
A Rapid Analytical Approach for Monitoring Pharmaceuticals in Hospital Wastewater—A DPX-Based Procedure with Environmentally-Friendly Extraction Phase Coupled to High Performance Liquid Chromatography–Diode Array/Fluorescence Detectors


Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Synthesis and Characterization of the Extraction Phases
2.3. Instrumentation and Chromatographic Conditions
2.4. Optimization of DPX Procedure
2.5. Evaluation of Extraction Phase
2.6. Optimization of Desorption Solvent
2.7. Optimization of Extraction and Desorption Steps
2.8. Evaluation of pH and Salting-Out Effect
2.9. Analytical Figures of Merit and Analysis of Hospital Wastewater Samples
3. Results and Discussion
3.1. Optimization of DPX Procedure
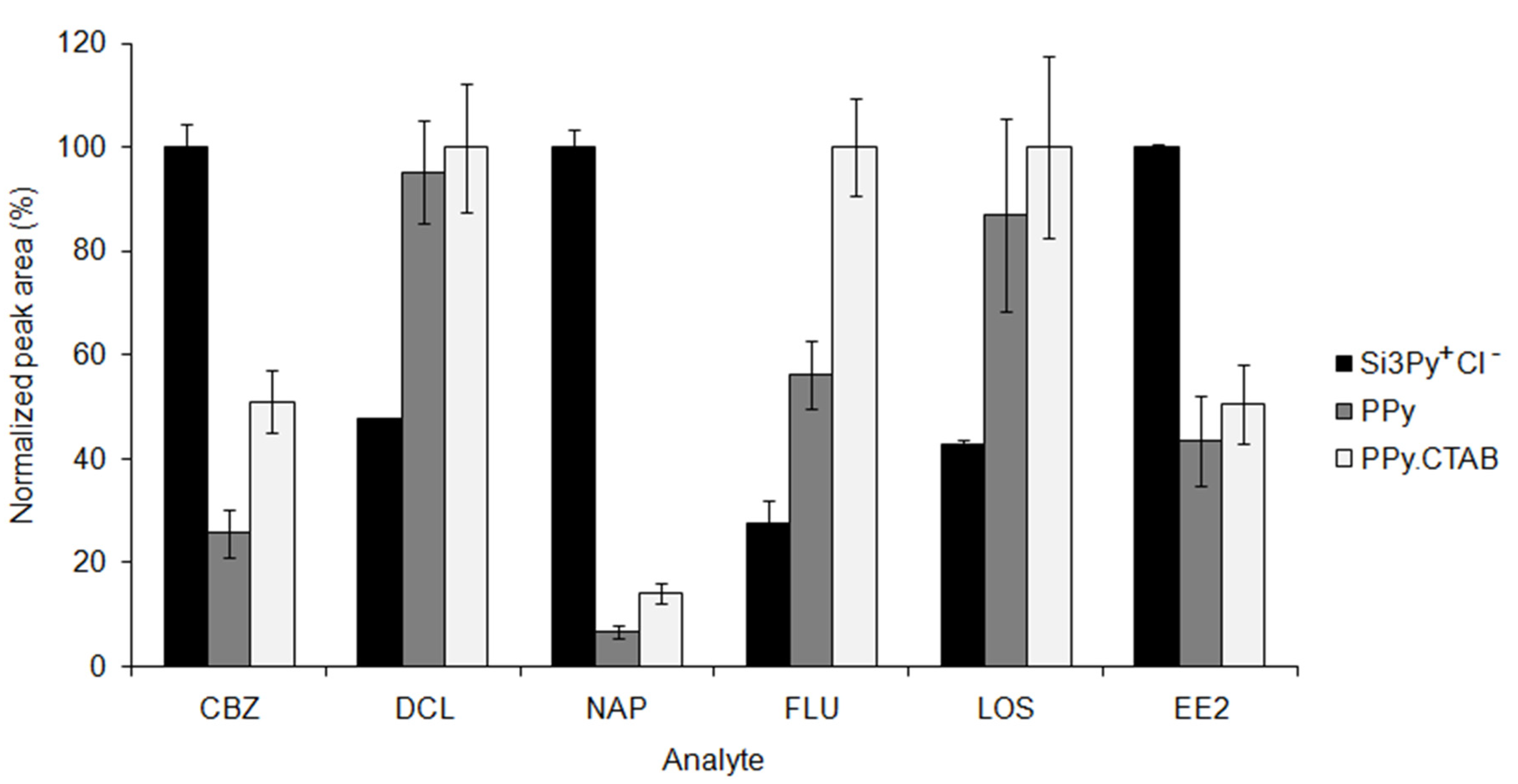
3.1.1. Evaluation of the Extraction Phase
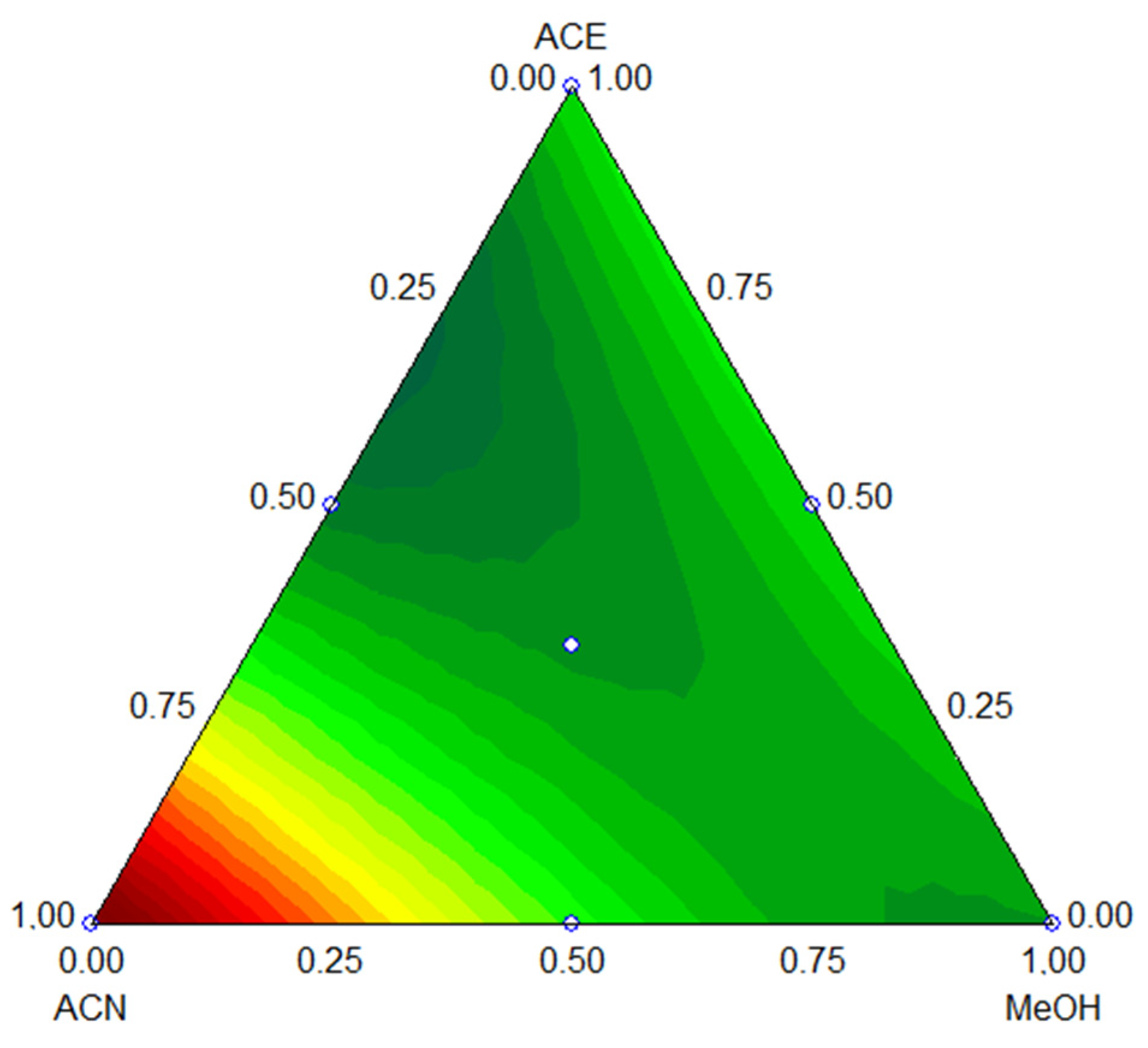
3.1.2. Optimization of the Desorption Solvent
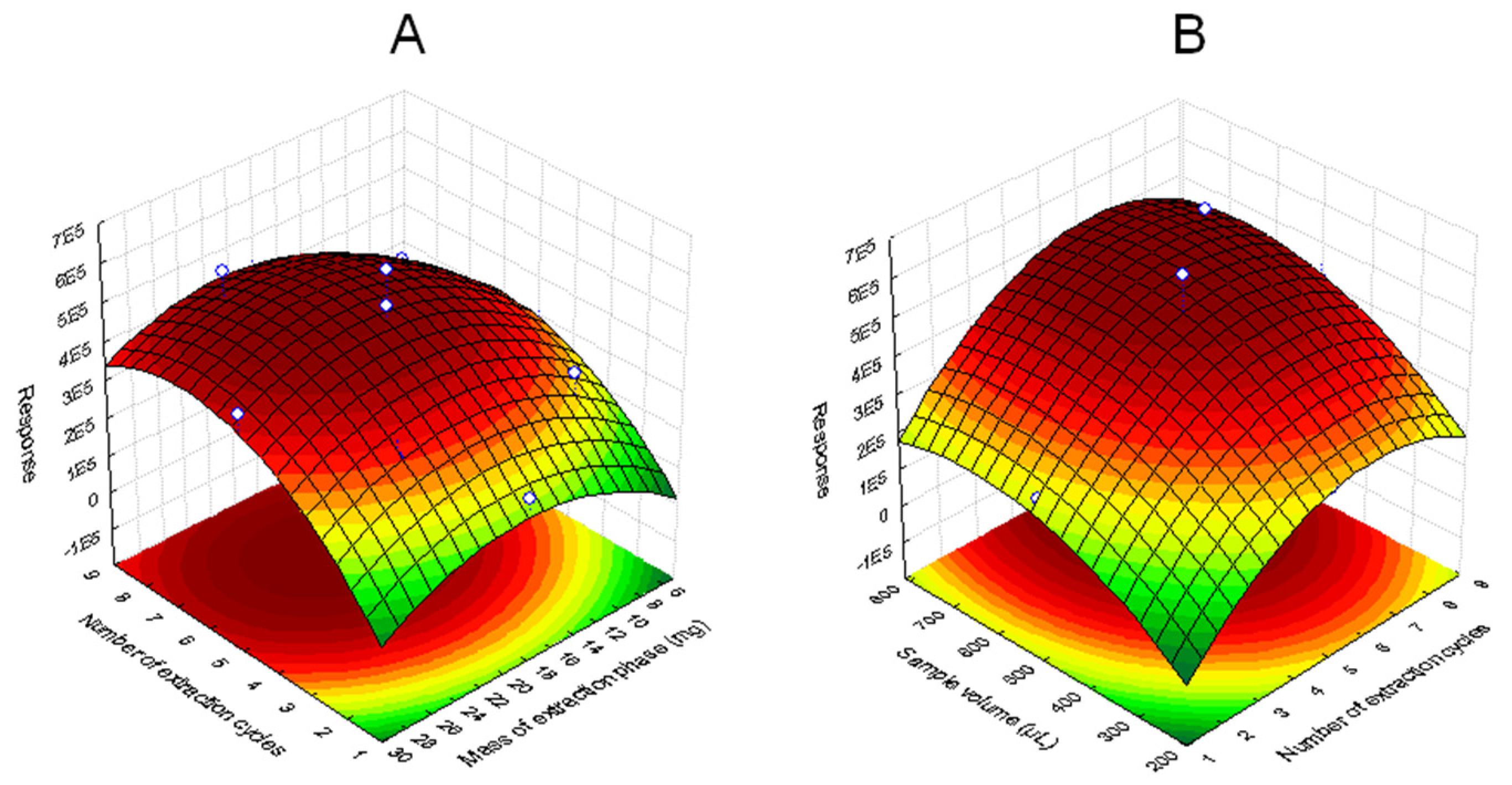
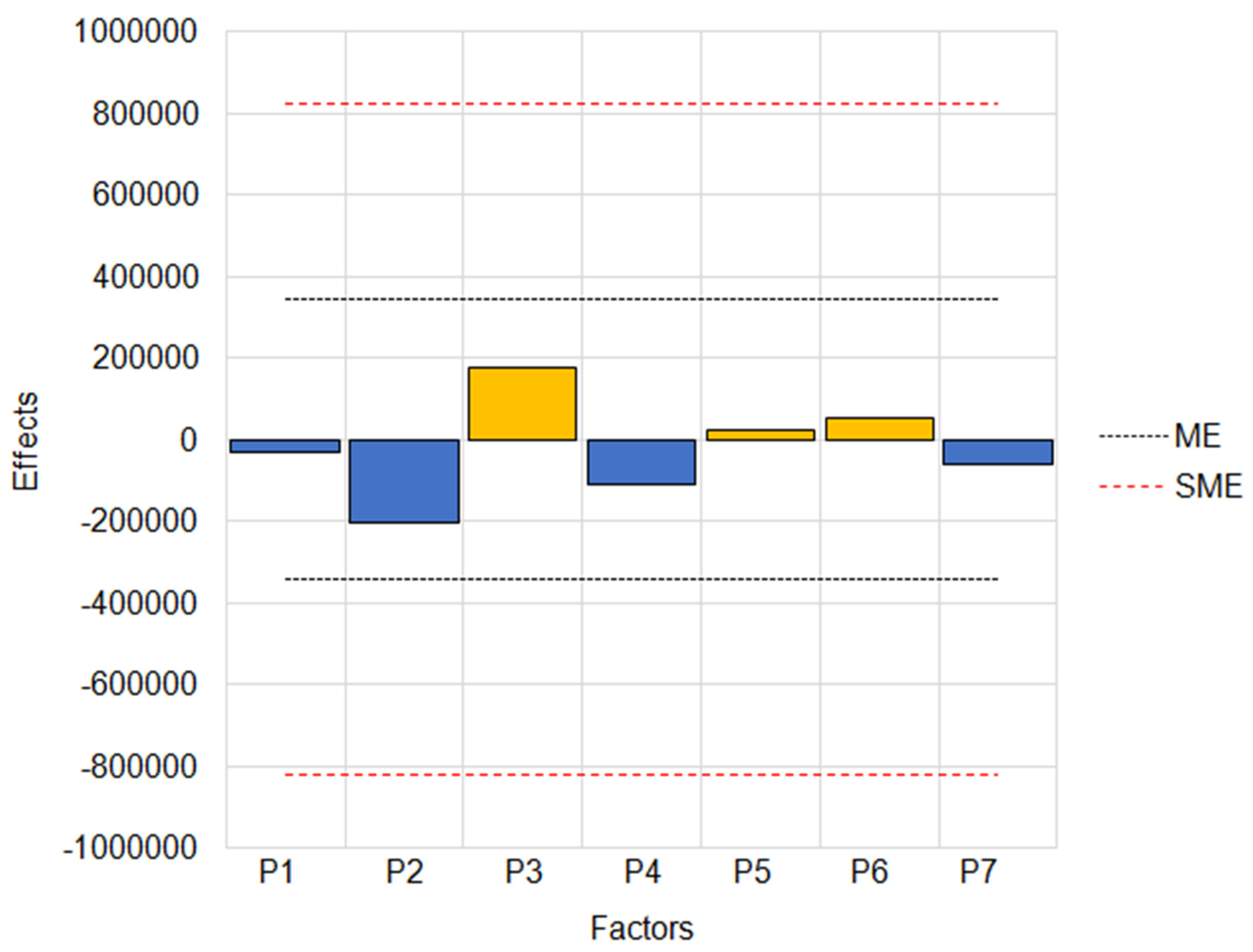
3.1.3. Optimization of Extraction Step
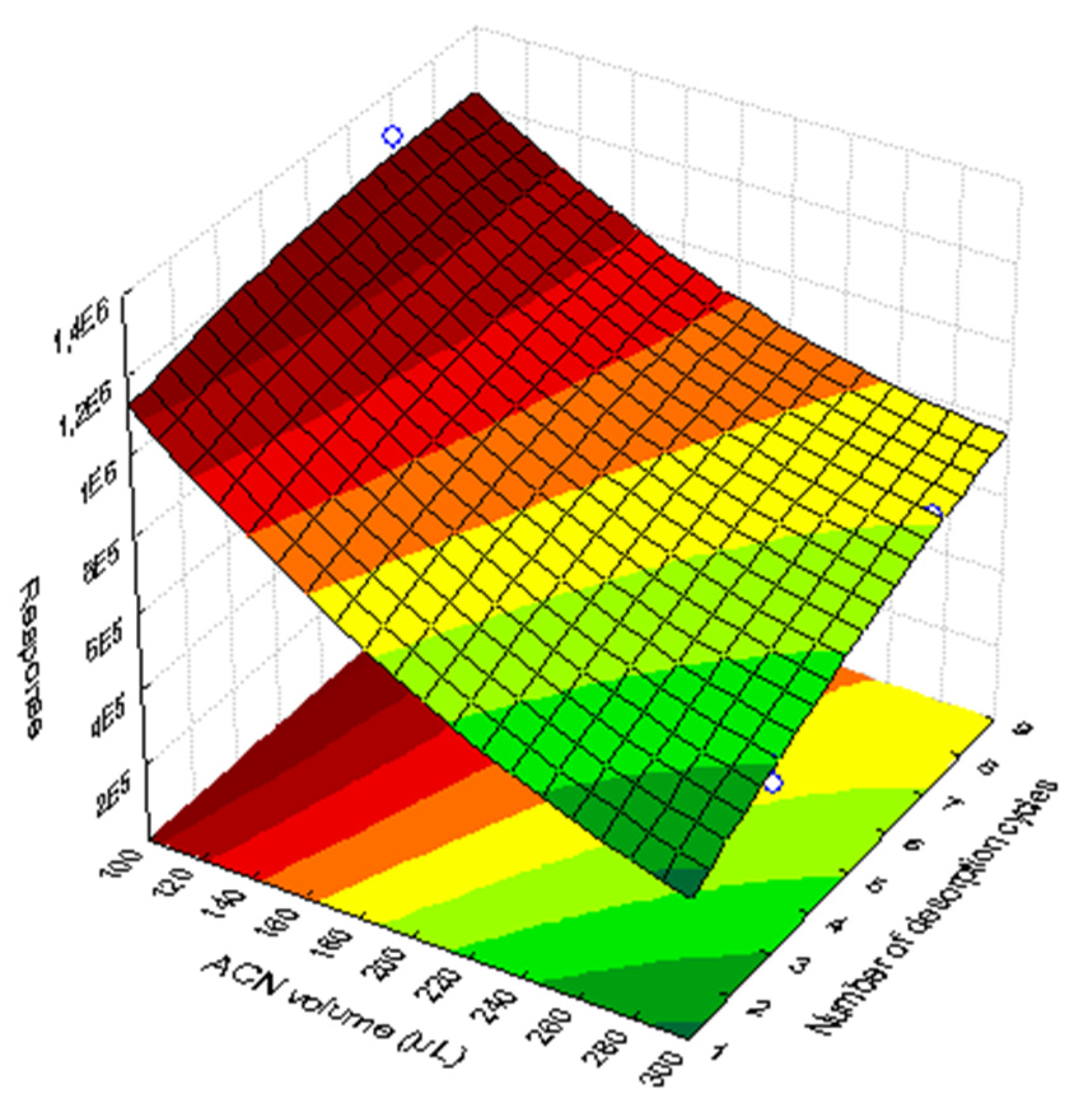
3.1.4. Optimization of the Desorption Step
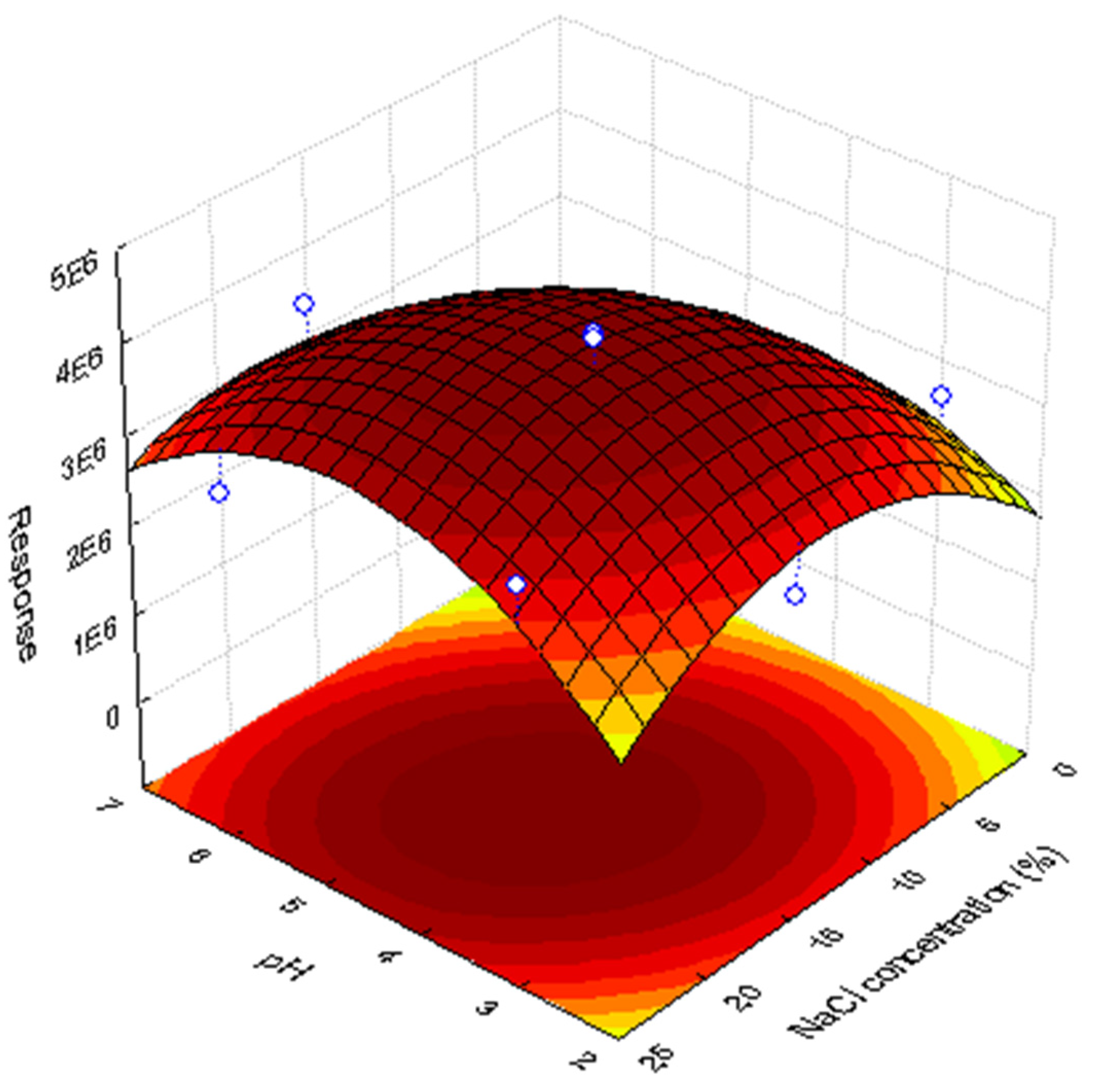
3.2. Evaluation of PH and Salting-Out Effect
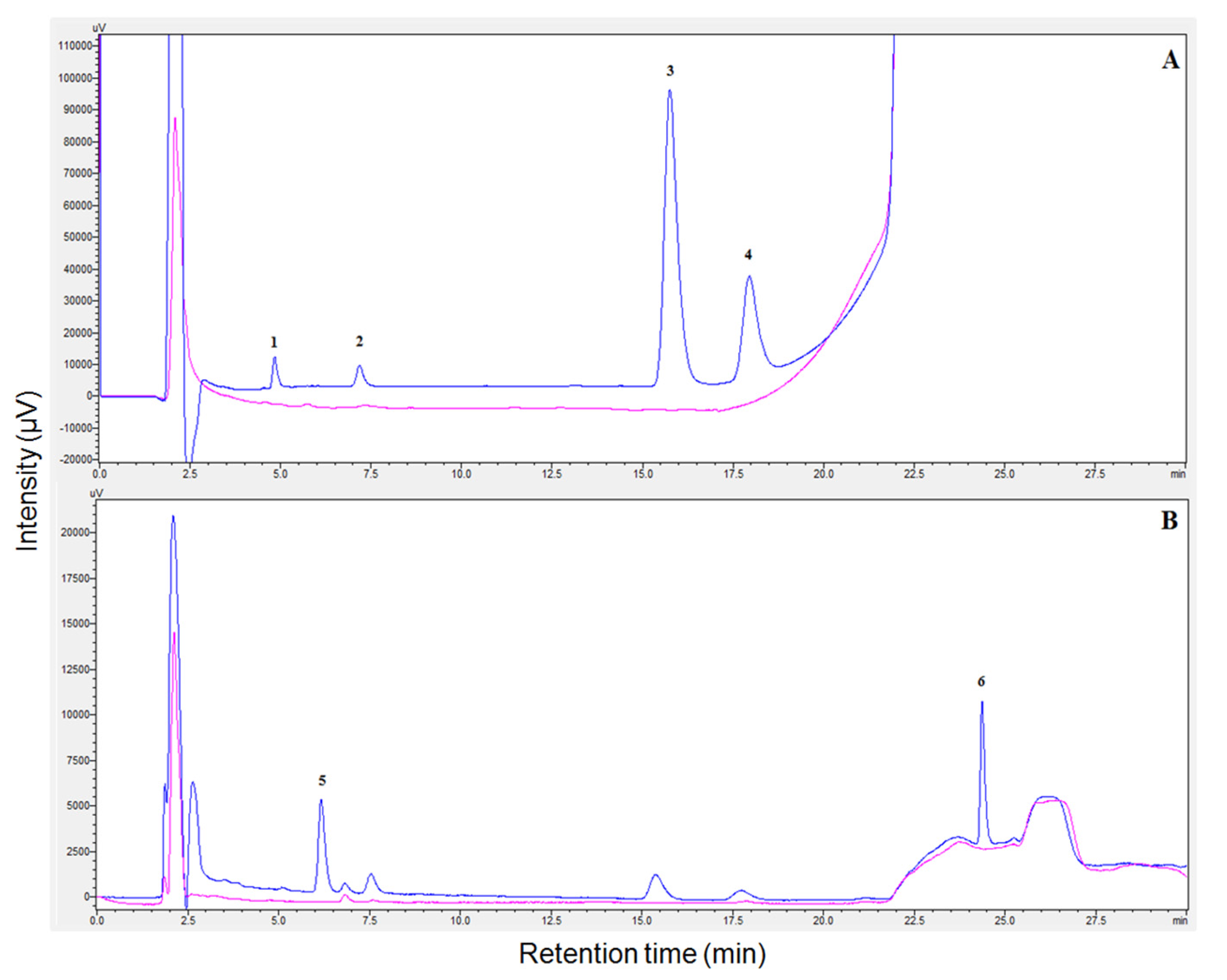
3.3. Analytical Figures of Merit and Analysis of Hospital Wastewater Samples
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, W.C. Occurrence, sources, and fate of pharmaceuticals in aquatic environment and soil. Environ. Pollut. 2014, 187, 193–201. [Google Scholar] [CrossRef]
- de Almeida, C.A.A.; Brenner, C.G.B.; Minetto, L.; Mallmann, C.A.; Martins, A.F. Determination of anti-anxiety and anti-epileptic drugs in hospital effluent and a preliminary risk assessment. Chemosphere 2013, 93, 2349–2355. [Google Scholar] [CrossRef]
- Gracia-Lor, E.; Sancho, J.V.; Serrano, R.; Hernandez, F. Occurrence and removal of pharmaceuticals in wastewater treatment plants at the Spanish Mediterranean area of Valencia. Chemosphere 2012, 87, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Al Aukidy, M.; Verlicchi, P.; Voulvoulis, N. A framework for the assessment of the environmental risk posed by pharmaceuticals originating from hospital effluents. Sci. Total Environ. 2014, 493, 54–64. [Google Scholar] [CrossRef]
- Azuma, T.; Arima, N.; Tsukada, A.; Hirami, S.; Matsuoka, R.; Moriwake, R.; Ishiuchi, H.; Inoiama, T.; Teranishi, Y.; Yamaoka, M.; et al. Detection of pharmaceuticals and phytochemicals together with their metabolites in hospital effluents in Japan, and their contribution to sewage treatment plant influents. Sci. Total Environ. 2016, 548, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Onesios, K.M.; Yu, J.T.; Bouwer, E.J. Biodegradation and removal of pharmaceuticals and personal care products in treatment systems: A review. Biodegradation 2019, 20, 441–466. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.J.; Wu, M.H.; Lei, J.Q.; Fu, C.; Gu, J.Z.; Xu, G. The fate and risk assessment of psychiatric pharmaceuticals from psychiatric hospital effluent. Ecotox. Environ. Saf. 2018, 150, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Marta, Z.; Bobaly, B.; Fekete, J.; Magda, B.; Imre, T.; Szabo, P.T. Simultaneous determination of ten nonsteroidal anti-inflammatory drugs from drinking water, surface water and wastewater using micro UHPLC-MS/MS with on-line SPE system. J. Pharmaceut. Biomed. 2018, 160, 99–108. [Google Scholar] [CrossRef]
- Puckowski, A.; Mioduszewska, K.; Lukaszewicz, P.; Borecka, M.; Caban, M.; Maszkowska, J.; Stepnowski, P. Bioaccumulation and analytics of pharmaceutical residues in the environment: A review. J. Pharmaceut. Biomed. 2016, 127, 232–255. [Google Scholar] [CrossRef]
- Babarahimi, V.; Talebpour, Z.; Haghighi, F.; Adib, N.; Vahidi, F. Validated determination of losartan and valsartan in human plasma by stir bar sorptive extraction based on acrylate monolithic polymer, liquid chromatographic analysis and experimental design methodology. J. Pharmaceut. Biomed. 2018, 153, 204–213. [Google Scholar] [CrossRef]
- Lima, D.L.D.; Silva, C.P.; Otero, M.; Esteves, V.I. Low cost methodology for estrogens monitoring in water samples using dispersive liquid-liquid microextraction and HPLC with fluorescence detection. Talanta 2013, 115, 980–985. [Google Scholar] [CrossRef]
- Wang, R.; Li, W.Q.; Chen, Z.L. Solid phase microextraction with poly(deep eutectic solvent) monolithic column online coupled to HPLC for determination of non-steroidal anti-inflammatory drugs. Anal. Chim. Acta 2018, 1018, 111–118. [Google Scholar] [CrossRef]
- Carasek, E.; Merib, J. Membrane-based microextraction techniques in analytical chemistry: A review. Anal. Chim. Acta 2015, 880, 8–25. [Google Scholar] [CrossRef] [PubMed]
- Bordin, D.C.; Alves, M.N.; de Campos, E.G.; De Martinis, B.S. Disposable pipette tips extraction: Fundamentals, applications and state of the art. J. Sep. Sci. 2016, 39, 1168–1172. [Google Scholar] [CrossRef]
- Brewer, W.E. Disposable Pipette Extraction. U.S. Patent 6566145B2, 9 February 2000. [Google Scholar]
- Chaves, A.R.; Moura, B.H.F.; Caris, J.A.; Rabelo, D.; Queiroz, M.E.C. The development of a new disposable pipette extraction phase based on polyaniline composites for the determination of levels of antidepressants in plasma samples. J. Chromatogr. 2015, 1399, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Corazza, G.; Merib, J.; Magosso, H.A.; Bittencourt, O.R.; Carasek, E. A hybrid material as a sorbent phase for the disposable pipette extraction technique enhances efficiency in the determination of phenolic endocrine-disrupting compounds. J. Chromatogr. A 2017, 1513, 42–50. [Google Scholar] [CrossRef]
- Mafra, G.; Spudeit, D.; Brognoli, R.; Merib, J.; Carasek, E. Expanding the applicability of cork as extraction phase for disposable pipette extraction in multiresidue analysis of pharmaceuticals in urine samples. J. Chromatogr. B 2018, 1102–1103, 159–166. [Google Scholar] [CrossRef]
- Mores, L.; da Silva, A.C.; Merib, J.; Dias, A.N.; Carasek, E. A natural and renewable biosorbent phase as a low-cost approach in disposable pipette extraction technique for the determination of emerging contaminants in lake water samples. J. Sep. Sci. 2019, 42, 1404–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turazzi, F.C.; Morés, L.; Carasek, E.; Merib, J.; de Oliveira Barra, G.M. A rapid and environmentally friendly analytical method based on conductive polymer as extraction phase for disposable pipette extraction for the determination of hormones and polycyclic aromatic hydrocarbons in river water samples using high-performance liquid chromatography/diode array detection. J. Environ. Chem. Eng. 2019, 7, 103–156. [Google Scholar]
- Mravcakova, M.; Boukerma, K.; Omastova, M.; Chehimi, M.M. Montmorillonite/polypyrrole nanocomposites. The effect of organic modification of clay on the chemical and electrical properties. Mat. Sci. Eng. C Mater. 2006, 26, 306–313. [Google Scholar] [CrossRef]
- Ramoa, S.; Barra, G.M.O.; Merlini, C.; Schreiner, W.H.; Livi, S.; Soares, B.G. Production of montmorillonite/polypyrrole nanocomposites through in situ oxidative polymerization of pyrrole: Effect of anionic and cationic surfactants on structure and properties. Appl. Clay Sci. 2015, 104, 160–167. [Google Scholar] [CrossRef]
- Dimpe, K.M.; Nomngongo, P.N. Current sample preparation methodologies for analysis of emerging pollutants in different environmental matrices. TrAC Trend. Anal. Chem. 2016, 82, 199–207. [Google Scholar] [CrossRef]
- Bletsou, A.A.; Jeon, J.; Hollender, J.; Archontaki, E.; Thomaidis, N.S. Targeted and non-targeted liquid chromatography-mass spectrometric workflows for identification of transformation products of emerging pollutants in the aquatic environment. TrAC Trend. Anal. Chem. 2015, 66, 32–44. [Google Scholar] [CrossRef] [Green Version]
- Pawlinsyn, J. Handbook of Solid Phase Microextraction; Chemical Industry Press: Beijing, China, 2009. [Google Scholar]
- Barwick, V.J. Preparation of Calibration Curves: A Guide to Best Practice. 2003. Available online: https://www.lgcgroup.com/media/1735/prepration-of-calibration-curves_a-guide-to-best-practice.pdf (accessed on 24 June 2021).
- AOAC. Official Methods of Analysis, Appendix F: Guidelines for Standard Method Performance Requirements. 2016. Available online: http://www.eoma.aoac.org/app_f.pdf (accessed on 29 November 2019).
- Mulholland, M.; Waterhouse, J. Investigation of the limitations of saturated fractional factorial experimental designs, with confounding effects for an HPLC ruggedness test. Chromatographia 1988, 25, 769–774. [Google Scholar] [CrossRef]
- Bokor, I.; Sdraulig, S.; Sanagou, M. Robustness testing of an in-situ caesium extraction unit. J. Environ. Radioact. 2017, 178–179, 426–429. [Google Scholar] [CrossRef]
- Hund, E.; Vander Heyden, Y.; Haustein, M.; Massart, D.L.; Smeyers-Verbeke, J. Comparison of several criteria to decide on the significance of effects in a robustness test with an asymmetrical factorial design. Anal. Chim. Acta 2000, 404, 257–271. [Google Scholar] [CrossRef]
- Ashfaq, M.; Noor, N.; Saif-Ur-Rehman, M.; Sun, Q.; Mustafa, G.; Nazar, M.F.; Yu, C.P. Determination of Commonly Used Pharmaceuticals in Hospital Waste of Pakistan and Evaluation of Their Ecological Risk Assessment. Clean-Soil Air Water 2017, 45, 1–10. [Google Scholar] [CrossRef]
- Pebdani, A.A.; Dadfarnia, S.; Shabani, A.M.H.; Khodadoust, S.; Haghgoo, S. Application of modified stir bar with nickel:zinc sulphide nanoparticles loaded on activated carbon as a sorbent for preconcentration of losartan and valsartan and their determination by high performance liquid chromatography. J. Chromatogr. A 2016, 1437, 15–24. [Google Scholar] [CrossRef]
- Rashvand, M.; Vosough, M. Graphene oxide-polyaniline nanocomposite as a potential sorbent for dispersive solid-phase extraction and determination of selected pharmaceutical and personal care products in wastewater samples using HPLC with a diode-array detector. Anal. Methods 2016, 8, 1898–1907. [Google Scholar] [CrossRef]
- Arismendi, D.; Becerra-Herrera, M.; Cerrato, I.; Richter, P. Simultaneous determination of multiresidue and multiclass emerging contaminants in waters by rotating-disk sorptive extraction-derivatization-gas chromatography/mass spectrometry. Talanta 2019, 201, 480–489. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Chemical Structure | pKa | log Ko/w |
|---|---|---|---|
| CBZ |  | 15.96 | 2.77 |
| DCL |  | 4.00 | 4.26 |
| NAP |  | 4.19 | 2.99 |
| FLU |  | 9.80 | 4.17 |
| LOS |  | 5.85 | 5.00 |
| EE2 |  | 10.33 | 3.90 |
| Analyte | Linear Equation | Linear Range (µg L−1) | Correlation Coefficient (r) | LOD (µg L−1) | LOQ (µg L−1) |
|---|---|---|---|---|---|
| CBZ | y = 645.03x − 731.96 | 5–100 | 0.9996 | 1.510 | 5.00 |
| DCL | y = 760.09x − 2089.9 | 5–100 | 0.9990 | 1.510 | 5.00 |
| NAP | y = 262281x − 21877 | 0.1–10 | 0.9996 | 0.030 | 0.10 |
| FLU | y = 9805.6x + 3435 | 1–50 | 0.9710 | 0.303 | 1.00 |
| LOS | y = 11535x − 5496.7 | 0.5–100 | 0.9991 | 0.151 | 0.50 |
| EE2 | y = 101441x − 13706 | 0.5–25 | 0.9994 | 0.151 | 0.50 |
| Analyte | Spiked Concentration (µg L−1) | Relative Recovery (%, n = 3) | Precision (RSD, %) | |
|---|---|---|---|---|
| Intra-Day (n = 3) | Inter-Day (n = 9) | |||
| CBZ | 5 | 118 | 6 | 15 |
| 25 | 127 | 13 | ||
| 100 | 97 | 13 | ||
| DCL | 5 | 86 | 8 | 19 |
| 25 | 94 | 19 | ||
| 100 | 86 | 7 | ||
| NAP | 0.10 | 110 | 19 | |
| 1 | 113 | 18 | ||
| 5 | 117 | 7 | 18 | |
| FLU | 1 | 83 | 5 | |
| 5 | 98 | 4 | 10 | |
| 25 | 115 | 7 | ||
| LOS | 0.50 | 120 | 1 | |
| 5 | 108 | 6 | 19 | |
| 100 | 80 | 12 | ||
| EE2 | 0.5 | 97 | 13 | |
| 5 | 118 | 12 | 17 | |
| 25 | 109 | 15 | ||
| Method | Analyte | Extraction Phase | Matrix | Linear Range (µg L−1) | LOQ (µg L−1) | Time of Sample Preparation | Solvent Volume |
|---|---|---|---|---|---|---|---|
| This work DPX and HPLC-DAD/FLD | CBZ | 20 mg of Si3Py+Cl− | Hospital wastewater | 5–100 | 5 | 6.5 min | 100 µL |
| DCL | 5–100 | 5 | |||||
| NAP | 0.1–10 | 0.1 | |||||
| FLU | 1–50 | 1 | |||||
| LOS | 0.5–100 | 0.5 | |||||
| EE2 | 0.5–25 | 0.5 | |||||
| LLE and HPLC-UV (Ashfaq et al. 2017) | DIC NAP | 40 mL of chloroform | Hospital wastewater | 300–10000 | 12 8 | - | 42 mL |
| SBSE and HPLC-UV (Pebdani et al. 2016) | LOS | 50 mg of Ni:ZnS-AC | Urine | 0.4–50 | 0.38 | Approximately 32 min | 280 µL |
| RDSE and GC-MS (Arismendi et al. 2019) | DIC EE2 | 40 mg of Oasis® HLB | Drinking water and natural waters (well and river) | 0.1–100 | 0.12 0.21 | Approximately 115 min | 10 mL |
| SPE and HPLC-DAD (Ngubane et al. 2019) | DCL NAP | 200 mg of Oasis® HLB | Estuary and seawater | 100–5000 | 0.036 0.025 | - | 16 mL |
| DSPE and HPLC-DAD (Rashvand and Vosough 2016) | CBZ DCL NAP | 43 mg of GO-PANI | Effluent from sewage Treatment Plants (STPs) | – | 1.12 0.36 0.15 | Approximately 21 min | 3.5 mL |
| SPE and HPLC-MS (Matongo et al. 2015) | CBZ | 60 mg of Oasis® HLB | Surface water and wastewater | 0.297–100 | 0.891 | Approximately 112 min | 21 mL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meneghini, V.; Corazza, G.; Magosso, H.A.; Merib, J.; Carasek, E. A Rapid Analytical Approach for Monitoring Pharmaceuticals in Hospital Wastewater—A DPX-Based Procedure with Environmentally-Friendly Extraction Phase Coupled to High Performance Liquid Chromatography–Diode Array/Fluorescence Detectors. Separations 2021, 8, 109. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8080109
Meneghini V, Corazza G, Magosso HA, Merib J, Carasek E. A Rapid Analytical Approach for Monitoring Pharmaceuticals in Hospital Wastewater—A DPX-Based Procedure with Environmentally-Friendly Extraction Phase Coupled to High Performance Liquid Chromatography–Diode Array/Fluorescence Detectors. Separations. 2021; 8(8):109. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8080109
Chicago/Turabian StyleMeneghini, Vanessa, Gabriela Corazza, Hérica A. Magosso, Josias Merib, and Eduardo Carasek. 2021. "A Rapid Analytical Approach for Monitoring Pharmaceuticals in Hospital Wastewater—A DPX-Based Procedure with Environmentally-Friendly Extraction Phase Coupled to High Performance Liquid Chromatography–Diode Array/Fluorescence Detectors" Separations 8, no. 8: 109. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8080109