
Optimization of Plant Extract Purification Procedure for Rapid Screening Analysis of Sixteen Phenolics by Liquid Chromatography


Abstract
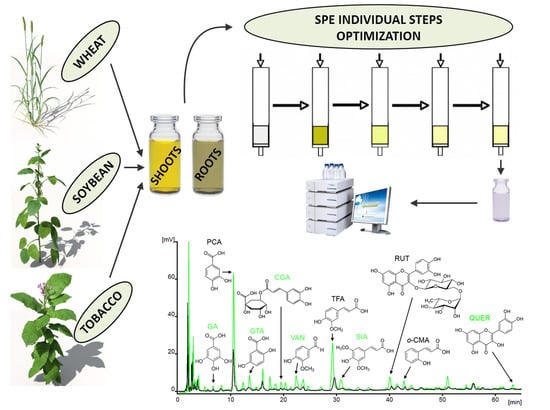
:
1. Introduction
2. Experimental
2.1. Chemicals
2.2. Preparation of Standard Solutions, Calibration Standards, and Model Mixtures
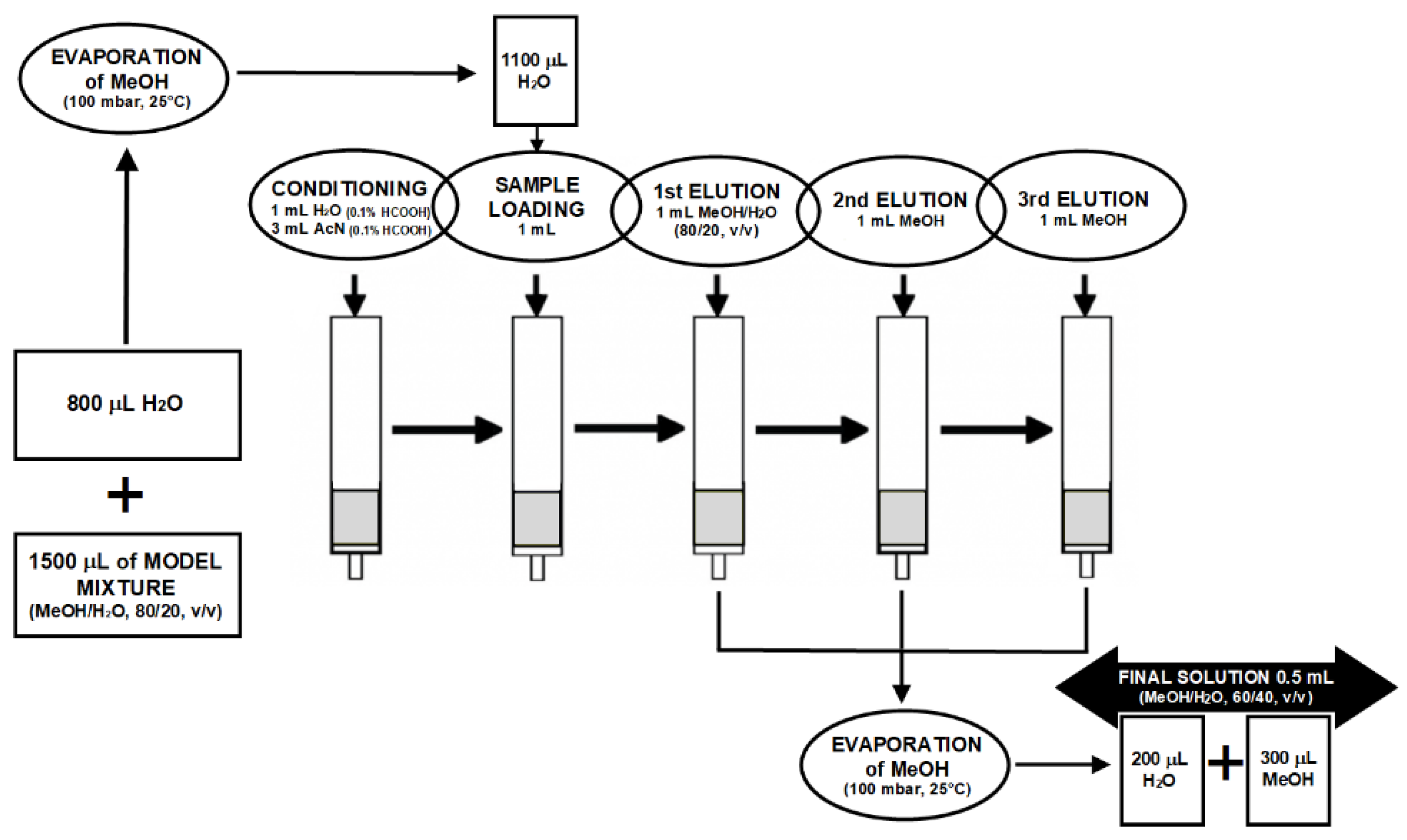
2.3. Preparation of SPE Columns
2.4. HPLC Analysis
2.5. Preparation of Plant Extracts
3. Results and Discussion
3.1. SPE Optimization
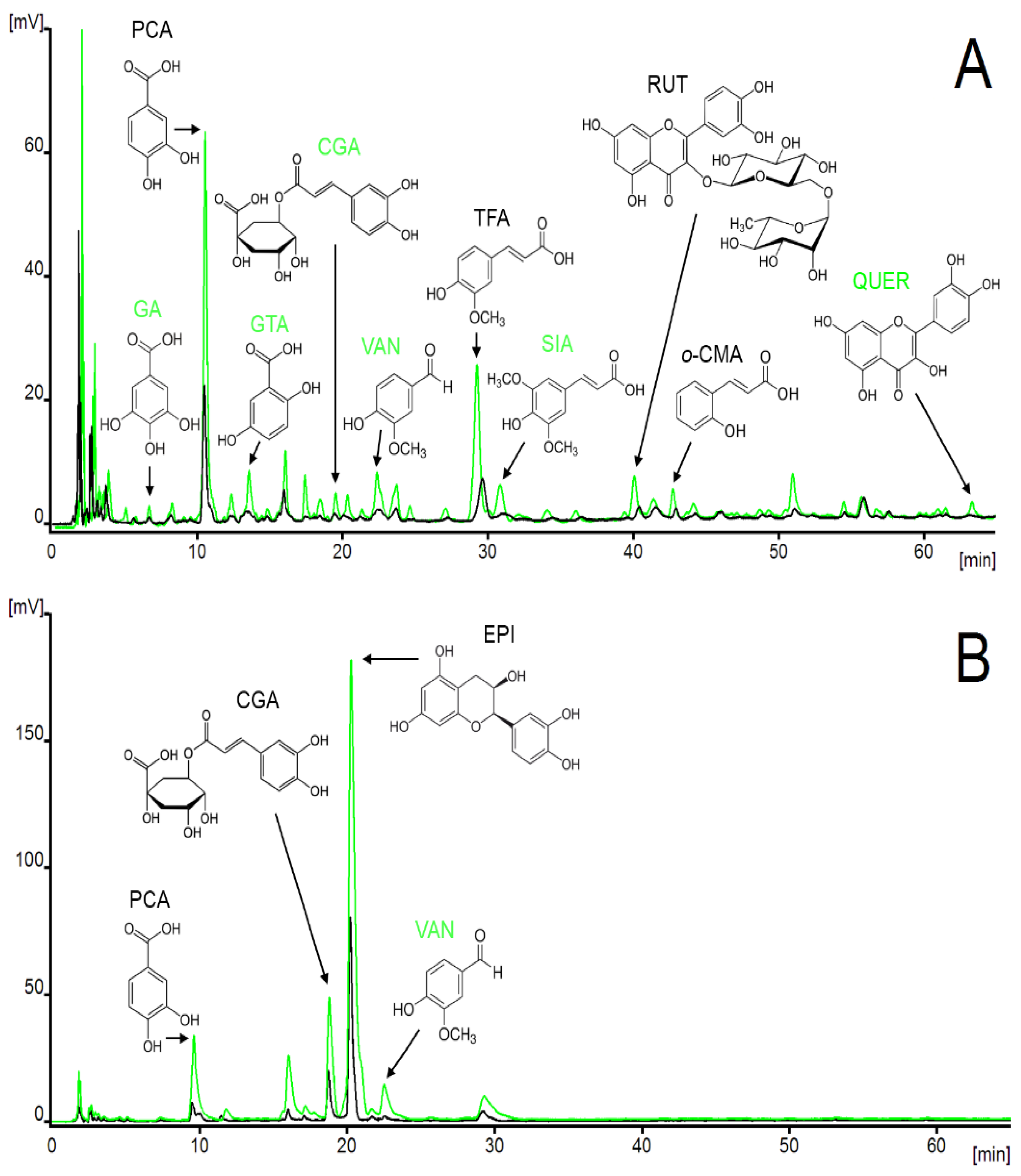
3.2. Application of SPE Method to Various Plant Extracts
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kumar, N.; Goel, N. Phenolic acids: Natural versatile molecules with promising therapeutic applications. Biotechnol. Rep. 2019, 24, e00370. [Google Scholar] [CrossRef]
- Kulichová, K.; Maliarová, M.; Sokol, J.; Lašáková, K.; Havrlentová, M. Determination of selected phenolic acid and majoritarian avenanthramides in different varieties of naked oats (Avena sativa L.) grown in Slovakia. Nova Biotechnol. Chim. 2018, 17, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Sokamte, T.A.; Mbougueng, P.D.; Tatsadjieu, N.L.; Sachindra, N.M. Phenolic compounds characterization and antioxidant activities of selected spices from Cameroon. S. Afr. J. Bot. 2019, 121, 7–15. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Y.; Liang, Y.; Liang, K.; Zhang, F.; Xu, T.; Wang, M.; Song, H.; Liu, X.; Lu, B. Determination of phenolic acid profiles by HPLC-MS in vegetables commonly consumed in China. Food Chem. 2019, 276, 538–546. [Google Scholar] [CrossRef]
- Ding, T.; Cao, K.; Fang, W.; Zhu, G.; Chen, C.; Wang, X.; Wang, L. Evaluation of phenolic components (anthocyanins, flavanols, phenolic acids, and flavonols) and their antioxidant properties of peach fruits. Sci. Hortic. 2020, 268, 109365. [Google Scholar] [CrossRef]
- Lukić, I.; Radeka, S.; Budić-Leto, I.; Bubola, M.; Vrhovsek, U. Targeted UPLC-QqQ-MS/MS profiling of phenolic compounds for differentiation of monovarietal wines and corroboration of particular varietal typicity concepts. Food Chem. 2019, 300, 125251. [Google Scholar] [CrossRef]
- Liu, Z.; Bruins, M.E.; De Bruijn, W.J.C.; Vincken, J.P. A comparison of the phenolic composition of old and young tea leaves reveals a decrease in flavanols and phenolic acids and an increase in flavonols upon tea leaf maturation. J. Food Compos. Anal. 2020, 86, 103385. [Google Scholar] [CrossRef]
- Esteban Muñoz, A.; Sánchez Hernández, S.; Recio Tolosa, A.; Pérez Burillo, S.; Olalla Herrera, M. Evaluation of differences in the antioxidant capacity and phenolic compounds of green and roasted coffee and their relationship with sensory properties. Food Sci. Technol. 2020, 128, 109457. [Google Scholar]
- Lin, D.; Xiao, M.; Zhao, J.; Li, Z.; Xing, B.; Li, X.; Kong, M.; Li, L.; Zhang, Q.; Liu, Y.; et al. An overview of plant phenolic compounds and their importance in human nutrition and management of type 2 diabetes. Molecules 2016, 21, 1374. [Google Scholar] [CrossRef]
- Abbas, M.; Saeed, F.; Anjum, F.M.; Afzaal, M.; Tufail, T.; Bashir, M.S.; Ishtiaq, A.; Hussian, S.; Suleria, H.A.R. Natural polyphenols: An overview. Int. J. Food Prop. 2017, 20, 1689–1699. [Google Scholar] [CrossRef] [Green Version]
- Ramesh Kumar, B. Application of HPLC and ESI-MS techniques in the analysis of phenolic acids and flavonoids from green leafy vegetables (GLVs). J. Pharm. Anal. 2017, 7, 349–364. [Google Scholar] [CrossRef]
- Domínguez-Rodríguez, G.; Marina, M.L.; Plaza, M. Strategies for the extraction and analysis of non-extractable polyphenols from plants. J. Chromatogr. A 2017, 1514, 1–15. [Google Scholar] [CrossRef]
- Tanase, C.; Coșarcă, S.; Muntean, D.L. A critical review of phenolic compounds extracted from the bark of woody vascular plants and their potential biological activity. Molecules 2019, 24, 1182. [Google Scholar] [CrossRef] [Green Version]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant. Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef]
- Márquez-García, B.; Ángeles Fernández-Recamales, M.; Córdoba, F. Effects of cadmium on phenolic composition and antioxidants activities of Erica andevalensis. J. Bot. 2012. [Google Scholar] [CrossRef] [Green Version]
- Ali, N.; Hadi, F. Phytoremediation of cadmium improved with the high production of endogenous phenolics and free proline contents in Parthenium hysterophorus plant treated exogenously with plant growth regulator and chelating agents. Environ. Sci. Pollut. Res. 2015, 22, 13305–13318. [Google Scholar] [CrossRef]
- Kuppusamy, P.; Lee, K.D.; Song, C.E.; Ilavenil, S.; Srigopalram, S.; Arasu, M.V.; Choi, K.C. Quantification of major phenolic and flavonoid markers in forage crop Lolium multiflorum using HPLC-DAD. Rev. Bras. Farmacogn. 2018, 28, 282–288. [Google Scholar] [CrossRef]
- Gong, L.; Chi, J.; Zhang, Y.; Wang, J.; Sun, B. In vitro evaluation of the bioaccessibility of phenolic acids in different whole wheats as potential prebiotics. Food Sci. Technol. 2019, 100, 435–443. [Google Scholar] [CrossRef]
- Ripari, V.; Baia, Y.; Gänzle, M.G. Metabolism of phenolic acids in whole wheat and rye malt sourdoughs. Food Microbiol. 2019, 77, 43–51. [Google Scholar] [CrossRef]
- Gregorová, Z.; Kováčik, J.; Klejdus, B.; Maglovski, M.; Kuna, R.; Hauptvogel, P.; Matušíková, I. Drought-induced respones of physiology, metabolites, and PR proteins in Triticum aestivum. J. Agric. Food Chem. 2015, 63, 8125–8133. [Google Scholar] [CrossRef]
- Mongkhonsin, B.; Nakbanpote, W.; Hokura, A.; Nuengchamnong, N.; Maneechai, S. Phenolic compounds responding to zinc and/or cadmium treatments in Gynura pseudochina (L.) DC. extracts and biomass. Plant Physiol. Biochem. 2016, 109, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Lattanzio, V.; Di Venere, D.; Linsalata, V.; Lima, G.; Ippolito, A.; Salerno, M. Antifungal activity of 2,5-dimethoxybenzoic acid on postharvest pathogens of strawberry fruits. Postharvest Biol. Technol. 1996, 9, 325–334. [Google Scholar] [CrossRef]
- Ponce, M.A.; Bompadre, M.J.; Scervino, J.M.; Ocampo, J.A.; Chaneton, E.J.; Godeas, A.M. Flavonoids, benzoic acids and cinnamic acids isolated from shoots and roots of Italian rye grass (Lolium multiflorum Lam.) with and without endophyte association and arbuscular mycorrhizal fungus. Biochem. Syst. Ecol. 2009, 37, 245–253. [Google Scholar] [CrossRef]
- Hidalgo, W.; Kai, M.; Schneider, B. 4-Methoxycinnamic acid—An unusual phenylpropanoid involved in phenylphenalenone biosynthesis in Anigozanthos preissii. Phytochemistry 2015, 117, 476–481. [Google Scholar] [CrossRef] [Green Version]
- Stalikas, C.D. Extraction, separation and detection methods for phenolic acids and flavonoids. J. Sep. Sci. 2007, 30, 3268–3295. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, D.R. Optimum nutrient solutions for plants. Science 1920, 52, 562–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naczk, M.; Shahidi, F. Phenolics in cereals, fruits and vegetables: Occurrence, extraction and analysis. J. Pharm. Biomed. Anal. 2006, 41, 1523–1542. [Google Scholar] [CrossRef] [PubMed]
- Veljković, J.N.; Pavlović, A.N.; Mitić, S.S.; Tošić, S.B.; Stojanović, G.S.; Kaličanin, B.M.; Stanković, D.M.; Stojković, M.B.; Mitić, M.N.; Brcanović, J.M. Evaluation of individual phenolic compounds and antioxidant properties of black, green, herbal and fruit tea infusions consumed in Serbia: Spectrophotometrical and electrochemical approaches. J. Food Nutr. Res. 2013, 52, 12–24. [Google Scholar]
- Zhao, H.; Zhang, H.; Yang, S. Phenolic compounds and its antioxidant activities in ethanolic extracts from seven cultivars of Chinese jujube. Food Sci. Hum. Well. 2014, 3, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Dimcheva, V.; Kaloyanov, N.; Karsheva, M.; Funeva-Peycheva, M.; Stoilova, N. HPLC-DAD method for simultaneous determination of natural polyphenols. Open J. Anal. Bioanal. Chem. 2019, 3, 39–43. [Google Scholar] [CrossRef] [Green Version]
- González-González, R.M.; Barragán-Mendoza, L.; Peraza-Campos, A.L.; Muñiz-Valencia, R.; Ceballos-Magaña, S.G.; Parra-Delgado, H. Validation of an HPLC-DAD method for the determination of plant phenolics. Rev. Bras. Farmacogn. 2019, 29, 689–693. [Google Scholar] [CrossRef]
- De Magalhães, B.E.A.; Santana, D.D.A.; Silva, I.M.D.J.; Minho, L.A.C.; Gomes, M.A.; Almeida, J.R.G.D.S.; Lopes dos Santos, W.N. Determination of phenolic composition of oilseed whole flours by HPLC-DAD with evaluation using chemometric analyses. Microchem. J. 2020, 155, 104683. [Google Scholar] [CrossRef]
- Bajkacz, S.; Baranowska, I.; Buszewski, B.; Kowalski, B.; Ligor, M. Determination of flavonoids and phenolic acids in plant materials using SLE-SPE-UHPLC-MS/MS method. Food Anal. Methods 2018, 11, 3563–3575. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Zhang, P.; Warner, R.D.; Shen, S.; Johnson, S.; Fang, Z. Comprehensive profiling of phenolic compounds by HPLC-DAD-ESI-QTOF-MS/MS to reveal their location and form of presence in different sorghum grain. Food Res. Int. 2020, 137, 109671. [Google Scholar] [CrossRef] [PubMed]
- Guzmán-Ortiz, F.A.; San Martín-Martínez, E.; Valverde, M.E.; Rodríguez-Aza, Y.; De, J.; Berríos, J.; Mora-Escobedo, R. Profile analysis and correlation across phenolic compounds, isoflavones and antioxidant capacity during germination of soybeans (Glycine max L.). CyTA J. Food. 2017, 15, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Torras-Claveria, L.; Jáuregui, O.; Codina, C.; Tiburcio, A.F.; Bastida, J.; Viladomat, F. Analysis of phenolic compounds by high-performance liquid chromatography coupled to electrospray ionization tandem mass spectrometry in senescent and water-stressed tobacco. Plant Sci. 2012, 182, 71–78. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | tR (min) | Detection (nm) | Regression Equation | LOD (ng/mL) | LOQ (ng/mL) |
|---|---|---|---|---|---|
| GA | 5.14 | 280 | y = 1.347x + 0.384 R2 = 0.995 | 238.9 | 796.4 |
| PCA | 10.11 | 260 | y = 1.774x + 0.338 R2 = 0.997 | 138.7 | 462.3 |
| GTA | 13.89 | 240 | y = 0.475x + 0.069 R2 = 0.993 | 425.9 | 1419.8 |
| CA | 16.49 | 325 | y = 1.249x + 0.341 R2 = 0.997 | 482.5 | 1608.5 |
| VA | 17.45 | 260 | y = 1.455x − 0.072 R2 = 0.995 | 157.6 | 525.2 |
| CGA | 18.66 | 325 | y = 1.778x + 0.544 R2 = 0.995 | 150.0 | 500.0 |
| EPI | 21.08 | 240 | y = 0.574x − 0.038 R2 = 0.998 | 462.7 | 1542.5 |
| VAN | 22.27 | 280 | y = 1.180x + 0.131 R2 = 0.997 | 436.9 | 1456.4 |
| p-CMA | 25.60 | 290 | y = 1.978x − 0.228 R2 = 0.995 | 207.3 | 691.1 |
| TFA | 29.89 | 325 | y = 1.143x + 0.151 R2 = 0.996 | 545.5 | 1818.3 |
| SIA | 31.71 | 325 | y = 1.433x + 0.096 R2 = 0.997 | 421.2 | 1404.0 |
| DMBA | 35.72 | 240 | y = 1.549x − 1.424 R2 = 0.993 | 131.7 | 439.1 |
| RUT | 39.99 | 260 | y = 0.709x + 0.023 R2 = 0.993 | 340.3 | 1134.4 |
| o-CMA | 43.54 | 280 | y = 1.289x + 0.036 R2 = 0.997 | 375.8 | 1252.6 |
| 4MCA | 60.04 | 290 | y = 1.709x–0.033 R2 = 0.995 | 211.9 | 706.3 |
| QUER | 61.92 | 260 | y = 0.747x–0.044 R2 = 0.997 | 299.5 | 998.5 |
| Conditioning Solvents | GA | PCA | GTA | CA | VA | CGA | EPI | VAN | p-CMA | TFA | SIA | DMBA | RUT | o-CMA | 4MCA | QUER |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 mL MeOH 3 mL H2O | 0 | 0 | 0 | 1.8 | 4.7 | 5.1 | 7.6 | 9.0 | 11.1 | 14.9 | 15.2 | 27.5 | 47.7 | 49.3 | 50.4 | 58.3 |
| 3 mL MeOH * 3 mL H2O * | 0 | 0 | 0 | 3.7 | 7.4 | 7.1 | 8.6 | 9.7 | 14.5 | 16.7 | 18.9 | 29.3 | 49.6 | 51.0 | 52.8 | 59.1 |
| 3 mL MeOH 1 mL H2O | 0 | 0 | 4.6 | 12.8 | 15.9 | 16.1 | 17.2 | 17.9 | 18.6 | 19.5 | 19.4 | 30.9 | 57.1 | 58.7 | 59.6 | 64.5 |
| 3 mL MeOH * 1 mL H2O * | 0 | 0 | 5.9 | 14.9 | 17.3 | 18.7 | 18.8 | 18.9 | 20.8 | 23.4 | 24.8 | 38.8 | 63.5 | 64.0 | 64.1 | 69.6 |
| 3 mL H2O 3 mL MeOH | 0 | 0 | 0 | 4.6 | 8.6 | 8.8 | 9.2 | 10.1 | 15.2 | 17.7 | 18.0 | 28.8 | 50.5 | 52.4 | 57.8 | 67.6 |
| 3 mL H2O * 3 mL MeOH * | 0 | 0 | 5.0 | 14.8 | 19.9 | 20.0 | 19.8 | 20.0 | 22.3 | 24.1 | 25.0 | 40.8 | 58.1 | 64.9 | 60.1 | 69.8 |
| 1 mL H2O 3 mL MeOH | 4.7 | 15.5 | 19.8 | 35.8 | 34.8 | 40.5 | 60.1 | 72.5 | 74.3 | 78.5 | 80.0 | 89.9 | 90.1 | 91.0 | 91.9 | 92.1 |
| 1 mL H2O * 3 mL MeOH * | 9.7 | 19.0 | 26.0 | 37.7 | 46.0 | 48.8 | 78.7 | 81.7 | 82.0 | 82.3 | 83.1 | 91.6 | 99.5 | 99.7 | 99.6 | 99.8 |
| 1 mL H2O 3 mL AcN | 19.4 | 45.1 | 95.8 | 98.1 | 98.8 | 99.3 | 99.5 | 99.5 | 99.6 | 99.7 | 99.6 | 99.8 | 99.7 | 99.7 | 99.6 | 99.7 |
| 1 mL H2O * 3 mL AcN * | 31.5 | 53.9 | 97.8 | 98.2 | 98.5 | 99.1 | 99.2 | 99.4 | 99.4 | 99.5 | 99.4 | 99.6 | 99.8 | 99.7 | 99.7 | 99.8 |
| Sample Solvent | GA | PCA | GTA | CA | VA | CGA | EPI | VAN | p-CMA | TFA | SIA | DMBA | RUT | o-CMA | 4MCA | QUER |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MeOH/H2O 80/20 (v/v) | 0 | 0 | 0 | 0 | 0 | 5.1 | 5.9 | 7.0 | 8.5 | 10.6 | 14.3 | 20.9 | 39.7 | 41.4 | 44.0 | 49.1 |
| MeOH/H2O 80/20 (v/v) * | 0 | 0 | 0 | 2.7 | 4.5 | 8.6 | 9.5 | 10.8 | 18.2 | 25.7 | 28.8 | 29.8 | 48.5 | 51.4 | 56.2 | 57.8 |
| MeOH/H2O 50/50 (v/v) | 0 | 0 | 8.6 | 9.6 | 17.6 | 14.0 | 16.3 | 19.6 | 25.1 | 29.9 | 34.7 | 37.8 | 56.3 | 58.9 | 60.6 | 63.5 |
| MeOH/H2O 50/50 (v/v) * | 0 | 0 | 14.6 | 14.5 | 22.3 | 19.7 | 22.2 | 24.6 | 30.8 | 34.4 | 39.8 | 43.2 | 60.4 | 62.6 | 63.1 | 68.3 |
| AcN/H2O 10/90 (v/v) | 10.1 | 20.4 | 40.2 | 98.2 | 98.5 | 98.3 | 98.5 | 98.7 | 98.7 | 99.0 | 99.2 | 99.4 | 99.4 | 99.7 | 99.6 | 99.8 |
| AcN/H2O 10/90 (v/v) * | 14.2 | 35.2 | 46.4 | 98.4 | 98.5 | 98.5 | 98.6 | 98.7 | 98.9 | 98.9 | 99.0 | 99.1 | 99.3 | 99.4 | 99.7 | 99.6 |
| AcN/H2O 5/95 (v/v) | 29.1 | 48.8 | 98.7 | 98.7 | 98.8 | 98.9 | 99.1 | 99.1 | 99.2 | 99.2 | 99.5 | 99.6 | 99.7 | 99.7 | 99.8 | 99.8 |
| AcN/H2O 5/95 (v/v) * | 31.5 | 53.9 | 97.8 | 98.2 | 98.5 | 99.1 | 99.2 | 99.4 | 99.4 | 99.5 | 99.4 | 99.6 | 99.8 | 99.7 | 99.7 | 99.8 |
| Analyte | Losses Caused by Overflowing after Sample Loading (%) | Recoveries (%) | |||
|---|---|---|---|---|---|
| First Elution MeOH/H2O (80/20, v/v) | Second Elution MeOH | Third Elution MeOH | Total | ||
| GA | 12.9 | 86.8 | 0 | 0 | 86.8 |
| PCA | 9.6 | 90.2 | 0 | 0 | 90.2 |
| GTA | 4.2 | 95.3 | 0 | 0 | 95.3 |
| CA | 3.4 | 80.2 | 16.0 | 0 | 96.2 |
| VA | 1.4 | 80.2 | 18.0 | 0 | 98.2 |
| CGA | 1.5 | 78.4 | 19.9 | 0 | 98.3 |
| EPI | 0 | 78.9 | 20.6 | 0 | 99.5 |
| VAN | 0 | 75.3 | 24.5 | 0 | 99.8 |
| p-CMA | 0 | 75.1 | 24.6 | 0 | 99.7 |
| TFA | 0 | 75.0 | 24.6 | 0 | 99.6 |
| SIA | 0 | 72.7 | 26.3 | 0 | 99.0 |
| DMBA | 0 | 70.9 | 22.5 | 5.9 | 99.3 |
| RUT | 0 | 68.6 | 25.9 | 5.2 | 99.7 |
| o-CMA | 0 | 66.6 | 26.7 | 6.4 | 99.7 |
| 4MCA | 0 | 64.2 | 27.0 | 8.4 | 99.6 |
| QUER | 0 | 58.8 | 30.4 | 10.6 | 99.8 |
| Concentration | GA | PCA | GTA | CA | VA | CGA |
|---|---|---|---|---|---|---|
| 25 μg/mL | 86.8 | 90.2 | 95.3 | 96.2 | 98.2 | 98.3 |
| 50 μg/mL | 86.3 | 89.1 | 94.5 | 95.5 | 97.9 | 98 |
| 75 μg/mL | 84.6 | 85.9 | 90.7 | 91.5 | 94.0 | 94.2 |
| 150 μg/mL | 74.5 | 77.6 | 80.6 | 80.9 | 83.5 | 85.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranušová, P.; Matušíková, I.; Nemeček, P. Optimization of Plant Extract Purification Procedure for Rapid Screening Analysis of Sixteen Phenolics by Liquid Chromatography. Separations 2021, 8, 13. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8020013
Ranušová P, Matušíková I, Nemeček P. Optimization of Plant Extract Purification Procedure for Rapid Screening Analysis of Sixteen Phenolics by Liquid Chromatography. Separations. 2021; 8(2):13. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8020013
Chicago/Turabian StyleRanušová, Petra, Ildikó Matušíková, and Peter Nemeček. 2021. "Optimization of Plant Extract Purification Procedure for Rapid Screening Analysis of Sixteen Phenolics by Liquid Chromatography" Separations 8, no. 2: 13. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8020013