
Quantitative Capillary Electrophoresis for Analysis of Extracellular Vesicles (EVqCE)
 ,
, 
Abstract
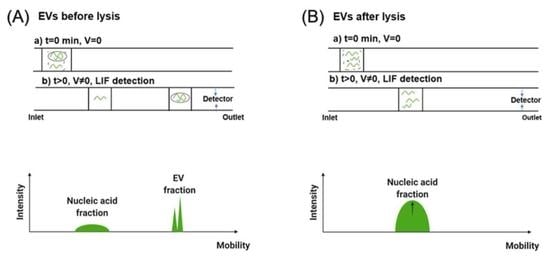
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Isolation of EVs
2.3. Nanoparticle Tracking Analysis (NTA)
2.4. Flow Cytometry
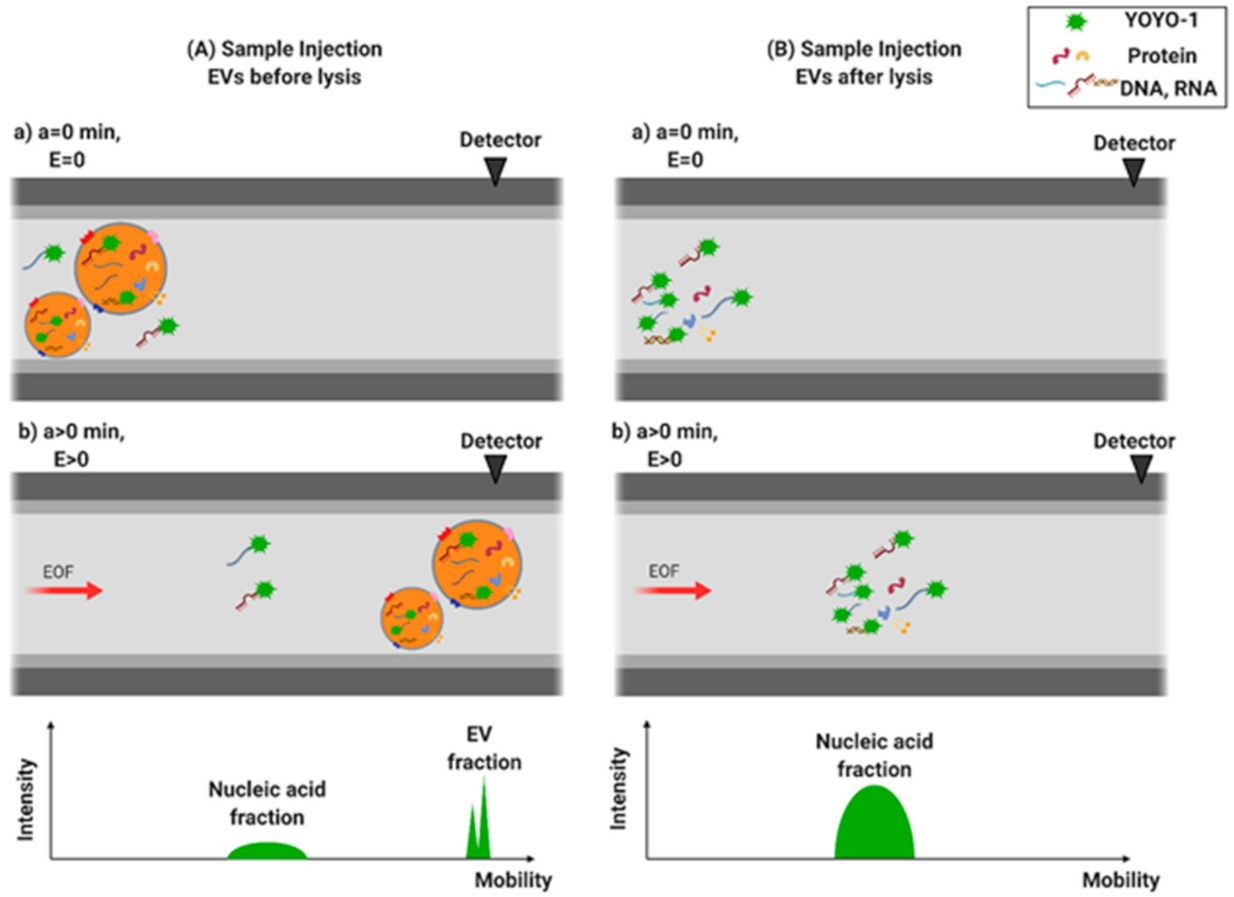
2.5. EV Staining and Lysis
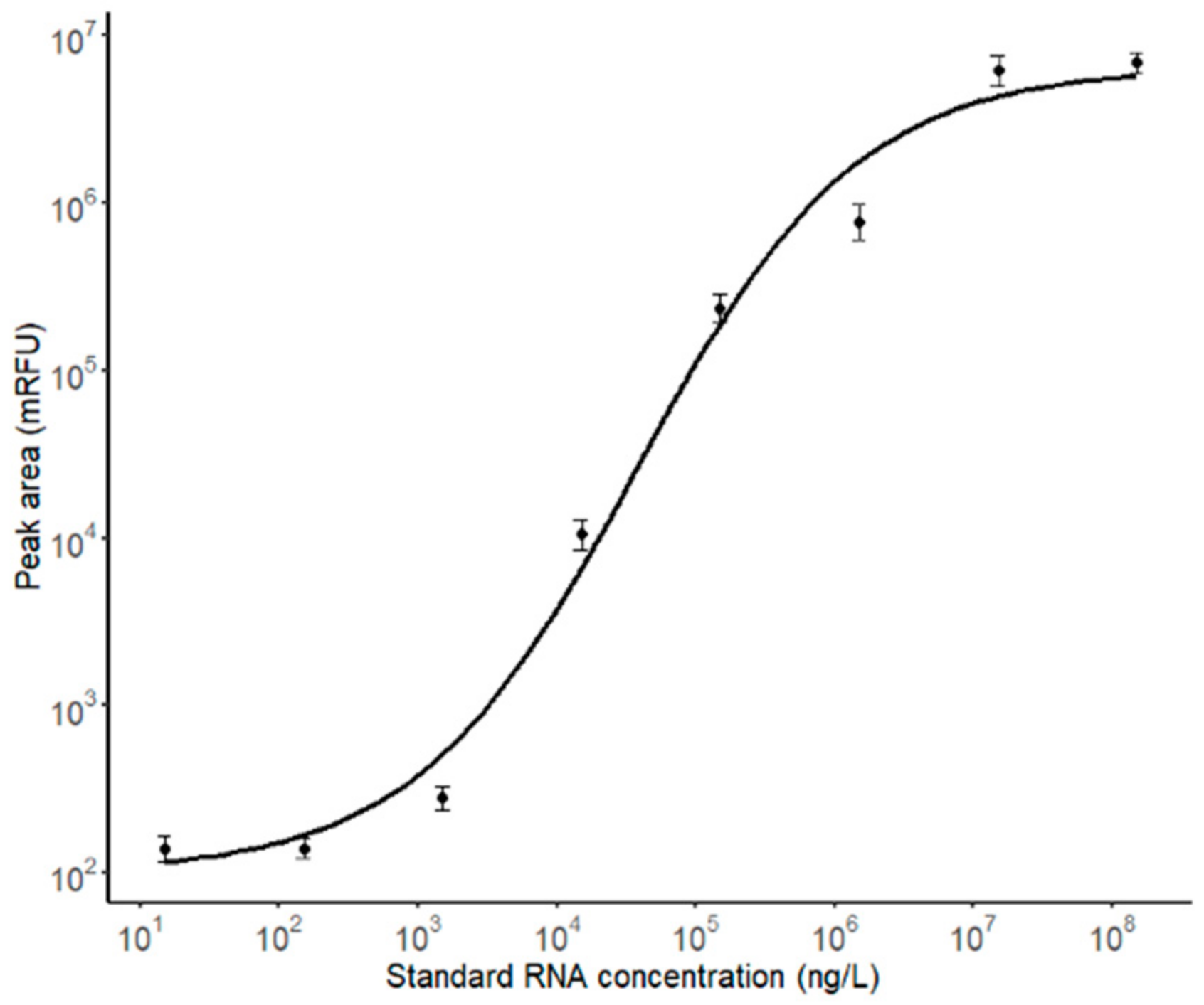
2.6. Calibration Curve
2.7. Capillary Electrophoresis
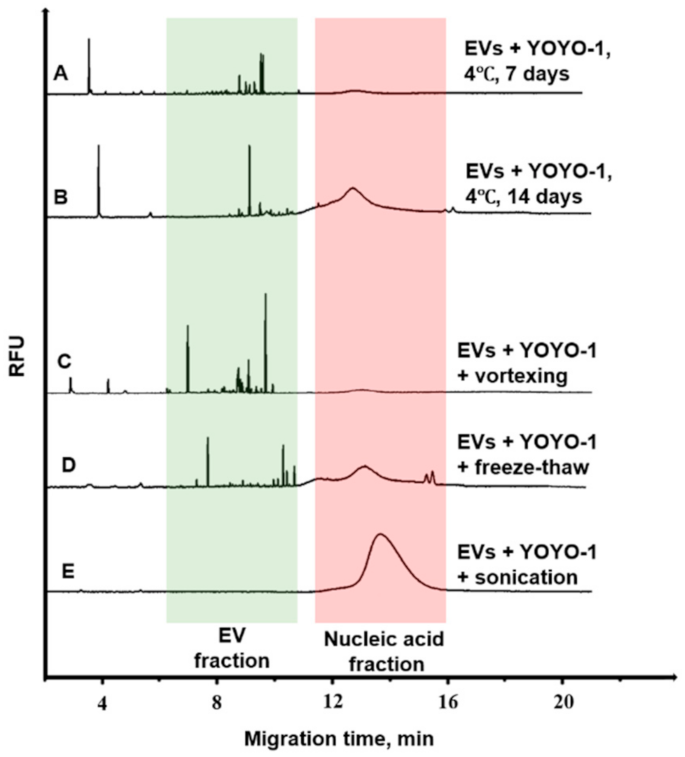
2.8. EV Degradation Conditions
3. Result and Discussion
3.1. Characterization of EVs by NTA and Flow Cytometry
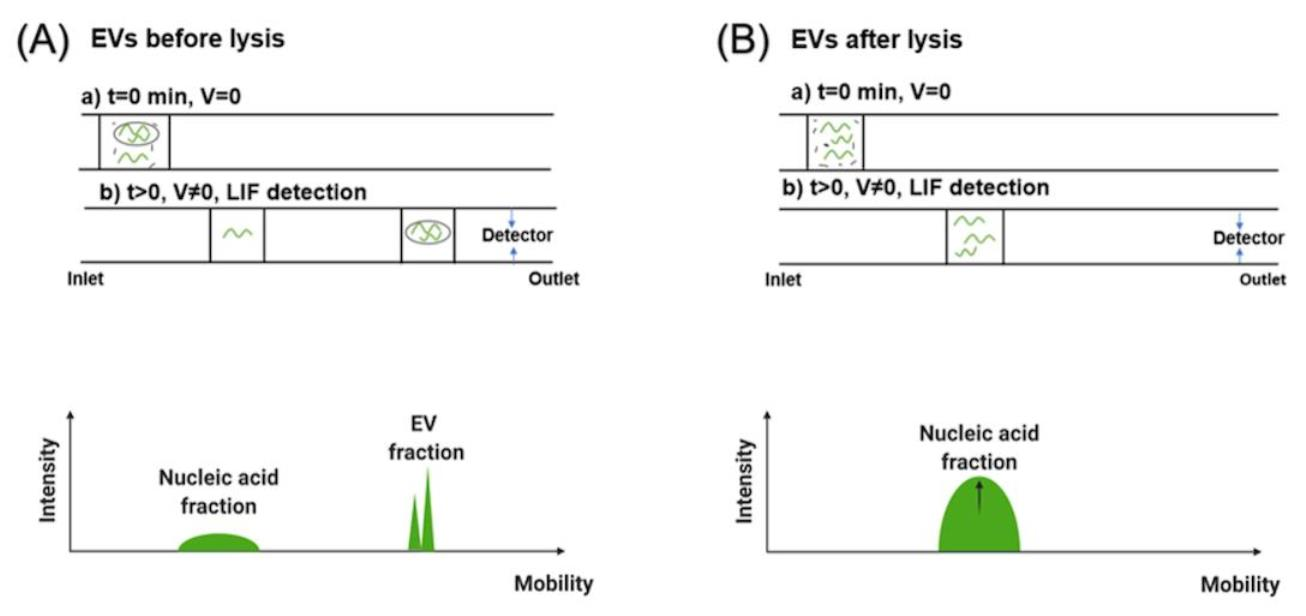
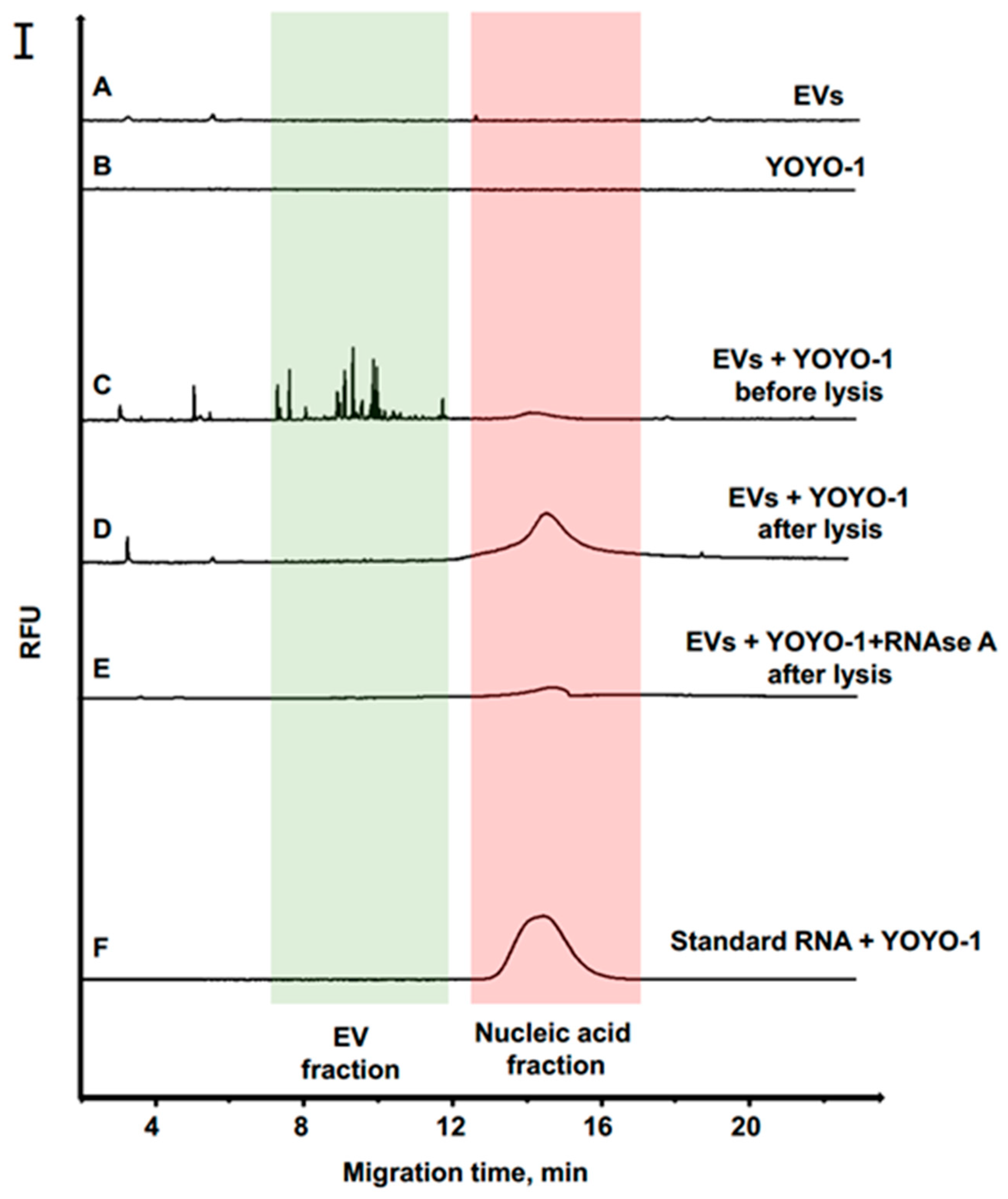
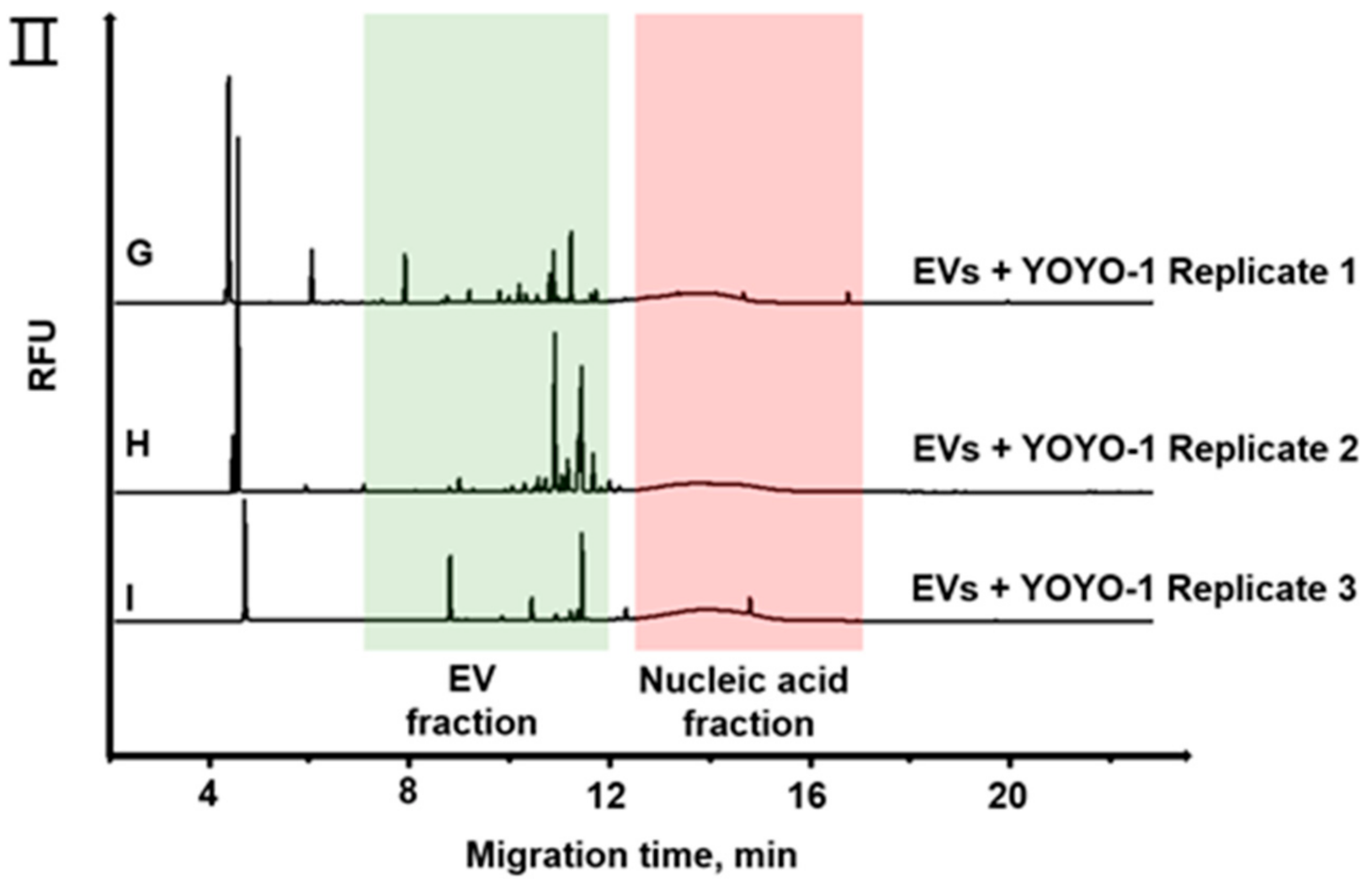
3.2. Separation of EVs by CE
3.3. RNA Quantification of EVs from Cancer Cells
3.4. Degradation Analysis of EVs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Akers, J.C.; Ramakrishnan, V.; Nolan, J.P.; Duggan, E.; Fu, C.-C.; Hochberg, F.H.; Chen, C.C.; Carter, B.S. Comparative analysis of technologies for quantifying extracellular vesicles (EVs) in clinical cerebrospinal fluids (CSF). PLoS ONE 2016, 11, e0149866. [Google Scholar] [CrossRef]
- Choi, D.S. Urinary extracellular vesicles for biomarker source to monitor polycystic kidney disease. Proteom. Clin. Appl. 2015, 9, 447–448. [Google Scholar] [CrossRef]
- György, B.; Szabó, T.G.; Pásztói, M.; Pál, Z.; Misják, P.; Aradi, B.; László, V.; Pállinger, E.; Pap, E.; Kittel, A. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oosthuyzen, W.; Sime, N.E.; Ivy, J.R.; Turtle, E.J.; Street, J.M.; Pound, J.; Bath, L.E.; Webb, D.J.; Gregory, C.D.; Bailey, M.A. Quantification of human urinary exosomes by nanoparticle tracking analysis. J. Physiol. 2013, 591, 5833–5842. [Google Scholar] [CrossRef]
- Rupert, D.L.; Claudio, V.; Lässer, C.; Bally, M. Methods for the physical characterization and quantification of extracellular vesicles in biological samples. Biochim. Biophys. Acta (BBA) Gen. Subj. 2017, 1861, 3164–3179. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.; Clayton, A. How pure are your vesicles? J. Extracell. Vesicles 2013, 2, 19861. [Google Scholar] [CrossRef]
- Witwer, K.W.; Buzás, E.I.; Bemis, L.T.; Bora, A.; Lässer, C.; Lötvall, J.; Nolte-‘t Hoen, E.N.; Piper, M.G.; Sivaraman, S.; Skog, J. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2013, 2, 20360. [Google Scholar] [CrossRef] [PubMed]
- Hartjes, T.A.; Mytnyk, S.; Jenster, G.W.; van Steijn, V.; van Royen, M.E. Extracellular vesicle quantification and characterization: Common methods and emerging approaches. Bioengineering 2019, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Pertierra, E.; Oliveira-Rodríguez, M.; Matos, M.; Gutiérrez, G.; Moyano, A.; Salvador, M.; Rivas, M.; Blanco-López, M.C. Extracellular vesicles: Current analytical techniques for detection and quantification. Biomolecules 2020, 10, 824. [Google Scholar] [CrossRef]
- Chia, B.S.; Low, Y.P.; Wang, Q.; Li, P.; Gao, Z. Advances in exosome quantification techniques. TrAC Trends Anal. Chem. 2017, 86, 93–106. [Google Scholar] [CrossRef]
- Mehdiani, A.; Maier, A.; Pinto, A.; Barth, M.; Akhyari, P.; Lichtenberg, A. An innovative method for exosome quantification and size measurement. JoVE (J. Vis. Exp.) 2015, 95, e50974. [Google Scholar] [CrossRef] [Green Version]
- Lane, R.E.; Korbie, D.; Anderson, W.; Vaidyanathan, R.; Trau, M. Analysis of exosome purification methods using a model liposome system and tunable-resistive pulse sensing. Sci. Rep. 2015, 5, 7639. [Google Scholar] [CrossRef] [Green Version]
- Im, H.; Shao, H.; Park, Y.I.; Peterson, V.M.; Castro, C.M.; Weissleder, R.; Lee, H. Label-free detection and molecular profiling of exosomes with a nano-plasmonic sensor. Nat. Biotechnol. 2014, 32, 490–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Wang, K.; Cui, J.; Liu, H.; Bu, X.; Ma, H.; Wang, W.; Gong, H.; Lausted, C.; Hood, L. Label-free quantitative detection of tumor-derived exosomes through surface plasmon resonance imaging. Anal. Chem. 2014, 86, 8857–8864. [Google Scholar] [CrossRef] [Green Version]
- Heider, S.; Metzner, C. Quantitative real-time single particle analysis of virions. Virology 2014, 462, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Gao, X. Nanoparticle counting: Towards accurate determination of the molar concentration. Chem. Soc. Rev. 2014, 43, 7267–7278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedmak, J.J.; Grossberg, S.E. A rapid, sensitive, and versatile assay for protein using Coomassie brilliant blue G250. Anal. Biochem. 1977, 79, 544–552. [Google Scholar] [CrossRef]
- Smith, P.E.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Moon, P.-G.; Lee, J.-E.; Cho, Y.-E.; Lee, S.J.; Chae, Y.S.; Jung, J.H.; Kim, I.-S.; Park, H.Y.; Baek, M.-C. Fibronectin on circulating extracellular vesicles as a liquid biopsy to detect breast cancer. Oncotarget 2016, 7, 40189. [Google Scholar] [CrossRef] [Green Version]
- Nolan, J.P.; Duggan, E. Analysis of individual extracellular vesicles by flow cytometry. In Flow Cytometry Protocols; Springer: Berlin/Heidelberg, Germany, 2018; pp. 79–92. [Google Scholar]
- Rupert, D.b.L.; Lässer, C.; Eldh, M.; Block, S.; Zhdanov, V.P.; Lotvall, J.O.; Bally, M.; Hook, F. Determination of exosome concentration in solution using surface plasmon resonance spectroscopy. Anal. Chem. 2014, 86, 5929–5936. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Dragovic, R.A.; Gardiner, C.; Brooks, A.S.; Tannetta, D.S.; Ferguson, D.J.; Hole, P.; Carr, B.; Redman, C.W.; Harris, A.L.; Dobson, P.J. Sizing and phenotyping of cellular vesicles using Nanoparticle Tracking Analysis. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, C.; Ferreira, Y.J.; Dragovic, R.A.; Redman, C.W.; Sargent, I.L. Extracellular vesicle sizing and enumeration by nanoparticle tracking analysis. J. Extracell. Vesicles 2013, 2, 19671. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, P.R.; Zheng, Y.; Fischer, R.; Heidasch, R.; Gardiner, C.; Evetts, S.; Hu, M.; Wade-Martins, R.; Turner, M.R.; Morris, J. Identification of distinct circulating exosomes in Parkinson’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Van Der Pol, E.; Hoekstra, A.; Sturk, A.; Otto, C.; Van Leeuwen, T.; Nieuwland, R. Optical and non-optical methods for detection and characterization of microparticles and exosomes. J. Thromb. Haemost. 2010, 8, 2596–2607. [Google Scholar] [CrossRef]
- Varga, Z.; Yuana, Y.; Grootemaat, A.E.; Van der Pol, E.; Gollwitzer, C.; Krumrey, M.; Nieuwland, R. Towards traceable size determination of extracellular vesicles. J. Extracell. Vesicles 2014, 3, 23298. [Google Scholar] [CrossRef]
- Akagi, T.; Ichiki, T. Microcapillary chip-based extracellular vesicle profiling system. In Extracellular Vesicles; Springer: Berlin/Heidelberg, Germany, 2017; pp. 209–217. [Google Scholar]
- Morani, M.; Mai, T.D.; Krupova, Z.; Defrenaix, P.; Multia, E.; Riekkola, M.-L.; Taverna, M. Electrokinetic characterization of extracellular vesicles with capillary electrophoresis: A new tool for their identification and quantification. Anal. Chim. Acta 2020, 1128, 42–51. [Google Scholar] [CrossRef]
- Tani, Y.; Kaneta, T. Indirect capillary electrophoresis immunoassay of membrane protein in extracellular vesicles. J. Chromatogr. A 2020, 1629, 461513. [Google Scholar] [CrossRef]
- Azizi, A.; Mironov, G.G.; Muharemagic, D.; Wehbe, M.; Bell, J.C.; Berezovski, M.V. Viral quantitative capillary electrophoresis for counting and quality control of RNA viruses. Anal. Chem. 2012, 84, 9585–9591. [Google Scholar] [CrossRef]
- Mironov, G.G.; Chechik, A.V.; Ozer, R.; Bell, J.C.; Berezovski, M.V. Viral quantitative capillary electrophoresis for counting intact viruses. Anal. Chem. 2011, 83, 5431–5435. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Mark, M.T.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermann, A.J.; Gorski, S.A.; Vogel, J. Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 2012, 10, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Abdelmohsen, K.; Mustapic, M.; Kapogiannis, D.; Gorospe, M. RNA in extracellular vesicles. Wiley Interdiscip. Rev. RNA 2017, 8, e1413. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quantification Method (Particles/mL) | MBA-MB-231 | NCl-H1975 | CCL-119 |
|---|---|---|---|
| EVqCE | (5.1 ± 0.2) × 109 | (1.4 ± 0.1) × 109 | (1.3 ± 0.4) × 109 |
| NTA | (4.9 ± 0.1) × 109 | (2.3 ± 0.2) × 109 | (2.6 ± 0.3)× 109 |
| Flow cytometry | (1.7 ± 0.3) × 109 | (3.6 ± 0.7) × 108 | (6.0 ± 0.8)× 108 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dou, Y.; Ren, L.; Kulabhusan, P.K.; Zaripov, E.A.; Berezovski, M.V. Quantitative Capillary Electrophoresis for Analysis of Extracellular Vesicles (EVqCE). Separations 2021, 8, 110. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8080110
Dou Y, Ren L, Kulabhusan PK, Zaripov EA, Berezovski MV. Quantitative Capillary Electrophoresis for Analysis of Extracellular Vesicles (EVqCE). Separations. 2021; 8(8):110. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8080110
Chicago/Turabian StyleDou, Yuchu, Lixuan Ren, Prabir Kumar Kulabhusan, Emil A. Zaripov, and Maxim V. Berezovski. 2021. "Quantitative Capillary Electrophoresis for Analysis of Extracellular Vesicles (EVqCE)" Separations 8, no. 8: 110. https://0-doi-org.brum.beds.ac.uk/10.3390/separations8080110