
Development of a Modified QuEChERS Procedure for the Isolation of Pesticide Residues from Textile Samples, Followed by GC–MS Determination


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Sample Preparation
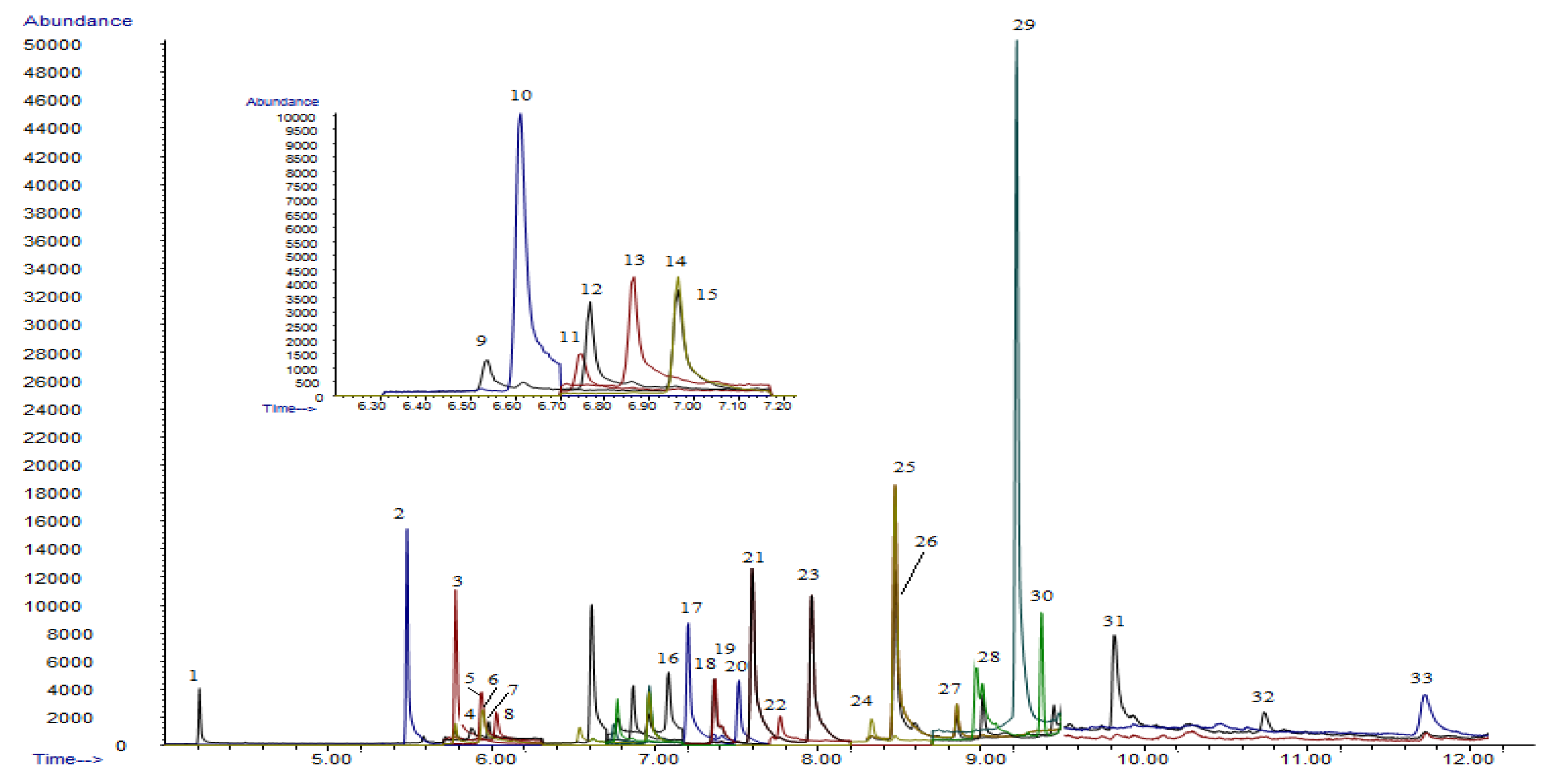
2.3. Instrument and Apparatus
2.4. Validation Process
3. Results
3.1. Modification of the QuEChERS Method
3.2. Sample Weight Selection
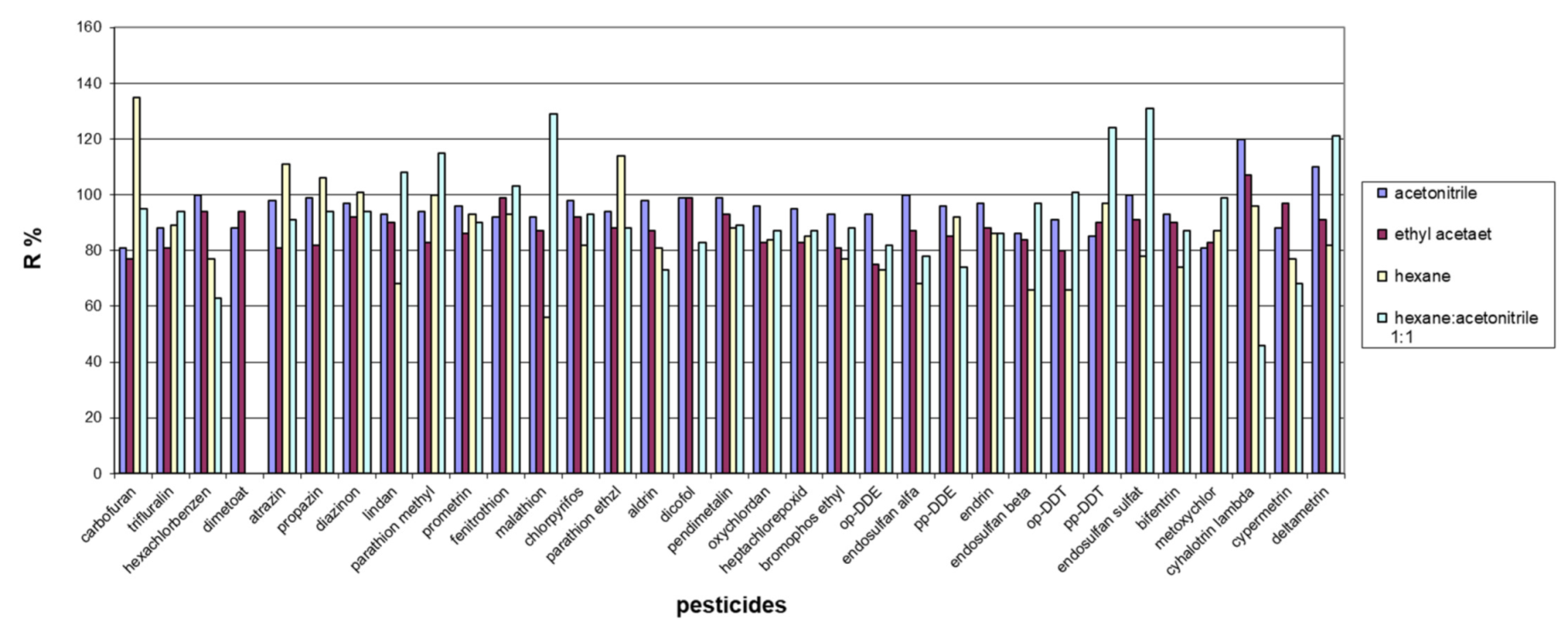
3.3. Selection of Extraction Solvent
3.4. Selection of an Appropriate Sorbent for Dispersive SPE
3.5. Method Validation
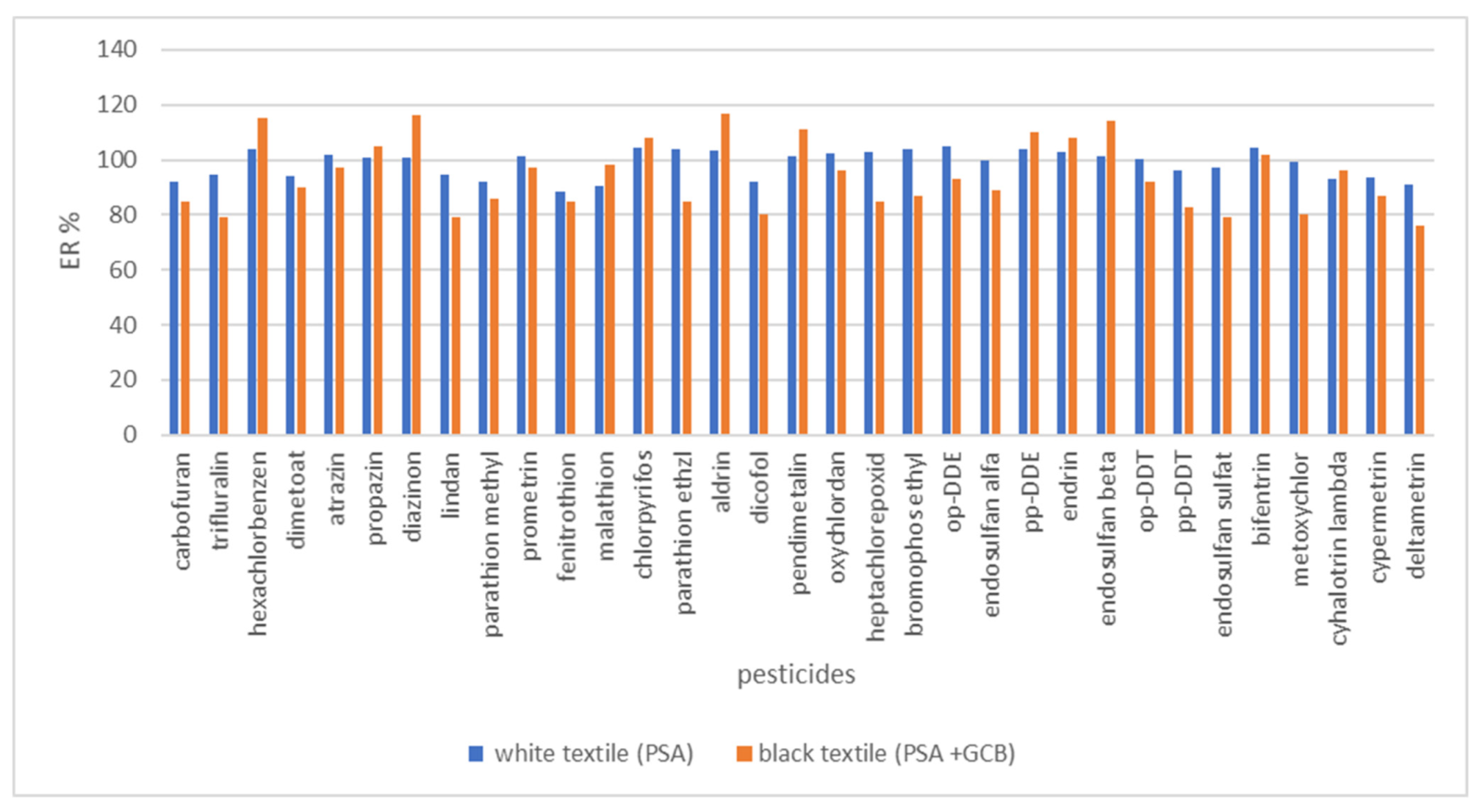
3.6. Real Sample Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Attallah, E.R.; Abdelwahed, M.H.; Abo-Aly, M.M. Development and validation of multi-residue method for determination of 412 pesticide residues in cotton fiber using GC-MS/MS and LC-MS/MS. J. Text. Inst. 2018, 109, 46–63. [Google Scholar] [CrossRef]
- Gordon, S.; Hsieh, Y.-L. Cotton: Science and Technology, 1st ed.; Woodhead Publishing Limited: Cambridge, UK, 2007; pp. 425–459. [Google Scholar]
- Reddy, N.; Yang, Y. Properties and potential applications of natural cellulose fibers from the bark of cotton stalks. Bioresour. Technol. 2009, 100, 3563–3569. [Google Scholar] [CrossRef] [PubMed]
- Kranthi, K.R.; Russell, D.A. Changing trends in cotton pest management. In Integrated Pest Management: Innovation-Development Process, 1st ed.; Pimentel, D., Peshin, R., Eds.; Springer Science+Business Media B.V.: Dordrecht, The Netherlands, 2009; pp. 499–541. [Google Scholar]
- Ma, M.; Dong, S.; Jin, W.; Zhang, C.; Zhou, W. Fate of the organophosphorus pesticide profenfos in cotton fiber. J. Environ. Sci. Health Part B 2019, 54, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.T.; Lu, C.M.; Li, H.; Zhang, Z.X.; Li, J. Determination of eleven organophosphorus pesticide residues in textiles by using HPLC-HRMS. Anal. Sci. 2017, 33, 1027–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Pan, X.; Venne, L.; Dunnum, S.; McMurry, S.T.; Cobb, G.P.; Anderson, T.A. Development of a method for the determination of 9 currently used cotton pesticides by gas chromatography with electron capture detection. Talanta 2008, 75, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Satyajeet, R.; Kumar, S.A.; Anshuman, S.; Sapna, Y.; Mohammed Haris, M.; Mohana Krishna Reddy, M. Comparative Evaluation of QuEChERS Method Coupled to DLLME Extraction for the Analysis of Multiresidue Pesticides in Vegetables and Fruits by Gas Chromatography-Mass Spectrometry. Food Anal. Methods 2016, 9, 2656–2669. [Google Scholar]
- Kim, J.; Kim, J.I.; Lee, C.W. Development and validation of a modified QuEChERS method coupled with LC-MS/MS to determine arbutin in pear peels. Food Sci. Biotechnol. 2016, 25, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Gracia, M.; Ascension, M.; Alba, M.; Cermeño, S.; Oliva, J.; Barba, A. Determination of Nine Fungicides in Grape and Wine Using QuEChERS Extraction and LC/MS/MS Analysis. J. AOAC Int. 2015, 98, 1745–1751. [Google Scholar]
- Tariq, M.I.; Afzal, S.; Hussain, I. Degradation and persistence of cotton pesticides in sandy loam soils from Punjab, Pakistan. Environ. Res. 2006, 100, 184–196. [Google Scholar] [CrossRef]
- Anastassiades, M.; Lehotay, S.J.; Stajnbaher, D.; Schenck, F.J. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and dispersive solid phase extraction for the determination of pesticide residues in produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Wang, S.; Bian, Y.; Chen, F.; Yu, G.; Gu, C.; Jiang, X. Dicofol application resulted in high DDTs residue in cotton fields from northern Jiangsu province, China. J. Hazard. Mater. 2008, 150, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Abaga, N.O.Z.; Alibert, P.; Dousset, S.; Savadogo, P.W.; Savadogo, M.; Sedogo, M. Insecticide residues in cotton soils of Burkina Faso and effects of insecticides on fluctuating asymmetry in honey bees (Apis mellifera Linnaeus). Chemosphere 2011, 83, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Blossom, S.; Gaganjyot, B. Monitoring of Insecticide Residues in Cotton Seed in Punjab, India. Bull. Environ. Contam. Toxicol. 2004, 73, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Barker, T.B.; Milivojevich, A. Quality by Experimental Design, 4th ed.; Chapman and Hall/CRC: Boca Raton, FL, USA, 2016; pp. 1–754. [Google Scholar]
- De Matos, E.M.C.; Ribeiro, L.C.; Prestes, O.D.; da Silva, J.A.G.; de Farias, B.S.; Pinto, L.A.; Zanella, R. Multiclass method for the determination of pesticide residues in oat using modified QuEChERS with alternative sorbent and liquid chromatography with tandem mass spectrometry. Food Anal. Methods 2019, 12, 2835–2844. [Google Scholar] [CrossRef]
- Walorczyk, S.; Drożdżyński, D. Improvement and extension to new analytes of a multi-residue method for the determination of pesticides in cereals and dry animal feed using gas chromatography–tandem quadrupole mass spectrometry revisited. J. Chromatogr. A 2012, 1251, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Rasche, C.; Fournes, B.; Dirks, U.; Speer, K.N. Multi-residue pesticide analysis (gas chromatography-tandem mass spectrometry detection)—Improvement of the quick, easy, cheap, effective, rugged, and safe method for dried fruits and fat-rich cereals-benefit and limit of a standardized apple purée calibration (screening). J. Chromatogr. A 2015, 1403, 21–31. [Google Scholar] [PubMed]
- Dors, G.C.; Primel, E.G.; Fagundes, C.A.A.; Mariot, C.H.P.; Badiale Furlong, E. Distribution of pesticide residues in rice grain and in its coproducts. J. Braz. Chem. Soc. 2011, 22, 1921–1930. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Pesticide | Retention Time [min] | Chemical Function | Fragment Ions [m/z] | Starting Time of SIM Group [min] | ||
|---|---|---|---|---|---|---|
| Carbofuran | 4.207 | Carbamate | 164 | 149 | 131 | 3.00 |
| Trifluralin | 5.499 | Dinitroanyline | 306 | 264 | 307 | |
| Hexachlorobenzene | 5.796 | OCP | 284 | 286 | 282 | 5.70 |
| Dimethoate | 5.894 | OPP | 87 | 93 | 164 | |
| Atrazin | 5.968 | Triazine | 200 | 215 | 202 | |
| Propazin | 5.979 | Triazine | 214 | 229 | 172 | |
| Diazinone | 6.025 | OPP | 179 | 137 | 152 | |
| Lindane | 6.042 | OPP | 181 | 183 | 109 | |
| Parathion-methyl | 6.579 | OPP | 263 | 125 | 211 | 6.30 |
| Prometrin | 6.642 | Triazine | 241 | 184 | 226 | |
| Fenitrothion | 6.768 | OPP | 277 | 125 | 260 | 6.74 |
| Malathion | 6.808 | OPP | 173 | 127 | 125 | |
| Chlorpyrifos | 6.899 | OPP | 197 | 314 | 199 | |
| Parathion-ethyl | 6.977 | OPP | 291 | 109 | 139 | |
| Aldrin | 6.997 | OCP | 263 | 261 | 265 | |
| Dicofol | 7.117 | OCP | 139 | 111 | 250 | |
| Pendimethalin | 7.242 | Dinitroanyline | 252 | 162 | 281 | 7.19 |
| Oxychlordane | 7.379 | OCP | 353 | 355 | 115 | |
| Heptachlor epoxide | 7.408 | OCP | 353 | 355 | 351 | |
| Bromophos-ethyl | 7.557 | OPP | 359 | 357 | 303 | |
| o,p-DDE | 7.642 | OCP | 246 | 248 | 318 | |
| p,p-DDE | 7.997 | OCP | 246 | 318 | 316 | 7.75 |
| Endosulfan-alfa | 7.808 | OCP | 241 | 239 | 195 | |
| Endrin | 8.368 | OCP | 263 | 265 | 281 | 8.15 |
| Endosulfan-beta | 8.471 | OCP | 237 | 239 | 216 | |
| o,p-DDT | 8.500 | OCP | 235 | 237 | 165 | |
| p,p-DDT | 8.883 | OCP | 235 | 237 | 165 | 8.80 |
| Endosulfan sulfate | 8.923 | OCP | 229 | 272 | 387 | |
| Bifenthrin | 9.243 | Pyrethroid | 181 | 165 | 166 | |
| Methoxychlor | 9.391 | OCP | 227 | 228 | 274 | |
| Cyhalothrin lambda | 9.746 | Pyrethroid | 181 | 197 | 208 | 9.56 |
| Cypermethrin | 10.797 | Pyrethroid | 163 | 165 | 181 | |
| Deltamethrin | 11.952 | Pyrethroid | 181 | 253 | 251 | |
| Name | ER% 10 µg/kg (RSD%) | ER% 50 µg/kg (RSD%) | ER% 250 µg/kg (RSD%) | LCL [µg/kg] | LOD [µg/kg] | LOQ [µg/kg] | MRL [µg/kg] |
|---|---|---|---|---|---|---|---|
| Carbofuran | 88 (6) | 91 (4) | 92 (2) | 5 | 0.980 | 3.240 | 100 |
| Trifluralin | 101 (2) | 86 (6) | 95 (1) | 0.5 | 0.057 | 0.190 | 10 |
| Hexachlorobenzene | 96 (7) | 97 (1) | 104 (1) | 5 | 0.750 | 2.470 | 20 |
| Dimethoate | 62 (13) | 96 (11) | 94 (7) | 5 | 1.400 | 4.620 | 10 |
| Atrazine | 97 (6) | 96 (4) | 102 (1) | 5 | 1.210 | 3.980 | 50 |
| Propazin | 98 (4) | 94 (3) | 101 (2) | 0.1 | 0.090 | 0.290 | |
| Diazinon | 100 (4) | 102 (9) | 101 (2) | 5 | 1.402 | 4.673 | 20 |
| Lindane | 70 (10) | 97 (9) | 95 (7) | 5 | 1.490 | 4.910 | 10 |
| Parathion methyl | 63 (13) | 90 (9) | 92 (7) | 5 | 1.360 | 4.500 | 20 |
| Promethryn | 100 (2) | 97 (3) | 101 (2) | 0.1 | 0.060 | 0.190 | |
| Fenitrothion | 70 (15) | 97 (9) | 88 (9) | 5 | 1.500 | 4.950 | 20 |
| Malathion | 64 (9) | 95 (8) | 90 (9) | 5 | 1.520 | 5.000 | 20 |
| Chlorpyrifos | 101 (3) | 101 (3) | 104 (1) | 0.1 | 0.060 | 0.190 | 300 |
| Parathion ethyl | 99 (2) | 98 (6) | 104 (2) | 1 | 0.149 | 0.495 | |
| Aldrin | 98 (3) | 95 (5) | 104 (1) | 5 | 0.980 | 3.240 | 20 |
| Dicofol | 122 (11) | 103 (9) | 92 (9) | 5 | 1.520 | 5.000 | 100 |
| Pendimethalin | 96 (5) | 92 (5) | 101 (4) | 5 | 1.320 | 4.350 | 50 |
| Oxychlordan | 98 (4) | 96 (2) | 102 (1) | 0.5 | 0.366 | 1.220 | |
| Heptachlorepoxid | 100 (1) | 96 (1) | 103 (2) | 0.5 | 0.366 | 1.220 | 10 |
| Bromophos ethyl | 101 (1) | 96 (2) | 104 (1) | 0.1 | 0.045 | 0.149 | 20 |
| O,p-DDE | 98 (3) | 97 (6) | 105 (2) | 0.1 | 0.035 | 0.118 | 50 |
| Endosulfan alfa | 70 (8) | 92 (4) | 100 (3) | 5 | 1.130 | 3.740 | 30 |
| P,p-DDE | 97 (1) | 95 (4) | 104 (1) | 0.5 | 0.161 | 0.538 | 50 |
| Endrin | 75 (9) | 96 (2) | 103 (1) | 5 | 1.390 | 4.600 | |
| Endosulfan beta | 100 (2) | 106 (5) | 101 (2) | 5 | 1.290 | 4.250 | 30 |
| O,p-DDT | 102 (3) | 98 (5) | 100 (6) | 5 | 1.260 | 4.170 | 50 |
| P,p-DDT | 99 (1) | 106 (10) | 96 (7) | 5 | 0.843 | 2.809 | 50 |
| Endosulfan sulfat | 72 (14) | 89 (13) | 97 (8) | 5 | 1.520 | 5.000 | 30 |
| Bifenthrin | 99 (1) | 95 (1) | 105 (1) | 0.5 | 0.128 | 0.427 | 500 |
| Methoxychlor | 94 (11) | 95 (15) | 99 (13) | 0.1 | 0.030 | 0.090 | 10 |
| Cyhalotrin lambda | 98 (3) | 95 (8) | 93 (8) | 1 | 0.280 | 0.910 | 200 |
| Cypermethrin | 96 (2) | 99 (7) | 93 (7) | 1 | 0.260 | 0.850 | 200 |
| Deltamethrin | 100 (4) | 102 (8) | 91 (5) | 1 | 0.270 | 0.900 | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hrouzková, S.; Szarka, A. Development of a Modified QuEChERS Procedure for the Isolation of Pesticide Residues from Textile Samples, Followed by GC–MS Determination. Separations 2021, 8, 106. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8080106
Hrouzková S, Szarka A. Development of a Modified QuEChERS Procedure for the Isolation of Pesticide Residues from Textile Samples, Followed by GC–MS Determination. Separations. 2021; 8(8):106. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8080106
Chicago/Turabian StyleHrouzková, Svetlana, and Agneša Szarka. 2021. "Development of a Modified QuEChERS Procedure for the Isolation of Pesticide Residues from Textile Samples, Followed by GC–MS Determination" Separations 8, no. 8: 106. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8080106