1. Introduction
As per the “International Association for Study of Pain (IASP)”, neuropathic pain is “pain caused by a lesion or disease of the somatosensory system” [
1]. According to recommendations for pharmacological management of neuropathic pain guidelines, the first-line treatment for neuropathic control is antidepressants, α2δ modulators of calcium channel (i.e., PGB and GBP) and finally topical lidocaine. The recommended second-line treatment is opioid analgesics (TRM) but in limited clinical cases they are preferred as first-line treatments [
2,
3].
In most cases of neuropathic pain, monotherapy prescribed drugs have side effects and limited efficacy. Hence, combination of different drugs is considered to enhance analgesic efficacy and minimize side effects as much as possible (as synergistic interaction permits dose reduction of the combined prescribed drugs) [
4]. Combination of pregabalin and tramadol is one of the most important drug combinations for treating neuropathic pain, especially for postherpetic neuralgia and painful polyneuropathy [
5,
6]. They are also prevalent drugs of abuse detected in forensic cases of crime and suicide.
Accordingly, this presented study introduces an analytical assay for routine simple therapeutic monitoring of the combination of TRM and PGB, as a preliminary clinical study to support future clinical pharmacokinetic studies.
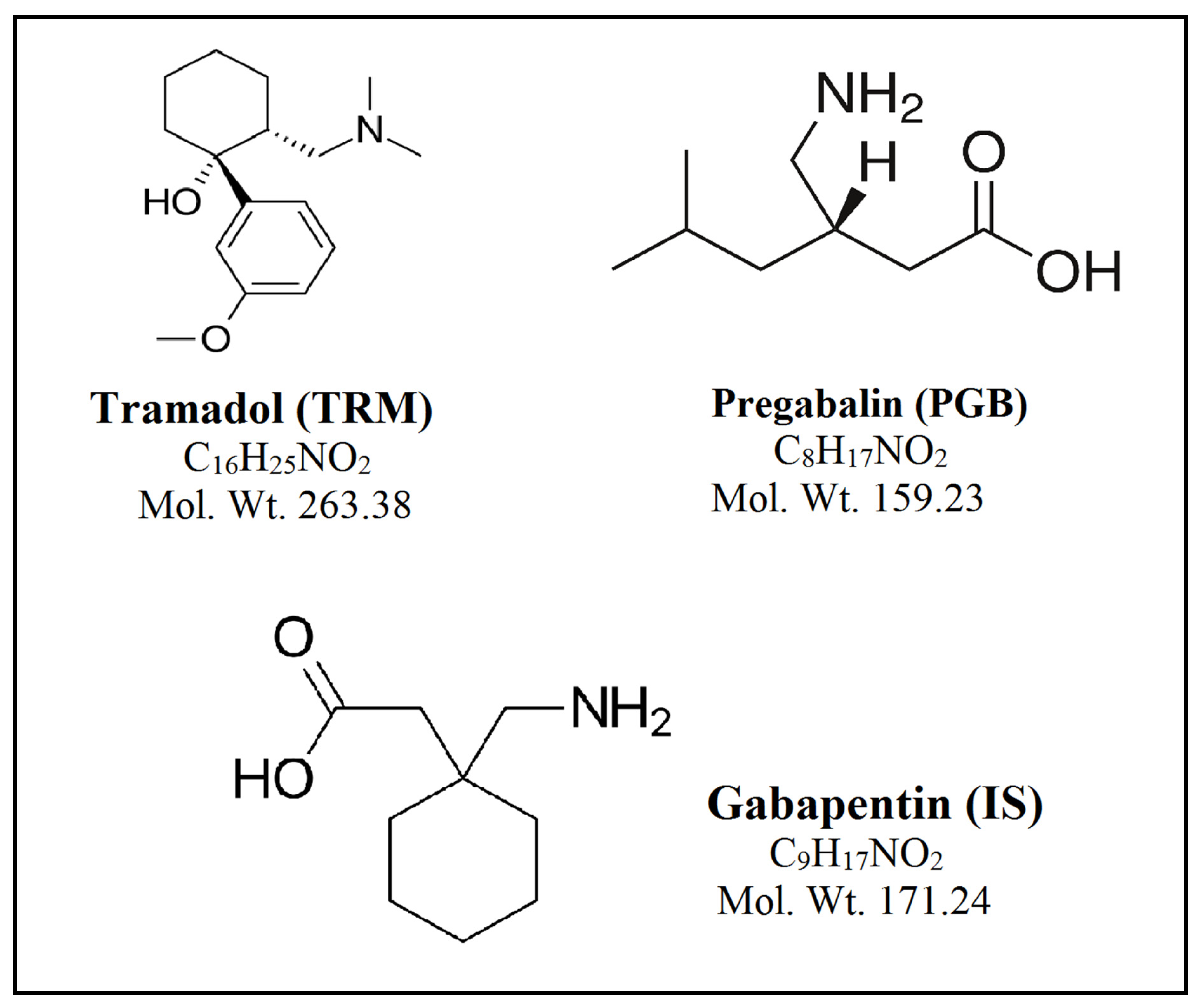
TRM, (1RS, 2RS)-2-[(dimethylamino)methyl] -1-(3-methoxyphenyl)cyclohexanol, is a centrally acting synthetic analgesic drug,
Figure 1. It is used globally for the treatment of moderate to severe pain, including neuropathic pain, as an alternative to opioid analgesics [
7]. TRM is considered a relatively safe drug with a low abuse potential and so low potential for dependence relative to morphine [
8]. Following oral administration, TRM is completely absorbed (<90%) with average bioavailability of nearly 70%. Ninety percent of oral TRM is eliminated in urine and 10% in feces, while 30% of a TRM oral dose is excreted as the unchanged drug in urine [
9].
The British Pharmacopoeia has described a non-aqueous titration technique for detecting TRM potentiometrically [
10]. The literature review revealed different analytical methods for TRM determination, either in biological fluids or pharmaceutical preparations, including HPLC [
11,
12,
13,
14], LC-MS [
15,
16,
17], GC-MS [
18,
19], TLC [
20], spectrophotometry [
21], ion-selective electrode [
22] and voltammetry [
23].
PGB, (S)-3-(amino methyl)-5-methylhexanoic acid, is a neurotransmitter γ-amino-butyric acid (GABA) structural analogue, which was used in common as an antiepileptic and analgesic drug,
Figure 1. The European Federation of Neurological Societies recommendations are using PGB as a first-line treatment for management of pain with patients suffering post-herpetic neuralgia, central neuropathic pain, and painful diabetic neuropathy [
24]. There are few reported analytical methods for assay of PGB in biological fluids and pharmaceuticals involving GC–MS [
25,
26], LC–MS [
27,
28,
29], HPLC [
30,
31,
32,
33], spectrophotometry [
34] and TLC [
35,
36]. After oral intake, PGB is rapidly absorbed with high bioavailability of more than 90% [
37]. It is not metabolized in humans and is almost entirely excreted unchanged in urine (<90%). Therapeutic plasma levels of PGB range between 0.3 and 14 mg/L [
38].
There are few reported methods for analysis of PGB in human plasma or human urine by HPLC after precolumn derivatization using different derivatizing agents [
30,
31,
32]. In practice, derivatization is considered a tedious technique which requires extra steps and strictly maintained conditions to obtain accurate estimation of the analyte [
27]. On the other hand, the LC separation of TRM using ultraviolet or fluorescence detection often requires surfactants or electrolyte buffers with its technical problems of crystal formation in connecting detector cells and tubes as well as damaging of pump seals; besides, they cause suppression of analytes’ ionization in MS/MS analysis [
17].
Though there are several reported methods for assay of TRM and PGB separately, there are not any published methods for their simultaneous determination. Hence, introduction of an analytical method that could analyze TRM and PGB simultaneously was a neat idea without the need for derivatization of PGB or using electrolyte buffers and surfactants for separation of TRM. LC–MS/MS, as an analytical method, has been widely trusted as a principal tool in the structural elucidation and quantitation of drugs due to its superior sensitivity, selectivity and efficiency. The proposed method offers advantages of sensitivity for analysis of TRM, whose therapeutic concentrations in plasma are low (70–592 ng mL
−1) [
39].
The objective of the presented study is to introduce the LC method for simultaneous quantification of the two analytes in pure form, spiked human plasma and human urine, using gabapentin (IS) as the internal standard and describing MS/MS detection with its high sensitivity and selectivity. Efficient sample preparation was required to achieve high method selectivity as an alternative to the time- and cost- consuming sample concentration and clean-up by solid-phase or liquid-liquid extraction. Accordingly, the work presented assayed the drugs in human spiked plasma and human urine samples by simple protein precipitation. Moreover, the method was applied for determination of TRM and PGB concentration in real human plasma and human urine samples taken from neuropathic pain patients treated by the drug therapy combination. A semi-quantitative greenness assessment was achieved using the eco-scale method for ensuring environmental safety of the proposed method [
40].
2. Material and Methods
2.1. Pure Standards
TRM was supplied by “Sigmatec Pharmaceutical Industries, El Monofeya, Egypt”. PGB was kindly provided by “Eva Pharma, Cairo, Egypt”. Gabapentin (IS) was kindly obtained from “Delta Pharma, Cairo, Egypt”. Their purities were found to be 99.60%, 99.92% and 99.70%, respectively, according to analysis certificates issued by the companies.
2.2. Chemicals and Reagents
HPLC-grade methanol and formic acid were purchased from “Sigma-Aldrich Chemie, Germany”.
Deionized water was from “SEDICO Pharmaceuticals Co., Cairo, Egypt”.
Cellulose acetate syringe filters of 0.45 μm pore size were from “Gemma Medical, Barcelona, Spain”.
Human plasma and human urine were kindly gifted by “Beni-Suef University Hospital, Beni-Suef, Egypt”.
2.3. LC-MS/MS Instrumentation and Chromatographic Conditions
UPLC MS/MS “(Waters 3100, Milford, MA, USA)” was used utilizing a well-managed binary solvent pump “(Acquity Ultra Performance LC, Milford, MA, USA)”. Samples were injected with “Acquity Ultra Performance LC auto-sampler”. The LC system was coupled with “Acquity Ultra Performance LC triple quadrupole mass spectrometer detector (Acquity Ultra Performance LC, Milford, MA, USA)”. For data processing and acquisition, “MassLynx software version 4.1 (Waters Corporation, Milford, MA, USA)” was used. Chromatographic separation was achieved on “Phenomenex Luna® Omega 1.6um polar C18 (LC 150 × 2.1 mm) column (Phenomenex, Torrance, CA, USA)”. Elution of the analytes was achieved at room temperature with methanol–water (70:30, v/v) containing 0.1 volume percentage of formic acid as the mobile phase at 0.3 mL/min flow rate with a total run time not exceeding 2.5 min. per injection.
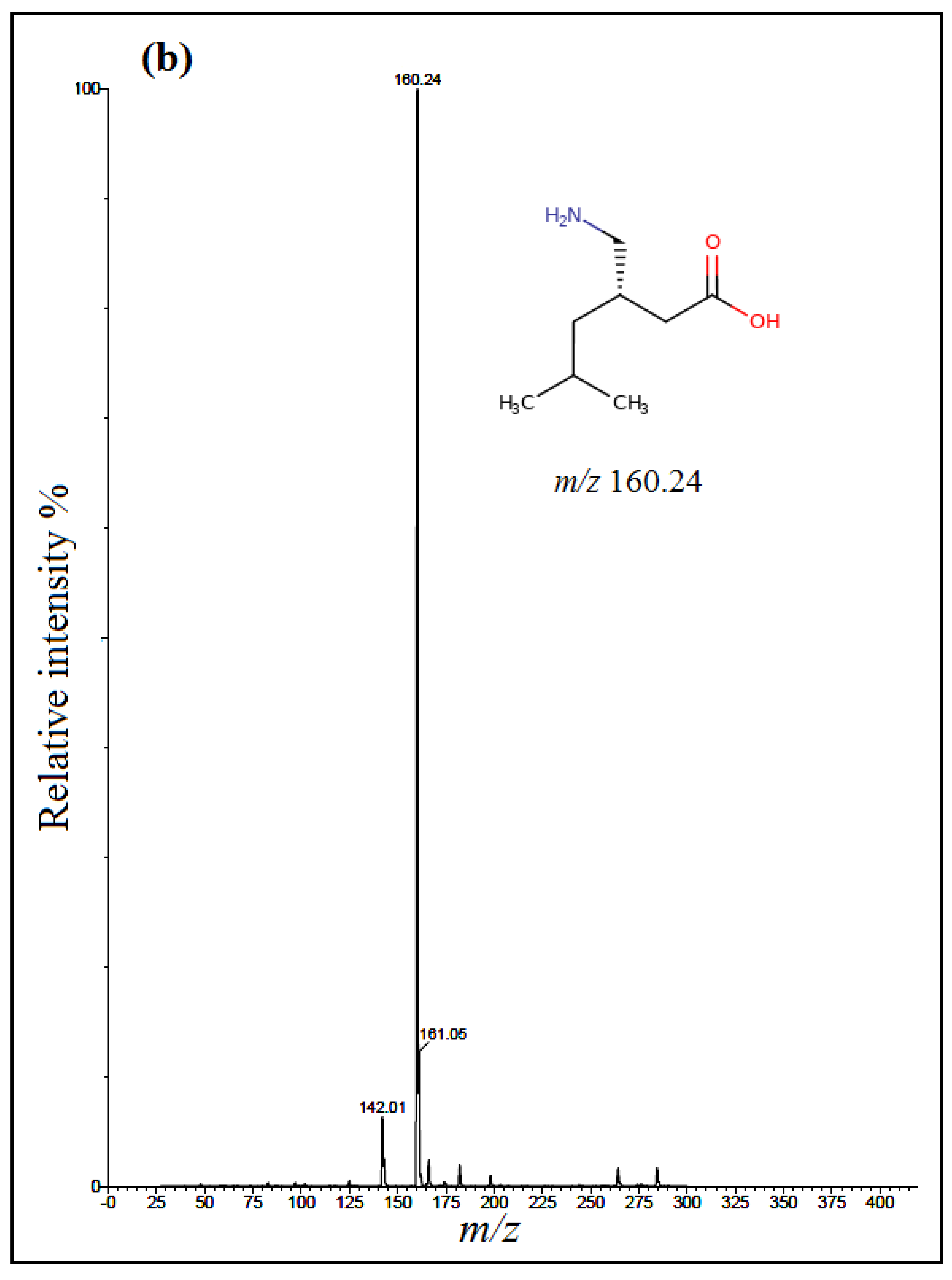
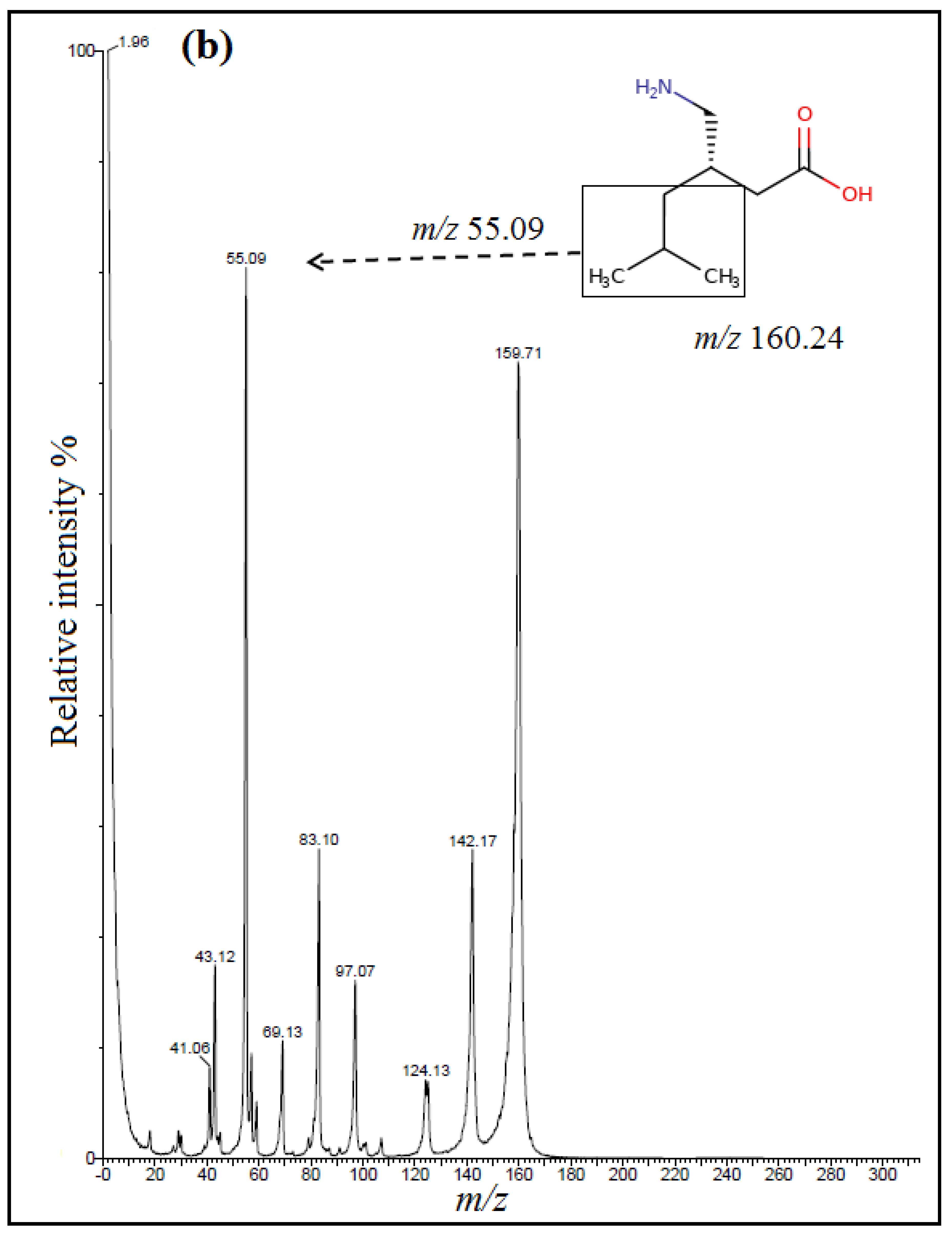
The detection of analytes was done using multiple-reaction mode (MRM) with an electrospray positive ionization (+ESI). Positive electrospray mass spectra showed intense [M + H]
+ ions at
m/z 263.89 for TRM,
m/z 160.24 for PGB and
m/z 171.95 for IS (
Figure 2); however, positively ionized fragments showed the product ions at
m/z 58.13 for TRM,
m/z 55.09 for PGB and
m/z 67.12 for IS (
Figure 3).
Collision energy and other drugs and instrumental parameters were optimized on their solutions using a concentration of 100 ng mL
−1. The instrument was programmed for a scan dwell time of 0.104 s. The optimized collision energies and other parameters are presented in
Table 1.
2.4. Standard Solutions
Stock standard solutions of TRM, PGB and IS (1 mg mL−1) were separately prepared in methanol and stored at −20 °C when not in use. Corresponding working solutions at 100 and 1 μg mL−1 were obtained by appropriate stock drug solution dilutions to prepare calibration curves and control samples. Drugs’ standard solutions were mixed to obtain final concentrations in ranges of 10–1000 ng mL−1 for TRM and PGB in pure form in mobile phase.
The three drugs’ concentration levels from calibration curves (low, medium and high) were separately prepared as control samples in biological matrices at concentrations of 30, 400 and 750 ng mL−1 for TRM and PGB.
2.5. Sample Preparation and Extraction Procedure
Standard calibration samples in biological matrices were daily prepared through spiking 450 µL drug-free human plasma or human urine and 50 µL of IS working solution with appropriate standard solutions of TRM and PGB to prepare six separate calibrators in the concentration ranges of 10–1000 ng mL−1 in human plasma and human urine (10, 50, 100, 250, 500, 1000 ng mL−1), and the concentration of IS in human plasma and human urine samples was 100 ng mL−1. Samples were vortexed for 30 s and then one mL of methanol was added to each sample, vortexed for 1 min, centrifuged at a speed of 3000 rpm for 30 min, filtered using 0.45 µm syringe filter, and 10 µL supernatant was injected into the column for analysis.
2.6. Method Validation
The proposed method study was validated as per FDA bioanalytical method validation guidelines [
41] regarding selectivity, sensitivity, linearity, precision, accuracy, extraction efficiency and stability.
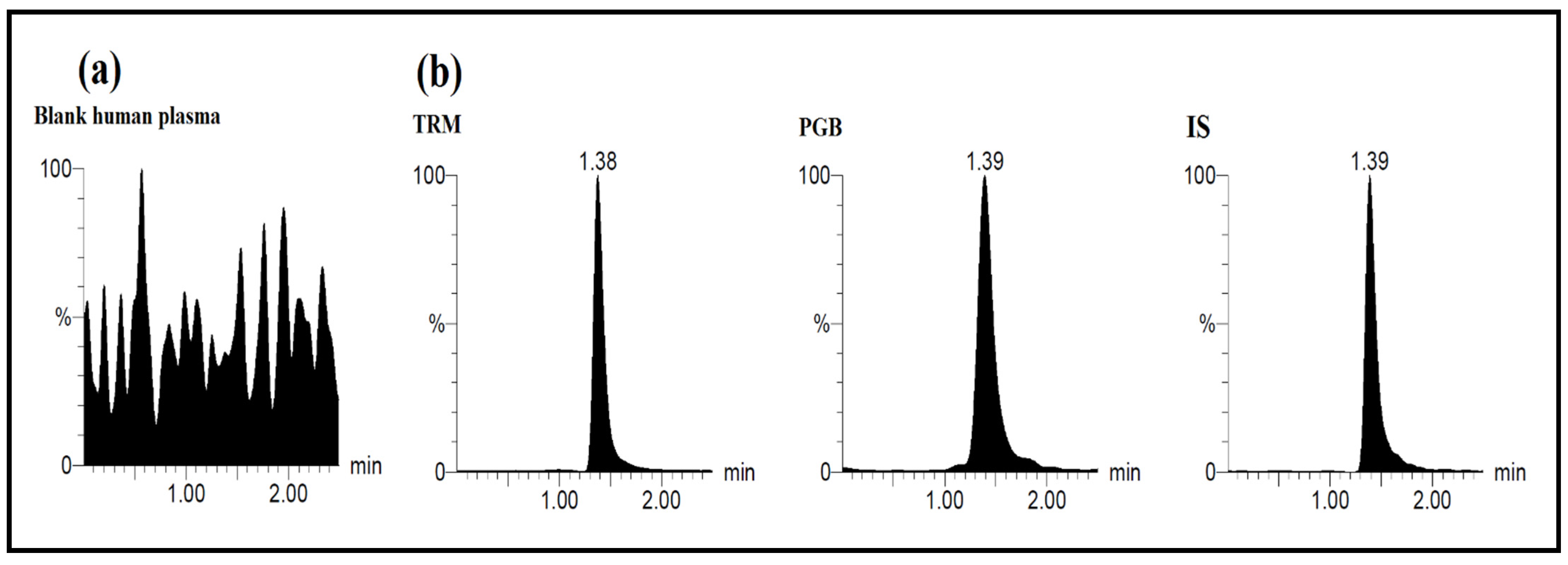
Assay selectivity was ascertained by assaying human blank plasma and urine samples to guarantee that no matrix endogenous interferences were encountered. Calibration standards of six concentrations of TRM and PGB (10, 50, 100, 250, 500, 1000 ng mL−1) were extracted and quantified. The peak area ratio of analytes/IS were calculated for establishment of calibration curves in spiked human plasma and human urine.
Linearity of analytes’ calibration curves was affirmed by drawing peak-area ratios versus the corresponding concentrations. Calibration curve was established and assayed along with the control samples.
Five sets of control samples were analyzed on five separate runs in a single day to evaluate the intra-day accuracy and precision (n = 5), while the inter-day data were estimated by analyzing each control sample in five successive days (n = 5). Method precision was calculated as (% RSD), whereas the accuracy was defined as the percent of target concentration of each analyte. Lower limit of quantification was determined at the lowest calibration standard, providing an accuracy and precision lower than 20% [
41].
Tramadol and PGB recoveries of control samples from human plasma and human urine were evaluated to express extraction efficiency of the proposed method. It was estimated by comparing extracted analytes and IS peak areas to those of non-extracted reference solutions at the same concentration level (n = 5).
Human plasma/human urine stability of TRM and PGB was investigated at high and low control concentration levels by comparing data of analyzed samples after and before exposure to stability conditions. Control samples were kept at room temperature for 4 h, at 4 °C for 24 h and at −20 °C for 15 days, then analyzed for estimating short- and long-term stability (n = 5). Three freeze–thaw cycles were performed for studying the drugs’ stability in control samples where control samples were kept at –20 °C for 24 h, then thawed at room temperature, then refrozen for 24 h until completion of the three cycles.
2.7. Clinical Application
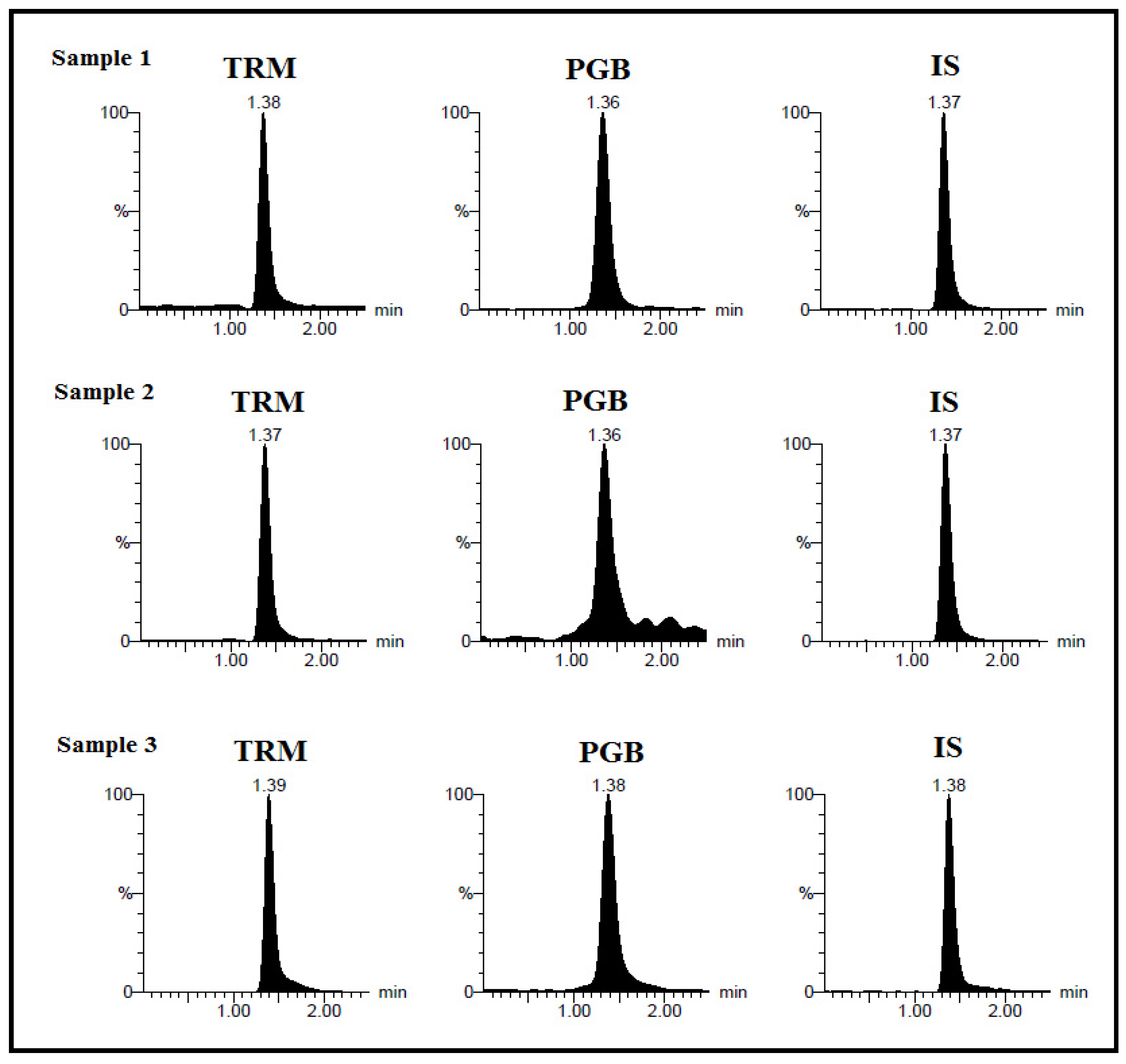
Following development and validation, the developed method’s applicability was tested by quantification of the two drugs in real human plasma and human urine samples of neuropathic patients treated with TRM and PGB followed by complete monitoring at “Beni-Suef University Hospital, Egypt”, with acceptance with serial no: REC-H-PhBSU-19001. The study was approved by the “ethics committee of the Faculty of Pharmacy, Beni-Suef University, Egypt”.
4. Conclusions
The presented work introduces the first analytical method for simultaneous quantitative assay of TRM and PGB, a binary mixture that is widely used in various cases of neuropathic pain and forensic cases. A sensitive, accurate and isocratic ESI–LC–MS/MS method for assaying TRM and PGB in pure form in spiked human plasma and human urine has been established. The procedure exhibited wide linearity of 10–1000 ng mL−1 with high sensitivity for both TRM and PGB. The method was found to be a stable and fast tool for bioanalytical study of the drugs with short total analysis time of 2.5 min.
These findings allowed its successful application for clinical study of real plasma and human urine samples of neuropathic pain patients treated by the drugs. However, the presented study is a preliminary one that supports future pharmacokinetics study; further studies on metabolism of tramadol and pregabalin and detection of their active metabolites should be pursued. This study proposed simple protein precipitation for sample preparation using methanol as a compatible solvent with LC–MS/MS, offering advantages of minimizing the probe molecule ionization effect on ESI–LC–MS/MS. The method was validated following FDA guidelines and proved to be reasonably suitable for future pharmacokinetic studies and therapeutic drug monitoring of TRM and PGB combinations in bioanalytical environments. Regarding green chemistry considerations, semi-quantitative eco-scale assessment was applied proving that the presented method is an acceptable green analytical method.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}