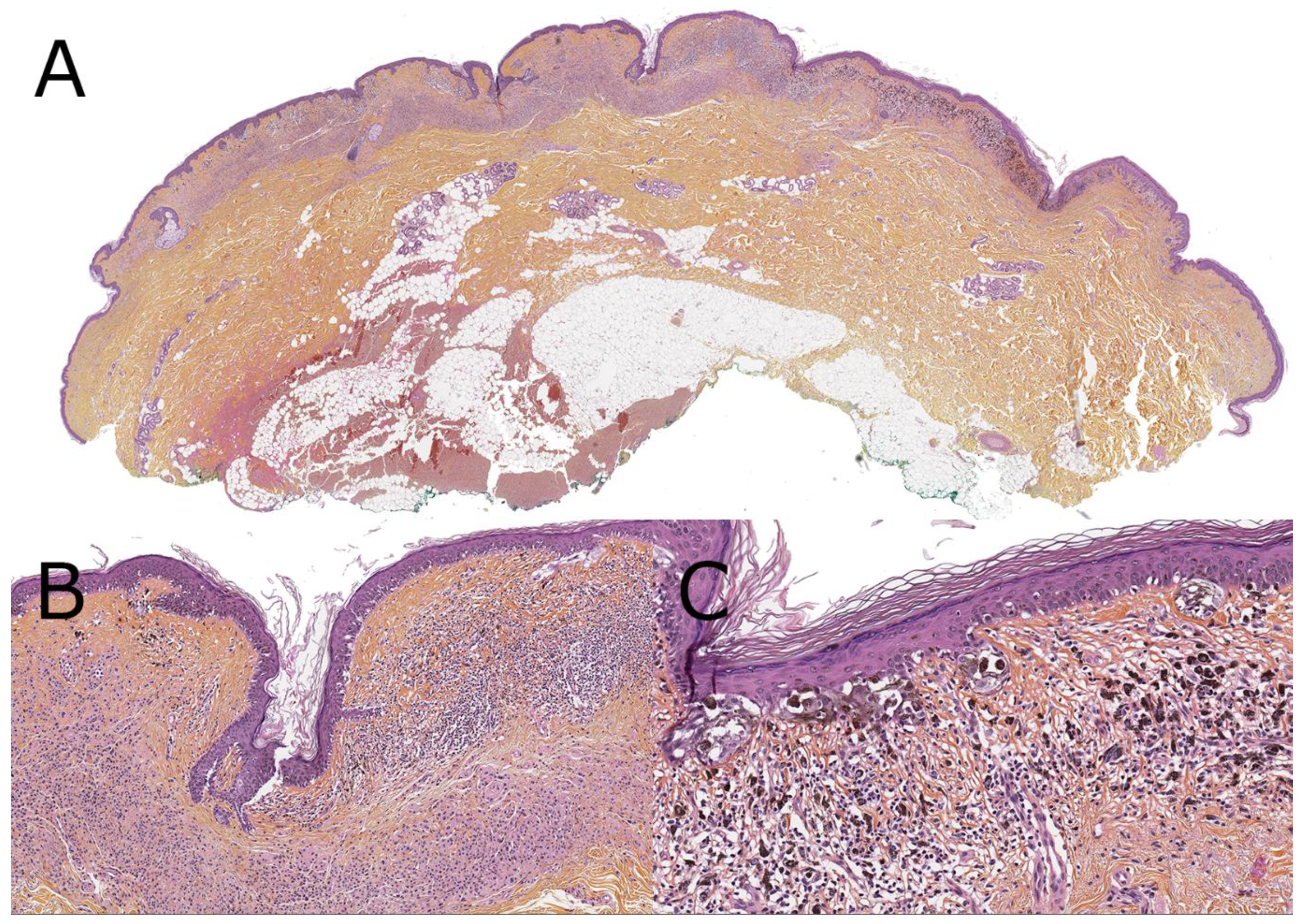
5. Nevoid Melanomas
6. Melanomas Arising in the Setting of a Germline Predisposition Syndrome
7. Unclassified Melanomas
8. Melanoma Risk in Children with Impaired Immunity
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Gamble, R.G.; Asdigian, N.L.; Aalborg, J.; Gonzalez, V.; Box, N.F.; Huff, L.S.; Barón, A.E.; Morelli, J.G.; Mokrohisky, S.T.; Crane, L.A.; et al. Sun damage in ultraviolet photographs correlates with phenotypic melanoma risk factors in 12-year-old children. J. Am. Acad. Dermatol. 2012, 67, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Duffy, D.L.; Lee, K.J.; Jagirdar, K.; Pflugfelder, A.; Stark, M.S.; McMeniman, E.K.; Soyer, H.P.; Sturm, R.A. High naevus count and MC1R red hair alleles contribute synergistically to increased melanoma risk. Br. J. Dermatol. 2019, 181, 1009–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannous, Z.S.; Mihm, M.C.; Sober, A.J.; Duncan, L.M. Congenital melanocytic nevi: Clinical and histopathologic features, risk of melanoma, and clinical management. J. Am. Acad. Dermatol. 2005, 52, 197–203. [Google Scholar] [CrossRef]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef]
- Elder, D.E.; Massi, D.; Scolyer, R.A.; Willemze, R. WHO Classification of Skin Tumours; International Agency for Research on Cancer: Lyon, France, 2018. [Google Scholar]
- Pappo, A.S.; McPherson, V.; Pan, H.; Wang, F.; Wang, L.; Wright, T.; Hussong, M.; Hawkins, D.; Kaste, S.C.; Davidoff, A.M.; et al. A prospective, comprehensive registry that integrates the molecular analysis of pediatric and adolescent melanocytic lesions. Cancer 2021. [Google Scholar] [CrossRef]
- Seynnaeve, B.; Lee, S.; Borah, S.; Park, Y.; Pappo, A.; Kirkwood, J.M.; Bahrami, A. Genetic and Epigenetic Alterations of TERT Are Associated with Inferior Outcome in Adolescent and Young Adult Patients with Melanoma. Sci. Rep. 2017, 7, 45704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahrami, A.; Barnhill, R.L. Pathology and genomics of pediatric melanoma: A critical reexamination and new insights. Pediatr. Blood Cancer 2018, 65, e26792. [Google Scholar] [CrossRef]
- Goto, K.; Pissaloux, D.; Durand, L.; Tirode, F.; Guillot, B.; de la Fouchardière, A. Novel three-way complex rearrangement of TRPM1-PUM1-LCK in a case of agminated Spitz nevi arising in a giant congenital hyperpigmented macule. Pigment. Cell Melanoma Res. 2020, 33, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; He, J.; Yelensky, R.; Esteve-Puig, R.; Botton, T.; Yeh, I.; Lipson, D.; Otto, G.; Brennan, K.; Murali, R.; et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat. Commun. 2014, 5, 3116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, I.; Busam, K.J.; McCalmont, T.H.; LeBoit, P.E.; Pissaloux, D.; Alberti, L.; de la Fouchardière, A.; Bastian, B.C. Filigree-like Rete Ridges, Lobulated Nests, Rosette-like Structures, and Exaggerated Maturation Characterize Spitz Tumors With NTRK1 Fusion. Am. J. Surg. Pathol. 2019, 43, 737–746. [Google Scholar] [CrossRef]
- Amin, S.M.; Haugh, A.M.; Lee, C.Y.; Zhang, B.; Bubley, J.A.; Merkel, E.A.; Verzì, A.E.; Gerami, P. A Comparison of Morphologic and Molecular Features of BRAF, ALK, and NTRK1 Fusion Spitzoid Neoplasms. Am. J. Surg. Pathol. 2017, 41, 491–498. [Google Scholar] [CrossRef]
- Bastian, B.C.; LeBoit, P.E.; Pinkel, D. Mutations and copy number increase of HRAS in Spitz nevi with distinctive histopathological features. Am. J. Pathol. 2000, 157, 967–972. [Google Scholar] [CrossRef] [Green Version]
- Donati, M.; Kastnerova, L.; Martinek, P.; Grossmann, P.; Sticová, E.; Hadravský, L.; Torday, T.; Kyclova, J.; Michal, M.; Kazakov, D.V. Spitz Tumors With ROS1 Fusions: A Clinicopathological Study of 6 Cases, Including FISH for Chromosomal Copy Number Alterations and Mutation Analysis Using Next-Generation Sequencing. Am. J. Dermatopathol. 2020, 42, 92–102. [Google Scholar] [CrossRef]
- Gerami, P.; Kim, D.; Compres, E.V.; Zhang, B.; Khan, A.U.; Sunshine, J.C.; Quan, V.L.; Busam, K. Clinical, morphologic, and genomic findings in ROS1 fusion Spitz neoplasms. Mod. Pathol. 2021, 34, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Pissaloux, D.; Kauer, F.; Huriet, V.; Tirode, F.; de la Fouchardière, A. GOPC-ROS1 mosaicism in agminated Spitz naevi: Report of two cases. Virchows Arch. 2021. [Google Scholar] [CrossRef]
- Wiesner, T.; Murali, R.; Fried, I.; Cerroni, L.; Busam, K.; Kutzner, H.; Bastian, B.C. A distinct subset of atypical Spitz tumors is characterized by BRAF mutation and loss of BAP1 expression. Am. J. Surg. Pathol. 2012, 36, 818–830. [Google Scholar] [CrossRef] [Green Version]
- Yeh, I.; de la Fouchardiere, A.; Pissaloux, D.; Mully, T.W.; Garrido, M.C.; Vemula, S.S.; Busam, K.J.; LeBoit, P.E.; McCalmont, T.H.; Bastian, B.C. Clinical, histopathologic, and genomic features of Spitz tumors with ALK fusions. Am. J. Surg. Pathol. 2015, 39, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastnerova, L.; Martinek, P.; Grossmann, P.; Steiner, P.; Vanecek, T.; Kyclova, J.; Ferak, I.; Zalud, R.; Slehobr, O.; Svajdler, P.; et al. A Clinicopathological Study of 29 Spitzoid Melanocytic Lesions With ALK Fusions, Including Novel Fusion Variants, Accompanied by Fluorescence In Situ Hybridization Analysis for Chromosomal Copy Number Changes, and Both TERT Promoter and Next-Generation Sequencing Mutation Analysis. Am. J. Dermatopathol. 2020, 42, 578–592. [Google Scholar]
- De la Fouchardière, A.; Tee, M.K.; Peternel, S.; Valdebran, M.; Pissaloux, D.; Tirode, F.; Busam, K.J.; LeBoit, P.E.; McCalmont, T.H.; Bastian, B.C.; et al. Fusion partners of NTRK3 affect subcellular localization of the fusion kinase and cytomorphology of melanocytes. Mod. Pathol. 2021, 34, 735–747. [Google Scholar] [CrossRef]
- Yeh, I.; Tee, M.K.; Botton, T.; Shain, A.H.; Sparatta, A.J.; Gagnon, A.; Vemula, S.S.; Garrido, M.C.; Nakamaru, K.; Isoyama, T.; et al. NTRK3 kinase fusions in Spitz tumours. J. Pathol. 2016, 240, 282–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, K.; Pissaloux, D.; Tirode, F.; de la Fouchardière, A. Spitz nevus with a novel TFG-NTRK2 fusion: The first case report of NTRK2-rearranged Spitz/Reed nevus. J. Cutan. Pathol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Newman, S.; Pappo, A.; Raimondi, S.; Zhang, J.; Barnhill, R.; Bahrami, A. Pathologic Characteristics of Spitz Melanoma With MAP3K8 Fusion or Truncation in a Pediatric Cohort. Am. J. Surg. Pathol. 2019, 43, 1631–1637. [Google Scholar] [CrossRef] [PubMed]
- Quan, V.L.; Zhang, B.; Zhang, Y.; Mohan, L.S.; Shi, K.; Wagner, A.; Kruse, L.; Taxter, T.; Beaubier, N.; White, K.; et al. Integrating Next Generation Sequencing with Morphology Improves Prognostic and Biologic Classification of Spitz Neoplasms. J. Investig. Dermatol. 2020, 140, 1599–1608. [Google Scholar] [CrossRef]
- Quan, V.L.; Zhang, B.; Mohan, L.S.; Shi, K.; Isales, M.C.; Panah, E.; Taxter, T.J.; Bealubier, N.; White, K.; Gerami, P. Activating Structural Alterations in MAPK Genes Are Distinct Genetic Drivers in a Unique Subgroup of Spitzoid Neoplasms. Am. J. Surg. Pathol. 2019, 43, 538–548. [Google Scholar] [CrossRef]
- Houlier, A.; Pissaloux, D.; Masse, I.; Tirode, F.; Karanian, M.; Pincus, L.B.; McCalmont, T.H.; LeBoit, P.E.; Bastian, B.C.; Yeh, I.; et al. Melanocytic tumors with MAP3K8 fusions: Report of 33 cases with morphological-genetic correlations. Mod. Pathol. 2020, 33, 846–857. [Google Scholar] [CrossRef]
- Donati, M.; Kastnerova, L.; Ptakova, N.; Michal, M.; Kazakov, D.V. Polypoid Atypical Spitz Tumor With a Fibrosclerotic Stroma, CLIP2-BRAF Fusion, and Homozygous Loss of 9p21. Am. J. Dermatopathol. 2020, 42, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Perron, E.; Pissaloux, D.; Neub, A.; Hohl, D.; Tartar, M.D.; Mortier, L.; Alberti, L.; de la Fouchardiere, A. Unclassified sclerosing malignant melanomas with AKAP9-BRAF gene fusion: A report of two cases and review of BRAF fusions in melanocytic tumors. Virchows Arch. 2018, 472, 469–476. [Google Scholar] [CrossRef]
- Wu, G.; Barnhill, R.L.; Lee, S.; Li, Y.; Shao, Y.; Easton, J.; Dalton, J.; Zhang, J.; Pappo, A.; Bahrami, A. The landscape of fusion transcripts in spitzoid melanoma and biologically indeterminate spitzoid tumors by RNA sequencing. Mod. Pathol. 2016, 29, 359–369. [Google Scholar] [CrossRef] [Green Version]
- VandenBoom, T.; Quan, V.L.; Zhang, B.; Garfield, E.M.; Kong, B.Y.; Isales, M.C.; Panah, E.; Igartua, C.; Taxter, T.; Beaubier, N.; et al. Genomic Fusions in Pigmented Spindle Cell Nevus of Reed. Am. J. Surg. Pathol. 2018, 42, 1042–1051. [Google Scholar] [CrossRef]
- Yeh, I.; Botton, T.; Talevich, E.; Shain, A.H.; Sparatta, A.J.; de la Fouchardiere, A.; Mully, T.W.; North, J.P.; Garrido, M.C.; Gagnon, A.; et al. Activating MET kinase rearrangements in melanoma and Spitz tumours. Nat. Commun. 2015, 6, 7174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donati, M.; Nosek, D.; Waldenbäck, P.; Martinek, P.; Jonsson, B.A.; Galgonkova, P.; Hawawrehova, M.; Berouskova, P.; Kastnerova, L.; Persichetti, P.; et al. MAP2K1-Mutated Melanocytic Neoplasms With a SPARK-Like Morphology. Am. J. Dermatopathol. 2021, 43, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Bartenstein, D.W.; Fisher, J.M.; Stamoulis, C.; Weldon, C.; Huang, J.T.; Gellis, S.E.; Liang, M.G.; Schmidt, B.; Hawryluk, E.B.; Bartenstein, D.W.; et al. Clinical features and outcomes of spitzoid proliferations in children and adolescents. Br. J. Dermatol. 2019, 181, 366–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lallas, A.; Kyrgidis, A.; Ferrara, G.; Kittler, H.; Apalla, Z.; Castagnetti, F.; Longo, C.; Moscarella, E.; Piana, S.; Zalaudek, I.; et al. Atypical Spitz tumours and sentinel lymph node biopsy: A systematic review. Lancet Oncol. 2014, 15, e178–e183. [Google Scholar] [CrossRef]
- Pol-Rodriquez, M.; Lee, S.; Silvers, D.N.; Celebi, J.T. Influence of age on survival in childhood spitzoid melanomas. Cancer 2007, 109, 1579–1583. [Google Scholar] [CrossRef]
- Lee, S.; Barnhill, R.L.; Dummer, R.; Dalton, J.; Wu, J.; Pappo, A.; Bahrami, A. TERT Promoter Mutations Are Predictive of Aggressive Clinical Behavior in Patients with Spitzoid Melanocytic Neoplasms. Sci. Rep. 2015, 5, 11200. [Google Scholar] [CrossRef] [Green Version]
- Davies, O.M.T.; Majerowski, J.; Segura, A.; Kelley, S.W.; Sokumbi, O.; Humphrey, S.R. A sixteen-year single-center retrospective chart review of Spitz nevi and spitzoid neoplasms in pediatric patients. Pediatr. Dermatol. 2020, 37, 1073–1082. [Google Scholar] [CrossRef]
- Raghavan, S.S.; Peternel, S.; Mully, T.W.; North, J.P.; Pincus, L.B.; LeBoit, P.E.; McCalmont, T.H.; Bastian, B.C.; Yeh, I. Spitz melanoma is a distinct subset of spitzoid melanoma. Mod. Pathol. 2020, 33, 1122–1134. [Google Scholar] [CrossRef] [PubMed]
- Kinsler, V. Satellite lesions in congenital melanocytic nevi—Time for a change of name. Pediatr. Dermatol. 2011, 28, 212–213. [Google Scholar] [CrossRef] [Green Version]
- Kinsler, V.A.; Thomas, A.C.; Ishida, M.; Bulstrode, N.W.; Loughlin, S.; Hing, S.; Chalker, J.; McKenzie, K.; Abu-Amero, S.; Slater, O.; et al. Multiple congenital melanocytic nevi and neurocutaneous melanosis are caused by postzygotic mutations in codon 61 of NRAS. J. Investig. Dermatol. 2013, 133, 2229–2236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polubothu, S.; McGuire, N.; Al-Olabi, L.; Baird, W.; Bulstrode, N.; Chalker, J.; Josifova, D.; Lomas, D.; O’Hara, J.; Ong, J.; et al. Does the gene matter? Genotype-phenotype and genotype-outcome associations in congenital melanocytic naevi. Br. J. Dermatol. 2020, 182, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Martins da Silva, V.; Martinez-Barrios, E.; Tell-Martí, G.; Dabad, M.; Carrera, C.; Aguilera, P.; Brualla, D.; Esteve-Codina, A.; Vicente, A.; Puig, S.; et al. Genetic Abnormalities in Large to Giant Congenital Nevi: Beyond NRAS Mutations. J. Investig. Dermatol. 2019, 139, 900–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltres, A.; Salhi, A.; Houlier, A.; Pissaloux, D.; Tirode, F.; Haddad, V.; Karanian, M.; Dahlouk, S.Y.; Boukendakdji, F.; Dahlouk, D.; et al. Malignant melanoma with areas of rhabdomyosarcomatous differentiation arising in a giant congenital nevus with RAF1 gene fusion. Pigment. Cell Melanoma Res. 2019, 32, 708–713. [Google Scholar] [CrossRef]
- Kinsler, V.A.; O’Hare, P.; Bulstrode, N.; Calonje, J.E.; Chong, W.K.; Hargrave, D.; Jacques, T.; Lomas, D.; Sebire, N.; Slater, O. Melanoma in congenital melanocytic naevi. Br. J. Dermatol. 2017, 176, 1131–1143. [Google Scholar] [CrossRef]
- Castilla, E.E.; da Graça Dutra, M.; Orioli-Parreiras, I.M. Epidemiology of congenital pigmented naevi: I. Incidence rates and relative frequencies. Br. J. Dermatol. 1981, 104, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Paradela, S.; Fonseca, E.; Prieto, V.G. Melanoma in children. Arch. Pathol. Lab. Med. 2011, 135, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Vourc’h-Jourdain, M.; Martin, L.; Barbarot, S. Large congenital melanocytic nevi: Therapeutic management and melanoma risk: A systematic review. J. Am. Acad. Dermatol. 2013, 68, 493–498. [Google Scholar] [CrossRef]
- Yélamos, O.; Arva, N.C.; Obregon, R.; Yazdan, P.; Wagner, A.; Guitart, J.; Gerami, P. A comparative study of proliferative nodules and lethal melanomas in congenital nevi from children. Am. J. Surg. Pathol. 2015, 39, 405–415. [Google Scholar] [CrossRef]
- Price, H.N.; Schaffer, J.V. Congenital melanocytic nevi—When to worry and how to treat: Facts and controversies. Clin. Dermatol. 2010, 28, 293–302. [Google Scholar] [CrossRef]
- Busam, K.J.; Shah, K.N.; Gerami, P.; Sitzman, T.; Jungbluth, A.A.; Kinsler, V. Reduced H3K27me3 Expression Is Common in Nodular Melanomas of Childhood Associated With Congenital Melanocytic Nevi But Not in Proliferative Nodules. Am. J. Surg. Pathol. 2017, 41, 396–404. [Google Scholar] [CrossRef]
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 2012, 150, 1135–1146. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, O.; Fraitag, S.; Hohl, D. 5-Hydroxymethylcytosine Expression in Proliferative Nodules Arising within Congenital Nevi Allows Differentiation from Malignant Melanoma. J. Investig. Dermatol. 2016, 136, 2453–2461. [Google Scholar] [CrossRef] [Green Version]
- Lacoste, C.; Avril, M.-F.; Frassati-Biaggi, A.; Dupin, N.; Chrétien-Marquet, B.; Mahé, E.; Bodemer, C.; Vergier, B.; Fouchardière, A.; Fraitag, S. Malignant Melanoma Arising in Patients with a Large Congenital Melanocytic Naevus: Retrospective Study of 10 Cases with Cytogenetic Analysis. Acta Derm. Venereol. 2015, 95, 686–690. [Google Scholar] [CrossRef] [Green Version]
- Feito-Rodríguez, M.; de Lucas-Laguna, R.; Bastian, B.C.; Leboit, P.; González-Beato, M.J.; López-Gutiérrez, J.C.; Requena, L.; Pizarro, A. Nodular lesions arising in a large congenital melanocytic naevus in a newborn with eruptive disseminated Spitz naevi. Br. J. Dermatol. 2011, 165, 1138–1142. [Google Scholar] [CrossRef]
- Macagno, N.; Etchevers, H.C.; Malissen, N.; Rome, A.; Hesse, S.; Mallet, S.; Degardin, N.; Gaudy, C. Reduced H3K27me3 Expression is Common in Nodular Melanomas of Childhood Associated With Congenital Melanocytic Nevi But Not in Proliferative Nodules. Am. J. Surg. Pathol. 2018, 42, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Hayes-Jordan, A.; Green, H.; Prieto, V.; Wolff, J.E. Unusual cases: Melanomatosis and nephroblastomatosis treated with hyperthermic intraperitoneal chemotherapy. J. Pediatr. Surg. 2012, 47, 782–787. [Google Scholar] [CrossRef]
- Cajaiba, M.M.; Benjamin, D.; Halaban, R.; Reyes-Múgica, M. Metastatic peritoneal neurocutaneous melanocytosis. Am. J. Surg. Pathol. 2008, 32, 156–161. [Google Scholar] [CrossRef]
- Faillace, W.J.; Okawara, S.H.; McDonald, J.V. Neurocutaneous melanosis with extensive intracerebral and spinal cord involvement. Report of two cases. J. Neurosurg. 1984, 61, 782–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswamy, V.; Delaney, H.; Haque, S.; Marghoob, A.; Khakoo, Y. Spectrum of central nervous system abnormalities in neurocutaneous melanocytosis. Dev. Med. Child Neurol. 2012, 54, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Mugica, M.; Chou, P.; Byrd, S.; Ray, V.; Castelli, M.; Gattuso, P.; Gonzalez-Crussi, F. Nevomelanocytic proliferations in the central nervous system of children. Cancer 1993, 72, 2277–2285. [Google Scholar] [CrossRef]
- Cassarino, D.S.; Fullen, D.R.; Sondak, V.K.; Duray, P.H. Metastatic nevoid melanoma in a 4 1/2-year-old child. J. Cutan. Pathol. 2003, 30, 647–651. [Google Scholar] [CrossRef]
- Knöpfel, N.; Martín-Santiago, A.; Del Pozo, L.J.; Saus, C.; Pascual, M.; Requena, L. Amelanotic naevoid melanoma in a 16-month-old albino infant. Clin. Exp. Dermatol. 2017, 42, 84–88. [Google Scholar] [CrossRef]
- Moyer, A.B.; Diwan, A.H. “Puffy shirt appearance”: Cell crowding at low magnification may represent nevoid melanoma. J. Cutan. Pathol. 2019, 46, 805–809. [Google Scholar] [CrossRef]
- Idriss, M.H.; Rizwan, L.; Sferuzza, A.; Wasserman, E.; Kazlouskaya, V.; Elston, D.M. Nevoid melanoma: A study of 43 cases with emphasis on growth pattern. J. Am. Acad. Dermatol. 2015, 73, 836–842. [Google Scholar] [CrossRef]
- Goldstein, A.M.; Chan, M.; Harland, M.; Hayward, N.K.; Demenais, F.; Bishop, D.T.; Azizi, E.; Bergman, W.; Scarrà, G.B.; Bruno, W.; et al. Features associated with germline CDKN2A mutations: A GenoMEL study of melanoma-prone families from three continents. J. Med. Genet. 2007, 44, 99–106. [Google Scholar] [CrossRef]
- Casula, M.; Paliogiannis, P.; Ayala, F.; De Giorgi, V.; Stanganelli, I.; Mandalà, M.; Colombino, M.; Manca, A.; Sini, M.C.; Caracò, C.; et al. Germline and somatic mutations in patients with multiple primary melanomas: A next generation sequencing study. BMC Cancer 2019, 19, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouenne, F.; de Beauchene, I.C.; Bollaert, E.; Avril, M.-F.; Caron, O.; Ingster, O.; Lecesne, A.; Benuisiglio, P.; Terrier, P.; Caumette, V.; et al. Germline CDKN2A/P16INK4A mutations contribute to genetic determinism of sarcoma. J. Med. Genet. 2017, 54, 607–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollipara, R.; Cooley, L.D.; Horii, K.A.; Hetherington, M.L.; Leboit, P.E.; Singh, V.; Zwick, D.L. Spitzoid melanoma in a child with Li-Fraumeni syndrome. Pediatr. Dev. Pathol. 2014, 17, 64–69. [Google Scholar] [CrossRef]
- Jacquemus, J.; Perron, E.; Pissaloux, D.; Alberti, L.; de la Fouchardière, A. Atypical cutaneous melanocytic tumours arising in two patients with Li-Fraumeni syndrome. Pathology 2017, 49, 801–805. [Google Scholar] [CrossRef]
- Schaff, L.R.; Marghoob, A.; Rosenblum, M.K.; Meyer, R.; Khakoo, Y. Malignant transformation of neurocutaneous melanosis (NCM) following immunosuppression. Pediatr. Dermatol. 2019, 36, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Berge, L.A.M.; Andreassen, B.K.; Stenehjem, J.S.; Heir, T.; Karlstad, Ø.; Juzeniene, A.; Ghiasvand, R.; Larsen, I.K.; Green, A.C.; Veierød, M.B.; et al. Use of Immunomodulating Drugs and Risk of Cutaneous Melanoma: A Nationwide Nested Case-Control Study. Clin. Epidemiol. 2020, 12, 1389–1401. [Google Scholar] [CrossRef] [PubMed]
- Fattouh, K.; Ducroux, E.; Decullier, E.; Kanitakis, J.; Morelon, E.; Boissonnat, P.; Sebbag, L.; Jullien, D.; Euvrard, S. Increasing incidence of melanoma after solid organ transplantation: A retrospective epidemiological study. Transpl. Int. 2017, 30, 1172–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Molecular Alteration | Function | Frequence | References |
|---|---|---|---|---|
| NTRK1 | Gene fusion | Receptor tyrosine kinase | Common | [10,11,12] |
| HRAS | Mutation amplification | Serine/Threonine kinase | Common | [13] |
| ROS1 | Gene fusion | Receptor tyrosine kinase | Common | [14,15,16,17] |
| ALK | Gene fusion | Receptor tyrosine kinase | Common | [10,12,18,19] |
| RET | Gene fusion | Receptor tyrosine kinase | Rare | [10] |
| NTRK3 | Gene fusion | Receptor tyrosine kinase | Common | [20,21] |
| NTRK2 | Gene fusion | Receptor tyrosine kinase | Rare | [22] |
| MAP3K8 | Gene fusion | Serine/Threonine kinase | Common | [23,24,25,26] |
| BRAF | Gene fusion | Serine/Threonine kinase | Uncommon | [12,25,27,28,29] |
| MET | Gene fusion | Receptor tyrosine kinase | Rare | [30,31] |
| ERBB4 | Gene fusion | Receptor tyrosine kinase | Rare | [24] |
| FGFR1 | Gene fusion | Receptor tyrosine kinase | Rare | [24] |
| LCK | Gene fusion | Tyrosine kinase | Rare | [9] |
| MAP2K1 | Missense mutation | Serine/Threonine kinase | Rare | [32] |
| MAP3K3 | Gene fusion | Serine/Threonine kinase | Rare | [24] |
| MERTK | Gene fusion | Receptor tyrosine kinase | Rare | [16] |
| PRKDC | Gene fusion | Serine/Threonine kinase | Rare | [24] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de la Fouchardière, A.; Boivin, F.; Etchevers, H.C.; Macagno, N. Cutaneous Melanomas Arising during Childhood: An Overview of the Main Entities. Dermatopathology 2021, 8, 301-314. https://0-doi-org.brum.beds.ac.uk/10.3390/dermatopathology8030036
de la Fouchardière A, Boivin F, Etchevers HC, Macagno N. Cutaneous Melanomas Arising during Childhood: An Overview of the Main Entities. Dermatopathology. 2021; 8(3):301-314. https://0-doi-org.brum.beds.ac.uk/10.3390/dermatopathology8030036
Chicago/Turabian Stylede la Fouchardière, Arnaud, Felix Boivin, Heather C. Etchevers, and Nicolas Macagno. 2021. "Cutaneous Melanomas Arising during Childhood: An Overview of the Main Entities" Dermatopathology 8, no. 3: 301-314. https://0-doi-org.brum.beds.ac.uk/10.3390/dermatopathology8030036