
Overview of Sample Preparation and Chromatographic Methods to Analysis Pharmaceutical Active Compounds in Waters Matrices


Laboratory of Bromatology and Water Quality, iMed.UL (Institute for Medicines and Pharmaceutical Sciences); Faculty of Pharmacy, University of Lisbon, 1649-003 Lisboa, Portugal
Separations 2021, 8(2), 16; https://0-doi-org.brum.beds.ac.uk/10.3390/separations8020016
Submission received: 12 January 2021
/
Revised: 31 January 2021
/
Accepted: 3 February 2021
/
Published: 9 February 2021
(This article belongs to the Special Issue Challenges and Trends in the Analysis of Compounds of Emerging Concern)
Abstract
:In the environment, pharmaceutical residues are a field of particular interest due to the adverse effects to either human health or aquatic and soil environment. Because of the diversity of these compounds, at least 3000 substances were identified and categorized into 49 different therapeutic classes, and several actions are urgently required at multiple steps, the main ones: (i) occurrence studies of pharmaceutical active compounds (PhACs) in the water cycle; (ii) the analysis of the potential impact of their introduction into the aquatic environment; (iii) the removal/degradation of the pharmaceutical compounds; and, (iv) the development of more sensible and selective analytical methods to their monitorization. This review aims to present the current state-of-the-art sample preparation methods and chromatographic analysis applied to the study of PhACs in water matrices by pinpointing their advantages and drawbacks. Because it is almost impossible to be comprehensive in all PhACs, instruments, extraction techniques, and applications, this overview focuses on works that were published in the last ten years, mainly those applicable to water matrices.

1. Introduction
The problem of water availability and quality is a fundamental issue of the 21st century. Over the past two decades, there has been increasing concern regarding several biologically active environmental contaminants in the aquatic environment. Many of these compounds belong to the group of so-called contaminants of emerging concern (CECs).
The CECs are naturally occurring, manufactured or human-made chemicals, or materials that have now been discovered or suspected in various environmental compartments and whose toxicity or persistence are likely to significantly alter a living being’s metabolism. Such potential CECs should remain “emerging” if the information is scarce in the scientific literature or poorly documented issues regarding the associated potential problems that they could cause [1]. The contamination of environmental compartments, such as surface water, groundwater, and soil with these chemicals shows some potential to pose risks to the environment or human health. Still, they are not yet subjected to regulatory criteria or norms for protecting human health or the environment [1,2].
The CECs include many micropollutants, among them certain pesticides and their degradation/transformation products, industrial chemicals (surfactants and surfactant residues, gasoline additives, brominated flame retardants, plasticizers, and perfluorinated compounds), disinfection by-products, personal care products (PCP), human and veterinary medicines and their metabolites, and nanomaterials, and so on. Some of these compounds also belong to the group of persistent organic pollutants (POPs) and endocrine-disrupting chemicals (EDCs) [3,4].
The impact of chronic exposure to these or other contaminants on humans, animals, vegetation, and aquatic species is a matter of concern for the scientific community and regulatory and supervisory entities, since they may not only compromise the functioning and maintenance of ecosystems, which is crucial in promoting the ecosystem services on which humanity depends, but also causing various public health problems [5,6]. Regarding human’s health, special attention has been given to drinking water and the quality and contamination of raw water (surface and groundwater) used for its production. Therefore, the presence of these contaminants is an environmental issue. It has been the backbone for the development of EU’s policy regarding the protection of water resources through the Water Framework Directives, which emphasize the need for well-developed monitoring programs as mechanisms for understanding the occurrence and fate of environmental contaminants, as well as assessing the risks that are associated with long-term exposure to low concentrations of these compounds or combined effects of mixtures. The guidelines will be proposed after the evaluation of these results, if necessary.
Because of the diversity of structures, physico-chemicals and biological properties, special attention has been given to pharmaceutical active compounds (PhACs), because, in contrast to conventional pollutants, they are deliberately designed to have a biological impact, even at low concentrations [7,8,9].
The sources of PhACs, their metabolites, and degradation products include: (i) effluents of pharmaceutical plants; (ii) runoff from agriculture, livestock and aquaculture; (iii) domestic and hospital effluents; and, (iv) municipal wastewater treatment plants (WWTPs). The presence of these target compounds in the environment can have adverse effects on aquatic and terrestrial organisms, and they can occur at any biological hierarchy level, such as cell, organ, organism, population, and ecosystem. These effects can be observed in concentrations in the order of ng/L for certain compounds [10,11].
It is difficult to predict which environmental and public health implications may arise from PhACs in freshwater ecosystems, since the individual concentrations that are usually found in the environment are lower than those that are able to cause direct adverse effects (acute toxicity). However, even trace concentrations have shown that they might have direct toxicity towards individual aquatic organisms [12,13,14,15].
Concerning drinking water, several risk assessments studies indicate that very low concentrations of pharmaceuticals are very unlikely to pose any risks to human health. Still, there are knowledge gaps in assessing the risks that are associated with long term, low-level exposures to pharmaceuticals and possible combined effects of chemical mixtures. The investigation of possible additive or synergistic effects of mixtures would be necessary for an accurate exposure assessment in order to determine whether there are any potential risks to human health, while considering sensitive subpopulations [16,17].
Because of these challenging questions, PhACs in the environment are reported in thousands of publications during the last decades and reviewed by many authors, demonstrating an increasing concern about them [18,19,20,21,22,23,24,25,26,27].
Efforts have been made to improve the knowledge and data available on sources of PhACs, and how pollution occurs to identify targeted and effective control options. The determination of these compounds in the various environmental matrices, such as water (wastewater, surface water, groundwater, and water for human consumption), soils, sediments, and biota, has grown and is subject to increasingly demanding legislative requirements (in the order of µg/L or ng/L, or ng/g for solid matrices).
In water policy, Directive 2013/39/EU refers for the first time to the contamination of water and soil by pharmaceutical waste as an environmental problem [28]. In Europe, to ensure the monitoring of compounds subject to possible hazards, such as emerging pollutants, and to guarantee a quality database to identify/prioritize substances, this directive identifies a set of priority substances in a watch list subject to update. The watch list mechanism was established to require the temporary monitoring of other substances for which evidence suggested a possible risk to or via the environment, in order to inform the selection of additional priority substances. The watch list should contain a maximum of 10 substances or groups of substances, indicating the matrices to be monitored as well as possible methods of analysis that do not involve high costs. Relative to PhACs, this first watch list includes a non-steroidal anti-inflammatory drug (diclofenac) and the hormones 17-β-estradiol (E2) and 17-α-ethinyl estradiol (EE2) to determine the concentration levels that facilitate the application of appropriate measures given the risk that these substances represent. Two other PhACs, carbamazepine and sulfamethoxazole, are also being studied for possible inclusion in this list.
The first watch list was adopted in the Decision 2015/495/EU of 20 March 2015 [29] and it includes seven PhACs: two natural hormones (E2 and estrone (E1)), one synthetic hormone (EE2), one nonsteroidal anti-inflammatory drugs (diclofenac), and three macrolide antibiotics (erythromycin, clarithromycin, and azithromycin). This directive was repealing by Decision 2018/840/EU [30] of 5 June 2018 and the substances mentioned above (EE2, E2, E1, azithromycin, clarithromycin, and erythromycin) were included, together with two other antibiotics, amoxicillin and ciprofloxacin. The Commission removed the diclofenac from the watch list due to the sufficient high-quality monitoring data available for diclofenac [30]. The inclusion of amoxicillin and ciprofloxacin is consistent with the European One Health Action Plan against Antimicrobial Resistance (AMR), which supports the use of the watch list to “improve knowledge of the occurrence and spread of antimicrobials in the environment” [31].
All of the evaluations and decisions were supported by the occurrence studies of these contaminants in the aquatic environment, where the high-quality data on their concentrations (quality of the analytical results) were essential. Besides, the knowledge of their concentration is essential as a starting point to apply more advanced treatments to improve their removal and, consequently, minimize their environmental risk [32].
Under this scenario, analytical chemistry plays an essential part in providing high-quality information by applying two different approaches: (i) target analysis and (ii) screening methods for the determination of non-target or unknown compounds (degradation products). Both of the approaches demand highly sophisticated techniques that enable the implementation of sensitive and selective methods to provide accurate data regarding the identification, confirmation, and quantification of compounds [33].
In this context, the analytical challenges, mainly in quantitative analysis, are focused on developing and validating new materials, strategies, and procedures to quickly meet the requirements for selectivity, sensitivity, speed, and green methods. Because of the constantly updating of international standards, guidelines, and recommendations, constant innovation is mandatory in both the pre-treatment procedures and analytical equipment to obtain reliable, true, and reproducible data [34,35].
Gas chromatography (GC) or liquid chromatography (LC) coupled to mass spectrometry (MS) or tandem mass spectrometry (MS/MS) are advanced methods that can determine target compounds to the nanogram per liter level or lower and they are commonly applied for the detection of PhACs in different water [36,37] and solid matrices, such as sediments, sludges, and biota [38,39,40]. Because of the polar characteristics of PhACs, the liquid chromatography is the most used [41,42,43,44]. Nevertheless, since the late 80s, liquid chromatography-mass spectrometry (LC–MS) has rapidly grown in popularity as a technique for environmental control.
Common mass analyzers applied in target analysis of these compounds are triple quadrupoles (QqQ) [45,46], ion-traps (IT) [38], and quadrupole-linear ion trap (QLIT-MS/MS) [37,47]. On the field of liquid chromatography coupled to tandem mass spectrometry, ultra-performance liquid chromatography (UPLC) has enabled the development of more sensitive, fast, and environmentally friendly methods for pharmaceuticals [10,48,49,50].
Improvements in the identification capability for target compounds have also been achieved by the introduction of high-resolution mass spectrometry (HRMS). The time-of-flight (TOF), quadrupole-TOF (QTOF), and Orbitrap-based mass spectrometry in combination with chromatographic techniques have resulted in a valuable tool, not only for qualitative, but also quantitative, analysis of target compounds [33]. Gómez et al. (2010) have described the advantage of using a QTOF analyzer in the identification, confirmation, and quantification of almost 400 contaminants (mainly pesticides and pharmaceuticals) in surface water and wastewater [51].
The analysis of transformation products has also become an important issue in environmental chemistry, as it has been found that they may be more toxic and/or persistent than the parent compounds, thus representing a higher risk to the environment and human health [52,53,54]. In this area, LC or GC coupled to HRMS has been widely applied in the identification of transformation products, with time-of-flight (TOF), quadrupole-TOF (QTOF), and Orbitrap mass spectrometers increasing in use. These techniques were used in the study of the behavior of PhACs, the identification of their transformation or degradation products in wastewaters, natural waters, and drinking waters [51,55,56].
Owing to the large-scale dilution of these contaminants in water sources (natural waters) or due to the matrix complexity, a preliminary sample preparation technique for concentration or/and clean-up is mandatory, such as liquid-liquid extraction (LLE) [57], solid-phase extraction (SPE) [10,27,58,59,60], solid-phase microextraction (SPME) [27,28], or stir bar sorptive extraction (SBSE) [61]. Currently, pharmaceuticals are usually extracted from water samples by off-line solid-phase extraction [10,46,61] and on-line solid phase extraction [44,62].
Several other methodologies namely microwave assisted extraction (MAE) [63,64], pressurized liquid extraction (PLE) [65], ultrasonic solvent extraction (USE) [66], accelerated solvent extraction (ASE) [67] and QuEChERS (Quick, Easy, Cheap, Effective, Rugged, and Safe) [68,69] have been developed as pre-treatment procedures for the determination of PhACs, metabolites, and degradation products in solid samples, such as sediments, sludge or sewage sludge, and biota.
All of the developed methods should be validated to ensure that every measurement performed will be close enough to the unknown real value for the target compound’s content in the sample. With this purpose, several items should be addressed: (i) applicability, fitness for purpose, and acceptability limits; (ii) specificity and selectivity; (iii) calibration study, involving the goodness of the fit of the calibration function and dynamic concentration range, sensitivity, detection, and quantification limits, as well as assessment for matrix effects (recovery studies); and, (iv) accuracy study, involving trueness, precision, and robustness, as well as the estimation of measurement uncertainty [70,71].
This study is an overview of the most applied methodologies. It summarizes data that were published in the past twenty years, mainly in the last ten years regarding the methods used to quantify PhACs worldwide, relative to pre-treatment and quantification methods, focused on water matrices, mostly environmental samples. There are hundreds of publications close to this subject, but 241 were selected for this purpose. The author searched the Web of Science and Scopus database of peer-reviewed literature and mainly selected studies involving water samples, environmental samples (surface and groundwater), wastewater, and drinking water.
2. Sample Preparation Methods
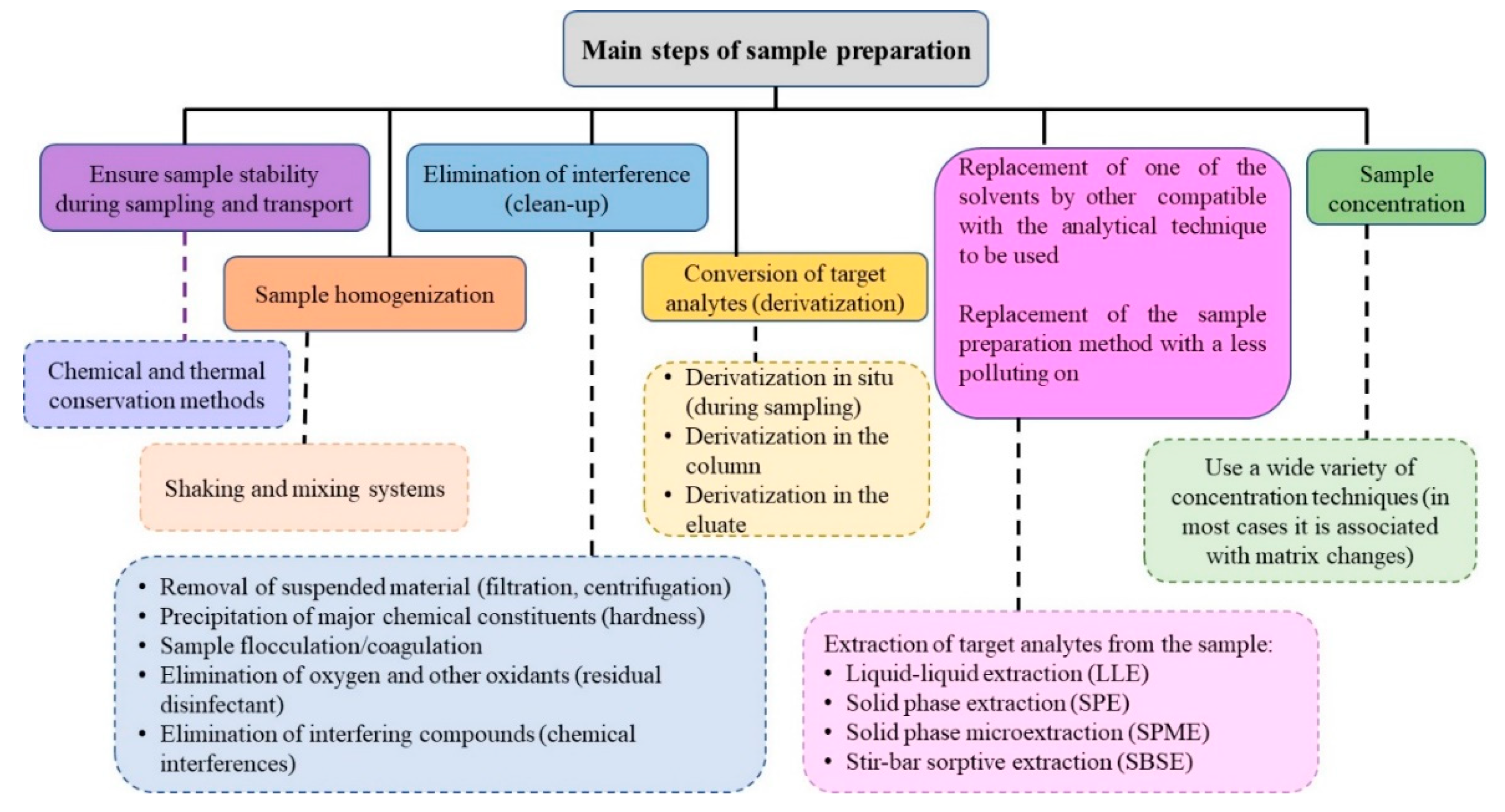
The main goal of the sample preparation (SP) methods is to transfer the target analytes from the matrix, in a more suitable way, to be analyzed by the selected analytical technique, using, in the case of trace analysis, strategies for the enrichment of the analytes, to gain sensitivity. Usually, several steps are required, involving up to about 80% of the analytical time. Figure 1 shows the main phases of the sample preparation methods. Regarding trace analysis, these steps can represent a significant source of errors that will affect the analytical result. There are two types of errors:(i) negative error, due to the analyte loss; and, (ii) positive error, which results from the addition of analyte to the sample or enrichment, or due to the presence of matrix interferents [72]. Any selected method should be minimized or overcome both errors.
The selection of the sample preparation method depends on several factors, namely, the type of matrix, the nature of the target compounds (volatility, polarity, solubility), their concentration in the matrix (mainly in trace or ultra-trace concentrations), the analytical method (gas chromatography, liquid chromatography, and their detectors), costs, ease of automation, and analysis time. Each method should be evaluated relative to their advantages and drawbacks [73].
Sample preparation has been subject to a continuous evolution that is mainly based on two approaches, discovery and development of new materials and improvements in technology, both using the principles of separation science [74].
For decades, liquid-liquid extraction (LLE) was the selected technique in the official methods of the EPA (Environmental Protection Agency) for the pre-concentration of organic compounds in aqueous samples. LLE is based on the partition of organic compounds between the aqueous sample and an immiscible organic solvent. The extraction efficiency depends on the physicochemical properties of analyte (solubility, pKa, log Pow, etc), solvent (polarity), sample, volume of the solvent, number of extractions, water: solvent ratio, as well as other parameters, such as pH and ionic strength of the sample. The solvents, in which the analyte is extracted, may be organic liquids, supercritical fluids, and superheated liquids [75]. Significant progress in LLE was made through the development of liquid–liquid microextraction techniques, such as single liquid microdrops, nanodrops, or picodrops extractions [74]. Other new LLE approaches include single liquid drop extraction with two phases, unsupported liquid membrane extraction with three phases, supported liquid membrane extraction (SLM) [74], microporous membrane liquid–liquid extraction (MMLLE) [76], and membrane-assisted solvent extraction (MASE) [77]. However, the enrichment factor of these techniques is not enough to quantify the organic compounds in trace or ultra-trace concentrations.
In the last two decades, the tendency to extract and analyze the largest number of compounds simultaneously to save time, expenses, and labor has led to the development of simple, fast, and generic sample treatments. Consequently, more “dirty” extracts and smaller recoveries are obtained, an accepted commitment for the analysis of multi-class compounds (multi-resides analysis) with different physical-chemical properties [78]. The selection of SP methods is based on the criteria of both the efficiency and its environmental impact, and SP has undergone considerable movement toward green sample preparation (GSP). In this perspective, new concepts have emerged that are allied to methodologies that use fewer volumes of organic solvents or even free of organic solvents, equally efficient, with less environmental impact, and less risk to public health. Among the various techniques, solid-phase extraction (SPE), solid-phase microextraction (SPME), and stir bar sorption extraction (SBSE) show a good recovery of target analytes (lower threshold limits), high precision, speed, and ease of automation. These techniques combine sample extraction, purification, and enrichment in a unique procedure [79]. Therefore, they are the most used techniques for monitored PhACs belonged to different pharmaceutical classes, both off-line [60,80,81,82] and on-line [44,62,83,84].
Because it is almost impossible to be comprehensive in the coverage of all pre-treatment techniques to the pharmaceutical’s analysis, the attention is focused on most used techniques, namely SPE, DSPE, SBSE and SPME.
Table 1 summarises many methods for PhACs analyzed in water matrices and the corresponding merit figures of SP procedure, chromatographic methods, and method validation. The table is at the end of methods before the validation section due to its size.
2.1. Solid-Phase Extraction
Conventional off-line solid-phase extraction (SPE) on cartridges [85,86] and discs [87] is a well-established technique that is routinely used for the extraction/concentration of several target compounds as well as for removing interfering components from matrix before chromatographic analysis. The technique is very versatile, and it is another criterion for its selection, due to the wide variety of sorbents chemistries available.
The SPE process shows five main stages: adsorbent conditioning, passing the sample, washing the adsorbent, drying the adsorbent, and eluting the analytes. When developing an SPE procedure, it is essential to correctly choose the packing material and the washing and elution solvents, according to the characteristics of the analytes and matrix. The final extract should also be compatible with the analytical technique used [72].
The extracts obtained by SPE technique are cleaner and the recoveries are higher. However, it has some disadvantages, namely: (i) clogging of the adsorbent layer by solid or fatty components present in the sample; (ii) extraction only of analytes dissolved in aqueous solvents; (iii) high variability due to different adsorbent capacity between sorbents batches; and, (iv) the breakthrough or limitation of the extractive capacity of the sorbent material (when he concentration of the analyte(s) is very high) [73,88].
Wastewater samples should be previously filtered and, in some cases, centrifuged in order to remove suspended matter that may clog SPE sorbents [48].
For some target compounds, it may be necessary to adjust the pH of the sample or other chemical corrections. Complexing agents (for example, Na2EDTA or citric acid) are usually added for complexing metal ions, which, otherwise, could bind to some analytes, such as tetracyclines and fluoroquinolones [89,90,91].
The sample pH adjustment can increase the affinity of some analytes for the SPE adsorbent, according to the type of adsorbent and the characteristics of the analytes [91,92,93].
The selection of adsorbent to analyze several PhACs belonging to different therapeutic classes is the critical step in SPE method development, and in most cases, the selected adsorbent is the best compromise, due different physical and chemical properties of the target compounds [10]. Based on the nature of target compounds, a careful choice of the sorbent allows for obtaining high recovery efficiencies and enrichment factors, which typically range between 20 and 1000 [78]. Relative to PhACs in drinking waters and clean waters, the typically factor is 500 and for wastewater the typical factors range between 20 and 100 [94]. These factors are a strong point of this technique.
The adsorbents that are used in SPE can be classified according to the interaction between the adsorbent material and the target compounds. The most common interactions are: (i) hydrophobic, nonpolar or reverse phase interactions; (ii) polar, hydrophilic or normal phase interactions; and, (iii) ionic interactions (cationic or anionic) [73].
There is a wide range of sorbents (packing materials) that can be used in the manufacture of solid phases for SPE. Some of them are of natural origins, such as graphitized carbon black (GCB), diatomaceous earth, alumina, silica, silica-based bonded phase (C18, C8), magnesium silicate (florisil), and cellulose, but others are synthetic polymers, such as the polystyrene-divinylbenzene and poly (divinylbenzene-co-N-vinylpyrrolidone). The last one has a high degree of porosity and, thus, a higher real active surface, allowing for a higher adsorption capacity. This new generation of polymers allows the extraction of a wide variety of analytes regardless of their nature, whether they are acidic, basic, and neutral, whether polar or nonpolar, and they are sometimes called HLB (Hydrophilic-Lipophilic-Balance) [74].
HLB cartridges consist of a hydrophobic component (polystyrene and/or divinylbenzene) and hydrophilic component (methacrylate, N-vinylpyrrolidone, and vinylamidazole). Unlike traditional SPE sorbents, where the reverse phases are made up of silica that can only retain non-polar or moderately polar compounds, this type of polymeric adsorbent can retain a wide range of acidic to basic compounds, with low to high polarity, being very useful in the development of methods that consist of compounds with different physicochemical properties. In addition, silica-based sorbents are unstable over a wide pH range and they contain free silanol groups, which can irreversibly bind to some analytes, such as tetracyclines [95].
The HLB sorbent is the SPE material that is most often (Table 1) used in the extraction of PhACs belonging to different therapeutic classes [10,23,45,48,60,96].
In the analysis of complex matrices, such as wastewater, the extraction/concentration and cleanup process requires passing the sample through more than one type of SPE material. One example of this approach is the use of an anion exchange adsorbent, followed by an HLB adsorbent for the determination of antibiotics, such as fluoroquinolones, sulfonamides, and trimethoprim [97]. Mixed-mode sorbents were developed to simplify this type of analysis. This application combines two or more functional groups into a single cartridge. This combination allows multiple retention interactions between the sorbent and the analytes, improved cleanup, better reproducibility, and recovery, leading to, overall, more sensitive, precise, and accurate analytical methods [74]. An example of this application is a SPE cartridge with a cation exchange resin and an HLB copolymer is the mixed-mode/cationic-exchange (MCX) [98,99], mixed-mode/anion-exchange (MAX) [26], and weak anion-exchange (WAX) [27]. These mixed-mode sorbents were used for reducing interference and matrix effects in the determination of PhACs in waters [92,98,100,101,102,103]. Unfortunately, the sorption capabilities of these cartridges are often limited when compared to the HLB cartridge for large volumes of water or complex environmental matrices [88].
The matrix’s complexity always affects the recovery of the PhACs, regardless of the sorbent used. The relative recoveries always show wide ranges of values due to the diversity of compounds under analysis (Table 1). This amplitude is higher for methods with a higher number of compounds, and when the number of therapeutic classes is also higher [37,44,51,96,99]. Sorbents with dual polarity (hydrophobic and hydrophilic) are the most versatile, so Oasis HLB is the most used sorbent [45,104,105]. For the analysis of PhACs belonging to the same therapeutic class, usually with similar polarity, more selective cartridges, both polar [26,82] and non-polar [106] are chosen. When mixed-mode sorbents, such as Oasis MAX and WAX, are used, the sample pH is a crucial issue because this parameter can affect the sorption adsorbent capacity to a greater degree. Different ionic forms of the PhACs and the sorbent can be present, depending on the sample pH [107]. Unfortunately, the sorption capabilities of these cartridges are often limited when compared to the HLB cartridge.
Regarding the use of an off-line or on-line SPE, one of the critical issues is the sample size, particularly the relative size of the water sample and injectable sample. This size depends on the expected concentration of PhACs in water samples. The enrichment factor should be high for drinking water with trace concentrations of PhACs. The water sample size is much larger than the injectable sample. Therefore, it is much more useful to opt for off-line SPE in order to obtain lower quantification limits. For wastewater, the enrichment factors can be lower, and the on-line SPE can be an advantageous option. However, coeluting of matrix interferences can be a significative drawback. In addition, on-line SPE coupled to ultra-performance liquid chromatography (UPLC) shows disadvantages because of the elevated back pressure that is generated by the high flow rates used in small particle size columns (<2 µm) (83).
The SPE technique has been applied to the analysis of PhACs that belong to several therapeutical classes (Table 1) in river waters [21,46,108], groundwaters [109], drinking waters [10,110], marine waters, and wastewaters [23,25,27,46,108], both wastewater influent and effluent.
For example, Gilart et al. (108) compared the selectivity and capacity of Oasis HLB, Oasis MAX, Oasis WAX, and a commercially available molecularly imprinted polymer (MIP) specific for non-steroidal anti-inflammatory drugs (NSAIDs) on the extraction of a group of 15 PhACs from wastewater. These four different commercial sorbents were tested to check which is more effective in preconcentrating and selectively extracting acidic PhACs from environmental waters. Although the recoveries that were obtained for the 15 PhACs by Oasis HLB with ultra-pure water and wastewater were similar (71–103%), this sorbent did not enable selective washing of real samples. Regarding the two mixed-mode sorbents, the washing step was able to eliminate all of the basic compounds, in contrast to ultra-pure-water experiments. The recoveries range between 60–100% (Oasis MAX) and 14–105% (Oasis WAX). The recoveries of PhACs in wastewater with MIP were slightly lower (45–102%) than those that were obtained with ultra-pure water, but this sorbent allowed for the selective extraction of acidic analytes from wastewater samples [107].
This technique still has several limitations, most of them being associated with the cartridge packing material, regardless of SPE’s advantages. Consequently, not only sorbents of new forms (fibers, bars) appeared, but, above all, alternative modes of extraction. In this last approach, the dispersive solid phase extraction (DSPE) and the magnetic solid phase extraction (MSPE) stand out.
2.1.1. Dispersive Solid-Phase Extraction
Dispersive SPE (DSPE) is based on the SPE methodology, but the sorbent is directly added to the sample without conditioning the clean-up, being easily carried out by shaking and centrifugation. After that, the sorbent with the analytes is washed with an appropriate solvent to recover analytes [111]. Because the contact surface between the sorbent and sample is higher, the extraction equilibrium is more quickly achieved, reducing the extraction time. The adsorbents do not need to be packed into the SPE cartridges and the problems of column blocking and high pressure often encountered in SPE are overcome. Furthermore, it is a more environmentally friendly method than standard solid-phase extraction, as the amount of sorbent and volume of solvent are lower. However, the crucial centrifugation/filtering step is difficult to automatize and become a critical hindrance in environmental analysis due to the high number of samples [78,112]. A noteworthy improvement of DSPE arises with magnetic solid-phase extraction (MSPE), where the centrifugation step is not necessary, in order to overcome this drawback.
The DSPE was applied to the analysis of antibiotics in mineral waters [113] and wastewaters [111], and natural hormones in river waters [111]. In the last study, a new organic adsorbent was prepared by the electrospinning method with polyacrylonitrile (PAN) and activated carbon. The activated carbon decorated PAN nanofibers showed LOQs that were between 0.53–2.17 µg/L. These sorbents can reusable, with satisfactory recoveries for more than ten uses.
2.1.2. Magnetic Solid-Phase Extraction
Briefly, the magnetic nanoparticles (MNPs) coated or hybridizated with other materials (magnetic sorbent) is added to the sample, vortexed and then separated from the solution under magnetic conditions. After separation, the material (containing the analytes) was dissolved in low amount of a suitable organic solvent to form the extract to be analysed [72,75,88,112].
MNPs have attracted a great deal of interest in the separation of different organic environmental contaminants due to their unique paramagnetic properties, high surface areas, customized surface modifications, and good dispersion in solution, which can affect the sensitivity and selectivity of the method [114]. Additionally, magnetic adsorbents can be readily recycled, which is economical and eco-friendly. The current trends are the development of new magnetic adsorbents with high adsorption capacity and selectivity [112,115].
Several materials have been used for preparation of the surface coatings of MNPs, such as silica, carbon nanomaterials, polymers, surfactants, and ionic liquids [113,116,117,118].
A tremendous increment in progress of MSPE is also associated with the use of carbon nanomaterials. Carbon exists in several allotropic forms, such as fullerenes, carbon nano tubes (CNTs), including single-walled CNTs (SWCNTs) and multi-wall CNTs (MWCNTs), carbon nanohorns, carbon nanocones, carbon nanodisks, carbon nanofibers, nanotube rings, graphene oxide (GO) and graphene (G), and diamonds. However, to date, from the analytical point of view, the applications have mainly been focused on the use of fullerenes, CNTs, and GO/G [119,120,121].
The magnetite (Fe3O4) nanoparticles coated with different materials have been used to extract different classes of pharmaceuticals in surface water. These MNPs were used to extract macrolide antibiotics in surface waters by MSPE [122].
Graphene was easily immobilized on silica-coated magnetite (designated Fe3O4@SiO2/graphene) and it was used as an adsorbent to extract six sulfonamide antibiotics (sulfapyridine, sulfamerazine, sulfameter, sulfachloropyridazine, and sulfadoxine) from surface water and wastewater samples [123].
A novel magnetic polyethyleneimine modified reduced graphene oxide (Fe3O4@PEI-RGO) was used as adsorbents for the extraction of polar non-steroidal anti- drugs (NSAIDs) from tap water, groundwater, and river water. When compared with Fe3O4@PEI and Fe3O4@PEI-GO, the Fe3O4@PEI-RGO showed a higher extraction efficiency for polar NSAIDs [114].
Gemfibrozil was extracted by MSPE using β-cyclodextrin-grafted graphene oxide (GO)/magnetite (Fe3O4) nano-hybrid [124].
Abdolmohammad & Talleb evaluated the use of MNPs composite Fe3O4@(Fe-benzene-1,3,5-tricarboxylic acid) as a sorbent for a higher group of lipid regulators (bezafibrate, clofibric acid, clofibrate, gemfibrozil, and fenofibrate) in water matrices [125].
Ethylenediamine-functionalized magnetic carbon nanotubes (EDA@Mag-CNTs) that were used as sorbent showed a high extraction capacity of the steroid hormone by MSPE procedure [126].
Several other coatings were evaluated as sorbent in MSPE for the extraction of PhACs that belong to different therapeutic classes [127,128].
The metal organic frameworks (MOFs) are another type of sorbents used in the extraction of organic compounds in water matrices. They show several advantageous properties, such as high porosity, tunable structures, ultra-high surface areas, outer-surface functionalization, and high thermal stability. Based on metal ion geometry and bridging ligands, MOFs can show different topologies and dimensionalities: one-dimensional, two-dimension or three-dimension MOFs (1D MOFs, 2D MOFs, and 3D MOFs). The 3D MOFs are highly porous and stable, since coordination bonds spread in three directions [78,112,129].
The analysis of 58 human and veterinary drugs (24 steroid hormones, 22 sulfonamides, and 12 quinolones) in ultra-trace concentrations in river water by MSPE were performed while using three-dimensional interconnected magnetic chemically modified graphene oxide (3D-Mag-CMGO) as adsorbent [130].
Recently, several automated MSPE approaches for the analysis of organic micro-pollutants and antibiotics have been reported [112].
Most of these adsorbent materials are under intensive research, unfortunately they are not commercially available. Thus, its potential use in routine analysis is still limited.
2.1.3. Molecularly Imprinted Polymers
The selectivity of the sorbent can also be achieved through the development of selective extraction phases that were obtained by molecularly imprinted polymers (MIPs), which are synthesized by the polymerization of functional and crosslinking monomers around a template analyte. After the elimination of template by extraction or chemical reaction, the cavities (binding sites) are exposed in the polymeric matrix. These cavities are complementary to the template in size, shape, and position of functional groups of the template analyte and its structurally related analytes [112,131,132].
The MIPs that are associated to SPE are also known in literature as MIP-SPE or MISPE, and they have been applied to the analysis of antidepressants, antibiotics, and beta-blockers [110,132,133]. To date, the potential of MISPE is still poorly explored in the analysis of PhACs in the environmental analysis.
It is difficult to separate MIPs with small particle sizes from aqueous samples when they are applied as the sorbent in SPE or SPME. Therefore, surface molecular imprinting polymerization onto MNPs to form magnetic MIPs (MMIPs) is usually applied to increase the number of imprinting or recognition sites (because of the high surface to volume ratio of MNPs) and mass transfer kinetics of the MIPs, as well as their simple magnetic separation from the sample solution [112].
2.2. Sorbent-Based Microextraction
The sorbent-based microextraction has led to a new generation of techniques with different and improved characteristics. Different solid-phase microextraction (SPME) techniques, including fibers, stir bar sorptive extraction (SBSE), thin-film microextraction (TFME), and automated techniques, such as in-tube solid-phase microextraction (IT-SPME) enhance the capacity of monitorization of a broad number of compounds that belong to different classes of organic contaminants, including CECs in complex water matrices. Some of these procedures are in automated configurations, on-line SPME, and IT-SPME. Although many of these automated techniques have been normally coupled with gas chromatography (GC), there are also some applications with liquid chromatography (LC) [137,138].
A clear advantage of the miniaturized devices is their portability, which significantly facilitates the implementation of on-site sampling and reduces errors that are associated with the sample transport and possible changes during storage [78].
Solid-phase microextraction (SPME) with fibers (usually referred as SPME) and stir-bar sorptive extraction (SBSE) have been the most used in water analysis.
The theory of SPME in both the direct immersion (DI) or in headspace technique (HS) was described in detail by Pawliszyn and coworkers [139]. Since then, many configurations have been successfully implemented, which can be classified into static and dynamic techniques. Static procedures are typically carried out in stirred samples, including fiber SPME, and they constitute the most common format for this technique.
The HS-SPME technique is usually applied in an equilibrated situation with the analytes being distributed between the fiber coating and HS gas present in a sealed vial. In theory, the HS-SPME has several advantages: the fiber does not contact with the sample, the background adsorption and matrix effects are reduced, which also enhances the life expectancy of SPME fiber. The extraction by SPME is influenced by several factors: sample matrix, stirring, temperature, sample volume, the size of the HS vial, the ratio of the HS to aqueous phase, and the position of the coated fiber in the HS, which can all affect the time that is required for the analyte to equilibrate between the HS vial contents and the SPME fiber coating [140,141]. The effects of these factors and advantages of HS-SPME versus DI-SPME have been studied by several authors utilizing many groups of compounds and many environmental, biological, and food samples [142,143,144].
As most SPME coatings are nonpolar, the technique is mostly applied to gas chromatography (thermic desorption) [141]. For the analysis of polar compounds, derivatization is required to enhance the selectivity and sensitivity of the SPME method [145,146].
The derivatization process can occur in the solution, in the coating phase, or in the GC injection port, and it is applicable to both techniques, immersion and headspace. The derivatization and extraction procedure can both be performed on a commercial autosampler that is coupled with GC [88].
The ionic liquids (ILs) and polymeric ionic liquids (PILs) overcome the drawback relative to the extraction of highly polar compounds from water matrices, and it has also quickly become a fast and cost-effective alternative technique to be coupled to LC. For LC analysis, these coatings applied to SPME should be highly robust when exposed to organic desorption solvents and not slough from the support material [147].
Other developments of novel polymeric sorbent materials, including graphene, molecularly imprinted polymers (MIPs), and metal-organic frameworks (MOFs), have also been developed for the extraction/enrichment of organic contaminants specifically for aqueous samples [112,138].
Hybrid materials polymerized from an IL monomer and an organic/inorganic crosslinker have been applied as SPME sorbent coatings for the analysis of several organic contaminants in aqueous samples by HPLC, some of them PhACs [106,148].
However, these novel materials were home-made. Therefore, they are not commercially available, and their use is limited.
Regarding water analysis, the SPME has been applied to the analysis of PhACs that belong to a wide range of therapeutic classes, such as NSAIDs [146,149], antiepileptic [149,150], lipid regulators [145], anti-diabetics [145], beta-blockers, anti-depressives [93], and antibiotics. In this last group, several classes were under study, namely tetracyclines [151], macrolides [152], and sulfonamides [62,153].
The number of PhACs that belong to a different therapeutic class analyzed by SPME techniques is much lower than the SPE technique, because this technique is not adequate for high samples volumes. On the other hand, the fiber’s fragility limits its use in complex matrices, mainly wastewaters. This limitation can be overcome in headspace SMPE, but it is limited to volatile compounds. Regarding PhACs, it implies the derivatization of the polar compounds and their analysis by gas chromatography [146].
The principle of SBSE is equal to SPME, but, due to higher volume of sorbent material, about 50—250 times larger than SPME, the sensitivity of this procedure is higher. After exposure to a sample, the stir bar that is covered with a thick film of polydimethylsiloxane (PDMS) is removed and the sorbed compounds are then either thermally desorbed, and analyzed by GC-MS or desorbed by an organic solvent, for improved selectivity or for interfacing to an LC subsequent system [154,155].
Because of the nonpolar character of sorbent, the SBSE has been mostly applied to the analysis of nonpolar or weakly polar compounds, and derivatization is a mandatory step for the analysis of very polar compounds [155].
The SBSE shows some drawbacks regarding the desorption step in GC applications that limits the automatization of the procedure, because the stir bar cannot be transposed directly in the injection port for thermally desorption. Therefore, it is a necessary specially designed thermal desorption unit, which is only available in sophisticated instrumentation. Furthermore, some target compounds can be lost during the manual transfer of stir bar to the desorption device with a loss of sensitivity gained [156]. Another limitation is the number of commercially available sorptive phases, such as polydimethylsiloxane (PDMS), ethylene glycol (EG)-silicone, carboxen, carbowax-divinylbenzene (CW-DVB), and polyacrylate (PA).
As observed for SPME, a variety of home-made stir bar coating have emerged, such as nanocarbon materials, functional monomers, metal-organic frameworks (MOFs), ionic-liquids (ILs), template imprinted polymers, and inorganic particles [135,157]. Some of them have been applied to the analysis of PhACS in complex water matrices [61,158,159].
Fan et al. [158] developed a novel IL-bonded sol–gel stir bar coating for SBSE of NSAIDs, followed by high-performance liquid chromatography-ultraviolet detection (HPLC-UV).
The stir bar was wrapped by a porous membrane (MPSBSE) to filter out the high molecular weight interferences (humic acid among others). A hydrophobic polytetrafluoroethylene (PTFE) membrane that was impregnated with methanol was employed to protect the C18 coated stir bar. The C18-MPSBSE was used for the direct determination of two common NSAIDs, ketoprofen and naproxen in complex water samples [160].
Molecular imprinted polymers and magnetic carbon nanotubes were combined in a stir bar for the enrichment of cefaclor and cefalexin in water samples [161].
A novel dual-template molecularly imprinted polymer (MIP)-coated stir bar was prepared for the analysis of environmental estrogens in complex samples. This dual template showed two different kinds of specific binding sites and three-dimensional cavities increasing the adsorption capacity of the target compounds [162].
A monolithic and hydrophilic stir bar coating based upon a copolymer of methacrylic acid and divinylbenzene copolymer, which was designed as poly(MAA-co-DVB), was synthesized and use for extraction of polar PhACs (paracetamol, caffeine, antipyrine, propranolol, carbamazepine, naproxen, and diclofenac) from river water and effluent wastewater from a treatment plant (WWTP) [163].
There is still a long way to go to put these materials available on the market, regardless of the selected sample preparation method and the new materials used. Selectivity was one of the first requirements under evaluation. The uniformity of new materials′ size, shape, and capacity are essential for their commercialization. The ease of automation is also a factor to consider, mainly in routine water analysis.
3. Analytical Instrumentation
3.1. Chromatography in Water Analysis
Monitoring environmental contaminants have grown rapidly and significantly in recent years, becoming a crucial analytical science area, developing new and innovative analytical techniques to identify and quantify pollutants in trace concentrations in the environment. Despite which sample preparation methods used for water sample enrichment, additional steps, including chromatography for separation and further quantification, can increase the data’s quality. Therefore, the interest in developing high performance, robust, and sensitive chromatographic methods has also increased. Consequently, new stationary phases, columns, and instruments have appeared, intending to simultaneously shorten the analysis time and obtain greater resolutions and sensitivities [78].
Currently, chromatography is considered to be the most used and versatile technique due to several factors: (i) the availability of very sensitive detection methods for all categories of chromatographic techniques; (ii) the relative speed of chromatographic separations; (iii) ease of use when compared to other analytical methods; and, (iv) the possibility of developing precise and trueness analytical methods.
The quantitative chromatographic analysis is used in all analytical chemistry areas, being essential in environmental sciences, namely in the monitoring of organic compounds in water matrices. Besides, the various organic parameters that are defined in the national and European legislation on the quality of water [28,29,164,165], pesticides, polycyclic aromatic hydrocarbons (PAHs), trihalomethanes, and alkyl halides, among others, are determined while using chromatographic techniques, as gas chromatography and liquid chromatography. These two techniques are also the techniques of choice in PhACs analysis.
The development of analytical methodologies for determining pharmaceuticals in environmental matrices, mainly in water, has boomed in the past years. In this context, the analytical challenges, primarily in quantitative analysis, had focused not only on the development of novel sample preparation methods (or new materials), but also on fast chromatographic methods, most of them being coupled to mass spectrometry (MS) [96,166,167].
Although gas chromatography has a high resolving power and it has been applied in the determination of PhACs, they are mostly polar and not very volatile or thermally unstable, which makes its direct determination by GC impossible, which implies the use of a derivatization step. Therefore, liquid chromatography (LC) has become the selected technique, as it allows the determination of PhACs from different therapeutic classes and with different physical-chemical properties after sample preparation techniques [97,168,169].
The main advantages of LC include the separation of a wide variety of compounds present in different samples, regardless of volatility, thermal stability, and polarity, in a few minutes, with high resolution, efficiency, and repeatability.
Literature shows the determination of PhACs in water samples by LC coupled with different detection techniques, such as ultraviolet and diode detectors [149,150] or fluorescence [20,60,110]. However, liquid chromatography coupled with mass spectrometry (MS) is the most used. The coupling of liquid chromatography with the mass spectrometer combines the advantages of chromatography (high selectivity and separation efficiency) with mass spectrometry (structural information, molar mass, and increased selectivity and detectability) [49].
HPLC has undergone some considerable improvements, especially in terms of innovative stationary phases and instrumentation. Short narrow-bore columns that are packed with fully porous sub-2 µm particles together with instruments exhibiting reduced extra-column volumes and able to withstand pressures up to 1000–1300 bar. This type of chromatography is known as ultra-high performance liquid chromatography (UHPLC) or ultra-performance liquid chromatography (UPLC). UPLC uses small diameter particles (typically 1.7 μm) in the stationary phase and short columns, which allow higher pressures and, ultimately, narrower LC peaks (5–10 s wide). In addition to providing narrow peaks and improved chromatographic separations, UPLC dramatically shortens the analysis times, often to 10 min. or less [49,86]. Therefore, it provides several advantageous characteristics, and the best ones are: employs less sample volume, provides faster analysis, less mobile phase consumption, higher resolution of chromatographic peaks, better detectability, and higher signal/noise ratio, which, in turn, decreases the limit of quantification (LOQ) [170,171,172].
Although the assortment of columns currently available for HPLC and UPLC is wide and varied, most methods being applied to PhACs analysis have been based primarily on the use of C18 reverse-phase (RP) columns (Table 1). However, columns C8 [3,90,104] and C16 [150] have also been used. The C16 is an RP-amide column used for the same separations as a C18, but shows higher wettability in high aqueous mobile phases. It is much more retentive for those PhACs that can interact by hydrophobic interactions and H-bonding with the amide group. Therefore, it is the right column for acidic PhACs.
The 1.7 µm and 1.8 µm UPLC BEH (Ethylene Bridged Hybrid) columns are the most used in UPLC. These columns allow a wide pH range (pH 1–12) due to the intrinsic chemical stability of hybrid particle technology. Therefore, they have been applied to the analysis of a great diversity of PhACs [37,45,94,101,173]. The UPLC HSS T3 columns have also been employed for PhACs analysis by UPLC in waters matrices. The HSS (High Strength Silica) particle is 100% silica ideally suited for the enhanced retention of polar compounds and metabolites by reversed-phase LC. This low-ligand density C18 column enables analytes to access the material’s pore structure more readily, providing a balanced retention of polar and hydrophobic molecules without the need for ion-pair reagents. The compatibility with 100% aqueous mobile phase and analysis of polar and non-polar compounds is an excellent advantage of these columns for PhACs analysis in water matrices [37,78,83,174,175].
The HPLC/UPLC with UV [123,125,127,176,177], DAD [111,114,134,150,161,162] and FLD [124] detection was used in the analysis of PhACs, but, in most applications, it is coupled with tandem mass spectrometry [37,45,98,101,178,179].
Mass spectrometry characteristics have raised it to an excellent position among analytical methods, mainly due to its sensitivity and detection limits. Regarding water matrices, optical detectors in LC methods for the analysis of PhACs are limited to wastewater, because its threshold limits are insufficient for measuring these target compounds at trace levels in groundwater and drinking water, even in surface waters. The enrichment factor of sample preparation method, even the large one used in the SPE technique (1000), is not enough to overcome their lower sensitivity. For example, the limits of quantification (LOQs) of 12 PhACs from four therapeutic classes by SPE-HPLC-UV-FLD (1000 mL of sample) ranged between 10–800 ng/L in surface waters, 10–850 ng/L in wastewater effluents, and 30–1100 ng/L in ranged between 10–800 ng/L in surface waters, 10–850 ng/L in wastewater effluents, and 30–1100 ng/L in wastewater influents [177]. The analysis of 90 PhACs of thirteen therapeutic classes by SPE-HPLC-MS/MS ranged between 0.06–196 ng/L in surface waters and 0.07–78 ng/L in wastewater effluents [46]. When considering the PhAC with the lowest LOQ in both techniques, the SPE-HPLC-UV-FLD method has a sensitivity approximately 150 times lower than that of the HPLC-MS/MS method, even using twice the sample volume.
The UPLC coupled to mass spectrometry has undoubtedly seen the most significant developments (almost at an exponential level) for applications in the field of research, but, above all, in routine analysis. This coupling (UPLC-MS) allowed for the increase of the sensitivity (as well as the precision and trueness) as phenomena of increased resolving power, reduction of ion suppression, and improvement of peak symmetry. Because of the narrow peaks produced by UPLC, the duty cycle should be fast, properties only available on the latest generations of MS devices. Therefore, specific analyzers are more readily compatible with UHPLC (e.g., QqQ or TOF/MS) than others (e.g., ion trap or Fourier-transform mass spectrometry) [180].
3.2. Mass Spectrometry: Interfaces and Analysers
Mass spectrometry (MS) is a widely used instrumental technique that is based on the ionization and fragmentation of sample molecules in the gas phase. MS has become the choice technique for the definitive identification of a wide variety of environment contaminants, including PhACs. In most cases, the separation of the compounds is achieved via chromatography (GC or LC) and the mass spectrometer is used as the detector. The separated compounds sequentially enter the mass spectrometer for the ionization, separation, and detection of the generated ions. The coupling of the chromatography system to the mass spectrometer allows for obtaining the retention time and mass spectral information for each separated compound, both of which can be compared with suitable reference standards [181].
The type of ionization in MS mainly depends on the internal energy transferred during the ionization process and on the physicochemical properties of the analyte to be ionized. Some ionization techniques are highly energetic and they produce extensive fragmentation. Other techniques are “smooth”, with little fragmentation and only produce ions of the molecular species. Electronic ionization (EI), chemical ionization (CI), and field ionization (FI) are techniques that can only be applied to gas-phase ionization and, consequently, to compounds that are sufficiently volatile and thermally stable. However, because most PhACs are thermally labile and low volatile, the molecules of these compounds must be directly extracted from the condensed phase (liquid or solid) to the gas. These sources of direct ionization are of two types: ion sources in the liquid phase and in the solid phase. The first are those used in association with liquid chromatography, where the analytes are in solution. The solution is introduced by nebulization in the form of drops in the source, where ions are produced at an atmospheric pressure and subsequently focused on the mass spectrometer through differential pumping systems. Electrospray ionization (ESI), atmospheric pressure chemical ionization (APCI), and atmospheric pressure photoionization (APPI) are sources of liquid phase ionization [181]. All of these ionization techniques have been applied to the analysis of PhACs.
Relative to pharmaceuticals determination, the ESI is the most used technique, but there are some analytical methods with APCI. Electrospray ionization proves to be a more sensitive technique than APCI; however, it is more susceptible to matrix effects, thus becoming a less robust technique [182,183].
There are few papers that report the use of APCI source for the ionization of moderate polar contaminants, such as PhACs, in water matrices. The target compounds under study belong to some therapeutical classes, such as anti-epileptic [184], antibiotics [185,186,187], NSAIDs [182], psychostimulants [188], benzodiazepines [183], and steroidal hormones [189,190].
Other technological solutions have also been studied to improve high throughput of environmental analysis by making conventional ionization interfaces more efficient. For example, the laser diode thermal desorption-atmospheric pressure chemical ionization (LDTD-APCI) apparatus has eliminated the use of a LC/GC step prior to detection, thereby reducing sample pre-treatment, analysis time, and cost, eliminating chromatography columns, and reducing solvent consumption, while increasing sample throughput [191,192]. In addition to these advantages, this technology also allows for the quantification in situ, which is essential in mapping contaminated areas and monitoring remediation effects. However, the direct analysis of heterogeneous and complex samples does not allow a precise and exact analysis of certain target compounds, mainly in trace concentration. Therefore, these techniques are most indicated to screen and control organic contaminants by semi-quantitative analysis [78].
During the ionization by electrospray, three kinds of ions are formed: molecular ions, protonated and deprotonated species, and adducts. The extent to which each of these ions is formed can be understood in terms of the balance between three essentially different processes, which occur inside the capillary: redox reactions (oxidation/reduction), which produce molecular ions, acid/base reactions (protonation/deprotonation), which result in the formation of protonated or deprotonated species, and coordination with cations (usually alkali metals) or anions (mainly chlorides), which leads to the formation of adducts. The ionization of most PhACs generally occurs through acid-base reactions since their redox potentials are low. In the case where the formation of ions is carried out by protonation/deprotonation, the pKa of the functional groups of the analytes is one of the main factors that influence the ionization process. In general, the alkaline compounds are ionized in the positive mode, giving rise to protonated ions at low pH values, and the acidic compounds ionize in the negative mode suffering deprotonation at high pH [193].
Single quadrupole mass spectrometry [90,176,194], especially tandem mass spectrometry (MS/MS) with triple quadrupole detection [10,46,48,109,126,132,195], QTOF (quadrupole-time of flight) [173,196], and quadrupole-ion-trap [91,197], have been the most used techniques in recent years, due to their high sensitivity and selectivity.
The tandem mass spectrometry (MS/MS) has been considered to be an identification and confirmation tool. The monitoring of the fragments provides more excellent discrimination than a single quadrupole (MS). This approach has several advantages, as it reduces the excessive sample preparation steps, decreases false positive and negative results in complex samples, allows for the identification of analytes with different physical-chemical characteristics at ultra-trace levels, results in less time and cost of analysis due to the reduced amount of reagents used in sample preparation and provides better detectability and selectivity.
Up to date, the residue analysis of PhACs in waters has been accomplished by LC–MS/MS in the selected reaction monitoring (SRM) mode or multiple reaction monitoring (MRM). The SRM mode has a severe limitation—the number of compounds that can be screened in a single run [86].
Up to 150–200 compounds (depending on the scan speed/dwell-time) can be analyzed in a run by LC–MS/MS in the SRM mode with a dedicated chromatographic method. Besides, when increasing the number of compounds included in the SRM method, the possibility of finding common or overlapped transitions for coeluting isobaric compounds increases. Another major limitation of these SRM methods is that they are blind to compounds not defined in the SRM method (non-target analysis). None or scarce information on possible non-target/unknown organic micro-contaminants or their degradation products is available when using these techniques [37].
Several hybrid systems have been used in the monitorization of PhACs in water matrices. Gros et al. [37] monitored 81 target compounds (PhACs and their metabolites) in wastewater influent and effluent, river, reservoir, sea, and tap waters using an ultra-high-performance liquid chromatography that was coupled to quadrupole linear ion trap tandem mass spectrometry (UPLC-QqLIT). The PhACs belong to 19 therapeutical classes. The main therapeutical classes under analysis were psychiatric drugs (15 compounds) and antibiotics (13 compounds). The method yielded detection limits between 0.04–20 ng/L in sea waters and 0.7–140 ng/L in wastewater influents. Thus, it is providing a reliable and robust tool that can be used for the routine analysis of multiple-class pharmaceuticals in aqueous samples. UPLC technology allowed for a fast separation of many pharmaceuticals, with improved sensitivity, altogether with a significant cost reduction in terms of time per analysis and solvent consumption. QqLIT allows for the application of the information dependent acquisition (IDA) function, where a targeted screening, monitoring one elected reaction monitoring (SRM) transition per compound, is performed in combination with an enhanced product ion scan (EPI). In this way, MS/MS spectra are achieved, which are afterwards matched with MS/MS spectra recorded in a library [37,43].
Gros et al. [174] also analyzed 53 antibiotic residues, covering various chemical groups and some of their metabolites in hospital and urban wastewaters, and river waters by UHPLC-QqQLIT. The method showed a good sensitivity with method quantification limit (MQL) between 1.44–44.6 ng/L for river waters and 9.81–272 ng/L for wastewater influents. The sensitivity, reduction of extraction volumes for all matrices, fast separation of a broad spectrum of antibiotics, and selectivity (due to tandem mass spectrometry) were the main advantages of this method. The method’s speediness was incredibly advantageous for detecting β-lactam antibiotics (penicillins and cephalosporins) that were challenging to analyze in multi-residue methods because of their instability in water.
3.3. High-Resolution Mass Spectrometry (HRMS)
The analysis of transformation products of CECs has become the subject of intense analysis in the field of environmental chemistry, given that the transformation products may be more toxic or persistent than the parent compounds, thus representing a greater risk for the environment and human health [53]. The LC or GC associated with high-resolution mass spectrometry (HRMS) has been widely applied in the identification of these transformation products [37]. In this context, the amoxicillin’s behavior in waste and surface water, which includes identifying its transformation products, was carried out using liquid chromatography that is associated with a QTOF [198].
While QTOF is more suitable for confirmation purposes, as well as for identification of unknown compounds or metabolites [43,199], because of their ability to provide exact mass measurements, QqLIT is appropriate for both quantitation and confirmation [96].
Calza et al. identified biotic and abiotic transformation products of clarithromycin and carbamazepine [200] and lincomycine [201] by liquid chromatography that was coupled to an Orbitrap analyzer.
Relative to these instruments, the last decade has witnessed a significant shift from using ion traps, single- and triple-quadrupole mass spectrometers towards employing mass spectrometers that provide an accurate mass of analytes, such as time-of-flight (TOF), Fourier transform ion cyclotron resonance (FT ICR), and Orbitrap detectors [202].
The HRMS instruments, such as TOF and Orbitrap, have revolutionized the research of CECs in the aquatic environment, due to their high sensitivity in full scan mode, their increased mass accuracy, and the capacity to distinguish the isotopic pattern. HRMS instruments can screen for unknowns, due to exact mass measurements. This unique characteristic distinguishes it to other mass spectrometry instruments [203].
In comparison to triple quadrupole mass spectrometers, which operate at unit resolution and generally in the selected reaction monitoring (SRM) or multiple reaction monitoring (MRM) modes for specific target analytes, TOF-mass spectrometers can acquire full-scan mass spectra at high resolution for all analytes without a loss in sensitivity. Because most of the TOF mass spectrometers have a resolution of at least 10,000 at full-width-half-maximum (FWHM) peak height, isotopic patterns are evident and empirical formulas, and chemical structures can be proposed for unknowns or confirmed for target analytes. This also makes it possible to use mass spectral libraries and enable the data file to be reinterrogated months later to find additional unknown contaminants [86].
Hybrid systems, such as HRMS hyphenated to a quadrupole, or linear ion trap (LTQ), such as the LTQ-Orbitrap, combine the tandem mass spectrometric capability that is associated with the LTQ with the high mass resolving power (up to 100,000 FWHM) and mass accuracy capability of the Orbitrap [202,203]. These hybrid configurations based on HRMS allow for the reliable interpretation of MS/MS spectra. They are valuable when dealing with complex environmental matrices, such as surface water or wastewater, where the co-elution of analytes with matrix interferences can result in ambiguous peaks [203,204].
This type of hybrid systems has also been used in the monitorization of PhACs in water matrices.
Bijlsma et al. [205] illustrated the potential of liquid chromatography coupled to a hybrid linear ion trap Fourier Transform Orbitrap mass spectrometer (LC-LTQ FT Orbitrap MS) for the simultaneous identification and quantification of 24 drugs of abuse and relevant metabolites in influent and effluent sewage water. The acquisition of full-scan accurate-mass data by Orbitrap, together with the simultaneous MS/MS measurements allowed by LTQ, is a powerful combination for confident identification and confirmation.
Abdallah et al. [206] show the UPLC-Q Exactive TM Orbitrap MS’s capability for simultaneous determination of 28 acidic and basic PhACs in a single run using rapid polarity switching of the electrospray ionisation source. The PhACs belong to seven therapeutical classes, such as antibiotics, antiseptics, beta-blockers, NSAIDs, narcotic analgesics, anti-hyperglycemic, and proton-pump inhibitors. The method applies rapid polarity switching in the heated ESI source for the simultaneous analysis of positive and negative ionised compounds in one chromatographic run of 16 min.
Althakafy et al. [207] optimized the conditions of ultra-high pressure liquid chromatography hyphenated with a quadrupole Orbitrap mass spectrometer (UHPLC- Q-Orbitrap-MS) to analyze 13 PhACs in water matrices. High-resolution MS and MS/MS analysis produce high selectivity and sensitivity based on the exact mass measurement of protonated and fragment ions using a narrow mass extraction window. The optimization approaches were performed to develop an acceptable method that allows for high resolution analysis of a range of PhACs in water samples with reliable quantification and confirmation of compound identities.
Bade et al. [208] compared the most popular HRMS instruments, quadrupole time-of-flight and linear trap quadrupole-Orbitrap, from two different laboratories. Wastewater (influent and effluent) and surface water samples from Spain and Italy were screened for 107 PhACs and abuse drugs (including 220 fragment ions). The sample results and findings of both instruments were compared, highlighting the advantages and drawbacks of the strategies that were applied in each case.
Chitescu et al. [209] applied LC-Q Exactive Orbitrap high resolution MS in both full scan (FS) MS and targeted MS/MS modes to study the occurrence of 67 pharmaceutical and antifungal residues in the Danube river.
Kalaboka et al. [210] screened 20 PhACs belonging to antibiotics, antipsychotics, anti-inflammatory drugs, plus acesulfame K with UHPLC–LTQ-Orbitrap MS in hospital and urban WWTP effluents. The method proved to be a powerful technique for the quantification and identification of analytes of interest in effluent matrix with excellent mass accuracies below 2 ppm and 5 ppm for positive and negative ionization, respectively.
However, low-resolution (LR) mass spectrometers continue to be the most used due to this equipment’s lowest price. Nevertheless, their properties make them suitable for high throughput targeted and screening approaches involving a significant number of PhACs [10,23,166,211]. Triple quadrupole has still preserved its market share due to the competitive price and unequalled performances in quantitative analysis [212,213,214].

{kind=link}
{kind=link}
Table 1.
Sample preparation methods and chromatographic methods for analysis of PhACs in water matrices.
Table 1.
Sample preparation methods and chromatographic methods for analysis of PhACs in water matrices.
| Matrix | Nº PhACs Therapeutical Classes | Extraction Configuration/Sample Volume | Phase System | Analytical Instrumentation | Stationary Phase/Column | Cal./R2 | Precision | Recovery | LOD/LOQ (ng/L) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Surface water | 1 PhACs (stimulant/caffeine) | LLE 1000 mL | --- | LC–APCI–MS | Luna HPLC (150 × 4.6 mm, 5 μm) | ISC 0.9997 | RSDr 8.3% RSDR 8.3% | 89% | LODs 4 | [188] |
| Surface waters (river and lakes) | 9 PhACs (lipid regulators, NSAIDs) | SPE | SDB-XC Empore disk | GC/MS after derivatization | DB5-MS | ESC | --- | Rel Rec 21–152% | --- | [21] |
| Wastewater and surface water | 10 PhACs (analgesic, lipid regulators, NSAIDs) | SPE WWE: 500 mL Surface water: 1000 mL | C18 Bondelut, (200 mg) Oasis HLB (60 mg) | GC-MS, after derivatization with MSTFA | HP5/MS (30 m × 0.2 5 mm × 0.25 μm) | ESC 0.9916–0.9999 | RSDr 5–13% | 53–94% (Oasis HLB) | --- | [215] |
| Wastewaters, surface waters, marine water and drinking waters | 18PhACs (antidepressant, antiepileptic, β-blockers, lipid regulators, NSAIDs, stimulants) | SPE | Oasis MCX (60 mg) | GC-MS, after derivatization with MSTFA | HP5/MS (30 m × 0.2 5 mm × 0.25 μm) | ISC | RSDr <20% | Rel Recoveries 54–120% | LODs WWE. 3.2–28.6 MW: 1.2–2.6 SW: 0.3–2.5 DW:0.1–1.5 | [216] |
| Tap water | 6 PhACs (NSAIDs) | On-line SPE 100 mL Off-line SPE 500 mL | LiChrospher RP-18e (500 mg) LiChrolut RP-18 (500 mg) | HPLC-DAD-MS | LiChrospher RP-18 (250 × 4.6 mm, 5 μm) | ESC 0.9992–0.9995 | RSDr On line: 0.2–6.4% Off-line: 3.3–10.4% | On line 96–104% Off-line 96.5–101.7 | On line LODS: 3.5–94 Off-line LODS: 20–950 | [217] |
| Drinking and Aquaculture Water | 6 PhACs Antibiotics | On-line SPE 25 mL | MIP | HPLC-DAD-FLD | Mediterranea Sea C18 (250 × 4.6 mm, 3 μm) | MMC >0.998 | RSDr 2–5% RSDR 2–6% | 62–102% | LODs DW: 1–11 AW: 1–12 | [110] |
| Surface water | 10 PhACs (analgesic, β-Blockers, Corticosteroids, NSAIDs) | SPE 1000 mL | Oasis HLB (500 mg) | HPLC-DAD-FLD | Purospher STAR C18e (125 × 3 mm, 5 μm) | ESC 0.9987–0.9999 | RSDr SW: 1.2–11.1% | Rec SW: 62–105% | LOQs SW: 6.5–3145 | [60] |
| Surface water and wastewater | 12 PhACs (antiepileptic, lipid regulators, NSAIDs, steroid hormones) | SPE 1000 mL | Strata X polymeric | HPLC-UV-FLD | Alltima C18 (250 × 4.6 mm, 5 μm) | ESC 0.9781–0.9980 | RSDr SW: 4–13% WWI: 7–15% WWE: 5–16% | SW: 86–104% WWI: 62–92% WWE: 65–100% | LOQs SW: 10–800 WWI: 30–1100 WWE: 10–850 | [177] |
| Wastewater | 65 PhACs (analgesic, antiepileptic, antibiotics, β-blockers, lipid regulators, natural hormones, NSAIDs, stimulants) | SPE 200 mL (acidic PhACs) 500 mL (neutral PhACs) | OASIS HLB (60 mg) (acidic PhACs) RP-C18ec cartridges (500 mg) (neutral PhACs) | HPLC-DAD-MS(ESI+) | Purospher® STAR RP-18 endcapped (5 µm) LiChroCART® 250-4 | ESC | RSDr, RSDR 1%–12% | Rel Rec WWI 64–104% | LOQs 4–95 | [23] |
| Surface water | 23 PhACs (analgesic/antipyretic, antibiotics, antiepileptic, antipsychotic, β-blockers, glucocorticoids, hormones, H2 receptor antagonist, lipid regulators, NSAIDs, stimulant) | SPE 1000 mL | SDB-disks | LC-UV/VIS-(ESI)-MS | Restek C18, (150 × 4.6 mm, 5 μm) | MMC >0.99 | RSDr 2.1–11.3% RSDR 5.9–21.5% | 46.8–92.1% | LOQs 11.1–354 | [176] |
| Surface water | 4 PhACs (estrogenic hormones) | SPE 2 L | C18 C18 + Florisil C18 + Florisil + NH2 | LC–ESI-MS | CAPCELL PAK C18 UG 120 (250 × 2.0 mm, 5 μm) | ---- | --- | 72–81% | LOQs 0.1–0.2 | [106] |
| Surface and wastewater water | 15 PhACs (analgetics, angiotensin converting enzyme inhibitors, angiotensin receptor antagonists, calcium antagonists, β-blockers, antidepressants, anticonvulsants, platelet antiaggregants, and cholesterol lowering agentes) | SPE 50 mL | Oasis Max | HPLC-MS/MS | Kinetex C18 (100 × 2.1 mm, 2.6 µm) | SAC >0.99 | <15% | 40–110% | LOQs 0.5–25 | [218] |
| Wastewater | 6 PhACs (analgesic, antiepileptic, antidepressants, stimulant) | SPE 100 mL | Oasis HLB (500 mg) | LC-APCI-MS/MS | Genesis C18 (150 × 3 mm, 4 μm) | ISC | RSDr WWI: 1–8% WWE:2–11% | Abs Rec WWI 79–161% WWE 75–178% Rel Rec WWI 84– 109% WWE 76–136% | --- | [183] |
| Surface, groundwater and drinking water | 8 PhACs (natural hormones, contraceptive hormones) | SPE 500 mL | Oasis HLB (200 mg) | HPLC-MS/MS | --- | ESC 0.9963–0.9998 | RSDr GW: 3.8–10% SW: 2.3–14% DW: 4.1–9.3% | GW: 41–77% SW: 40–95% DW: 54–82% | MDLs 0.69–11 | [59] |
| Surface and wastewater | 39 PhACs (corticosteroids) | SPE SW, WWE: 1000 mL WWI: 500 mL | In-house SPE Env + (150 mg), Strata-X-CW (100 mg), Strata-X-AW (100 mg) and Oasis HLB (200 mg) | HPLC-MS/MS | Poroshell 120 EC18 (100 ×2.1 mm, 2.7 μm) | --- | --- | 36–100% | LODs 0.5– 8 | [108] |
| Surface water and wastewater | 90 PhACs (analgesics, antidiabetic, antipropulsive, psychiatric drugs, antihistamine, anti-parkinson, antibiotics, anti-ulcer, antihypertensives, diuretics, antidiabetic, contraceptives, hormon therapy, anti-cancer) | SPE 100 mL | Oasis HLB (500 mg) | HPLC-MS/MS | Fully endcapped C18 Hypersil GOLD aQ (50 × 2.1 mm, 5 μm) GOLD Phenyl (50 × 2.1 mm, 3 μm) Porous grafite Hypercarb (50 × 2.1 mm, 5 μm) | ISC >0.980 | RSDr SW: 1–56% WWE: 2.7–50% | Rel Rec SW: 5–132% WWE: 35–246% | LOQs SW: 0.06–196 WWE: 0.07–78 | [46] |
| Surface and wastewater | 50 PhACs (antibiotics) | SPE SW: 1000 mL WWE: 500 mL WWI: 200 mL | Oasis HLB (500 mg) | HPLC-(ESI)-MS/MS | Eclipse Plus-C18 (100 mm × 2.1 mm, 1.8 μm) | ISC >0.995 | RSDr 0.63–9.67% RSDR 2.74–21.3% | SW: 49–292% WWE: 61–188% WWI: 32–446% | MQLs SW: 0.63–4.43 WWE: 1.42–9.52 WWI: 2.35–20 | [219] |
| Groundwater and surface water | 11 PhACs (antibiotics) | SPE 1000 mL | Oasis HLB (60 mg) | LC-(ESI)-MS/MS | Luna C8 (100 × 4.6-mm, 3 μm) | --- | --- | 84 - 130% | --- | [90] |
| Wastewater | 11 PhACs (analgesics, antibiotics, anxiolytics and hipnotics, lipid regulators, NSAIDs) | SPE WWI: 50 mL WWE: 100 mL | Oasis MAX (500 mg) | LC-(ESI)-MS/MS | Pursuit UPS C18 (2.1 × 50 mm, 2.4 µm) | ESC 0.9926–0.9992 | RSDr WWI: 7.2%–23% WWE: 5.9%–13% | Rel Rec WWI: 65.2–75.2% WWE: 64.1–88.2% | MQLs WWE: 1.4–200 WWI: 1.4–204 | [26] |
| Wastewater | 23 PhACs (antibiotics, antidepressant, antiepileptic, antiulcer, antihypertensives, lipid regulators, NSAIDs, stimulant) | SPE 250 mL | Oasis HLB (500 mg) | LC-MS/MS | SunFireTM C18 (100 × 3.0 mm, 3.5 μm) | --- | --- | --- | --- | [32] |
| Wastewater | 23 PhACs (analgesic, antibiotics, Antidepressives, diuretics, antipyretic, antiulcer, anxiolytics, cardiotonics, lipid regulators, NSAIDs) | SPE WWI: 50 mL WWE: 100 mL | LC sílica (500 mg) LC Florisil (1 g) GCB (1 g) LC SAX (500 mg) LC-NH2, WAX (500 mg) Oasis MAX (500 mg) | HPLC–(ESI)–MS/MS | Pursuit UPS C18 (50 × 2.1 mm, 2.4 µm) | ISC 0.9823–0.9988 | MAX cartridges RDSr WWI: 5%-17% WWE: 3%-18% RDSR WWI: 4%–15% WWE: 6%–26% | MAX cartridges LSL WWI:34–72% WWE: 3–90% MSL WWI:12–95% WWE: 12–129% HSL WWI:10–95% WWE: 9–176% | MAX cartridges MQLS WWI:3–19 WWE: 3–208 | [27] |
| Wastewater | 17 PhACs (analgesics, β-blockers, lipid regulators, NSAIDs, H2 histamine receptor antagonista, illicit drugs) | SPE WWE: 50 mL WWI: 25 mL | SCX (200 mg) Novel in-house SPE sorbents 2 Different sorbents: A and B | LC–(ESI)-MS/MS | Ascentis®Express C18 with Fused-Core technology (100 × 4.6 mm, 2.4 µm) | ESC ≥0.996 | RSDr and RSDR 16–23% | Sorbent A WWE: 19–106% WWI: 31–107% Sorbent B WWE: 43–98% WWI: 24–97% | Sorbent B LOQs WWE: 5–10 WWI: 10–20 | [220] |
| Wastewater, groundwater, and surface water | 8 PhACs (antibiotics, antiepileptic, β-blockers) | SPE WWI, WWE, SW, and GW: 100, 250, 500 and 1000 mL | Oasis HLB (60 mg) | HPLC-(ESI)-MS/MS | Zorbax XDB-C18 (50 × 2.1 mm, 5 μm) | ESC >0.99 | --- | Abs Rec GW: 40–97% SW:24–111% WWI: 18–121% WWE:52–105% Rel Rec GW: 48–121% SW: 19–118% WWI: 18–152% WWE: 59–127% | LOQs GW: 1–10 SW: 1–24 WWE:1.4–29 WWI: 3.5–163 | [109] |
| Drinking water | 27 PhACs (antibiotics, cardiotonic, neuroleptics, hormones, NSAIDs) | SPE 200 mL | Oasis HLB | UPLC-MS/MS | Acquity C18 (BEH) (100 × 2.1 mm, 1.7μm) | SAC | --- | - -- | --- | [80] |
| Wastewater and surface water | 5 PhACs (β-blockers) | SPE 250 mL | CNW MCX (60 mg) | HPLC-(ESI)-MS/MS | Chirobiotic T (150 mm × 2.1 mm, 5 μm) | ESC, ≥0.998 | RSDr <6.9% | Rel Rec SW 70.3%–99.4% WWI 59.5–97.9% | LOQs WWI 0.066–0.227 SW 0.059–0.227 | [82] |
| Wastewater | 15 PhACs (analgesics, NSAIDs, β-blockers, antiepileptics, antidepressants, lipid regulators) | SPE 250 mL MISEP 250 mL | Oasis HLB (150 mg) Oasis WAX (150 mg) Oasis MAX (150 mg) Affinilute MIP—NSAIDs (150 mg) | LC–(ESI)-MS/MS | Fused-CoreTM Ascentis Express C18 (100 × 4.6 mm, 2.7 μm) | ESC ≥0.996 | RSDr <15% (Rec > 25%) | WWE Oasis HLB 71–103% Oasis MAX 60–100% Oasis WAX 14–105% MIP 45–102% | MISEP LODs 0.5–2.0 | [107] |
| Groundwater, surface water, and wastewater | 14 PhACs (antibiotics, stimulant) | SPE 500 mL | Oasis HLB (500 mg) | Ion trap HPLC-MS/MS | Beta Basic C18 (100 × 2.1 mm, 3 µm) | ISC | RSDr 1.0–16% | Rel Rec (0.25 µg/L) GW:51–120% SW: 74–127% 82–126% | LOQs 100–650 | [92] |
| Wastewater | 6 PhACs (lipid regulators, NSAIDs) | SPE (MISEP) WWE: 50 mL WWI: 10 mL | Affinilute MIP—NSAIDs (150 mg) | LC–(ESI)-MS/MS | Fused-CoreTM Ascentis Express C18 (100 mm × 4.6 mm, 2.7 μm) | MMC ≥0.987 | RSDr <19% | WWE: 62–102% WWI: 69%–103% | LODSs 50–100 | [81] |
| Drinking water, surface water and reclaimed waters | 31 PhACs (antibiotics) | On-line SPE 1 mL, 5 mL and 10 mL | HyperSep retain PEP (porous polystyrene divinylbenzene, (20 × 3.0 mm, 12 μm) Hypersil gold aQ (polar endcapped C18, (20 ×2.1 mm, 12 μm) Hypercarb (porous graphitic carbon, 20 ×2.1 mm, 7 μm) | HPLC-(ESI)-MS/MS | Hypersil Gold C18 (50 × 2.1 mm, 1.9 μm) | ISC >0.99 | RSDr, RSDR <20% | 50–150% | LODSs 1.2–63 | [103] |
| Surface, groundwater and drinking water | 31 PhACs (analgesics, antibiotics, anticonvulsant, anti-depressants, β-blockers, corticosteroid, lipid regulators, nsaids, psychostimulant, sexual hormones) | SPE 500 mL | Oasis HLB (200 mg) | UPLC-(ESI)-MS/MS | Acquity BEH C18 (50 x 2.1 mm, 1.7 μm) | MMC 0.9951–0.9987 | RSDr <15% RSDR ≤20% | S: 43–111% GW: 31–120% DW: 49–88% | LOQs SW: 0.030–5.2 GW: 0.040–5.0 DW: 0.03–1.7 | [10] |
| Surface water and wastewater | 7 PhACs (antidepressants) | SPE (MISEP) 100 mL | MIP | UPLC-(ESI)-MS/MS | BEH C18 (50 × 2.1 mm, 1.7 μm) | ISC ≥0.992 | RSDr <15% | SW: 6–94% WWI: 6–90% WWE: 14–93% | MDLs SW: 0.3–12 WWI: 0.3–14 WWE: 0.3–11 | [132] |
| Wastewater | 16 PhACs (analgesic, antibiotics, antidepressant, antiepileptic, β-blockers, lipid regulators, NSAIDs, stimulant) | SPE WWE: 200 mL WWI: 100 mL | Oasis HLB (200 mg) | UPLC-MS/MS | UPLC BEH C18 (50 × 2.1 mm, 1.7 μm) | ISC >0.99 | RSDr 7.9% RSDR 10.4% | WWI: 70% WWE: 81% | LOQs WWE: 0.3–5.4 WWI: 1.0–20.2 | [179] |
| Surface and wastewater | 50 PhACs (antibiotics, antiulcer, cardiovascular, lipid regulators, NSAIDs, psychiatric drugs) | SPE | Oasis HLB (60 mg) | UHPLC-MS/MS | Acquity HSS T3 (100 × 2.1 mm, 1.8 µm) | ISC | <15% | Abs Rec 24–53% Rel Rec 48–147% | LOQs 2–170 | [175] |
| Surface water and wastewater | 20 PhACs (analgesic, anti-anxiety, antiarrhythmic, antiepileptic, antihistamine, contraceptives, corticosteroids, lipid regulators, stimulant, NSAIDs, steroid hormones) | SPE 1000 mL | Oasis HLB (200 mg) | UHPLC-MS/MS | ZORBAX Eclipse Plus C18 (50 × 2.1 mm, 1.8 μm) | ISC >0.99 | RSDr SW: 1.4–5.4% WWE: 3.2–21.6% | SW: 39–121% WW: 38–141% | MRLs 0.1–15 | [221] |
| Surface water and wastewater | 5 PhACs (antibiotics, steroid hormones) | 0n-line SPE 10 mL | Hypersil GOLD aQ (20 × 2.1 mm, 12 μm) | UHPLC-MS/MS | Kinetex Biphenyl (100 × 2.1 mm, 1.7 μm) Kinetex EVO C18 (100 × 2.1 mm, 1.7 μm) | ISC >0.99 | RSDr 2.3–8% RSDR 3.6–15% | 98.8–102% | LOQs WWE: 0.42–1.9 WWI: 0.49–1.9 | [222] |
| Surface water and wastewater | 20 PhACs (analgesics, anti-inflamatories, ansiolitics, Antidepressants, anti-ulcer, cardiovasculars, lipid regulators, psychiatric drugs) | SPE 100 mL | Oasis HLB (200 mg) | UHPLC-MS/MS | Acquity UPLC BEH C18 (50 × 2.1 mm, 1.7 μm) | ISC >0.99 | RSDr SW: 2–22% WWE: 2–18% WWI: 2–24% | Rel Rec SW: 27–117% WWE: 66–120% WWI: 55–124% | LOQs SW: 0.2–44 WWE: 3.6–85 WWI: 13–974 | [45] |
| Surface, groundwater, and wastewater | 73 PhACs (analgesic, antibiotics, antidepressants, anti-diabetic, antiepileptic, antihypertensive, β-blockers, diuretic, histamine H2 receptor antagonists, lipid regulators, NSAIDs, anti-cancer) | SPE SW, GW: 500 mL WWE: 200 mL WWI: 100 mL | Oasis HLB (60 mg) | UHPLC-MS/MS | BEH C18 (100 × 2.1 mm, 1.7 µm) | ISC >0.99 | RSDr 0.2–5% RSDR 0.1–10% | 50–150% | MDLs 0.01–8.98 | [94] |
| Surface and wastewater | 8 PhACs (antiepileptic, cardiovascular, lipid regulators, NSAIDs) | SPE | Oasis HLB (60 mg) | UHPLC-MS/MS | UPLC BEH C18 (50 × 2.1 mm, 1.7 µm) | ISC >0.98 | --- | 20–130% | LOQs 0.2–280 | [223] |
| Wastewater (efluente) | 33 PhACs (amphetamines, antidepressants, β-blockers) | SPE 50 mL | Oasis HLB (60 mg) | UPLC-MS/MS | Chirobiotic V (250 × 2.1 mm, 5 µm) (chiral separations) | ESC ≥0.997 | RSDr 2.8–28.2% RSDR 5–27.3% | Rel Rec 25.9–240.6% Abs Rec 46.9–142% | MQLs 0.09–109 | [64] |
| Surface water | 28 PhACs (analgesics, antibiotics, antidepressants, antiepileptics, β-blockers, bronchodilators, lipid regulators, histamine-2-blockers, calcium channel blockers, angiotensin-II antagonists, NSAIDs) | SPE | Oasis MCX | UPLC–MS/MS | ACQUITY UPLC BEH C18 (100 × 1 mm, 1.7 μm) | ISC | RSDr 3.1–23.9% (10 ng/L) RSDR 7.1–24.8% (10 ng/L) | Rel Rec 7.3–121% Abs Rec 5.2–131% | MQLs 0.3–50 | [98] |
| Surface and wastewater | 3 PhACs (antibiotics) | SPE 120 mL | Oasis HLB (60 mg) | LC–(ESI)-IT-MS/MS | Xterra MS C18 (50 × 2.1 mm, 2.5 μm) | --- | RSDr SW: 3.2–11.7% WWI: 8–18% | SW: 78.6–104.9% WWI: 70.1–99.5% | MDLs 30–70 | [197] |
| Surface and wastewater | 48 PhACs (analgesic, antibiotics, Anticoagulants, antidepressants, antidiabetic, antiepileptic Antihypertensive, β-blockers, diuretic, corticosteroides, histamine H2 receptor antagonists, hormones, lipid regulators, NSAIDs) | SPE 500 mL | Oasis MCX (150 mg) | UPLC-(ESI)-MS/MS | ACQUITY UPLC BEH C18 (100 × 1 mm, 1.7 μm) | ISC >0.99 | RSDr SW:3–25% WWE: 2–27% | SW: 48–123% WWE: 43–157% | LOQs SW: 3.4–85 WWE: 1.6–39 | [101] |
| Wastewater and surface water | 53 PhACs (antibiotics) | SPE 50 mL | Oasis HLB (60 mg) | UHPLC–(ESI)-QqLIT | Acquity HSS T3 (50 × 2.1 mm, 1.8 μm) | ISC 0.9878–1.0000 | RSDr 1–16% RSDR 4–30% | HWW: 30–176% SW: 20–121% WWI: 720–163% WWE: 20–180% | MQLs HWW: 3.97–164 SW: 1.44–44.6 WWI: 9.81–272 WWE: 4.93–183 | [174] |
| Surface and wastewater | 73 PhACs (analgesic, antibiotics, anti- cancer, antidiabetic, antidepressants, antiepileptic, antihypertensive, β-blockers, diuretic, histamine H2 receptor antagonists, lipid regulators, NSAIDs) | SPE SW: 500 mL WWI: 100 mL WWE: 200 mL | Oasis HLB (60 mg) Oasis MCX (150 mg) | LC-ESI-(QqLIT) MS/MS | Purospher Star RP-18 endcapped (125 × 2.0 mm, 5 μm) | ISC 0.9870–1.0000 | RSDr SW: 1–25% WWI: 2–21% WWE: 1–15% | Rel Rec SW: 10–194% WWI: 21–148% WWE: 30–121% | LOQs SW: 0.2–13 WWE: 0.6–28 WWI: 1–62 | [96] |
| Wastewater, groudwater and surface water | 19 PhACs (antibiotic/sulfonamides) | On-line SPE WWI: 5 mL WWE: 15 mL GW: 40 mL SW: 15 mL | Oasis HLB Hysphere C18 EC PRLPs cartridges Oasis HLB was selected | HPLC-QqLIT-MS/MS | Atlantis C18 (150 × 2.1 mm, 3 μm) | MMC WWI: 0.9948–0.9999 WWI: 0.9948–0.9999 SW: 0.9991–0.9999 GW: 0.9962–0.9999 | RSDr WWI: 1.5–10.3% WWE: 1.2–12.9% GW: 1.8–14.8% SW:1.0–25.6% RSDR <10% | Osais HLB 5–125% | MDLs WWI: 0.05–7.84 WWE: 0.01–6.90 SW: 0.02–4.52 GW: 0.02–5.13 | [62] |
| Wastewater, surface water, sea water and drinking water | 90 PhACs (analgesics, antibiotics, NSAIDs, anticoagulant, antidiabetic, β-Blockers, antihelmintics, antihypertensives, antiplatelet, diuretics, histamine H1 and H2 receptor antagonists, lipid regulators, prostatic hyperplasia, psychiatric drugs, sedation and muscle relaxation statin drugs, glucocorticoids, anti-asthma, tranquilizer, X-ray contrast agentes) | SPE MW: 200 mL WWI: 25 mL WWE: 50 mL DW: 500 mL SW: 100 mL | Sea waters Oasis HLB (200 mg) Other waters Oasis MCX (60 mg) | UPLC-QqLIT | PI Acquity HSS T3 (50 × 2.1 mm, 1.8 μm) NI Acquity BEH C18 (50 × 2.1 mm, 1.7 μm) | ISC | RSDr MW: 1.5–20% WWI: 2.0–33.7% WWE: 1.0–20% DW: 0.3–23% SW: 1.0–27.8% | MW: 30–147% WWI: 50–150% WWE: 40–146% DW: 12–157% SW: 30–158% | MQLs MW: 0.04–20 WWI: 0.7–140 WWE: 0.6–51 DW: 0.1–20 SW: 0.2–50.7 | [37] |
| Surface waters | 17 PhACs (antidepressant, antiepileptic, β-blockers, lipid regulators, NSAIDs Stimulants) | On-line SPE 0.5 mL | Oasis HLB, (2.1 × 20 mm, 25 μm) | UPLC-MS/MS | Acquity HSS T3 (150 × 2.1 mm, 1.7 μm) | ISC | --- | --- | LOQs 20–70 | [83] |
| Surface and drinking water | 28 PhACs (antibiotics, anticholinergic, antidepressant, antidiabetic, antifungal, anti-inflammatories, anti-ulcer agente, β-blockers, corticosteroids, histamine H2 receptor antagonists, lipid regulatorss, stimulant) | SPE 1000 mL | C18 (500 mg) | HPLC-ESI-microOTOF-QII | Shim-pack XR-ODS C18 (50 x 2.0 mm, 2.0 μm) | ESC 0.9907–0.9986 | RSDr 0.07–14.6% RSDR 0.85–22% | S: 9–293% DW: 4–286% | MQLs SW: 2.9–460 DW: 1.5–470 | [224,225] |
| Drinking water, groundwate, surface water, and wastewater | 100 PhACs (analgesic, antibiotics, antidepressants, antiepileptic, β-blockers, lipid regulators, NSAIDs, stimulant) | SPE 100 mL | Oasis HLB (200 mg) | HPLC-QTOF-MS | Zorbax Eclipse XDB-C8 (150 × 4.6 mm, 3.5 μm) | MMC >0.98 | RSDr 2–7% RSDR 5–12% | 65–105% | LODs 5–500 | [104] |
| Surface water and wastewater | 7 PhACs (antimycotic drugs) | SPE 500 mL | Oasis MCX | HPLC-Q-TOF-MS | ZORBAX Eclipse XBD C18 (100 × 2 mm, 3.5 μm) | ISC 0.9980–0.9999 | --- | SW: 84–104% WWE:71–109% WWI: 80–90% | LOQs 2–15 | [226] |
| Surface, groundwater, and sewage water | 74 PhACs (analgesics, anti-inflammatories, antibiotics, antidiabetic, antihypertensives, barbiturates, β-agonists, diuretics, histamine H2 receptor antagonists, lipid regulators and cholesterol lowering stain drugs, psychiatric drugs, anti-cancer drugs) | 0n-line SPE 2.5 mL | HySphere Resin GP PRLP-s Oasis HLB (1, 2.5 and 5 mL) | LC-ESI-(QqLIT) MS/MS | Purospher Star RP-18 endcapped (125 × 2.0 mm, 5 μm) | ISC 0.9637–1.0000 | RSDr 0.34–51% RSDR 0.51–89% | Abs Rec GW: 8.9–179% SW: 6.3–165% WWE: 3.3–174% WWI: 2.2–134% Rel Rec GW: 39–328% SW: 42–287% WWE: 12–535% WWI: 7.6–600% | MQLs GW: 0.11–260 SW: 0.02–78 WWE: 0.02–805 WWI: 0.01–1278 | [44] |
| Surface and wastewater | 105 PhACs (analgesics, antiepileptic anti-inflammatories, antibiotics, bronchodilatadors, β-blockers, diuretics, hormones, lipid regulators, psychiatrics, ulcer healings) | SPE 100 mL | Oasis HLB (200 mg) | HPLC-Q-TOF-MS | ZORBAX Eclipse Plus C18 (50 × 4.6 mm, 1.8 μm) | MMC >0.998 | <10% | 50–130% | LOQs 0.7–592.4 | [99] |
| Wastewater | 67 PhACs (illicit drugs, prescription drugs with potential for abuse and metabolites) | SPE 50 mL | Oasis MCX | UHPLC-QTOF-MS | UPLC BEH C18 (50 × 2.1 mm, 1.7 µm) | --- | --- | --- | IDLs 10–3500 | [227] |
| Surface and Wastewater | 29 PhACs (analgesics/anti-inflammatories, antibiotics, anti-ulcer, β-blockers, lipid regulators, histamine H2 receptor antagonist, psychiatric) | SPE SW, WWE, and WWI: 500, 200, and 100 mL | Oasis HLB (60 mg) | UPLC-QTOF-MS | Acquity C18 (50 ×2.1 mm, 1.7 μm) | RSDr 0.5–5.3% RSDR 2.1–9.1% | --- | MDLs 10–500 | [173] | |
| Surface, drinking, and groundwater. | 13 PhACs (analgesic, antibiotics, antiepileptic, lipid regulators, NSAIDs) | SPE 100 mL | Oasis-MCX (150 mg) | LC-(ESI)-QTOF-MS | RP-18 (100 × 2.1 mm, 3.5 μm) | MMC 0.995–0.999 | RSDr 2.0–12% (100 ng/L) RSDR 5–44% (25 ng/L) 13–34% (100 ng/L) | 63–195% | LOQs 5–25 | [91] |
| Wastewater | 160 PhACs (metabolites) | SPE 100 mL | Oasis HLB (60 mg) | UHPLC-QTOF-MS | UPLC BEH C18 (100 ×2.1 mm, 1.7 μm) | --- | --- | --- | --- | [105] |
| Surface and wastewaters | 87 PhACs (analgesics, antibiotics, antiepileptic, antipyretics, β-blockers, chemotherapy drugs, corticosteroids, diuretics, lipid regulators NSAIDs, psychiatric drugs) | SPE WWE: 200 mL SW: 400 mL | Oasis HLB (200 mg) | LC–(ESI)-QTOF-MS/MS | RP XDB-C18 (50 × 4.6 mm, 1.8 μm) | MMC 0.9000–0.9987 | RSDr 2–23% | 22–127% | MDLs <5–50 | [51] |
| Surface water, and influent and effluent wastewater | 22 PhACs (drugs of abuse stimulant, tranquilising drugs) | Direct injection | --- | Screening by HPLC- (ESI) QqQLIT-MS/MS Confirmation by LC–MS/MS | ZorbaxEclipse XDB, RP C8 (150 × 4.6 mm, 5 μm) | MMC R > 0.99 | RSDr, RSDR 4%–16% | --- | IDLs (LOQs) SW: 0.5–600 WWI: 10–700 WWE: 10–600 | [3] |
| Wastewater | 13 PhACs (antibiotic, antidepressant, anti-diabetic, lipid regulators, stimulant) | SPE 30 mL | Oasis HLB (60 mg) | UHPLC- Q-Orbitrap-MS | Agilent Extend-C18 (50 × 2.1 mm, 1.8 μm) | ESC 0.9875–0.9993 | --- | 95–101% | LOQs 40–2500 | [207] |
| Wastewater, surface water and drinking water | 27 PhACs (analgesic, antibiotics, antidepressant, antiepileptic, β-blockers, corticosteroids, lipid regulators, NSAIDs, steroid hormones, stimulant) | SPE 250 mL | Oasis MC X | UPLC-Q ExactiveTM Orbitrap-MS | Accucore RP-MS (100 x 2.1 μm) | ISC 0.9619–0.9990 | RSDr 2.0–7.7% RSDR 3.9–7.6% | 76–104% | MQLs 2.7–83.8 | [206] |
| Milli Water | 17 PhACs (antibiotic/sulfonamides) | --- | --- | UPLC- LTQ/Orbitrap MS/MS | --- | --- | --- | --- | --- | [55] |
| Marine waters | 11 PhACs (antibiotics) | SPE disks 1500 mL | Bakerbond Speedisk (H2O-Philic DVB) | HPLC-(ESI)-QqQIT HPLC-(ESI)-MS/MS | Eclipse XDB C18 (150 × 4.6 mm, 5 μm) | ESC 0.9984–1.0000 | RSDr 0.1–6.0 | Abs Rec 37.5–117% | LOQs 0.5–50 | [87] |
| Wastewater | 8 PhACs (steroid hormones) | SPE | RP Strata-X (surface-modified styrene divinylbenzene polymer) (200 mg) | LDTD-APCI-MS/MS | --- | MMC | RSDr 5–14% RSDR 5–17% | 77–121% | MDLs 13–42 | [190] |
| Wastewater | 9 PhACs (antibiotic, antiepileptic, NSAIDs, steroid hormones, stimulant) | SPE 250 mL | Strata ABW cartridge (500 mg) | LDTD-APCI-MS/MS | --- | MMC ≥0.991 | RSDr, RSDR <15 % | Rel Rec 76–106% | LODs 30–122 | [191] |
| Wastewater | 1 PhACs (NSAIDs/diclofenac) | --- | --- | LDTD-APCI-MS/MS | --- | ESC >0.999 | RSDr 7.1% RSDR 9.2 | 98.2–104.6% | MDLs 300 | [182] |
| Wastewater | 1 PhACs (antiepileptic/ carbamazepine) | --- | --- | LDTD-APCI-MS/MS | --- | ESC >0.999 | RSDr 8% RSDR 11% | 98–113% | LODs 12 | [184] |
| Wastewater | 3 PhACs (antibiotics) | DSPE | Activated carbon-decorated PAN nanofibers | HPLC-DAD | RP C-18 | ESC 0.9998 | RSDr <3.6% | 90–99%. | LODs 530–2170 | [111] |
| Mineral waters | 11 PhACs (antibiotic/sulfonamides) | DSPE 250 mL | MWCNTs m- MWCNTs) | UPLC-DAD | Hypersil Gold C18 (100 × 2.1 mm, 1.9 μm) | MMC 0.995–0.997 | RSDr 0.9–3.8% RSDR 1.7–2.9 | MWCNTs 40–100% m- MWCNTs 32 and 53% | LOQs 27–89 | [113] |
| Surface water | 4 PhACs (natural hormones) | DSPE 250 mL | mag-MFMIP Fe3O4/Poly(Stc- MPS)/SiO2 | HPLC-FLD | XDB-C18 | ESC 0.996–0.9999 | RSDr <7.0% | 72–102% | LODs 2.5–5.8 | [118] |
| Wastewater | 4 PhACs (NSAIDs) | MMSPD 1 mL | SiO2, C8, Magnetite particles Fe3O4 | HPLC-UV | Microsorb 100-C18 (150 × 4.6 mm, 5 μm) | --- | RSDr 1.0–2.1% RSDR 1.4–2.0% | >90% | LOQs 3000–5000 | [127] |
| Tap water, groundwater, and river water | 6 PhACs (NSAIDs) | MSPE 10 mL | Fe3O4@PEI-RGO (10 mg) | HPLC-DAD | BDS Hypersil C18 (250 × 4.6 mm, 5 μm) | ESC 0.9972–0.9986 | RSDr 0.39–6.67% RSDR 0.70%–8.75%, | 91.2–101.1% | LOQs 1000 | [114] |
| Surface water | 7 PhACs (antibiotics) | MSPE 3 mL | EMMIPs (10 mg) | HPLC-DAD | Ultimate XB-C18 (250 × 2.5 mm, 5 μm) | ESC 0.999–0.9999 | RSDr 0.9%–7.3%. | 79.3–92.4% | LOQs 100–310 | [134] |
| Surface water and wastewater | 6 PhACs (antibiotics/ sulfonamides) | MSPE | Fe3O4@SiO2/G (0.3 mg) | HPLC-UV | Inertsil® ODS-4 (150 × 4.6 mm, 5 μm) | --- | RSDr 3.3%–10.7% RSDR 4.6–9.8% | SW 76.4–104% WWI 74.2–89.3% | LOQs 320–530 | [123] |
| Wastewater | 1 PhACs (lipid regulator/gemfibrozil) | MSPE 200 mL | β-CD/ Fe3O4/GO NP (150 mg) | HPLC-FLD | --- | ESC 0.9989 | RSDr 1.09% RSDR 2.67% | 96.0–104.0% | LOQs 3 | [124] |
| Marine water, wastewater and drinking water | 5 PhACs (lipid regulators) | MSPE 10 mL | Fe3O4@Fe-BTC MMOF (5 mg) | HPLC-UV-Vis | Microsorb-MV 100-8 C18 (250 × 4.6 mm, 2.5 μm) | ESC 0.9992–0.996 | RSDr MW: 2.6–5.4% WW: 1.8–7.2% DW: 2.2–3.2% | MW 80.1–99.4% WWI 59.7–90% DW 83–100.5% | LOQs 460×103–725×103 | [125] |
| Surface water | 3 PhACs (antibiotics/macrolids) | MSPE 50 mL | Ol-coated Fe3O4MNP (50 mg) | LC-(ESI)-MS/MS | Kinetex XB-C18 (100 × 3 mm, 2.6 μm) | MMC | RSDr <6.0% RSDR <13% | 54–117% | LOQs 34–77 | [122] |
| Surface water | 24 PhACs (steroid hormones) | MSPE 100 mL | EDA@Mag-CNTs (50 mg) | UFLC-MS/MS | Shimpack XR-ODS II (150 × 2.0 mm, 2.2 μm) | ISC 0.9980–1.0000 | RSDr 1.6–6.4% RSDR 2.6–8.1% | 82.1–113%. | LOQs 0.02–1.0 | [126] |
| Mineral, tap and wastewater using | 12 PhACs (estrogenic hormones) | MSPE 25 mL | Fe3O4@poly(dopamine) (60 mg) | UFLC-(ESI)-MS/MS | X-Bridge C18 (100 mm × 4.6 mm, 3.5 μm) | ISC 0.9943–0.9991 | RSDr 3–15% | 70–119% | LOQs 10–1100 | [128] |
| Surface waters | 58 PhACs (antibiotics, steroid hormones) | MSPE 100 mL | 3D-Mag-CMGO (100 mg) | UFLC-(ESI)-MS/MS | Shimpack XR-ODS II (150 mm × 2.0 mm, 2.2 μm) | MMC 0.9981–0.9991 | RSDr 1.7–2.2% RSDR 7.3–9.0% | 78–109% | LOQs 0.10–2.09 | [130] |
| Surface water | 2 PhACs (antibiotics) | SBSE 25 mL | MIP MWCNTs | HPLC-DAD | ZORBAX C18 (4.6 × 150 mm, 5 μm) | ESC 0.9929–0.9945 | RSDr 5.1–5.5% RSDR 6.4–6.5% | 86.5–98.6% | LOQs 10×103–12×103 | [161] |
| Surface water | 4 PhACs (estrogen hormones) | SBSE 15 mL | dual-templates MIP | HPLC-DAD | C18 (250 × 4.6 mm, 5 μm) | ESC 0.9978–0.9992 | RSDr 2.0–5.5% | 62.8–98.0% | LOQs 1000–5000 | [162] |
| Surface and wastewater | 7 PhACs (analgesic, antiepileptic, β-blockers, NSAIDs, stimulants) | SBSE 100 mL | poly(MAA-co-DVB | LC-(ESI)-MS/MS | Kinetex C18 (100 × 4.6 mm, 2.6 μm) | ESC >0.999 | SW RSDr 1–7% WWE RSDr 3–19% | SW: 11–90% WWE: 10–85% | LODs 10--50 | [163] |
| Wastewater | 12 PhACs (analgesics, antibiotics, antipyretics, β-blockers, lipid regulators) | SBSE 50 mL | 32 µL EG Silicone Twister® 25 µL Acrylate Twister® | LC-(ESI)MS/MS | Kromasil 100 C18 (150 × 4.6 mm, 5 μm) | ESC, >0.997 | RSDr <15% (Rec > 16%) | PA: 1 - 43% EG silicone: 1–80% | LODSs 150–5000 | [61] |
| Tap water, groundwater, surface, and wastewater | 3 PhACs (NSAIDs) | DI-SPME 20 mL | PEG-g-MWCNTs) | GC-FID | CP-Sil 24CB - WCOT Fused silica (30 m × 0.32 mm × 0.25 μm) | ESC 0.9985–0.9992 | RSDr 5.9–8.1% RSDR 7.2–9.1% | 84–107%. | LOQs 50–70 | [143] |
| Surface water | 8 PhACs (analgesics, lipid regulators, NSAIDs) | On-line HS-SPME Derivatization with DMS 10 mL | PDMS–DVB, 65 μm | GC-MS | HP5MS (30 m × 0.25 mm × 0.25 μm) | ESC 0.994–0.999 | RSDr 0.6–12.3% | 92.5–110.8% | LOQs 0.34–4.13 | [145] |
| Tap water and wastewater | 6 PhACs (NSAIDs) | HS_SPME Derivatization with DMS 6 mL | PDMS 100 μm | GC-MS | HP5MS (30 m × 0.25 mm × 0.25 μm) | ISC 0.9920–0.9980 | RSDr 7.9–17.2% | 76–133% | LODs 0.3–2.9 | [146] |
| Surface water | 8 PhACs (antiepileptic, NSAIDs) | On-line SPME | PDMS–DVB, 65 μm | HPLC-DAD | Discovery RP-Amide C16 (150 × 4.6 mm, 5 μm) | ESC 0.994–0.999 | RSDr 4.4–8.2% RSDr 5.1–9.3% | 71.6–122.8% | LOQs 1000–4000 | [150] |
| Wastewater | 17 PhACs (analgesics, ansiolitics, Antibiotics, antidepressants, antiepileptic, antipsychotic, β-blockers, NSAIDs) | On-line dual-SPME 2 ×2 mL (pH=3, pH=11) | CW/TPR (50 µm) | LC–(ESI)-ITMS | Mediterranea Sea18) C18 (100 × 2.1 mm, 3 μm) | ISC 0.988–0.998 | RSDr 6.2–10.2% RSDR 7.4–13.6% | 89.2–109.7% | LOQs 0.005–0.01 | [93] |
| Wastewater | 9 PhACs (antibiotics) | SPME 1.5 mL | CW/TPR (50 µm) | HPLC-MS/MS | ZORBAX Eclipse XDB C18 (250 × 4.6 mm, 5 μm) | SAC WWI 0.9080–0.9992 WWE 0.9336–0.9987 | RSDr 7–50% | --- | LOQs WWI:9.2–1380 WWE: 14–260 | [152] |
| Tap water and surface water | 6 PhACs (hormones) | SPME 20 mL | Home made MMF/AMED | HPLC-DAD | Hypersil BDS C18 (250 × 4.6 mm, 5 μm) | ESC 0.9910–0.9980 | RSDr 3.5–5.0% RSDR 5.1–6.0% | 75.6–118% | LODs 130–340 | [148] |
| Wastewater | 16 PhACs (analgesic, antibiotic, antidepressant, lipid regulators, NSAIDs, stimulant) | SPME 10 mL | FPSE (FG)@PEG300 | UHPLC–LTQ Orbitrap MS | Hypersil Gold C18 analytical (100 × 2.1 mm, 1.9 μm) | ESC 0.9928–0.9989 | RSDr 8% RSDR 11% | 83–114% | LOQs 9.3–447.7 | [210] |
Legend: (Abs Rec) Abs Recovery; (Acrylate Twister®) polyacrylate (PA) with a proportion of polyethyleneglycol (PEG); (APCI) atmospheric pressure chemical ionization; (β-CD) β-cyclodextrin; (BEH) ethylene bridged hybrid; (Cal.) calibration; (CW/TPR) carbowax/templated resin; (DAD) diode array detector; (3D-Mag-CMGO) modified graphene oxide; (DMS) dimethyl sulfate; (DSPE) dispersive solid phase extraction; (DW) drinking water (tap water); (EDA@Mag-CNTs)ethylenediamine-functionalized magnetic carbon nanotubes; (EG Silicone Twister®) PEG modified silicone; (EMMIPs) epitope magnetic molecularly imprinted polymers; (ESC) external standard calibration; (ESI) electrospray ionization; (FPSE) fabric phase sorptive extraction; (Fe3O4) magnetite; (Fe3O4@PEI-RGO) polyethyleneimine modified reduced graphene oxide; (Fe3O4@Fe-BTC) Fe3O4@Fe-(benzene-1,3,5-tricarboxylic acid); (FLD) fluorescence detector; (G) graphene; (GC) gas cromatography; (GCB) graphitised carbon black; (GO) graphene oxide; (GW) groundwater; (HLB) hydrophilic-lipophilic-balance; (HPLC) high performance liquid chromatography; (HSL) high spiking level; (HWW) hospital wastewater; (IDL) instrumental detection limit; (ISC) internal standard calibration (isotopically labelled internal standard as surrogates); (LC) liquid chromatography; (LDTD/APCI) laser diode thermal desorption/atmospheric pressure chemical ionization; (LLE) liquid-liquid extraction; (LOD) limit of detection; (LOQ) limit of quantification; (LSL) low spiking level; (LTQ) linear trap quadrupole; (PEG-g-MWCNTs) poly(ethylene glycol) (PEG) grafted multi-walled carbon nanotubes; (MS) mass spectrometry; (MW) marine water (sea water); (MAX) mixed-anion exchange; (MCX) mixed-mode/cationic-exchange; (MDL) method detection limit; (MQL) method quantification limit; (MRL) method reporting limit; (MIP) molecularly imprinted polymer; (MMIP) magnetic MIP; (MMC) matrix matched calibration; (m-MFMIP) magnetic multi-functional molecularly imprinted polymer; (MISEP) molecularly imprinted SPE; (MMF/AMED) multiple monolithic fiber/ poly (1-allyl-3-methylimidazolium bis (trifluoro methyl) sulfonyl)imide-co-ethylene dimethacrylate); (MMOF) magnetic metal-organic framework; (m-MWCNTs) magnetic- multi- walled carbon nanotubes; (MNPs) Magnetic nanoparticles; (MMSPD) magnetic matrix solid phase dispersion; (MS/MS) triple quadrupole; (MSL) medium spiking level; (MSTFA) N-methyl-N-(trimethylsylil)trifluoroacetamide; (MWCNTs) multi- walled carbon nanotubes; (NI) negative electrospray ionization; (NP) nanoparticles; (Ol) oleate; (NSAIDs) Nonsteroidal anti-inflammatory drugs (PAN) polyacrylonitrile nanofibers; (PI) positive electrospray ionization; (poly(MAA-co-DVB) carbowax/templated resin 50 µm (CW/TPR), copolymer of methacrylic acid and divinylbenzene; (QqLIT) quadrupole-linear ion trap; (QqLIT) triple quadrupole–linear ion trap; (Rel Rec) Rel Recovery; (Rec) recovery; (RSDr) relative standard deviation under repeatability conditions (intra-day precision); (RSDR) relative standard deviation under intermediation precision (inter-day precision); (SAC) standard addition calibration; (SAX) strong anion exchange; (SCX) strong cation-exchange; (Sorbent A) AMPSA/HEMA/PETRA (20/60/20): SCX polymer obtained by copolymerisation of three monomers: 2-acrylamido-2-methylpropanesulphonic acid (AMPSA), 2-hydroxyethyl methacrylate (HEMA) and pentaerythritol triacrylate (PETRA); (Sorbent B) (HEMA/DVB (50/50): SCX polymer obtained by post-modification with sulphuric acid (H2SO4) of a copolymer based on HEMA and divinylbenzene (DVB); (SBSE) stir bar sorptive extraction; (SPE) solid phase extraction; (SPME) solid phase microextraction (SW) surface water; (TOF) time-of-flight; (U(H)PLC) ultra(-high) performance liquid chromatography; (UFLC) ultra-fast liquid chromatograph; (UV) ultra-violet; (UV-Vis) ultra-violet visible; (WAX) weak anion exchange; (WW) wastewater; (WWE) wastewater effluent; (WWI) wastewater influent.
4. Methods Validation
Analytical methods should be well characterized to define their application area and reliability, by meeting the specifications related to the intended use of the analytical results, according to current international recommendations [228]. This process, by which the method proves its “fitness for purpose”, is known as validation [229].
The validation should be started only after an initial period of familiarization with the method to be validated. Operators acquire knowledge regarding the conditions of handling and storing samples, preparation of reagents, carrying out tests, quality control procedures and interpretation of obtained results. The methods can be standard or internal. Regarding standardized methods, it is assumed that they have been adequately validated, are subject to periodic updating, and are recognized by the national or international authority [230].
Relative to an investigation, new methods are implemented or developed. Therefore, it is necessary to define which assays and statistical tests to apply, in order to assess whether the method applies to a given matrix. The quality and credibility of the results depend on the results of these tests.
Any method is a process that involves manipulations that are likely to accumulate errors (systematic or random) and, in some situations, can significantly change the result. The errors that are associated with analytical measurements can have different origins [230,231]: (i) random measurement errors (affect repeatability); (ii) effect of the run-in (considered as resulting from the bias of a run and the random variation of several runs); (iii) effect of the laboratory (viewed as the bias of a single laboratory); (iv) bias of the method; and, (v) matrix effect.
The error component that is associated with repeatability includes contributions of volumetric, gravimetric, and heterogeneous errors of the test material, and they are evaluated based on the dispersion of the results from the replicate measurement. The running effect contributes with an additional effect of variation between days, associated, for example, with the change of the operator, change of reagent lots, recalibration of the equipment, and environmental modifications (for example, change of temperature). The laboratory’s impact result from factors that are related to variations in standards, reagents, equipment, and environmental conditions. Differences between laboratories are assessed through collaborative studies between laboratories or participation in interlaboratory trials [230,231].
The validation requirements should be specified in directives, standards, or guides, which should be followed whenever possible. For example, the validation of methods for water analysis must follow a strategy in line with the ones that are used by International Standard Organization (ISO) [232,233,234], International Union of Pure and Applied Chemistry (IUPAC) [230], or EURACHEM [231], which ensure that the terminology applied to the validation, together with the statistical treatment, is interpreted in agreement with the concerning sector [229].
The minimum requirements for a new or developed method include the study and knowledge of the following parameters: (i) linearity/working range; (ii) analytical thresholds (detection and quantification limits); (iii) repeatability; (iv) reproducibility; (v) accuracy; and, vi) uncertainty [232]. It is necessary to define the acceptance criteria for each of these parameters, which varies with the matrix under analysis, the target compound’s concentration, or the developed method. Usually, these acceptance criteria are, in national or community legislation, applicable to that matrix and analyte. In the absence of such standards, it is usual to adopt others by analogy. For example, if the monitoring of PhACs in water is not part of the legislation, the acceptance criteria of other organic compounds analyzed by the same type of methods and whose concentration in the samples is in the same order of magnitude is assumed. Usually, the acceptance criteria for different analytes are similar if the analytical methods and samples’ concentrations are similar. A brief evaluation of the validation parameters is done, focusing on the main problems or limitations that are found in the literature.
Target compounds analyzed by the same or different methods show different linear profile when analyzed by other researchers. After the linearity study, the working range must be defined. According to ISO 8466-1 [232], the linear range should be evaluated in any quantification procedure, and several statistical parameters were defined, namely, determination coefficient (R2), coefficient of variation of the method (CVm), and Fisher Snedecor test (Mandel test). The R2 is a good correlation indicator but is not a linear indicator. However, most researchers report linear studies based on R2 and the other parameters are not quantified. Only a few reviews [10,59] write the Mandel test. Usually, the PG and tabulated F values are missing.
Another problem is the acceptable R2. Some publications report values lower or equal to 0.98. To fit the requirement of PG lower than F (0.05; 1; N-3), the R2 should be higher than 0.995, and the CVm should be lower than 5% [232]. In addition, some calibration curves have a broad range of concentration values, being higher than one magnitude order and not adjusted to the concentration of target compounds in samples. Some authors use the same calibration curve to quantify the target compounds in different matrix samples with very different concentrations. Therefore, the concentration range is not well adjusted to both samples. For example, PhACs in drinking water or wastewater [224,225], or surface water and wastewater [45]. Nevertheless, the calibration curve’s first concentration is not represented by the limit of quantification for each target compound.
The matrix interference is a great problem in the quantification of PhACs in trace concentration in complex matrices. The quantification by external standard method is not adequate, due to matrix interferences. This is particularly relevant in quantitative analysis of environmental samples using chromatography coupled to mass spectrometry due to the suppression or the enhancement of the analyte signals in the presence of matrix components. The standard addition calibration or the matrix matched calibration is often used. Even in these conditions, instrumental analysis has problems of response linearity over wide concentration ranges, especially in the analysis of complex matrices, such as surface or wastewater, especially when the matrix components can act differently, depending on the level of analyte concentration [78,235].
Standard addition calibration (SAC) is the most suitable method for compensating matrix effects, but it is time-consuming and laborious [10,48]. Therefore, other methods have been proposed, such as internal standard calibration (ISC) and matrix-matched calibration (MMC). The most versatile approach is to use isotopically labeled standards (ISC) [166,167,236]. This approach has been considered the best option for compensating signal suppression or enhancement in mass spectrometry. It uses isotope-labeled analogues for quantitative analysis. In LC-MS/MS analysis, it is expected that the ionization of the target analytes and the isotope-labeled analogues will be suppressed or enhanced to the same extent, as the analogues should have the same chromatographic retention times and ionization characteristics as the analytes (184). However, these standards may be expensive, and they are often not commercially available for environmental analysis. Sometimes it is possible to use structurally similar unlabeled compounds as internal standards that do not coelute with the analytes of interest [235].
Concerning the limit of detection (LOD) and limit of quantification (LOQ), most of the publications report their values that are based on a signal-to-noise ratio (S/N) of 3 and 10, respectively [10,96,222]. Few documents show the LOD and LOQ based on the standard deviation of the method (Sm), LOD = 3 × Sm and LOQ = 10 × Sm, respectively. Sm is the ratio between the relative standard deviation of the standard addition calibration curves (Sy/x) and its slope (b) [23,60].
The confirmation of the LOQ with a standard control with the same concentration value for the target compound was not performed for most of the studies. Besides, when used, it has a concentration higher than the LOQ.
Most of the studies evaluated the precision under repeatability and reproducibility conditions. The instrumental trueness is also assessed, but the trueness of the global method, pre-treatment and chromatographic method, is not always considered with certified reference materials (CRM) or spiked samples with appropriate concentration ranges [27].
Matrix effects, extraction efficiency, and absolute recovery of the developed analytical methods should always be determined [109,175,178].
Usually, the matrix effect is evaluated by recovery studies. However, the spiked concentrations are sometimes too high when compared to concentration values that were obtained in real samples. Some authors reported recoveries around 100% determined with surrogates. However, these values should have been reported as relative recoveries. This approach did not well evaluate the extraction efficiency. Although the recoveries higher than 70% are desirable, lower recoveries, even those lower 40% have been acceptable when their repeatability was good, to obtain as much information as possible regarding the presence of PhACs in environmental waters. When the ones corrected the SPE recoveries with the corresponding surrogates, the percentage of compounds with relative recovery around 100% increased significantly [44,64,221].
Notwithstanding the uncertainty estimate should be a necessary component of an analytical result, it is still a severe problem, especially in monitoring the presence of pharmaceuticals in the environmental waters [87].
There are several possible approaches to determine the uncertainty [237]: (i) bottom-up; (ii) fitness-for-purpose; (iii) top-down; (iv) validation-based; and, (v) robustness-based–based on robustness tests from interlaboratory studies. However, there are few papers relative to the estimation of expanded uncertainty of the methods for determining PhACs in environmental water samples.
Vera-Candioti et al. [150] reported expanded uncertainties between 8.5% to 29.0% for NSAIDs and antiepileptic drugs in river water by SPE- LC-DAD.
Henriques et al. [59] compared the expanded uncertainties that were obtained by bottom-up approach (method 1) and the top-bottom approach (method 2) in the quantification of endocrine disrupters in natural and drinking waters by SPE-LC-MS/MS [238,239]. The estimation of uncertainty by method 1 was between 18% and 26%, while, by method 2, it was between 15% and 30%. In method 1, the calibration component of uncertainty had the highest contribution to the expanded uncertainty. In method 2, there were no statistically significant differences between the contribution of the variability component and the accuracy component to the expanded uncertainty (p value = 0.60).
Kot-Wasik et al. [217] compared the expanded uncertainties of on-line and off-line SPE coupled to LC-DAD-MS technique to determine NSAIDs. The uncertainties ranged from 15% to 37% with on-line SPE and 19% to 64%with off-line SPE.
Borecka et al. [87] showed a procedure to facilitate the introduction of uncertainty estimation in chromatographic measurements to the analysis of pharmaceutical residues in environmental sample waters. The proposed method is in line with the Guide to the Expression of Uncertainty in Measurement (GUM) [239]. The uncertainties ranged from 4.2% to 69.7% for eleven antibiotics in marine waters by SPE disks-LC–MS/MS. This study demonstrates that the expanded uncertainty can be a powerful tool for optimizing an analytical method, since it allows for seeing where authors should concentrate their efforts to improve the reliability of their results [87].
The differences of expanded uncertainties can be explained by the different approaches to estimate uncertainty by these authors as well as the different quantification methods. The calibration component had the highest contribution for the expanded uncertainty [59]. When internal standards are used in calibration, they can reduce the value of uncertainty, but do not eliminate it completely [237]. When the concentration used to calculate the uncertainty is close to the LOD, the obtained uncertainty is higher [237]. Accordingly, this parameter should never be excluded when calculating the expanded uncertainty in trace analysis [87].
5. Conclusions
Medicines are chemicals or compounds used to cure, halt, or prevent diseases, ease symptoms, or help diagnose illnesses. Advances in therapies have enabled curing many diseases and save lives. Therefore, new pharmacologically active substances are being continuously developed to overcome old and new pathologies. However, their use in human and veterinary medicine has been growing awareness about the potential toxicity of pharmaceutical compounds in the environment, especially in water. Several analytical approaches have been reported in the literature, and this overview illustrated the most procedures that were applied in sample preparation, chromatographic, and detection methods to meet the requirements and demands of environmental water analysis.
This led to the evolution of methodologies for monitor the PhACs in trace concentrations while using new detectors, the development and implementation of more selective sample preparation methods, and the use of hyphenated techniques, such as chromatography coupled to mass spectrometry.
In the last decades, sample preparation has been at the forefront of analytical methods, to meet specific requirements: simplicity, speed, reduction of extraction and waste solvents, automation, safety, and cost reduction. The developed methods were mostly generic and comprehensive to satisfy large scale or concrete analyses requirements to comply with the intended quantification. New materials and extraction modes have been developed continuously, and rapid advances are expected, especially concerning nanostructured sorbents and their disposition in more advantageous forms (for example, fibers, bars).
Regarding sample preparation, different extractive materials for use as sorbents have been synthesized, including magnetic nanoparticles, molecularly imprinted polymers, ionic liquids, carbonaceous nanomaterials, and their many possible combinations to improve the development of these methodologies and increase the extraction’s efficiency.
It is necessary to highlight incorporating these materials in off-line and on-line sample preparation techniques that were coupled with the chromatographic analysis of PhACs in water samples. This combination has provided crucial advantages, such as the speed of analysis, total automation and miniaturization of systems, low cost and time, reduction of toxic organic solvents, samples’ volume, sorbents for extraction methods, and greater separation efficiencies.
PhACs were primarily determined by GC-MS and evolved into HPLC-MS, but LC-MS/MS is currently the primary choice for its analysis, due to the greater sensitivity.
The field of MS has broadened from the low-resolution to the high-resolution stage and from the electron-impact to the electrospray ionization mode to detect and quantify these target pollutants at much lower concentrations.
Target-compound techniques are insufficient for monitoring unknown compounds, typically environmental transformation products. In this regard, TOF and QTOF mass spectrometers, as well as Orbitrap mass spectrometers, can give valuable information, such as accurate mass measurement for the identification of metabolites and degradation products.
The analysis of PhACs in environmental samples is still a complicated and demanding task, and analysts face many problems that are associated with the need to perform reliable identification and quantification of these target compounds, especially in complex matrices. In any scientific research, crucial decisions are based on analytical information, which is based on analytical measurements, and it is assumed that reliable methods obtained them, hence the importance of quality assurance and quality control systems. For this reason, the validation of analytical techniques is crucial. In the validation, the estimate of the analytical measurement’s uncertainty is one of the less determined tools. However, the uncertainty is a basic characteristic of any measurement because it is always present at every step of a procedure.
In the future, research will focus on improving the extraction efficiency, selectivity, and speediness by developing new coated or parked materials and new designs for quantifying trace concentrations of PhACs from complex matrices, automating the devices if possible.
Finally, more research into these new methods for the detection and quantification of several PhACs in environmental waters is required. In addition, the validation of these new techniques and commercialization of new materials as sorbents require attention.
Funding
No funding was obtained for this review paper.
Conflicts of Interest
The author declares no conflict of interests.
References
- Sauve, S.; Desrosiers, M. A review of what is an emerging contaminant. Chem. Cent. J. 2014, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, M.O.; Moreira, N.F.F.; Ribeiro, A.R.; Pereira, M.F.R.; Silva, A.M.T. Occurrence and removal of organic micropollutants: An overview of the watch list of EU Decision 2015/495. Water Res. 2016, 94, 257–279. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Ucles, S.; Hernando, M.; Fernandez-Alba, A. Development of a solvent-free method for the simultaneous identification/quantification of drugs of abuse and their metabolites in environmental water by LC-MS/MS. Talanta 2011, 85, 157–166. [Google Scholar] [CrossRef]
- Deblonde, T.; Hartemann, P. Environmental impact of medical prescriptions: Assessing the risks and hazards of persistence, bioaccumulation and toxicity of pharmaceuticals. Public Health 2013, 127, 312–317. [Google Scholar] [CrossRef]
- Grizzetti, B.; Lanzanova, D.; Liquete, C.; Reynaud, A.; Cardoso, A. Assessing water ecosystem services for water resource management. Environ. Sci. Policy 2016, 61, 194–203. [Google Scholar] [CrossRef]
- Grizzetti, B.; Liquete, C.; Antunes, P.; Carvalho, L.; Geamana, N.; Giuca, R.; Leone, M.; McConnell, S.; Preda, E.; Santos, R.; et al. Ecosystem services for water policy: Insights across Europe. Environ. Sci. Policy 2016, 66, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Daughton, C.; Ternes, T. Pharmaceuticals and personal care products in the environment: Agents of subtle change? Environ. Health Perspect. 1999, 107, 907–938. [Google Scholar] [CrossRef]
- Daughton, C. Pharmaceuticals as environmental pollutants: The ramifications for human exposure. In International Encyclopedia of Public Health; Academic Press: Oxford, UK, 2008. [Google Scholar] [CrossRef]
- Kummerer, K. Pharmaceuticals in the environment. Ann. Rev. Environ. Resour. 2010, 35, 57–75. [Google Scholar] [CrossRef] [Green Version]
- Gaffney, V.D.J.; Cardoso, V.V.; Rodrigues, A.; Ferreira, E.; Benoliel, M.J.; Almeida, C.M.M. Analysis of pharmaceutical compounds in waters by SPE-UPLC-ESI-MS/MS. Química Nova 2014, 37, 138–149. [Google Scholar] [CrossRef] [Green Version]
- Kummerer, K. The presence of pharmaceuticals in the environment due to human use—Present knowledge and future challenges. J. Environ. Manag. 2009, 90, 2354–2366. [Google Scholar] [CrossRef]
- Fong, P.; Molnar, N. Norfluoxetine induces spawning and parturition in estuarine and freshwater bivalves. Bull. Environ. Contam. Toxicol. 2008, 81, 535–538. [Google Scholar] [CrossRef]
- Franzellitti, S.; Buratti, S.; Valbonesi, P.; Fabbri, E. The mode of action (MOA) approach reveals interactive effects of environmental pharmaceuticals on Mytilus galloprovincialis. Aquat. Toxicol. 2013, 140, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Franzellitti, S.; Buratti, S.; Capolupo, M.; Du, B.; Haddad, S.P.; Chambliss, C.K.; Brooks, B.W.; Fabbri, E. An exploratory investigation of various modes of action and potential adverse outcomes of fluoxetine in marine mussels. Aquat. Toxicol. 2014, 151, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Koutsogiannaki, S.; Franzellitti, S.; Fabbri, E.; Kaloyianni, M. Oxidative stress parameters induced by exposure to either cadmium or 17β-estradiol on Mytilus galloprovincialis hemocytes. The role of signaling molecules. Aquat. Toxicol. 2014, 146, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, V.; Mota-Filipe, H.; Pinto, R.; Thiemermann, C.; Loureiro, M.; Cardoso, V.; Benoliel, M.; Almeida, C. Chemical and biochemical characterization and in vivo safety evaluation of pharmaceuticals in drinking water. Environ. Toxicol. Chem. 2016, 35, 2674–2682. [Google Scholar] [CrossRef]
- WHO. Pharmaceuticals in Drinking-Water; WHO Press: Geneva, Switzerland, 2011. [Google Scholar]
- Bu, Q.; Wang, B.; Huang, J.; Deng, S.; Yu, G. Pharmaceuticals and personal care products in the aquatic environment in China: A review. J. Hazard. Mater. 2013, 262, 189–211. [Google Scholar] [CrossRef]
- Grenni, P.; Ancona, V.; Caracciolo, A. Ecological effects of antibiotics on natural ecosystems: A review. Microchem. J. 2018, 136, 25–39. [Google Scholar] [CrossRef]
- Patrolecco, L.; Rauseo, J.; Ademollo, N.; Grenni, P.; Cardoni, M.; Levantesi, C.; Luprano, M.; Caracciolo, A. Persistence of the antibiotic sulfamethoxazole in river water alone or in the co-presence of ciprofloxacin. Sci. Total Environ. 2018, 640, 1438–1446. [Google Scholar] [CrossRef]
- Boyd, G.; Palmeri, J.; Zhang, S.; Grimm, D. Pharmaceuticals and personal care products (PPCPs) and endocrine disrupting chemicals (EDCs) in stormwater canals and Bayou St. John in New Orleans, Louisiana, USA. Sci. Total Environ. 2004, 333, 137–148. [Google Scholar] [CrossRef]
- Wen, Z.H.; Chen, L.; Meng, X.Z.; Duan, Y.P.; Zhang, Z.S.; Zeng, E.Y. Occurrence and human health risk of wastewater-derived pharmaceuticals in a drinking water source for Shanghai, East China. Sci. Total Environ. 2014, 490, 987–993. [Google Scholar] [CrossRef]
- Salgado, R.; Noronha, J.; Oehmen, A.; Carvalho, G.; Reis, M. Analysis of 65 pharmaceuticals and personal care products in 5 wastewater treatment plants in Portugal using a simplified analytical methodology. Water Sci. Technol. 2010, 62, 2862–2871. [Google Scholar] [CrossRef]
- Salgado, R.; Pereira, V.J.; Carvalho, G.; Soeiro, R.; Gaffney, V.; Almeida, C.; Cardoso, V.V.; Ferreira, E.; Benoliel, M.J.; Ternes, T.A.; et al. Photodegradation kinetics and transformation products of ketoprofen, diclofenac and atenolol in pure water and treated wastewater. J. Hazard. Mater. 2013, 244, 516–527. [Google Scholar] [CrossRef] [PubMed]
- De Jesus Gaffney, V.; Almeida, C.M.M.; Rodrigues, A.; Ferreira, E.; Benoliel, M.J.; Cardoso, V.V. Occurrence of pharmaceuticals in a water supply system and related human health risk assessment. Water Res. 2015, 72, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.; Silva, L.; Meisel, L.; Lino, C.; Pena, A. Environmental impact of pharmaceuticals from Portuguese wastewaters: Geographical and seasonal occurrence, removal and risk assessment. Environ. Res. 2015, 136, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Sousa, M.; Goncalves, C.; Cunha, E.; Hajslova, J.; Alpendurada, M. Cleanup strategies and advantages in the determination of several therapeutic classes of pharmaceuticals in wastewater samples by SPE-LC-MS/MS. Anal. Bioanal. Chem. 2011, 399, 807–822. [Google Scholar] [CrossRef] [PubMed]
- European Union. Directive 2013/39/EU of the European Parliament and of the Council of 12 august 2013. Off. J. Eur. Union 2013, 226, 13–15. [Google Scholar]
- European Union. Commission implementing decision (EU) 2015/495 of 20 march 2015 establishing a watch list of substances for Union-wide monitoring in the field of water policy pursuant to Directive 2008/105/EC of the European Parliament and of the Council. Off. J. Eur. Union 2015, 78, 40–42. [Google Scholar]
- European Union. Commission implementing decision (EU) 2018/840 of 5 june 2018 establishing a watch list of substances for Union-wide monitoring in the field of water policy pursuant to Directive 2008/105/EC of the European Parliament and of the Council and repealing Commission Implementing Decision (EU) 2015/495. Off. J. Eur. Union 2018, 141, 9–12. [Google Scholar]
- European Community. Communication from the Commission of the Council and the European Parliament. In A European One Heath Action Plan against Antimicrobial Resistance (AMR); European Community: Brussels, Belgium, 2017. [Google Scholar]
- Afonso-Olivares, C.; Sosa-Ferrera, Z.; Santana-Rodríguez, J.J. Occurrence and environmental impact of pharmaceutical residues from conventional and natural wastewater treatment plants in Gran Canaria (Spain). Sci. Total Environ. 2017, 599, 934–943. [Google Scholar] [CrossRef]
- Agüera, A.; Martínez Bueno, M.J.; Fernández-Alba, A.R. New trends in the analytical determination of emerging contaminants and their transformation products in environmental waters. Environ. Sci. Pollut. Res. Int. 2013, 20, 3496–3515. [Google Scholar] [CrossRef]
- Merone, G.M.T.; Locatelli, A.; D’Ovidio, M.; Rosato, C.; de Grazia, E.; Santavenere, U.; Rossi, F.; Savini, S.F. Analytical chemistry in the 21st century: Chalenges, solutions, and future perspectives of complex matrices quantitative analysis in biological/clinical field. Analytica 2020, 1, 44–59. [Google Scholar] [CrossRef]
- Patnaik, P. Handbook of Environmental Analysis: Chemical Pollutants in Air, Water, Soil, and Solid Wastes; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Loos, R.; Carvalho, R.; Antonio, D.; Cornero, S.; Locoro, G.; Tavazzi, S.; Paracchini, B.; Ghiani, M.; Lettieri, T.; Blaha, L.; et al. EU-wide monitoring survey on emerging polar organic contaminants in wastewater treatment plant effluents. Water Res. 2013, 47, 6475–6487. [Google Scholar] [CrossRef]
- Gros, M.; Rodríguez-Mozaz, S.; Barceló, D. Fast and comprehensive multi-residue analysis of a broad range of human and veterinary pharmaceuticals and some of their metabolites in surface and treated waters by ultra-high-performance liquid chromatography coupled to quadrupole-linear ion trap tandem mass spectrometry. J. Chromatogr. A 2012, 1248, 104–121. [Google Scholar] [CrossRef] [PubMed]
- Jelic, A.; Petrovic, M.; Barceló, D. Multi-residue method for trace level determination of pharmaceuticals in solid samples using pressurized liquid extraction followed by liquid chromatography/quadropole-linear ion trap mass spectrometry. Talanta 2009, 80, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Chiaia-Hernandez, A.; Krauss, M.; Hollender, J. Screening of lake sediments for emerging contaminants by liquid chromatography atmospheric pressure photoionization and electrospray ionization coupled to high resolution mass spectrometry. Environ. Sci. Technol. 2013, 47, 976–986. [Google Scholar] [CrossRef]
- Barron, L.; Tobin, J.; Paull, B. Multi-residue determination of pharmaceuticals in sludge and sludge enriched soils using pressurized liquid extraction, solid phase extraction and liquid chromatography with tandem mass spectrometry. J. Environ. Monit. 2008, 10, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Hernández, F.; Sancho, J.V.; Ibáñez, M.; Abad, E.; Portolés, T.; Mattioli, L. Current use of high-resolution mass spectrometry in the environmental sciences. Anal. Bioanal. Chem. 2012, 403, 1251–1264. [Google Scholar] [CrossRef] [PubMed]
- Farré, M.; Kantiani, L.; Petrovic, M.; Pérez, S.; Barceló, D. Achievements and future trends in the analysis of emerging organic contaminants in environmental samples by mass spectrometry and bioanalytical techniques. J. Chromatogr. A 2012, 1259, 86–99. [Google Scholar] [CrossRef]
- Petrovic, M.; Farré, M.; de Alda, M.L.; Perez, S.; Postigo, C.; Köck, M.; Radjenovic, J.; Gros, M.; Barcelo, D. Recent trends in the liquid chromatography-mass spectrometry analysis of organic contaminants in environmental samples. J. Chromatogr. A 2010, 1217, 4004–4017. [Google Scholar] [CrossRef] [PubMed]
- López-Serna, R.; Pérez, S.; Ginebreda, A.; Petrović, M.; Barceló, D. Fully automated determination of 74 pharmaceuticals in enctrvironmental and waste waters by online solid phase extraction-liquid chromatography-electrospray-tandem mass speometry. Talanta 2010, 83, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Lor, E.; Sancho, J.V.; Hernández, F. Simultaneous determination of acidic, neutral and basic pharmaceuticals in urban wastewater by ultra high-pressure liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2010, 1217, 622–632. [Google Scholar] [CrossRef]
- Grabic, R.; Fick, J.; Lindberg, R.H.; Fedorova, G.; Tysklind, M. Multi-residue method for trace level determination of pharmaceuticals in environmental samples using liquid chromatography coupled to triple quadrupole mass spectrometry. Talanta 2012, 100, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Fontela, M.; Galceran, M.T.; Ventura, F. Fast liquid chromatography-quadrupole-linear ion trap mass spectrometry for the analysis of pharmaceuticals and hormones in water resources. J. Chromatogr. A 2010, 1217, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, V.; Cardoso, V.; Cardoso, E.; Teixeira, A.; Martins, J.; Benoliel, M.; Almeida, C. Occurrence and behaviour of pharmaceutical compounds in a Portuguese wastewater treatment plant: Removal efficiency through conventional treatment processes. Environ. Sci. Pollut. Res. 2017, 24, 14717–14734. [Google Scholar] [CrossRef]
- Richardson, S. Environmental mass spectrometry: Emerging contaminants and current issues. Anal. Chem. 2012, 84, 747–778. [Google Scholar] [CrossRef] [PubMed]
- Petrović, M.; Barceló, D. LC-MS for identifying photodegradation products of pharmaceuticals in the environment. Trends Anal. Chem. 2007, 26, 486–493. [Google Scholar] [CrossRef]
- Gómez, M.J.; Gómez-Ramos, M.M.; Malato, O.; Mezcua, M.; Férnandez-Alba, A.R. Rapid automated screening, identification and quantification of organic micro-contaminants and their main transformation products in wastewater and river waters using liquid chromatography-quadrupole-time-of-flight mass spectrometry with an accurate-mass database. J. Chromatogr. A 2010, 1217, 7038–7054. [Google Scholar] [CrossRef]
- Gaffney, V.D.; Cardoso, V.V.; Benoliel, M.J.; Almeida, C.M.M. Chlorination and oxidation of sulfonamides by free chlorine: Identification and behaviour of reaction products by UPLC-MS/MS. J. Environ. Manag. 2016, 166, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Escher, B.I.; Fenner, K. Recent advances in environmental risk assessment of transformation products. Environ. Sci. Technol. 2011, 45, 3835–3847. [Google Scholar] [CrossRef]
- Küster, A.; Alder, A.C.; Escher, B.I.; Duis, K.; Fenner, K.; Garric, J.; Hutchinson, T.H.; Lapen, D.R.; Péry, A.; Römbke, J.; et al. Environmental risk assessment of human pharmaceuticals in the European Union: A case study with the β-blocker atenolol. Integr. Environ. Assess. Manag. 2010, 6, 514–523. [Google Scholar] [CrossRef]
- Xia, B.; Liu, X.; Gu, Y.; Zhang, Z.; Wang, H.; Ding, L.; Zhou, Y. Non-target screening of veterinary drugs using tandem mass spectrometry on smart mass. J. Am. Soc. Mass Spectrom. 2013, 24, 789–793. [Google Scholar] [CrossRef]
- Beccaria, M.; Cabooter, D. Current developments in LC-MS for pharmaceutical analysis. Analyst 2020, 145. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Gong, Z.; Kelly, B.C. Rapid analysis of pharmaceuticals and personal care products in fish plasma micro-aliquots using liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2015, 1383, 104–111. [Google Scholar] [CrossRef]
- Ferhi, S.; Bourdat-Deschamps, M.; Daudin, J.; Houot, S.; Nelieu, S. Factors influencing the extraction of pharmaceuticals from sewage sludge and soil: An experimental design approach. Anal. Bioanal. Chem. 2016, 408, 6153–6168. [Google Scholar] [CrossRef]
- Henriques, M.; Cardoso, V.; Rodrigues, A.; Ferreira, E.; Benoliel, M.; Almeida, C. Experimental and statistical validation of several endocrine disrupters by solid-phase extraction, liquid chromatography tandem mass spectrometry. Water Resour. Prot. 2010, 2, 818–829. [Google Scholar] [CrossRef] [Green Version]
- Baranowska, I.; Kowalski, B. An analytical procedure for the determination of different therapeutic drugs in surface waters. Water Sci. Technol. 2009, 60, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Gilart, N.; Miralles, N.; Marce, R.; Borrull, F.; Fontanals, N. Novel coatings for stir bar sorptive extraction to determine pharmaceuticals and personal care products in environmental waters by liquid chromatography and tandem mass spectrometry. Anal. Chim. Acta 2013, 774, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Galan, M.; Diaz-Cruz, M.; Barcelo, D. Determination of 19 sulfonamides in environmental water samples by automated on-line solid-phase extraction-liquid chromatography-tandem mass spectrometry (SPE-LC-MS/MS). Talanta 2010, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, I.; Javad, S.; Yousaf, Z.; Iqbal, S.; Jabeen, K. Review: Microwave assisted extraction of phytochemicals an efficient and modern approach for botanicals and pharmaceuticals. Pak. J. Pharm. Sci. 2019, 32, 223–230. [Google Scholar]
- Evans, S.E.; Davies, P.; Lubben, A.; Kasprzyk-Hordern, B. Determination of chiral pharmaceuticals and illicit drugs in wastewater and sludge using microwave assisted extraction, solid-phase extraction and chiral liquid chromatography coupled with tandem mass spectrometry. Anal. Chim. Acta 2015, 882, 112–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunez, M.; Borrull, F.; Pocurull, E.; Fontanals, N. Pressurized liquid extraction followed by liquid chromatography with tandem mass spectrometry to determine pharmaceuticals in mussels. J. Sep. Sci. 2016, 39, 741–747. [Google Scholar] [CrossRef]
- Löffler, D.; Ternes, T.A. Determination of acidic pharmaceuticals, antibiotics and ivermectin in river sediment using liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2003, 1021, 133–144. [Google Scholar] [CrossRef]
- Sun, H.; Ge, X.; Lv, Y.; Wang, A. Application of accelerated solvent extraction in the analysis of organic contaminants, bioactive and nutritional compounds in food and feed. J. Chromatogr. A 2012, 1237, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.; Albino, S.; Silva, S.; Cravo, A.; Cardoso, V.; Benoliel, M.; Almeida, C. Development of a multiresidue method for the determination of 24 pharmaceuticals in clams by QuEChERS and liquid chromatography-triple quadrupole tandem mass spectrometry. Food Anal. Methods 2019, 12, 838–851. [Google Scholar] [CrossRef]
- Núñez, M.; Borrull, F.; Fontanals, N.; Pocurull, E. Determination of pharmaceuticals in bivalves using QuEChERS extraction and liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 3841–3849. [Google Scholar] [CrossRef] [PubMed]
- González, A.G.; Herrador, M.Á. A practical guide to analytical method validation, including measurement uncertainty and accuracy profiles. Trends Anal. Chem. 2007, 26, 227–238. [Google Scholar] [CrossRef]
- González, A.G.; Herrador, M.A.; Asuero, A.G. Intra-laboratory assessment of method accuracy (trueness and precision) by using validation standards. Talanta 2010, 82, 1995–1998. [Google Scholar] [CrossRef]
- Wasik, A.; Kot-Wasik, A.; Namiesnik, J. New trends in sample preparation techniques for the analysis of the residues of pharmaceuticals in environmental samples. Curr. Anal. Chem. 2016, 12, 280–302. [Google Scholar] [CrossRef]
- Ali, I.; Suhail, M.; Alharbi, O.; Hussain, I. Advances in sample preparation in chromatography for organic environmental pollutants analyses. J. Liq. Chromatogr. Relat. Technol. 2019, 42, 137–160. [Google Scholar] [CrossRef]
- Moldoveanu, S. Solutions and challenges in sample preparation for chromatography. J. Chromatogr. Sci. 2004, 42, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Maciel, E.; de Toffoli, A.; Lancas, F. Recent trends in sorption-based sample preparation and liquid chromatography techniques for food analysis. Electrophoresis 2018, 39, 1582–1596. [Google Scholar] [CrossRef]
- Hyötyläinen, T.; Riekkola, M.L. Sorbent- and liquid-phase microextraction techniques and membrane-assisted extraction in combination with gas chromatographic analysis: A review. Anal. Chim. Acta 2008, 614, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Iparraguirre, A.; Navarro, P.; Rodil, R.; Prieto, A.; Olivares, M.; Etxebarria, N.; Zuloaga, O. Matrix effect during the membrane-assisted solvent extraction coupled to liquid chromatography tandem mass spectrometry for the determination of a variety of endocrine disrupting compounds in wastewater. J. Chromatogr. A 2014, 1356, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Perez-Fernandez, V.; Rocca, L.; Tomai, P.; Fanali, S.; Gentili, A. Recent advancements and future trends in environmental analysis: Sample preparation, liquid chromatography and mass spectrometry. Anal. Chim. Acta 2017, 983, 9–41. [Google Scholar] [CrossRef]
- Tranchida, P.; Maimone, M.; Purcaro, G.; Dugo, P.; Mondello, L. The penetration of green sample-preparation techniques in comprehensive two-dimensional gas chromatography. Trends Anal. Chem. 2015, 71, 74–84. [Google Scholar] [CrossRef]
- Cimetiere, N.; Soutrel, I.; Lemasle, M.; Laplanche, A.; Crocq, A. Standard addition method for the determination of pharmaceutical residues in drinking water by SPE-LC-MS/MS. Environ. Technol. 2013, 34, 3031–3041. [Google Scholar] [CrossRef] [Green Version]
- Gilart, N.; Marcé, R.M.; Fontanals, N.; Borrull, F. A rapid determination of acidic pharmaceuticals in environmental waters by molecularly imprinted solid-phase extraction coupled to tandem mass spectrometry without chromatography. Talanta 2013, 110, 196–201. [Google Scholar] [CrossRef]
- Xu, J.; Sun, H.; Zhang, Y.; Alder, A.C. Occurrence and enantiomer profiles of β-blockers in wastewater and a receiving water body and adjacent soil in Tianjin, China. Sci. Total Environ. 2019, 650, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Togola, A.; Baran, N.; Coureau, C. Advantages of online SPE coupled with UPLC/MS/MS for determining the fate of pesticides and pharmaceutical compounds. Anal. Bioanal. Chem. 2014, 406, 1181–1191. [Google Scholar] [CrossRef] [Green Version]
- Huntscha, S.; Singer, H.P.; McArdell, C.S.; Frank, C.E.; Hollender, J. Multiresidue analysis of 88 polar organic micropollutants in ground, surface and wastewater using online mixed-bed multilayer solid-phase extraction coupled to high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2012, 1268, 74–83. [Google Scholar] [CrossRef]
- Diaz-Cruz, M.; de Alda, M.; Barcelo, D. Environmental behavior and analysis of veterinary and human drugs in soils, sediments and sludge. Trends Anal. Chem. 2003, 22, 340–351. [Google Scholar] [CrossRef]
- Richardson, S.; Ternes, T. Water analysis: Emerging contaminants and current issues. Anal. Chem. 2011, 83, 4614–4648. [Google Scholar] [CrossRef]
- Borecka, M.; Białk-Bielińska, A.; Siedlewicz, G.; Kornowska, K.; Kumirska, J.; Stepnowski, P.; Pazdro, K. A new approach for the estimation of expanded uncertainty of results of an analytical method developed for determining antibiotics in seawater using solid-phase extraction disks and liquid chromatography coupled with tandem mass spectrometry technique. J. Chromatogr. A 2013, 1304, 138–146. [Google Scholar] [CrossRef]
- Daniels, K.; Park, M.; Huang, Z.; Jia, A.; Flores, G.; Lee, H.; Snyder, S. A review of extraction methods for the analysis of pharmaceuticals in environmental waters. Crit. Rev. Environ. Sci. Technol. 2020, 50, 2271–2299. [Google Scholar] [CrossRef]
- Kim, S.; Carlson, K. Quantification of human and veterinary antibiotics in water and sediment using SPE/LC/MS/MS. Anal. Bioanal. Chem. 2007, 387, 1301–1315. [Google Scholar] [CrossRef]
- Lindsey, M.E.; Meyer, T.M.; Thurman, E.M. Analysis of trace levels of sulfonamide and tetracycline antimicrobials in groundwater and surface water using solid-phase extraction and liquid chromatography/mass spectrometry. Anal. Chem. 2001, 73, 4640–4646. [Google Scholar] [CrossRef]
- Stolker, A.A.M.; Nielsing, W.; Hogendoorn, E.A.; Versteegh, J.F.M.; Fuchs, R.; Brinkman, U.A.T. Liquid chromatography with triple-quadrupole or quadrupole-time of flight mass spectrometry for screening and confirmation of residues of pharmaceuticals in water. Anal. Bioanal. Chem. 2004, 378, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Batt, A.L.; Aga, D.S. Simultaneous analysis of multiple classes of antibiotics by ion trap LC/MS/MS for assessing surface water and groundwater contamination. Anal. Chem. 2005, 77. [Google Scholar] [CrossRef] [PubMed]
- Unceta, N.; Sampedro, M.C.; Abu Bakar, N.K.; Gómez-Caballero, A.; Goicolea, M.A.; Barrio, R.J. Multi-residue analysis of pharmaceutical compounds in wastewaters by dual solid-phase microextraction coupled to liquid chromatography electrospray ionization ion trap mass spectrometry. J. Chromatogr. A 2010, 1217, 3392–3399. [Google Scholar] [CrossRef]
- López-Serna, R.; Petrović, M.; Barceló, D. Development of a fast instrumental method for the analysis of pharmaceuticals in environmental and wastewaters based on ultra high performance liquid chromatography (UHPLC)-tandem mass spectrometry (MS/MS). Chemosphere 2011, 85, 1390–1399. [Google Scholar] [CrossRef] [PubMed]
- Seifrtová, M.; Nováková, L.; Lino, C.; Pena, A.; Solich, P. An overview of analytical methodologies for the determination of antibiotics in environmental waters. Anal. Chim. Acta 2009, 649, 158–179. [Google Scholar] [CrossRef] [PubMed]
- Gros, M.; Petrovic, M.; Barcelo, D. Tracing pharmaceutical residues of different therapeutic classes in environmental Waters by using liquid chromatography/quadrupole-linear ion trap mass spectrometry and automated library searching. Anal. Chem. 2009, 81, 898–912. [Google Scholar] [CrossRef]
- Wong, C.S.; MacLeod, S.L. JEM spotlight: Recent advances in analysis of pharmaceuticals in the aquatic environment. J. Environ. Monit. 2009, 11, 923–936. [Google Scholar] [CrossRef]
- Kasprzyk-Hordern, B.; Dinsdale, R.M.; Guwy, A.J. Multi-residue method for the determination of basic/neutral pharmaceuticals and illicit drugs in surface water by solid-phase extraction and ultra performance liquid chromatography-positive electrospray ionisation tandem mass spectrometry. J. Chromatogr. A 2007, 1161, 132–145. [Google Scholar] [CrossRef]
- Robles-Molina, J.; Lara-Ortega, F.J.; Gilbert-López, B.; García-Reyes, J.F.; Molina-Díaz, A. Multi-residue method for the determination of over 400 priority and emerging pollutants in water and wastewater by solid-phase extraction and liquid chromatography-time-of-flight mass spectrometry. J. Chromatogr. A 2014, 1350, 30–43. [Google Scholar] [CrossRef]
- Kasprzyk-Hordern, B.; Dinsdale, R.; Guwy, A. The removal of pharmaceuticals, personal care products, endocrine disruptors and illicit drugs during wastewater treatment and its impact on the quality of receiving waters. Water Res. 2009, 43, 363–380. [Google Scholar] [CrossRef] [PubMed]
- Batt, A.L.; Kostich, M.S.; Lazorchak, J.M. Analysis of ecologically relevant pharmaceuticals in wastewater and surface water using selective solid-phase extraction and UPLC-MS/MS. Anal. Chem. 2008, 80, 5021–5030. [Google Scholar] [CrossRef] [PubMed]
- Ido, A.; Hiromori, Y.; Meng, L.; Usuda, H.; Nagase, H.; Yang, M.; Hu, J.; Nakanishi, T. Occurrence of fibrates and their metabolites in source and drinking water in Shanghai and Zhejiang, China. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panditi, V.R.; Batchu, S.R.; Gardinali, P.R. Online solid-phase extraction-liquid chromatography-electrospray-tandem mass spectrometry determination of multiple classes of antibiotics in environmental and treated waters. Anal. Bioanal. Chem. 2013, 405, 5953–5964. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Thurman, E.M. Analysis of 100 pharmaceuticals and their degradates in water samples by liquid chromatography/quadrupole time-of-flight mass spectrometry. J. Chromatogr. A 2012, 1259, 148–157. [Google Scholar] [CrossRef]
- Hernández, F.; Ibáñez, M.; Gracia-Lor, E.; Sancho, J.V. Retrospective LC-QTOF-MS analysis searching for pharmaceutical metabolites in urban wastewater. J. Sep. Sci. 2011, 34, 3517–3526. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, H.; Chang, H. Improved method for analyzing estrogens in water by liquid chromatography-electrospray mass spectrometry. J. Chromatogr. A 2005, 1070, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Gilart, N.; Marcé, R.M.; Borrull, F.; Fontanals, N. Determination of pharmaceuticals in wastewaters using solid-phase extraction-liquid chromatography-tandem mass spectrometry. J. Sep. Sci. 2012, 35, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Ammann, A.A.; Macikova, P.; Groh, K.J.; Schirmer, K.; Suter, M.J. LC-MS/MS determination of potential endocrine disruptors of cortico signalling in rivers and wastewaters. Anal. Bioanal. Chem. 2014, 406, 7653–7665. [Google Scholar] [CrossRef] [PubMed]
- Vieno, N.; Tuhkanen, T.; Kronberg, L. Analysis of neutral and basic pharmaceuticals in sewage treatment plants and in recipient rivers using solid phase extraction and liquid chromatography-tandem mass spectrometry detection. J. Chromatogr. A 2006, 1134, 101–111. [Google Scholar] [CrossRef]
- Rodriguez, E.; Navarro-Villoslada, F.; Benito-Pena, E.; Marazuela, M.; Moreno-Bondi, M. Multiresidue determination of ultratrace levels of fluoroquinolone antimicrobials in drinking and aquaculture water samples by automated online molecularly imprinted solid phase extraction and liquid chromatography. Anal. Chem. 2011, 83, 2046–2055. [Google Scholar] [CrossRef]
- Mogolodi Dimpe, K.; Nomngongo, P.N. Application of activated carbon-decorated polyacrylonitrile nanofibers as an adsorbent in dispersive solid-phase extraction of fluoroquinolones from wastewater. J. Pharm. Anal. 2019, 9, 117–126. [Google Scholar] [CrossRef]
- Wu, A.; Zhao, X.; Wang, J.; Tang, Z.; Zhao, T.; Niu, L.; Yu, W.; Yang, C.; Fang, M.; Lv, H.; et al. Application of solid-phase extraction based on magnetic nanoparticle adsorbents for the analysis of selected persistent organic pollutants in environmental water: A review of recent advances. Crit. Rev. Environ. Sci. Technol. 2020. [Google Scholar] [CrossRef]
- Herrera-Herrera, A.V.; Hernández-Borges, J.; Afonso, M.M.; Palenzuela, J.A.; Rodríguez-Delgado, M. Comparison between magnetic and non magnetic multi-walled carbon nanotubes-dispersive solid-phase extraction combined with ultra-high performance liquid chromatography for the determination of sulfonamide antibiotics in water samples. Talanta 2013, 116, 695–703. [Google Scholar] [CrossRef]
- Li, N.; Chen, J.; Shi, Y. Magnetic polyethyleneimine functionalized reduced graphene oxide as a novel magnetic sorbent for the separation of polar non-steroidal anti-inflammatory drugs in waters. Talanta 2019, 191, 526–534. [Google Scholar] [CrossRef]
- Capriotti, A.; Cavaliere, C.; La Barbera, G.; Montone, C.; Piovesana, S.; Lagana, A. Recent applications of magnetic solid-phase extraction for sample preparation. Chromatographia 2019, 82, 1251–1274. [Google Scholar] [CrossRef]
- Liu, D.; Huang, Z.; Li, M.; Li, X.; Sun, P.; Zhou, L. Construction of magnetic bifunctional beta-cyclodextrin nanocomposites for adsorption and degradation of persistent organic pollutants. Carbohydr. Polym. 2020, 230. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Huang, Z.; Li, M.; Sun, P.; Yu, T.; Zhou, L. Novel porous magnetic nanospheres functionalized by beta-cyclodextrin polymer and its application in organic pollutants from aqueous solution. Environ. Pollut. 2019, 250, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; He, Q.; Wang, L.; Wang, X.; Dong, Q.; Huang, C. Preparation of magnetic multi-functional molecularly imprinted polymer beads for determining environmental estrogens in water samples. J. Hazard. Mater. 2013, 252, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Soto, J.M.; Cárdenas, S.; Valcárcel, M. Evaluation of carbon nanocones/disks as sorbent material for solid-phase extraction. J. Chromatogr. A 2009, 1216, 5626–5633. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Chen, L.; Li, J.; Liu, D.; Chen, L. Recent advances in solid-phase sorbents for sample preparation prior to chromatographic analysis. Trends Anal. Chem. 2014, 59, 26–41. [Google Scholar] [CrossRef]
- Li, X.; Zhu, G.; Luo, Y.; Yuan, B.; Feng, Y. Synthesis and applications of functionalized magnetic materials in sample preparation. Trends Anal. Chem. 2013, 45, 233–247. [Google Scholar] [CrossRef]
- Pérez, R.A.; Albero, B.; Férriz, M.; Tadeo, J.L. Analysis of macrolide antibiotics in water by magnetic solid-phase extraction and liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2017, 146, 79–85. [Google Scholar] [CrossRef]
- Luo, Y.; Shi, Z.; Gao, Q.; Feng, Y. Magnetic retrieval of graphene: Extraction of sulfonamide antibiotics from environmental water samples. J. Chromatogr. A 2011, 1218, 1353–1358. [Google Scholar] [CrossRef]
- Abdolmohammad-Zadeh, H.; Talleb, Z. Magnetic solid phase extraction of gemfibrozil from human serum and pharmaceutical wastewater samples utilizing a beta-cyclodextrin grafted graphene oxide-magnetite nano-hybrid. Talanta 2015, 134, 387–393. [Google Scholar] [CrossRef]
- Pena-Mendez, E.; Mawale, R.; Conde-Gonzalez, J.; Socas-Rodriguez, B.; Havel, J.; Ruiz-Perez, C. Metal organic framework composite, nano-Fe3O4@Fe-(benzene-1,3,5-tricarboxylic acid), for solid phase extraction of blood lipid regulators from water. Talanta 2020, 207. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, Y.; Zhan, P.; Chen, X.; Pan, S.; Jin, M. Fast determination of 24 steroid hormones in river water using magnetic dispersive solid phase extraction followed by liquid chromatography-tandem mass spectrometry. Environ. Sci. Pollut. Res. 2016, 23, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Arteaga, K.; Rodriguez, J.; Miranda, J.; Medina, J.; Barrado, E. Determination of non-steroidal anti-inflammatory drugs in wastewaters by magnetic matrix solid phase dispersion-HPLC. Talanta 2010, 80, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Socas-Rodríguez, B.; Hernández-Borges, J.; Salazar, P.; Martín, M.; Rodríguez-Delgado, M. Core-shell polydopamine magnetic nanoparticles as sorbent in micro-dispersive solid-phase extraction for the determination of estrogenic compounds in water samples prior to high-performance liquid chromatography-mass spectrometry analysis. J. Chromatogr. A 2015, 1397, 1–10. [Google Scholar] [CrossRef]
- Sajid, M.; Nazal, M.K.; Ihsanullah, I. Novel materials for dispersive (micro) solid-phase extraction of polycyclic aromatic hydrocarbons in environmental water samples: A review. Anal. Chim. Acta 2021, 1141, 246–262. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, Y.; Qiu, Q.; Zhu, Y.; Min, J.; Jin, M. A fast and high throughput LC-MS/MS method for the determination of 58 human and veterinary drugs in river water. Anal. Methods 2017, 9, 4228–4233. [Google Scholar] [CrossRef]
- Wackerlig, J.; Schirhagl, R. Applications of molecularly imprinted polymer nanoparticles and their advances toward industrial use: A review. Anal. Chem. 2016, 88, 250–261. [Google Scholar] [CrossRef]
- Demeestere, K.; Petrovic, M.; Gros, M.; Dewulf, J.; Van Langenhove, H.; Barcelo, D. Trace analysis of antidepressants in environmental waters by molecularly imprinted polymer-based solid-phase extraction followed by ultra-performance liquid chromatography coupled to triple quadrupole mass spectrometry. Anal. Bioanal. Chem. 2010, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, X.; Lu, W.; Wu, X.; Li, J. Molecular imprinting: Perspectives and applications. Chem. Soc. Rev. 2016, 45, 2137–2211. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, C.; Li, X.; Liu, L. Preparation and application of epitope magnetic molecularly imprinted polymers for enrichment of sulfonamide antibiotics in water. Electrophoresis 2017, 38, 2462–2467. [Google Scholar] [CrossRef]
- Rebelo, P.; Costa-Rama, E.; Seguro, I.; Pacheco, J.G.; Nouws, H.P.A.; Cordeiro, M.N.D.S.; Delerue-Matos, C. Molecularly imprinted polymer-based electrochemical sensors for environmental analysis. Biosens. Bioelectron. 2021, 172, 112719. [Google Scholar] [CrossRef] [PubMed]
- Wackerlig, J.; Lieberzeit, P. Molecularly imprinted polymer nanoparticles in chemical sensing—Synthesis, characterisation and application. Sens. Actuators B Chem. 2015, 207, 144–157. [Google Scholar] [CrossRef]
- Ouyang, G. Handbook of Solid Phase Microextraction; Pawliszyn, J., Ed.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 251–290. [Google Scholar]
- Montesdeoca-Esponda, S.; Torres-Padrón, M.E.; Sosa-Ferrera, Z.; Santana-Rodríguez, J.J. Analytical Separation Science; Jared, L., Anderson, A.B., Pino, V., Stalcup, A., Eds.; Wiley: Hoboken, NJ, USA, 2015; pp. 1897–1927. [Google Scholar]
- Pawliszyn, J. Solid Phase Microextraction, Theory and Practice; Wiley-VCH: New York, NY, USA, 1997. [Google Scholar]
- Almeida, C.; Boas, L. Analysis of BTEX and other substituted benzenes in water using headspace SPME-GC-FID: Method validation. J. Environ. Monit. 2004, 6, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Simoes, N.G.; Cardoso, V.V.; Ferreira, E.; Benoliel, M.J.; Almeida, C.M.M. Experimental and statistical validation of SPME-GC-MS analysis of phenol and chlorophenols in raw and treated water. Chemosphere 2007, 68, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Castells, P.; Santos, F.; Galceran, M. Solid-phase extraction versus solid-phase microextraction for the determination of chlorinated paraffins in water using gas chromatography—Negative chemical ionisation mass spectrometry. J. Chromatogr. A 2004, 157–162. [Google Scholar] [CrossRef]
- Sarafraz-Yazdi, A.; Amiri, A.; Rounaghi, G.; Eshtiagh-Hosseini, H. Determination of non-steroidal anti-inflammatory drugs in water samples by solid-phase microextraction based sol-gel technique using poly(ethylene glycol) grafted multi-walled carbon nanotubes coated fiber. Anal. Chim. Acta 2012, 720, 134–141. [Google Scholar] [CrossRef]
- López-Serna, R.; Marín-de-Jesús, D.; Irusta-Mata, R.; García-Encina, P.A.; Lebrero, R.; Fdez-Polanco, M.; Muñoz, R. Multiresidue analytical method for pharmaceuticals and personal care products in sewage and sewage sludge by online direct immersion SPME on-fiber derivatization—GCMS. Talanta 2018, 186, 506–512. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Zhu, F.; Jiang, R.; Zhou, S.; Zhu, D.; Liu, H.; Ouyang, G. Determination of eight pharmaceuticals in an aqueous sample using automated derivatization solid-phase microextraction combined with gas chromatography-mass spectrometry. Talanta 2015, 136, 198–203. [Google Scholar] [CrossRef]
- Araujo, L.; Wild, J.; Villa, N.; Camargo, N.; Cubillan, D.; Prieto, A. Determination of anti-inflammatory drugs in water samples, by in situ derivatization, solid phase microextraction and gas chromatography-mass spectrometry. Talanta 2008, 75, 111–115. [Google Scholar] [CrossRef]
- Yu, H.; Merib, J.; Anderson, J.L. Crosslinked polymeric ionic liquids as solid-phase microextraction sorbent coatings for high performance liquid chromatography. J. Chromatogr. A 2016, 1438, 10–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, M.; Yu, J.; Huang, X.; Li, H.; Lin, L.; Yuan, D. Monitoring of selected estrogen mimics in complicated samples usingpolymeric ionic liquid-based multiple monolithic fiber solid-phasemicroextraction combined with high-performance liquidchromatography. J. Chromatogr. A 2015, 1385, 12–19. [Google Scholar] [CrossRef]
- Gil García, M.D.; Cañada Cañada, F.; Culzoni, M.J.; Vera-Candioti, L.; Siano, G.G.; Goicoechea, H.C.; Martínez Galera, M. Chemometric tools improving the determination of anti-inflammatory and antiepileptic drugs in river and wastewater by solid-phase microextraction and liquid chromatography diode array detection. J. Chromatogr. A 2009, 1216, 5489–5496. [Google Scholar] [CrossRef] [PubMed]
- Vera-Candioti, L.; Gil García, M.D.; Martínez Galera, M.; Goicoechea, H.C. Chemometric assisted solid-phase microextraction for the determination of anti-inflammatory and antiepileptic drugs in river water by liquid chromatography-diode array detection. J. Chromatogr. A 2008, 1211, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Lock, C.M.; Chen, L.; Volmer, D.A. Rapid analysis of tetracycline antibiotics by combined solid phase microextraction/high performance liquid chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 1999, 13, 1744–1754. [Google Scholar] [CrossRef]
- McClure, E.L.; Wong, C.S. Solid phase microextraction of macrolide, trimethoprim, and sulfonamide antibiotics in wastewaters. J. Chromatogr. A 2007, 1169, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, V.K.; Terry, K.A.; Toito, J. Determination of sulfonamide antibiotics in wastewater: A comparison of solid phase microextraction and solid phase extraction methods. J. Chromatogr. A 2006, 1131, 1–10. [Google Scholar] [CrossRef]
- Sánchez-Rojas, F.; Bosch-Ojeda, C.; Cano-Pavó, J.M. A review of stir bar sorptive extraction. Chromatographia 2009, 69, S69–S84. [Google Scholar] [CrossRef]
- He, M.; Chen, B.; Hu, B. Recent developments in stir bar sorptive extraction. Anal. Bioanal. Chem. 2014, 406, 2001–2026. [Google Scholar] [CrossRef]
- Camino-Sánchez, F.J.; Rodríguez-Gómez, R.; Zafra-Gómez, A.; Santos-Fandila, A.; Vílchez, J.L. Stir bar sorptive extraction: Recent applications, limitations and future trends. Talanta 2014, 130, 388–399. [Google Scholar] [CrossRef]
- Hashemi, S.H.; Kaykhaii, M. Nanoparticle coatings for stir bar sorptive extraction, synthesis, characterization and application. Talanta 2021, 221, 121568. [Google Scholar] [CrossRef]
- Fan, W.; Mao, X.; He, M.; Chen, B.; Hu, B. Development of novel sol-gel coatings by chemically bonded ionic liquids for stir bar sorptive extraction--application for the determination of NSAIDS in real samples. Anal. Bioanal. Chem. 2014, 406, 7261–7273. [Google Scholar] [CrossRef] [PubMed]
- Gilart, N.; Marcé, R.M.; Cormack, P.A.; Fontanals, N.; Borrull, F. Development of new polar monolithic coatings for stir bar sorptive extraction. J. Sep. Sci. 2014, 37, 2225–2232. [Google Scholar] [CrossRef]
- Mao, X.; He, M.; Chen, B.; Hu, B. Membrane protected C. J. Chromatogr. A 2016, 1472, 27–34. [Google Scholar] [CrossRef]
- Peng, J.; Liu, D.; Shi, T.; Tian, H.; Hui, X.; He, H. Molecularly imprinted polymers based stir bar sorptive extraction for determination of cefaclor and cefalexin in environmental water. Anal. Bioanal. Chem. 2017, 409, 4157–4166. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Yang, Z.; Liu, Z. Development of dual-templates molecularly imprinted stir bar sorptive extraction and its application for the analysis of environmental estrogens in water and plastic samples. J. Chromatogr. A 2014, 1358, 52–59. [Google Scholar] [CrossRef]
- Bratkowska, D.; Fontanals, N.; Cormack, P.A.; Borrull, F.; Marcé, R.M. Preparation of a polar monolithic stir bar based on methacrylic acid and divinylbenzene for the sorptive extraction of polar pharmaceuticals from complex water samples. J. Chromatogr. A 2012, 1225, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ministério do Ambiente. Decreto-Lei n.º 236/1998, de 1 de Agosto de 1998, I Série—A; Ministério do Ambiente, do Ordenamento do Território e do Desenvolvimento Regional, Qualidade da Água: Lisbon, Portugal, 1998.
- Ministério do Ambiente. Decreto-Lei n.º 306/2007, de 27 de Agosto, Diário da República, 1.ª série, n.º 164, 27 de Agosto de 2007; Ministério do Ambiente, do Ordenamento do Território e do Desenvolvimento Regional, Qualidade da Água: Lisbon, Portugal, 2007.
- Petrovic, M.; Hernando, M.; Diaz-Cruz, M.; Barcelo, D. Liquid chromatography-tandem mass spectrometry for the analysis of pharmaceutical residues in environmental samples: A review. J. Chromatogr. A 2005, 1067, 1–14. [Google Scholar] [CrossRef]
- Gros, M.; Petrovic, M.; Barcelo, D. Multi-residue analytical methods using LC-tandem MS for the determination of pharmaceuticals in environmental and wastewater samples: A review. Anal. Bioanal. Chem. 2006, 386, 941–952. [Google Scholar] [CrossRef]
- Ternes, T. Analytical methods for the determination of pharmaceuticals in aqueous environmental samples. Trends Anal. Chem. 2001, 20, 419–434. [Google Scholar] [CrossRef]
- Ternes, T.A.; Bonerz, M.; Herrmann, N.; Loffler, D.; Keller, E.; Lacida, B.B.; Adler, A.C. Determination of pharmaceuticals, iodinated contrast media and musk fragrances in sludge by LC/tandem MS and GC/MS. J. Chromatogr. A 2005, 1067, 213–223. [Google Scholar] [CrossRef]
- O’Mahony, J.; Clarke, L.; Whelan, M.; O’Kennedy, R.; Lehotay, S.J.; Danaher, M. The use of ultra-high pressure liquid chromatography with tandem mass spectrometric detection in the analysis of agrochemical residues and mycotoxins in food—Challenges and applications. J. Chromatogr. A 2013, 1292, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Van de Steene, J.C.; Lambert, W.E. Comparison of matrix effects in HPLC-MS/MS and UPLC-MS/MS analysis of nine basic pharmaceuticals in surface waters. J. Am. Soc. Mass Spectrom. 2008, 19, 713–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churchwell, M.I.; Twaddle, N.C.; Meeker, L.R.; Doerge, D.R. Improving LC-MS sensitivity through increase in chromatographic performance: Comparisons of UPLC-ES/MS/MS to HPLC-ES/MS/MS. J. Chromatogr. B 2005, 825, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Petrovic, M.; Gros, M.; Barcelo, D. Multi-residue analysis of pharmaceuticals in wastewater by ultra-performance liquid chromatography-quadrupole-time-of-flight mass spectrometry. J. Chromatogr. A 2006, 1124, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Gros, M.; Rodríguez-Mozaz, S.; Barceló, D. Rapid analysis of multiclass antibiotic residues and some of their metabolites in hospital, urban wastewater and river water by ultra-high-performance liquid chromatography coupled to quadrupole-linear ion trap tandem mass spectrometry. J. Chromatogr. A 2013, 1292, 173–188. [Google Scholar] [CrossRef] [Green Version]
- Gracia-Lor, E.; Sancho, J.V.; Hernández, F. Multi-class determination of around 50 pharmaceuticals, including 26 antibiotics, in environmental and wastewater samples by ultra-high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2011, 1218, 2264–2275. [Google Scholar] [CrossRef]
- Nannou, C.; Kosma, C.; Albanis, T. Occurrence of pharmaceuticals in surface waters: Analytical method development and environmental risk assessment. Int. J. Environ. Anal. Chem. 2015, 95, 1242–1262. [Google Scholar] [CrossRef]
- Patrolecco, L.; Ademollo, N.; Grenni, P.; Tolomei, A.; Caracciolo, A.B.; Capti, S. Simultaneous determination of human pharmaceuticals in water samples by solid phase extraction and HPLC with UV- fluorescence detection. Microchem. J. 2013, 107, 165–171. [Google Scholar] [CrossRef]
- Gracia-Lor, E.; Sancho, J.; Serrano, R.; Hernandez, F. Occurrence and removal of pharmaceuticals in wastewater treatment plants at the Spanish Mediterranean area of Valencia. Chemosphere 2012, 87, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Sui, Q.; Huang, J.; Deng, S.; Yu, G. Rapid determination of pharmaceuticals from multiple therapeutic classes in wastewater by solid-phase extraction and ultra-performence liquid chromatography tandem mass spectrometry. Chin. Sci. Bull. 2009, 54, 4633–4643. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Aller, M.; Gurny, R.; Veuthey, J.L.; Guillarme, D. Coupling ultra high-pressure liquid chromatography with mass spectrometry: Constraints and possible applications. J. Chromatogr. A 2013, 1292, 2–18. [Google Scholar] [CrossRef] [PubMed]
- De Hoffman, E.; Stroobant, V. Mass Spectrometry—Principles and Applications; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar]
- Lonappan, L.; Pulicharla, R.; Rouissi, T.; Brar, S.K.; Verma, M.; Surampalli, R.Y.; Valero, J.R. Diclofenac in municipal wastewater treatment plant: Quantification using laser diode thermal desorption—Atmospheric pressure chemical ionization—Tandem mass spectrometry approach in comparison with an established liquid chromatography-electrospray ionization-tandem mass spectrometry method. J. Chromatogr. A 2016, 1433, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Metcalfe, C.D. Characterizing and compensating for matrix effects using atmospheric pressure chemical ionization liquid chromatography-tandem mass spectrometry: Analysis of neutral pharmaceuticals in municipal wastewater. Anal. Chem. 2008, 80, 2010–2017. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, D.P.; Brar, S.K.; Tyagi, R.D.; Picard, P.; Surampalli, R.Y. Carbamazepine in municipal wastewater and wastewater sludge: Ultrafast quantification by laser diode thermal desorption-atmospheric pressure chemical ionization coupled with tandem mass spectrometry. Talanta 2012, 99, 247–255. [Google Scholar] [CrossRef]
- Horimoto, S.; Mayumi, T.; Aoe, K.; Nishimura, N.; Sato, T. Analysis of beta-lactam antibiotics by high performance liquid chromatography-atmospheric pressure chemical ionization mass spectrometry using bromoform. J. Pharm. Biomed. Anal. 2002, 30, 1093–1102. [Google Scholar] [CrossRef]
- Pfeifer, T.; Tuerk, J.; Bester, K.; Spiteller, M. Determination of selected sulfonamide antibiotics and trimethoprim in manure by electrospray and atmospheric pressure chemical ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2002, 16, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Doerge, D.R.; Bajic, S. Multiresidue determination of quinolone antibiotics using liquid chromatography coupled to atmospheric-pressure chemical ionization mass spectrometry and tandem mass spectrometry. Rapid Commun. Mass Spectrom. 1995, 9, 1012–1016. [Google Scholar] [CrossRef]
- Gardinali, P.R.; Zhao, X. Trace determination of caffeine in surface water samples by liquid chromatography—Atmospheric pressure chemical ionization—Mass spectrometry (LC-APCI-MS). Environ. Int. 2002, 28, 521–528. [Google Scholar] [CrossRef]
- Matić, I.; Grujić, S.; Jauković, Z.; Laušević, M. Trace analysis of selected hormones and sterols in river sediments by liquid chromatography-atmospheric pressure chemical ionization-tandem mass spectrometry. J. Chromatogr. A 2014, 1364, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Fayad, P.B.; Prévost, M.; Sauvé, S. Laser diode thermal desorption/atmospheric pressure chemical ionization tandem mass spectrometry analysis of selected steroid hormones in wastewater: Method optimization and application. Anal. Chem. 2010, 82, 639–645. [Google Scholar] [CrossRef]
- Boisvert, M.; Fayad, P.B.; Sauvé, S. Development of a new multi-residue laser diode thermal desorption atmospheric pressure chemical ionization tandem mass spectrometry method for the detection and quantification of pesticides and pharmaceuticals in wastewater samples. Anal. Chim. Acta 2012, 754, 75–82. [Google Scholar] [CrossRef]
- Wu, J.; Hughes, C.S.; Picard, P.; Letarte, S.; Gaudreault, M.; Lévesque, J.F.; Nicoll-Griffith, D.A.; Bateman, K.P. High-throughput cytochrome P450 inhibition assays using laser diode thermal desorption-atmospheric pressure chemical ionization-tandem mass spectrometry. Anal. Chem. 2007, 79, 4657–4665. [Google Scholar] [CrossRef]
- Crotti, S.; Isak, I.; Traldi, P. Liquid Chromatgraphy; Salvatore Fanali, P.R.H., Colin, F., Poole, P., Schoenmakers, D.L., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 307–331. [Google Scholar]
- Alonso, S.G.; Catalá Maroto, R.R.; Gil, J.L.R.; Miguel, A.G.; Valcárcel, Y. Pollution by psychoactive pharmaceuticals in the Rivers of Madrid metropolitan area (Spain). Environ. Int. 2010, 36, 195–201. [Google Scholar] [CrossRef]
- Pozo, O.; Guerrero, C.; Sancho, J.; Ibanez, M.; Pitarch, E.; Hogendoorn, E.; Hernandez, F. Efficient approach for the reliable quantification and confirmation of antibiotics in water using on-line solid-phase extraction liquid chromatography/tandem mass spectrometry. J. Chromatogr. A 2006, 1103, 83–93. [Google Scholar] [CrossRef]
- Boix, C.; Ibanez, M.; Sancho, J.; Parsons, J.; de Voogt, P.; Hernandez, F. Biotransformation of pharmaceuticals in surface water and during waste water treatment: Identification and occurrence of transformation products. J. Hazard. Mater. 2016, 302, 175–187. [Google Scholar] [CrossRef]
- Yang, S.; Carlson, K.H. Solid-phase extraction-high-performance liquid chromatography-ion trap mass spectrometry for analysis of trace concentrations of macrolide antibiotics in natural and wastewater matrices. J. Chromatogr. A 2004, 1038, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Parada, A.; Agüera, A.; Gómez-Ramos, M.D.M.; García-Reyes, J.F.; Heinzen, H.; Fernández-Alba, A.R. Behavior of amoxicillin in wastewater and river water: Identification of its main transformation products by liquid chromatography/electrospray quadrupole time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2011, 25, 731–742. [Google Scholar] [CrossRef]
- Bueno, M.J.; Agüera, A.; Hernando, M.D.; Gómez, M.J.; Fernández-Alba, A.R. Evaluation of various liquid chromatography-quadrupole-linear ion trap-mass spectrometry operation modes applied to the analysis of organic pollutants in wastewaters. J. Chromatogr. A 2009, 1216, 5995–6002. [Google Scholar] [CrossRef]
- Calza, P.; Medana, C.; Padovano, E.; Giancotti, V.; Baiocchi, C. Identification of the unknown transformation products derived from clarithromycin and carbamazepine using liquid chromatography/high-resolution mass spectrometry. Rapid Commun. Mass Spectrom. 2012, 26, 1687–1704. [Google Scholar] [CrossRef] [PubMed]
- Calza, P.; Medana, C.; Padovano, E.; Dal Bello, F.; Baiocchi, C. Identification of the unknown transformation products derived from lincomycin using LC-HRMS technique. J. Mass Spectrom. 2012, 47, 751–759. [Google Scholar] [CrossRef]
- Makarov, A.; Scigelova, M. Coupling liquid chromatography to Orbitrap mass spectrometry. J. Chromatogr. A 2010, 1217, 3938–3945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bade, R.; Causanilles, A.; Emke, E.; Bijlsma, L.; Sancho, J.V.; Hernandez, F.; de Voogt, P. Facilitating high resolution mass spectrometry data processing for screening of environmental water samples: An evaluation of two deconvolution tools. Sci. Total Environ. 2016, 569, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Hogenboom, A.C.; van Leerdam, J.A.; de Voogt, P. Accurate mass screening and identification of emerging contaminants in environmental samples by liquid chromatography-hybrid linear ion trap Orbitrap mass spectrometry. J. Chromatogr. A 2009, 1216, 510–519. [Google Scholar] [CrossRef] [Green Version]
- Bijlsma, L.; Emke, E.; Hernández, F.; de Voogt, P. Performance of the linear ion trap Orbitrap mass analyzer for qualitative and quantitative analysis of drugs of abuse and relevant metabolites in sewage water. Anal. Chim. Acta 2013, 768, 102–110. [Google Scholar] [CrossRef]
- Abou-Elwafa Abdallah, M.; Nguyen, K.H.; Ebele, A.J.; Atia, N.N.; Ali, H.R.H.; Harrad, S. A single run, rapid polarity switching method for determination of 30 pharmaceuticals and personal care products in waste water using Q-Exactive Orbitrap high resolution accurate mass spectrometry. J. Chromatogr. A 2019, 1588, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Althakafy, J.T.; Kulsing, C.; Grace, M.R.; Marriott, P.J. Liquid chromatography—Quadrupole Orbitrap mass spectrometry method for selected pharmaceuticals in water samples. J. Chromatogr. A 2017, 1515, 164–171. [Google Scholar] [CrossRef]
- Bade, R.; Rousis, N.I.; Bijlsma, L.; Gracia-Lor, E.; Castiglioni, S.; Sancho, J.V.; Hernandez, F. Screening of pharmaceuticals and illicit drugs in wastewater and surface waters of Spain and Italy by high resolution mass spectrometry using UHPLC-QTOF MS and LC-LTQ-Orbitrap MS. Anal. Bioanal. Chem. 2015, 407, 8979–8988. [Google Scholar] [CrossRef] [Green Version]
- Chitescu, C.; Oosterink, E.; de Jong, J.; Stolker, A. Ultrasonic or accelerated solvent extraction followed by U-HPLC-high mass accuracy MS for screening of pharmaceuticals and fungicides in soil and plant samples. Talanta 2012, 88, 653–662. [Google Scholar] [CrossRef]
- Kalaboka, M.; Chrimatopoulos, C.; Jiménez-Holgado, C.; Boti, V.; Sakkas, V.; Albanis, T. Exploring the efficiency of UHPLC-orbitrap MS for the determination of 20 pharmaceuticals and acesulfame K in hospital and urban wastewaters with the aid of FPSE. Separations 2020, 7, 46. [Google Scholar] [CrossRef]
- Verlicchi, P.; Al Aukidy, M.; Galletti, A.; Petrovic, M.; Barceló, D. Hospital effluent: Investigation of the concentrations and distribution of pharmaceuticals and environmental risk assessment. Sci. Total Environ. 2012, 430, 109–118. [Google Scholar] [CrossRef]
- Al Aukidy, M.; Verlicchi, P.; Jelic, A.; Petrovic, M.; Barcelo, D. Monitoring release of pharmaceutical compounds: Occurrence and environmental risk assessment of two WWTP effluents and their receiving bodies in the Po Valley, Italy. Sci. Total Environ. 2012, 438, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Gros, M.; Petrovic, M.; Ginebreda, A.; Barcelo, D. Removal of pharmaceuticals during wastewater treatment and environmental risk assessment using hazard indexes. Environ. Int. 2010, 36, 15–26. [Google Scholar] [CrossRef]
- Petrovic, M.; De Alda, M.; Diaz-Cruz, S.; Postigo, C.; Radjenovic, J.; Gros, M.; Barcelo, D. Fate and removal of pharmaceuticals and illicit drugs in conventional and membrane bioreactor wastewater treatment plants and by riverbank filtration. Philos. Trans. A Math. Phys. Eng. Sci. 2009, 367, 3979–4003. [Google Scholar] [CrossRef] [PubMed]
- Togola, A.; Budzinski, H. Analytical development for analysis of pharmaceuticals in water samples by SPE and GC-MS. Analytical and Bio. Anal. Chem. 2007, 388, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Togola, A.; Budzinski, H. Multi-residue analysis of pharmaceutical compounds in aqueous samples. J. Chromatogr. A 2008, 1177, 150–158. [Google Scholar] [CrossRef]
- Kot-Wasik, A.; Dębska, J.; Wasil, A.; Namiesnik, J. Determination of non-steroidal anti-inflammatory drugs in natural waters using off-line and on-line SPE followed by LC coupled with DAD-MS. Chromatographia 2006, 64, 13–21. [Google Scholar] [CrossRef]
- Tarcomnicu, I.; van Nuijs, A.L.; Simons, W.; Bervoets, L.; Blust, R.; Jorens, P.G.; Neels, H.; Covaci, A. Simultaneous determination of 15 top-prescribed pharmaceuticals and their metabolites in influent wastewater by reversed-phase liquid chromatography coupled to tandem mass spectrometry. Talanta 2011, 83, 795–803. [Google Scholar] [CrossRef]
- Zhou, L.J.; Ying, G.G.; Liu, S.; Zhao, J.L.; Chen, F.; Zhang, R.Q.; Peng, F.Q.; Zhang, Q.Q. Simultaneous determination of human and veterinary antibiotics in various environmental matrices by rapid resolution liquid chromatography-electrospray ionization tandem mass spectrometry. J. Chromatogr. A 2012, 1244, 123–138. [Google Scholar] [CrossRef]
- Gilart, N.; Cormack, P.A.; Marcé, R.M.; Fontanals, N.; Borrull, F. Selective determination of pharmaceuticals and illicit drugs in wastewaters using a novel strong cation-exchange solid-phase extraction combined with liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2014, 1325, 137–146. [Google Scholar] [CrossRef]
- Anumol, T.; Merel, S.; Clarke, B.O.; Snyder, S.A. Ultra high performance liquid chromatography tandem mass spectrometry for rapid analysis of trace organic contaminants in water. Chem. Cent. J. 2013, 7, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusmaroli, L.; Insa, S.; Petrovic, M. Development of an online SPE-UHPLC-MS/MS method for the multiresidue analysis of the 17 compounds from the EU “Watch list”. Anal. Bioanal. Chem. 2018, 410, 4165–4176. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Lor, E.; Martínez, M.; Sancho, J.V.; Peñuela, G.; Hernández, F. Multi-class determination of personal care products and pharmaceuticals in environmental and wastewater samples by ultra-high performance liquid-chromatography-tandem mass spectrometry. Talanta 2012, 99, 1011–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, A.V.; Couto, C.F.; Lebron, Y.A.R.; Moreira, V.R.; Foureaux, A.F.S.; Reis, E.O.; Santos, L.V.S.; de Andrade, L.H.; Amaral, M.C.S.; Lange, L.C. Occurrence and risk assessment of pharmaceutically active compounds in water supply systems in Brazil. Sci. Total Environ. 2020, 746, 141011. [Google Scholar] [CrossRef]
- Reis, E.O.; Foureaux, A.F.S.; Rodrigues, J.S.; Moreira, V.R.; Lebron, Y.A.R.; Santos, L.V.S.; Amaral, M.C.S.; Lange, L.C. Occurrence, removal and seasonal variation of pharmaceuticals in Brasilian drinking water treatment plants. Environ. Pollut. 2019, 250, 773–781. [Google Scholar] [CrossRef]
- Casado, J.; Rodríguez, I.; Ramil, M.; Cela, R. Selective determination of antimycotic drugs in environmental water samples by mixed-mode solid-phase extraction and liquid chromatography quadrupole time-of-flight mass spectrometry. J. Chromatogr. A 2014, 1339, 42–49. [Google Scholar] [CrossRef]
- Hernández, F.; Bijlsma, L.; Sancho, J.V.; Díaz, R.; Ibáñez, M. Rapid wide-scope screening of drugs of abuse, prescription drugs with potential for abuse and their metabolites in influent and effluent urban wastewater by ultrahigh pressure liquid chromatography-quadrupole-time-of-flight-mass spectrometry. Anal. Chim. Acta 2011, 684, 87–97. [Google Scholar] [CrossRef]
- EURACHEM. The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method Validation and Related Topics. 1998. Available online: http://www.eurachem.org/guides/mval.htm (accessed on 9 January 2020).
- ISO/IEC 17025: 2017 (E). General Requirements for the Competence of Testing and Calibration Laboratories; International Organization for Standardization (ISO): Geneva, Switzerland, 2017. [Google Scholar]
- Thompson, M.; Ellison, S.; Wood, R. Harmonized guidelines for single-laboratory validation of methods of analysis—(IUPAC technical report). Pure Appl. Chem. 2002, 74, 835–855. [Google Scholar] [CrossRef]
- EURACHEM. Guide to Quality in Analytical Chemistry. 2002. Available online: http://www.eurachem.org/guides/accr.htm (accessed on 9 January 2020).
- ISO 8466-1. Part 1: Statistical evaluation of the linear calibration function. In Water Quality—Calibration and Evaluation of Analytical Methods and Estimation of Performance Characteristics; International Organization for Standardization (ISO): Geneva, Switzerland, 1990. [Google Scholar]
- ISO. Accuracy (Trueness and Precision) of Measurement Methods and Results—Part 1: General Principles and Definitions; ISO 5725-1; International Organization for Standardization (ISO): Geneva, Switzerland, 1994. [Google Scholar]
- ISO. Statistics—Vocabulary and Symbols—Part 2: Applied statistics; ISO 3534-2; International Organization for Standardization (ISO): Geneva, Switzerland, 2006. [Google Scholar]
- Stuber, M.; Reemtsma, T. Evaluation of three calibration methods to compensate matrix effects in environmental analysis with LC-ESI-MS. Anal. Bioanal. Chem. 2004, 378, 910–916. [Google Scholar] [CrossRef]
- Petrovic, M.; Elijarrat, E.; Lopez de Alda, M.J.; Barceló, D. Endocrine disrupting compounds and other emerging contaminants in the environment: A survey on new monitoring strategies and occurence data. Anal. Bioanal. Chem. 2004, 378, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Konieczka, P.; Namieśnik, J. Estimating uncertainty in analytical procedures based on chromatographic techniques. J. Chromatogr. A 2010, 1217, 882–891. [Google Scholar] [CrossRef]
- EA (European Co-Operation for Accreditation). EA Guidelines on the Expression of Uncertainty in Quantitative Testing; EA-4/16; EA: Amsterdam, The Netherlands, 2003. [Google Scholar]
- EURACHEM/CITAC. Quantifying Uncertainty in Analytical Measurement. Guide CG 4, 2nd ed. 2000. Available online: http://www.eurachem.org/guides/ptguide2000.pdf (accessed on 27 July 2019).
Figure 1.
Main steps of sample preparation methods. Legend: Steps (—) and procedures to be implemented (− − −) [72].
Figure 1.
Main steps of sample preparation methods. Legend: Steps (—) and procedures to be implemented (− − −) [72].

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Almeida, C.M.M. Overview of Sample Preparation and Chromatographic Methods to Analysis Pharmaceutical Active Compounds in Waters Matrices. Separations 2021, 8, 16. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8020016
AMA Style
Almeida CMM. Overview of Sample Preparation and Chromatographic Methods to Analysis Pharmaceutical Active Compounds in Waters Matrices. Separations. 2021; 8(2):16. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8020016
Chicago/Turabian StyleAlmeida, Cristina M. M. 2021. "Overview of Sample Preparation and Chromatographic Methods to Analysis Pharmaceutical Active Compounds in Waters Matrices" Separations 8, no. 2: 16. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8020016
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.