A New Cytotoxic Dimeric Sesquiterpene Isolated from Inula racemosa Hook. f. (Root): In Vitro and In Silico Analyses



 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Instruments
2.3. Chemicals and Reagents
2.4. Preparation of Methanolic Extract
2.5. Fractionation and Isolation of Phytoconstituents
2.6. Cytotoxicity Assay of Compound 1
2.6.1. Cell Culture and Treatments
2.6.2. Cytotoxicity Assay
2.7. Molecular Docking and Molecular Dynamics Simulation
2.8. Statistical Analysis
3. Results and Discussion
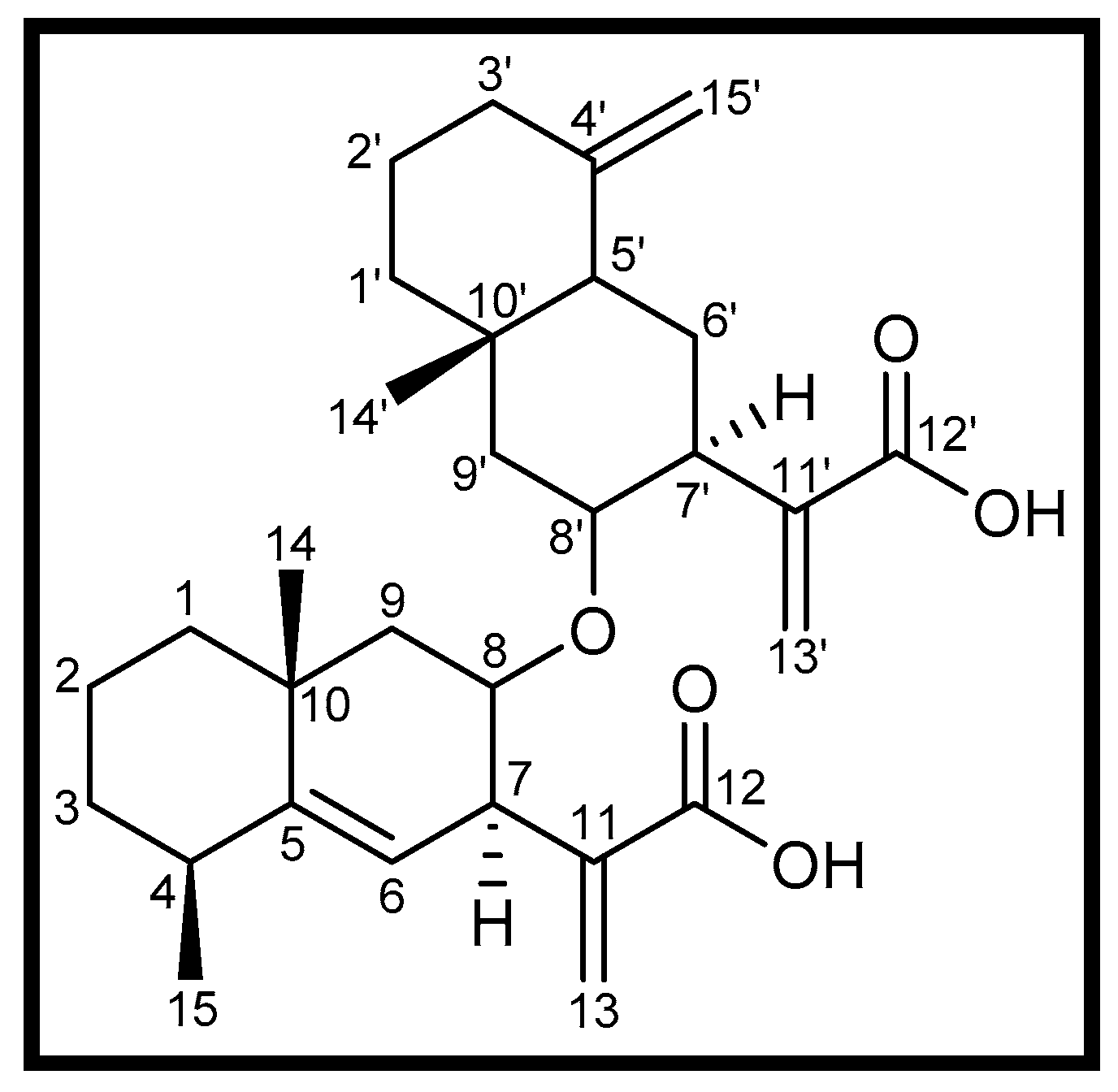
3.1. Isolation of Compound 1
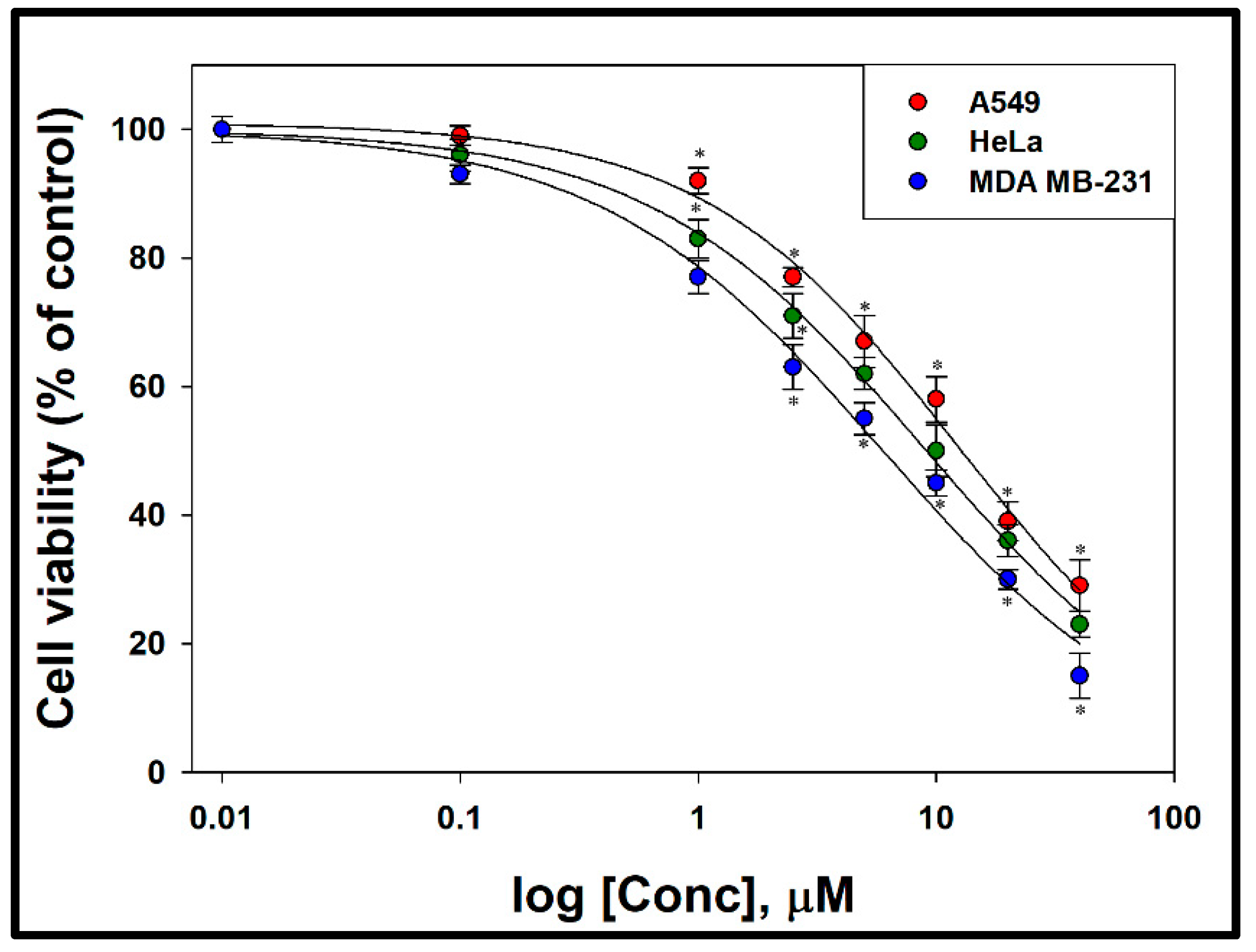
3.2. Cytotoxic Effect of Compound 1
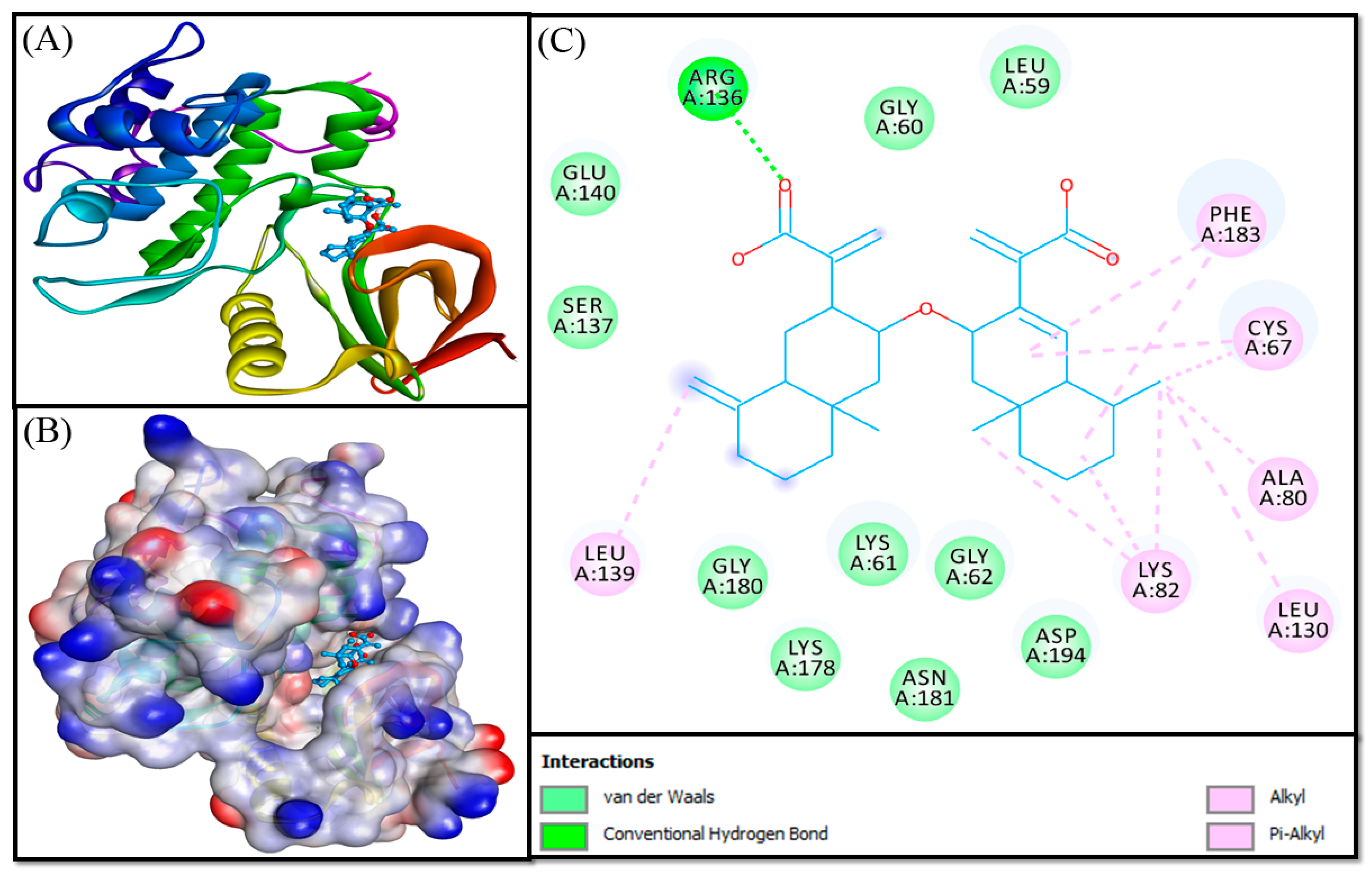
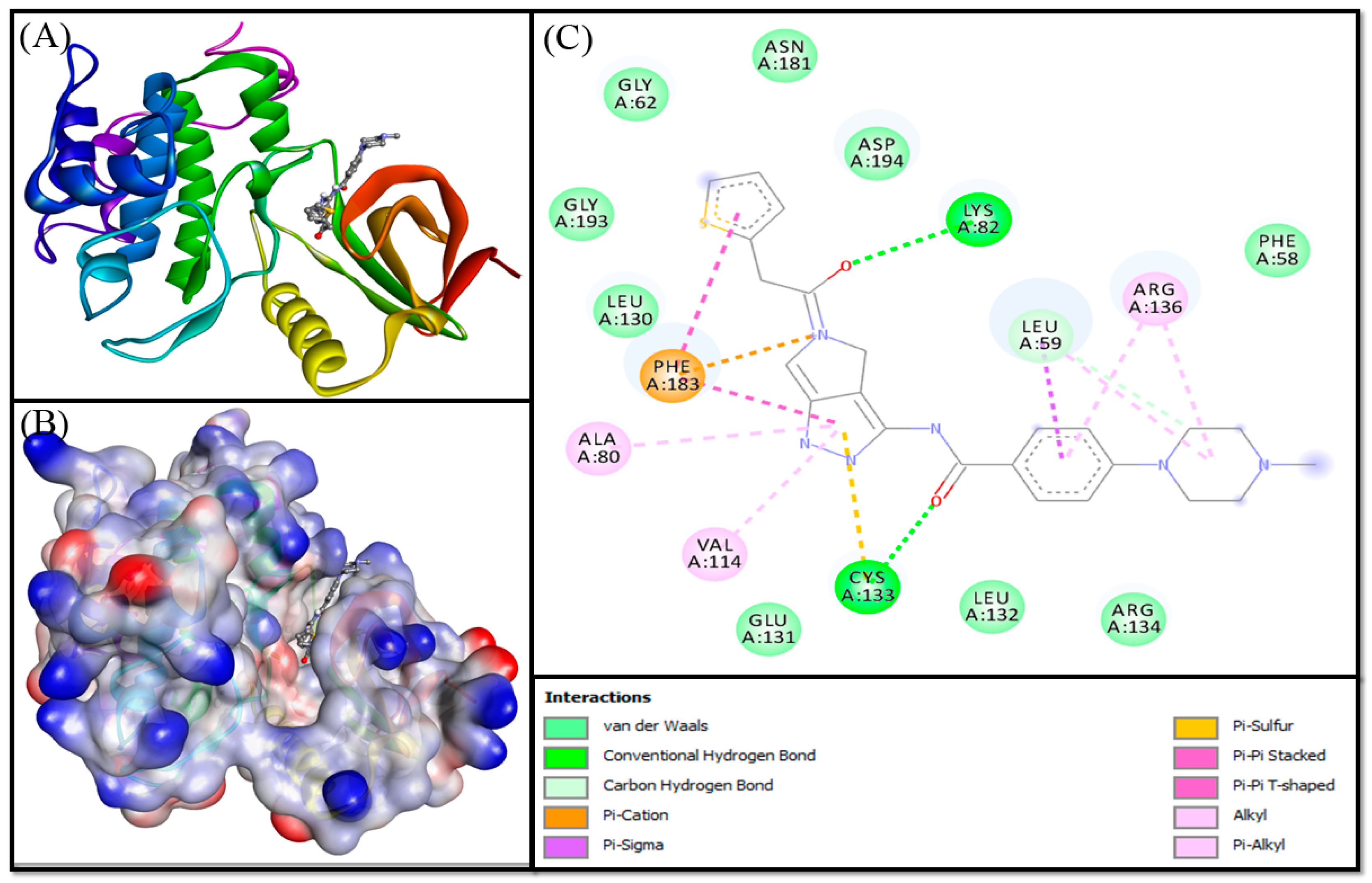
3.3. Molecular Docking Analysis
3.4. Molecular Dynamics Simulation Analysis
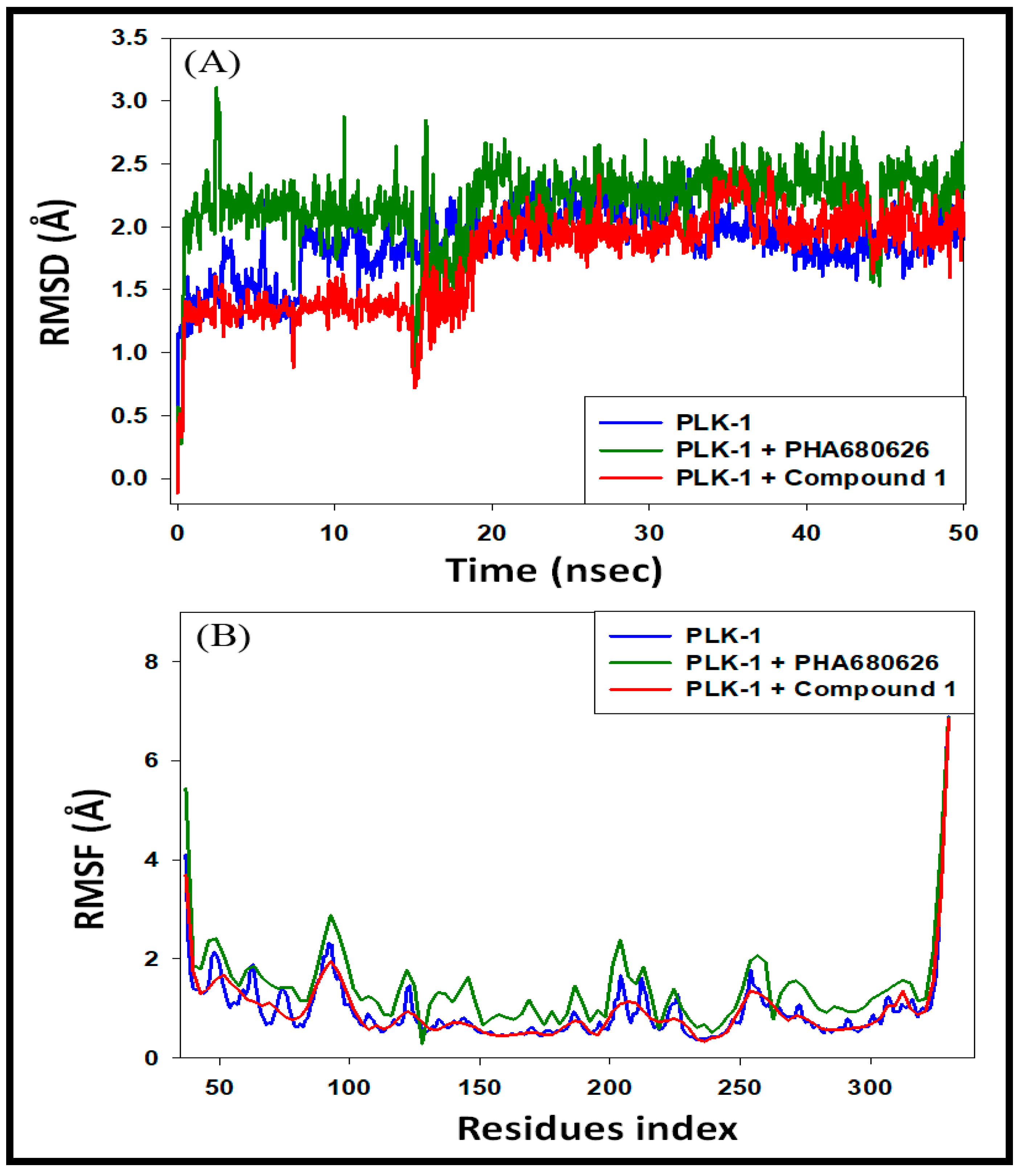
3.4.1. Analysis of RMSD and RMSF
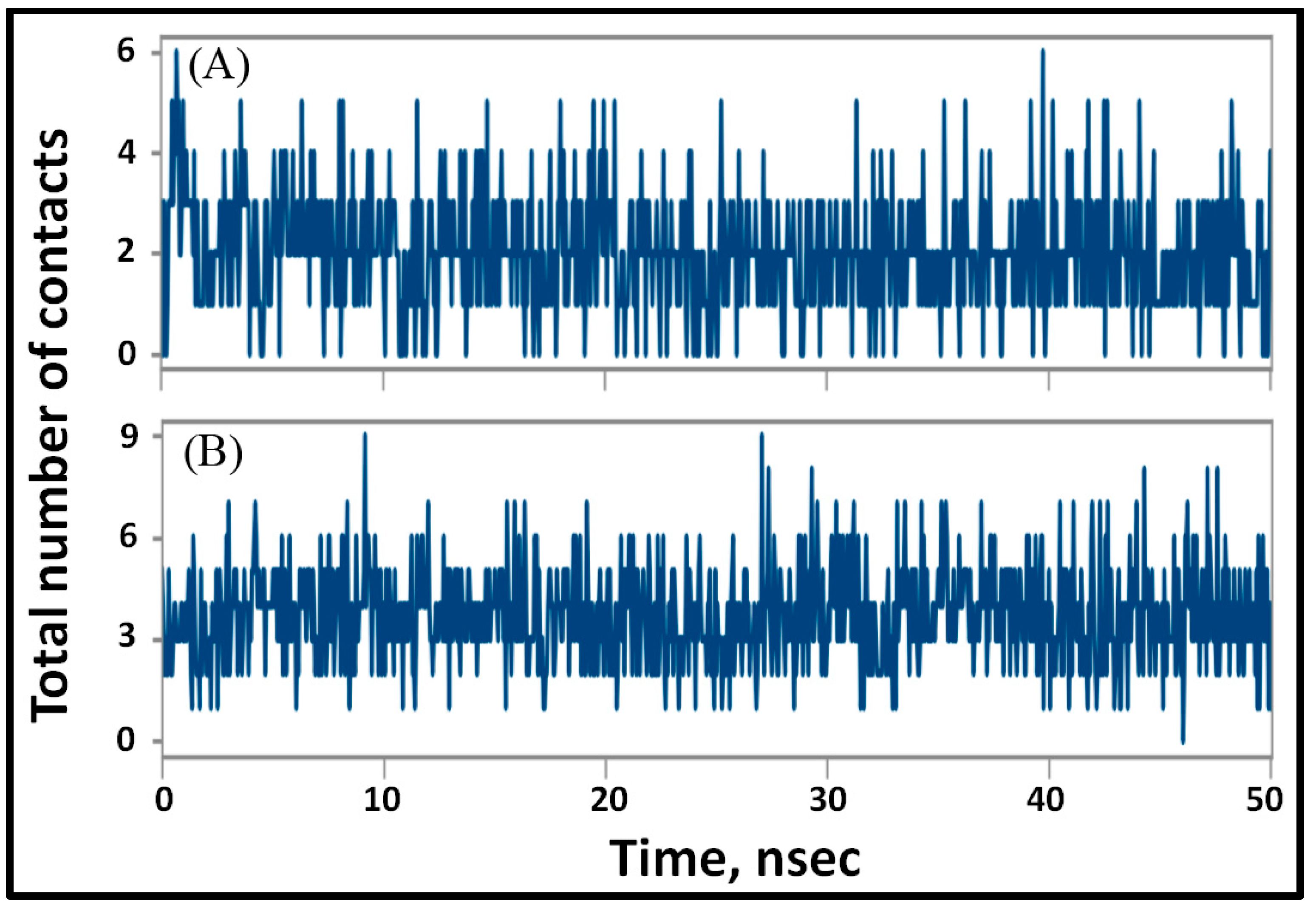
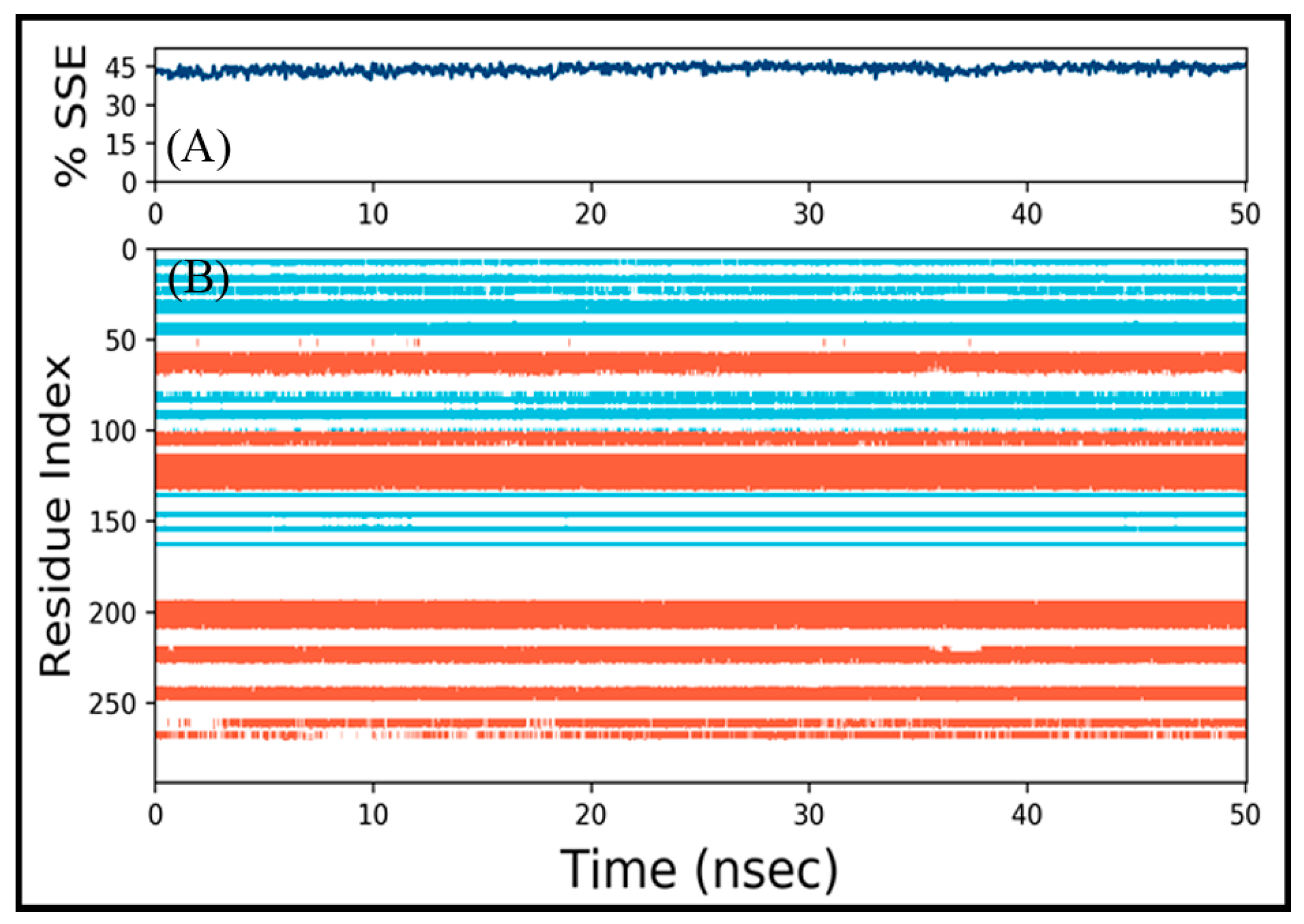
3.4.2. Analysis of Structural Changes
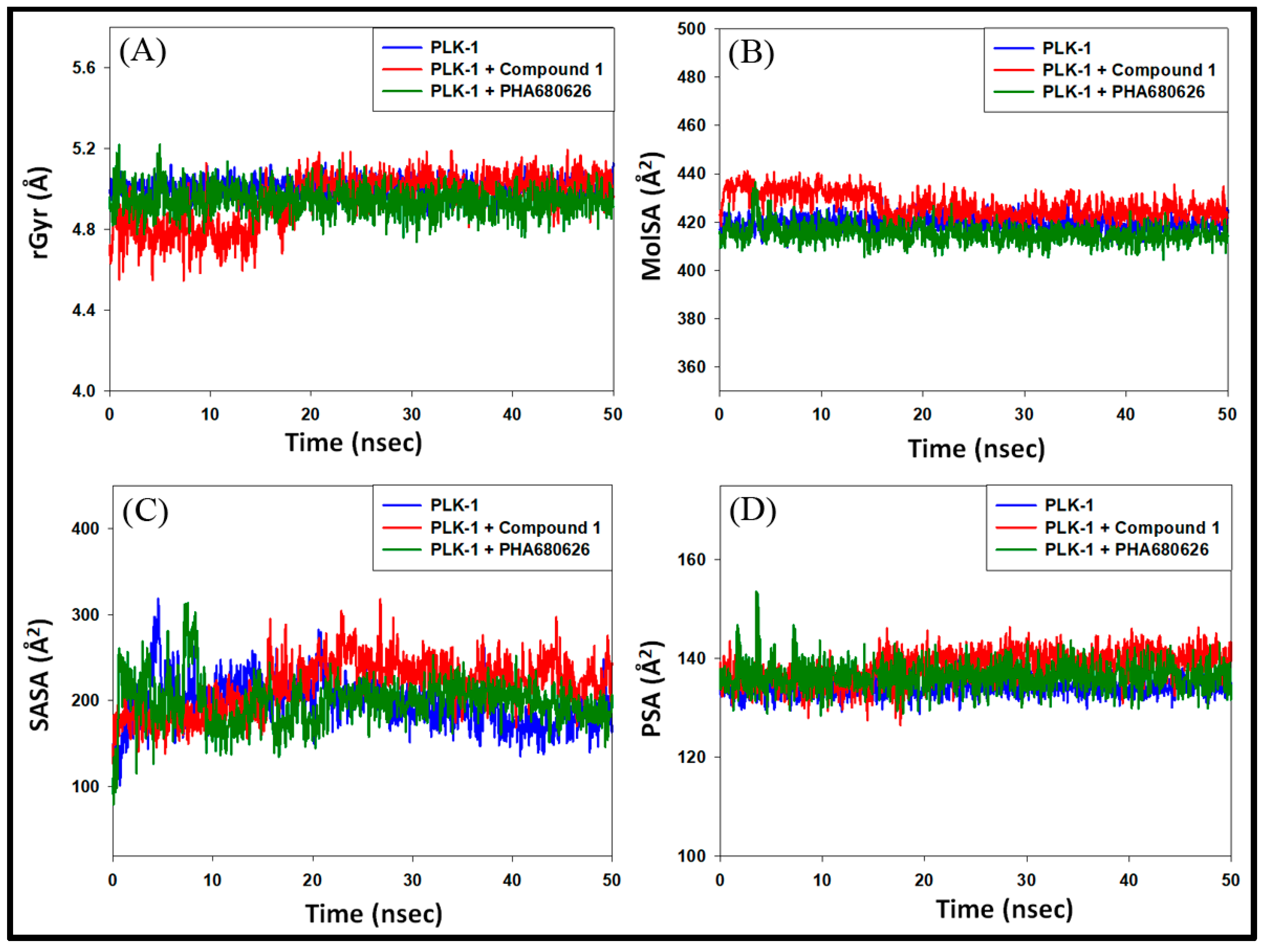
3.4.3. Analysis of Radius of Gyration (rGyr) and Surface Areas
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sotillo, W.S.; Villagomez, R.; Smiljanic, S.; Huang, X.; Malakpour, A.; Kempengren, S.; Rodrigo, G.; Almanza, G.; Sterner, O.; Oredsson, S. Anti-cancer stem cell activity of a sesquiterpene lactone isolated from Ambrosia arborescens and of a synthetic derivative. PLoS ONE 2017, 12, e0184304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.S.; Burke, T.R., Jr.; Park, J.E.; Bang, J.K.; Lee, E. Recent advances and new strategies in targeting PLK-1 for anticancer therapy. Trends Pharm. Sci. 2015, 36, 858–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, N.; Miyazaki, T.; Fujisawa, K.; Nasu, K.; Hamanaka, R.; Miyakawa, I. Expression of polo-like kinase in ovarian cancer is associated with histological grade and clinical stage. Cancer Lett. 2001, 164, 41–49. [Google Scholar] [CrossRef]
- Weichert, W.; Kristiansen, G.; Winzer, K.-J.; Schmidt, M.; Gekeler, V.; Noske, A.; Müller, B.-M.; Niesporek, S.; Dietel, M.; Denkert, C. Polo-like kinase isoforms in breast cancer: Expression patterns and prognostic implications. Virchows Arch. 2005, 446, 442–450. [Google Scholar] [CrossRef]
- Kneisel, L.; Strebhardt, K.; Bernd, A.; Wolter, M.; Binder, A.; Kaufmann, R. Expression of polo-like kinase (PLK1) in thin melanomas: A novel marker of metastatic disease. J. Cutan. Pathol. 2002, 29, 354–358. [Google Scholar] [CrossRef]
- Gach, K.; Długosz, A.; Janecka, A. The role of oxidative stress in anticancer activity of sesquiterpene lactones. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2015, 388, 477–486. [Google Scholar] [CrossRef]
- Firdous, Q.; Bhat, M.F.; Hussain, M.M. Ethnopharmacology, Phytochemistry and Biological activity of inula racemosa hook. f: A review. Int. J. Res. Ayurveda Pharm. 2018, 9, 95–102. [Google Scholar] [CrossRef]
- Arumugam, P.; Murugan, M. Antimutagenic and Antiapoptotic Effects of Aqueous Root Extract of Inula racemosa Hook. f. on 4-NQO-Induced Genetic Damage in Mice. ISRN Pharmacol. 2013, 2013, 1–5. [Google Scholar] [CrossRef]
- Bohlmann, F.; Mahanta, P.K.; Jakupovic, J.; Rastogi, R.C.; Natu, A.A. New sesquiterpene lactones from Inula species. Phytochemistry 1978, 17, 1165–1172. [Google Scholar] [CrossRef]
- Nguyen, S.T.; Nguyen, H.T.-L.; Truong, K.D. Comparative cytotoxic effects of methanol, ethanol and DMSO on human cancer cell lines. Biomed. Res. Ther. 2020, 7, 3855–3859. [Google Scholar] [CrossRef]
- Alajmi, M.F.; Rehman, T.; Hussain, A.; Rather, G.M. Pharmacoinformatics approach for the identification of Polo-like kinase-1 inhibitors from natural sources as anti-cancer agents. Int. J. Biol. Macromol. 2018, 116, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.T.; Ahmed, S.; Khan, A.U. Interaction of meropenem with ’N’ and ’B’ isoforms of human serum albumin: A spectroscopic and molecular docking study. J. Biomol. Struct. Dyn. 2016, 34, 1849–1864. [Google Scholar] [CrossRef] [PubMed]
- Brańka, A.C. Nosé-Hoover chain method for nonequilibrium molecular dynamics simulation. Phys. Rev. E 2000, 61, 4769–4773. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Bhat, S.A.; Rehman, T.; Hassan, I.; Tabrez, S.; Alajmi, M.F.; Hussain, A.; Husain, F.M.; Alamery, S.F. Rutin attenuates negatively charged surfactant (SDS)-induced lysozyme aggregation/amyloid formation and its cytotoxicity. Int. J. Biol. Macromol. 2018, 120, 45–58. [Google Scholar] [CrossRef]
- AlShabib, N.A.; Khan, J.M.; Malik, A.; AlSenaidy, M.A.; Rehman, M.T.; AlAjmi, M.F.; AlSenaidy, A.M.; Husain, F.M.; Khan, R.H. Molecular insight into binding behavior of polyphenol (rutin) with beta lactoglobulin: Spectroscopic and computational studies. J. Mol. Liq. 2018, 269, 511–520. [Google Scholar] [CrossRef]
- Rehman, T.; Shamsi, H.; Khan, A.U. Insight into the Binding Mechanism of Imipenem to Human Serum Albumin by Spectroscopic and Computational Approaches. Mol. Pharm. 2014, 11, 1785–1797. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Qin, J.J.; Jin, H.Z.; Fu, J.J.; Hu, J.J.; Wang, Y.; Yan, S.K. Bioactive dimeric sesquiterpenes from Inula japonica thumb. Bioorg. Med. Chem. Lett. 2009, 19, 710–713. [Google Scholar] [CrossRef]
- Qin, J.J.; Jin, H.Z.; Zhu, X.J.; Fu, J.J.; Hu, X.Z.; Liu, X.H.; Zhu, Y.; Yan, K.S.; Zhang, D.W. Neojaponicone A, a bioactive sesquiterpene lactone dimer with an unprecedented carbon skeleton from Inula japonica. Chem. Commun. 2011, 47, 1222–1224. [Google Scholar] [CrossRef]
- Qin, J.-J.; Jin, H.; Zhu, J.; Fu, J.; Hu, X.; Liu, X.; Zhu, Y.; Yan, S.; Zhang, W.-D. Japonicones E–L, Dimeric Sesquiterpene Lactones fromInula japonica Thunb. Planta Med. 2009, 76, 278–283. [Google Scholar] [CrossRef]
- Xu, X.Y.; Sun, P.; Guo, D.A.; Liu, X.; Liu, J.H.; Hu, L.H. Cytotoxic sesquiterpene lactone dimmers isolated from Inula japonica. Fitoterapia 2015, 101, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Kothe, M.; Kohls, D.; Low, S.; Coli, R.; Cheng, A.C.; Jacques, S.L.; Johnson, T.L.; Lewis, C.; Loh, C.; Nonomiya, J.; et al. Structure of the Catalytic Domain of Human Polo-like Kinase 1. Biochemistry 2007, 46, 5960–5971. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δH (mult., J/Hz) | δC Type | Position | δH (mult., J/Hz) | δC Type |
|---|---|---|---|---|---|
| 1 | 2.18, 2.16 | 41.8 CH2 | 1′ | 1.31, 1.01 | 36.9 CH2 |
| 2 | 1.25 | 27.5 CH2 | 2′ | 1.23 | 16.8 CH2 |
| 3 | 2.31 dt (9.2, 1.5) | 32.8 CH2 | 3′ | 2.10 dd (10.6, 1.9) | 41.4 CH2 |
| 4 | 2.43 m | 37.7 CH | 4′ | - | 149.1 C |
| 5 | - | 149.0 C | 5′ | 1.83 d (9.0) | 46.2 CH |
| 6 | 5.13 d (2.9) | 118.8 CH | 6′ | 1.50, 1.37 | 22.7 CH2 |
| 7 | 3.55 m | 39.5 CH | 7′ | 2.95 m | 40.6 CH |
| 8 | 4.81 m | 76.6 CH | 8′ | 4.48 m | 77.0 CH |
| 9 | 1.72, 1.54 | 42.2 CH2 | 9′ | 1.74, 1.57 | 42.7 CH2 |
| 10 | - | 34.3 C | 10′ | - | 34.3 C |
| 11 | - | 139.9 C | 11′ | - | 142.2 C |
| 12 | - | 170.7 C | 12′ | - | 170.8 C |
| 13 | 6.18 s, 5.60 s | 121.8 CH2 | 13′ | 6.10 s, 5.57 s | 120.2 CH2 |
| 14 | 0.80 s | 17.7 CH3 | 14′ | 1.17 s | 28.7 CH3 |
| 15 | 1.08 d (5.4) | 22.6 CH3 | 15′ | 4.75 s, 4.42 s | 106.7 CH2 |
| Donor-Acceptor Pair | Distance (Å) | Type of Interaction | Docking Energy (kcal mol−1) | Docking Affinity, Kd (M−1) |
|---|---|---|---|---|
| Control (PHA-680626) | ||||
| LYS82:HZ3—Lig:O | 2.7055 | Conventional Hydrogen Bond | −10.000 | 2.16 × 107 |
| CYS133:HN—Lig:O | 2.0328 | Conventional Hydrogen Bond | ||
| Lig:C—A:LEU59:O | 3.3471 | Carbon Hydrogen Bond | ||
| Lig:N—A:PHE183 | 4.6259 | Electrostatic (Pi-Cation) | ||
| LEU59:CD1—Lig | 3.7176 | Hydrophobic (Pi-Sigma) | ||
| LEU59:CD2—Lig | 3.6603 | Hydrophobic (Pi-Sigma) | ||
| CYS133:SG—Lig | 4.9895 | Pi-Sulfur bond | ||
| Lig—A:PHE183 | 3.6552 | Hydrophobic (Pi-Pi Stacked) | ||
| Lig—A:PHE183 | 5.7048 | Hydrophobic (Pi-Pi T-shaped) | ||
| LEU59—Lig | 4.8864 | Hydrophobic (Alkyl) | ||
| ARG136—Lig | 5.2156 | Hydrophobic (Alkyl) | ||
| Lig—A:ALA80 | 4.552 | Hydrophobic (Pi-Alkyl) | ||
| Lig—A:VAL114 | 4.7727 | Hydrophobic (Pi-Alkyl) | ||
| Lig—A:ARG136 | 4.8572 | Hydrophobic (Pi-Alkyl) | ||
| Compound 1 | ||||
| ARG136:HE—Lig:O | 1.9478 | Conventional Hydrogen Bond | −8.930 | 3.54 × 106 |
| ARG136:HH21—Lig:O | 2.8755 | Conventional Hydrogen Bond | ||
| CYS67—Lig | 4.4491 | Hydrophobic (Alkyl) | ||
| LYS82—Lig | 5.0996 | Hydrophobic (Alkyl) | ||
| Lig:C—A:CYS67 | 4.0577 | Hydrophobic (Alkyl) | ||
| Lig:C—A:LYS82 | 4.5342 | Hydrophobic (Alkyl) | ||
| Lig:C—A:LEU130 | 4.5272 | Hydrophobic (Alkyl) | ||
| Lig:C—A:LYS82 | 4.4063 | Hydrophobic (Alkyl) | ||
| Lig:C—A:LEU139 | 5.0879 | Hydrophobic (Alkyl) | ||
| PHE183—Lig | 4.8694 | Hydrophobic (Pi-Alkyl) | ||
| PHE183—Lig | 5.0389 | Hydrophobic (Pi-Alkyl) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyagi, R.; Alam, P.; Rehman, M.T.; AlAjmi, M.F.; Hussain, A.; Amin, S.; Mujeeb, M.; Mir, S.R. A New Cytotoxic Dimeric Sesquiterpene Isolated from Inula racemosa Hook. f. (Root): In Vitro and In Silico Analyses. Separations 2021, 8, 2. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8010002
Tyagi R, Alam P, Rehman MT, AlAjmi MF, Hussain A, Amin S, Mujeeb M, Mir SR. A New Cytotoxic Dimeric Sesquiterpene Isolated from Inula racemosa Hook. f. (Root): In Vitro and In Silico Analyses. Separations. 2021; 8(1):2. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8010002
Chicago/Turabian StyleTyagi, Rama, Perwez Alam, Md. Tabish Rehman, Mohamed Fahad AlAjmi, Afzal Hussain, Saima Amin, Mohd. Mujeeb, and Showkat R. Mir. 2021. "A New Cytotoxic Dimeric Sesquiterpene Isolated from Inula racemosa Hook. f. (Root): In Vitro and In Silico Analyses" Separations 8, no. 1: 2. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8010002