
Chalcogen···Chalcogen Bonding in Molybdenum Disulfide, Molybdenum Diselenide and Molybdenum Ditelluride Dimers as Prototypes for a Basic Understanding of the Local Interfacial Chemical Bonding Environment in 2D Layered Transition Metal Dichalcogenides




Abstract
:
1. Introduction
2. Computational Details and Model Systems
3. Results and Discussion
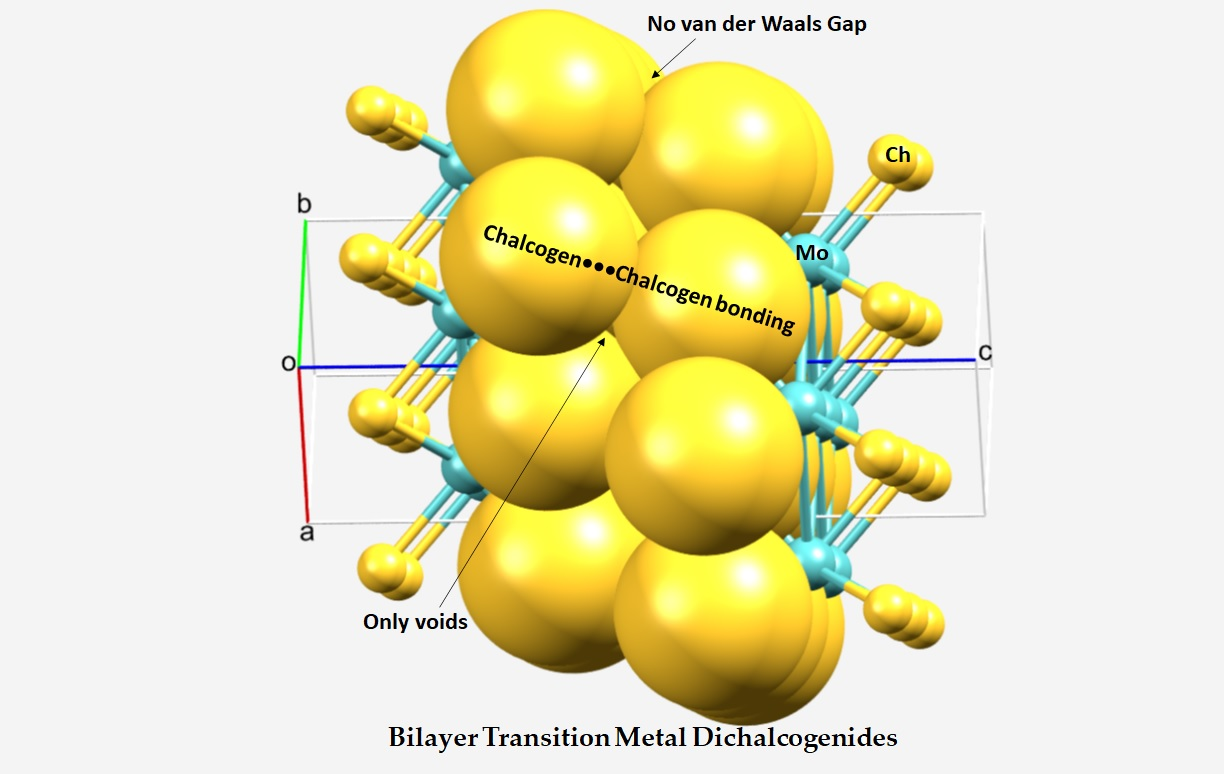
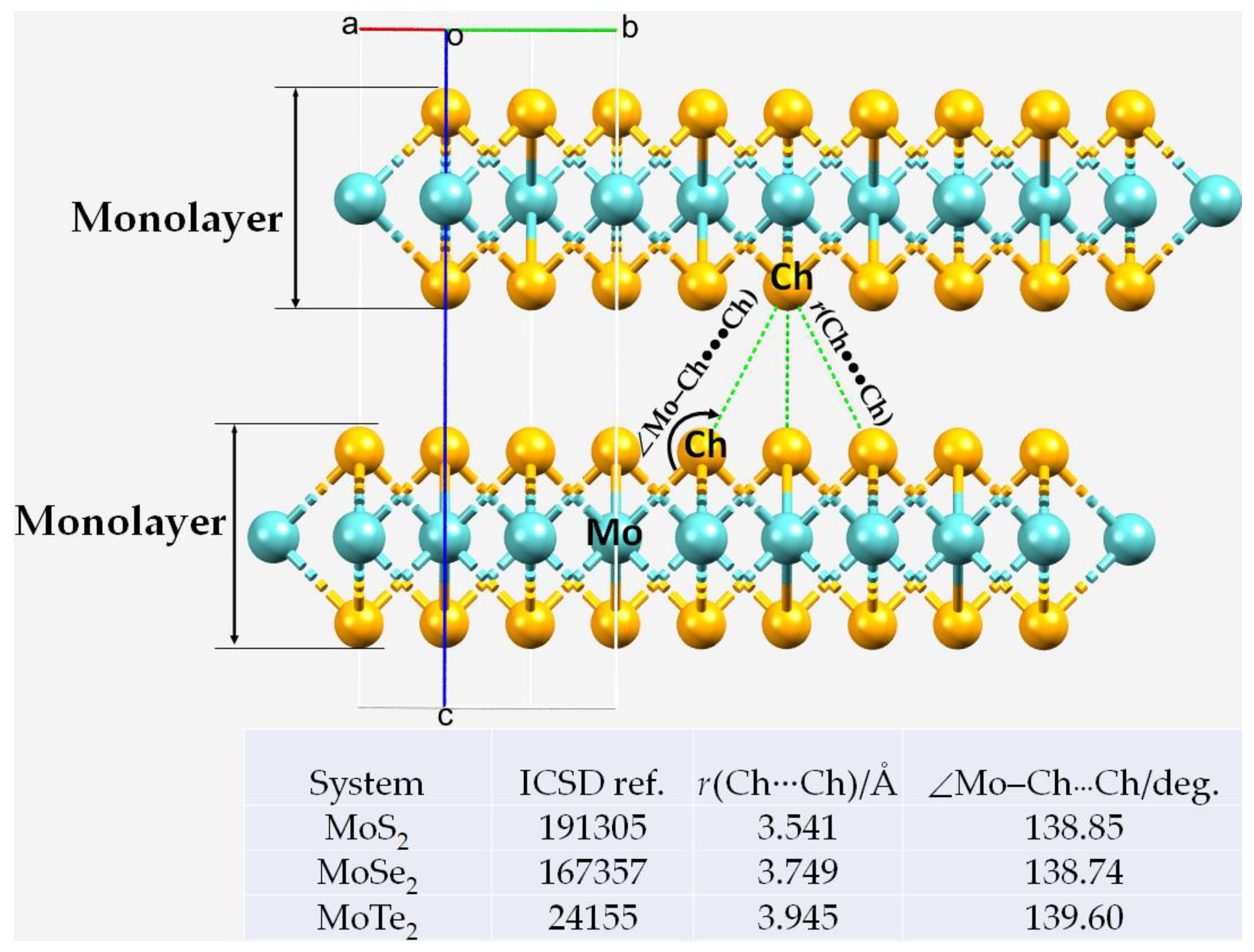
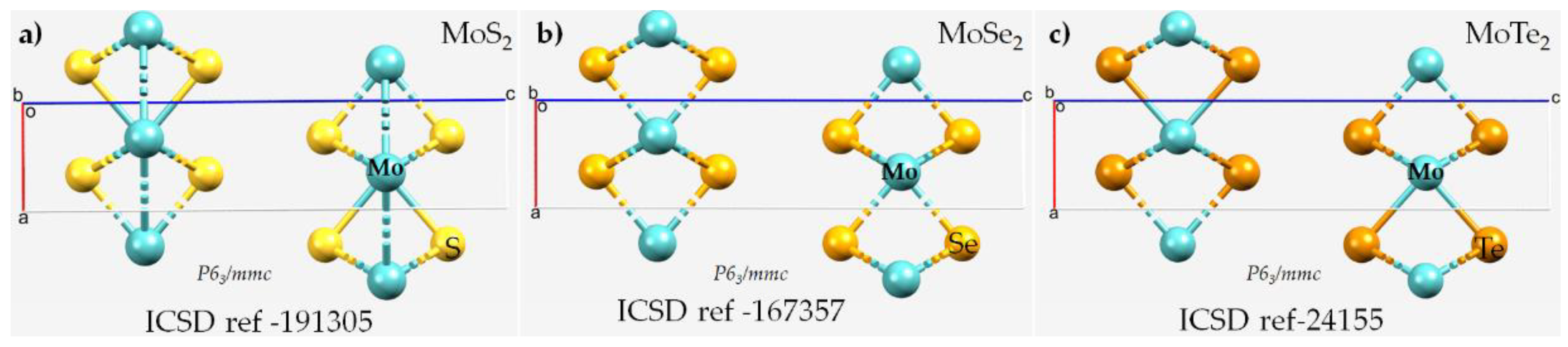
3.1. Interfacial Geometry of the 2H-MoCh2 Crystals and Comparison with Theory

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Symmetry | Space Group | a = b | c | α = β | γ | V | ρ |
|---|---|---|---|---|---|---|---|---|
| MoS2 | Hexagonal | P63/mmc | 3.168 | 12.466 | 90 | 120 | 108.38 | 4.91 |
| (3.169) | (12.324) | (90) | (120) | (107.18) | (4.96) | |||
| MoSe2 | Hexagonal | P63/mmc | 3.296 | 13.181 | 90 | 120 | 124.02 | 6.80 |
| (3.29) | (12.93) | (90) | (120) | (121.21) | (6.96) | |||
| MoTe2 | Hexagonal | P63/mmc | 3.508 | 14.197 | 90 | 120 | 151.31 | 7.71 |
| (3.518) | (13.974) | (90) | (120) | (149.79) | (7.786) |
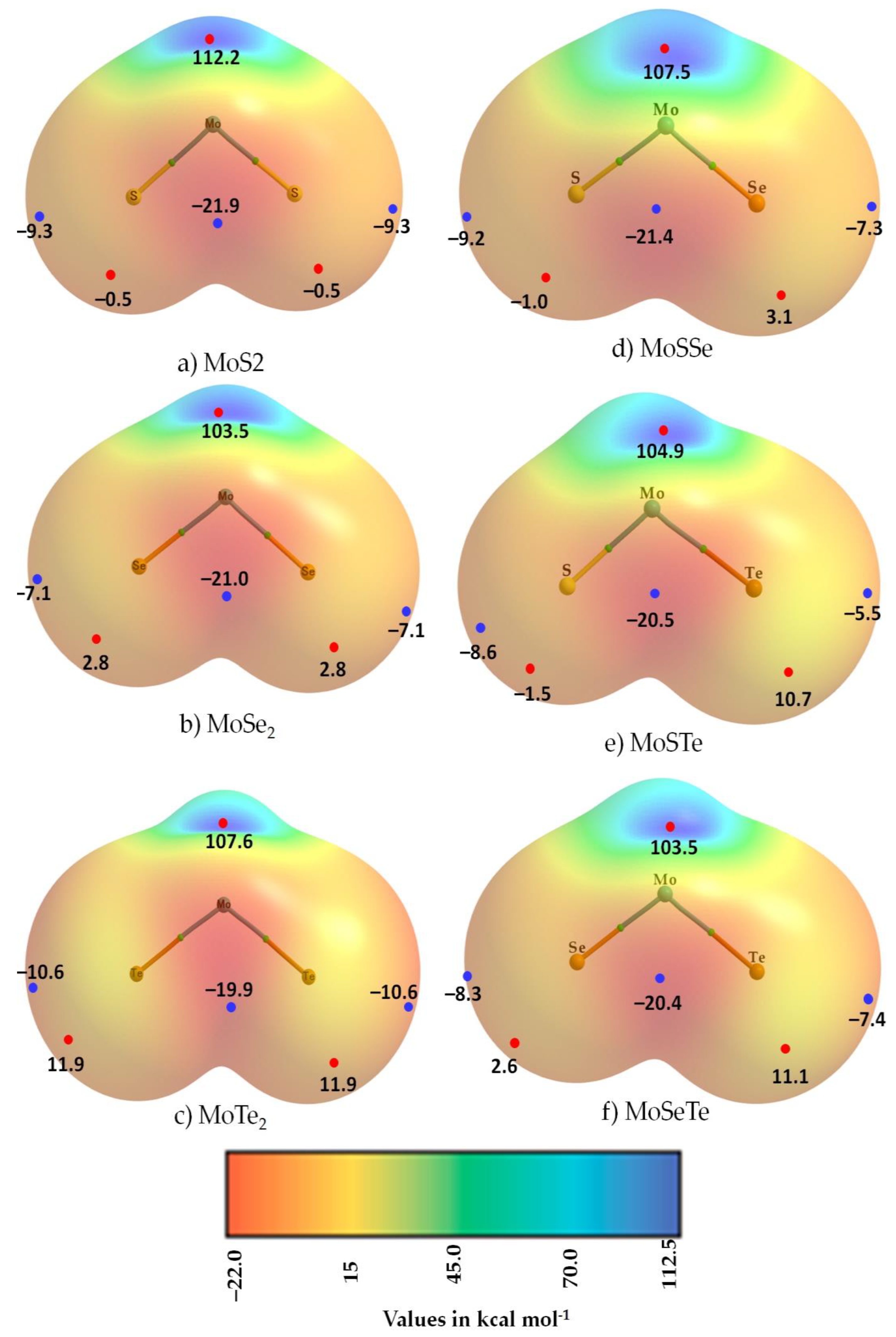
3.2. The Molecular Electrostatic Surface Potential
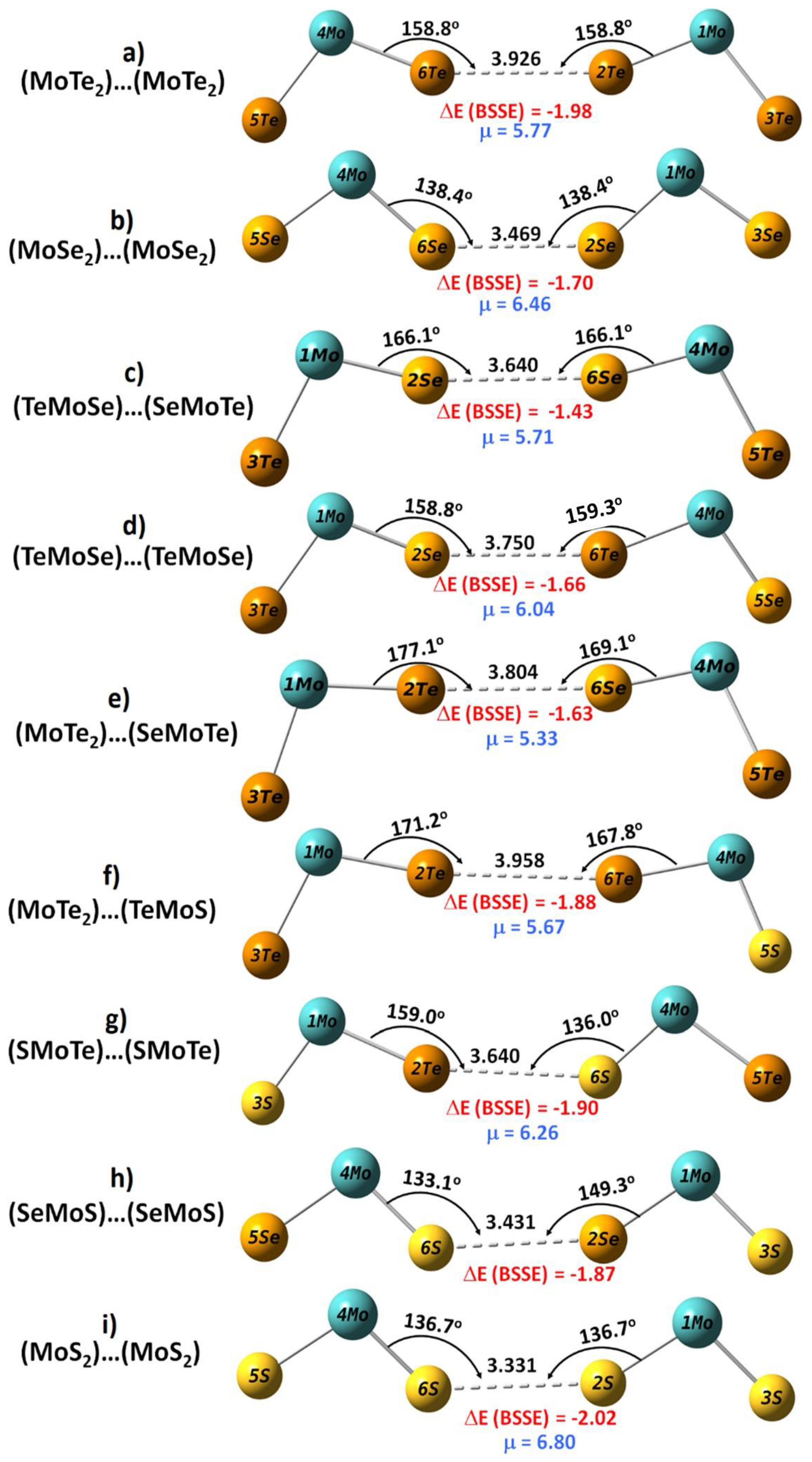
3.3. Intermolecular Geometry of Dimers
3.4. Energy Stability
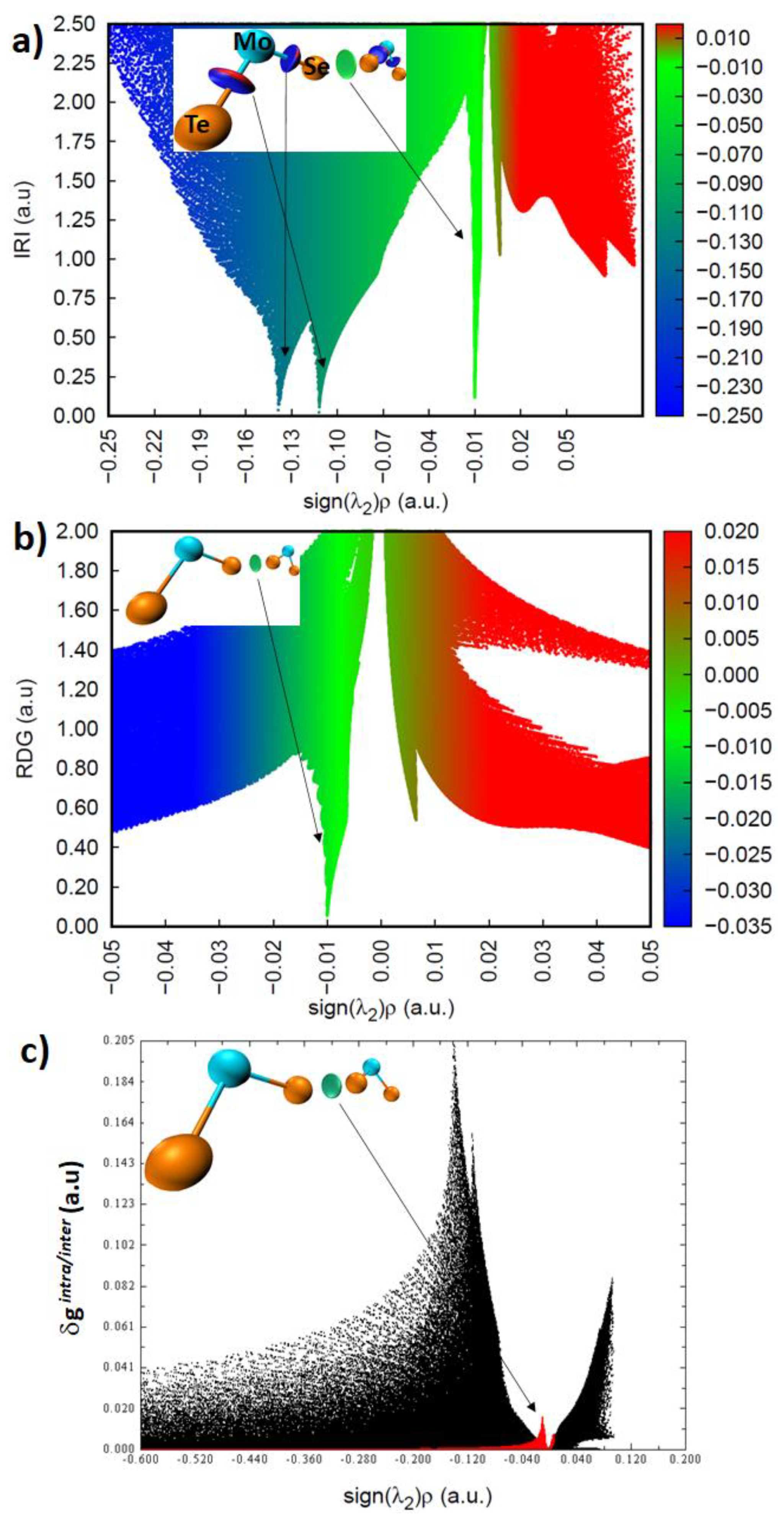
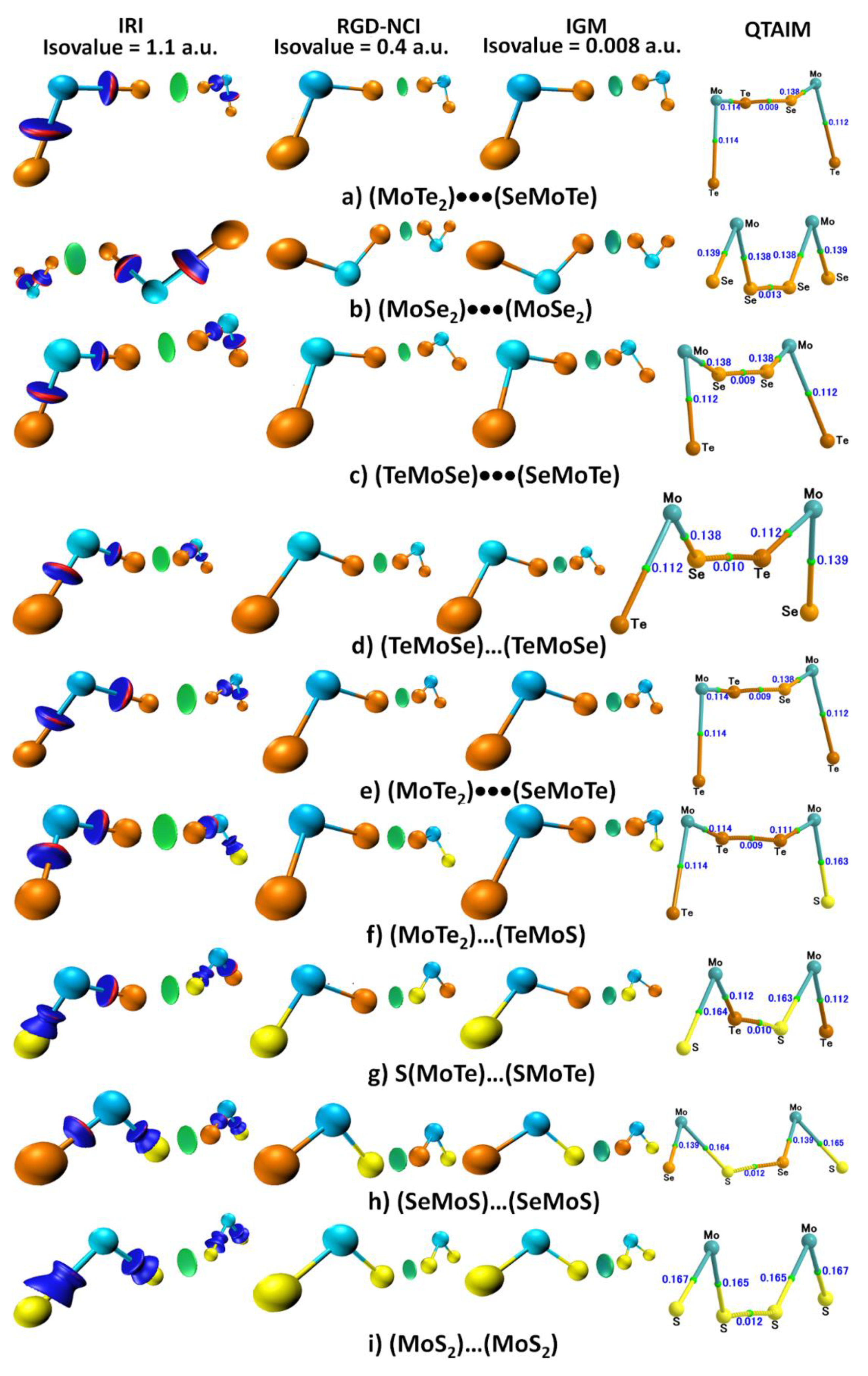
3.5. Isosurface and Bond Path Topological Properties of Charge Density
3.6. Nature of Second-Order Hyper-Conjugative Charge Transfer Delocalization between the Monomers in the (MoCh2)2 and (MoChCh′2)2 (Ch, Ch′ = S, Se and Te) Dimer Geometries
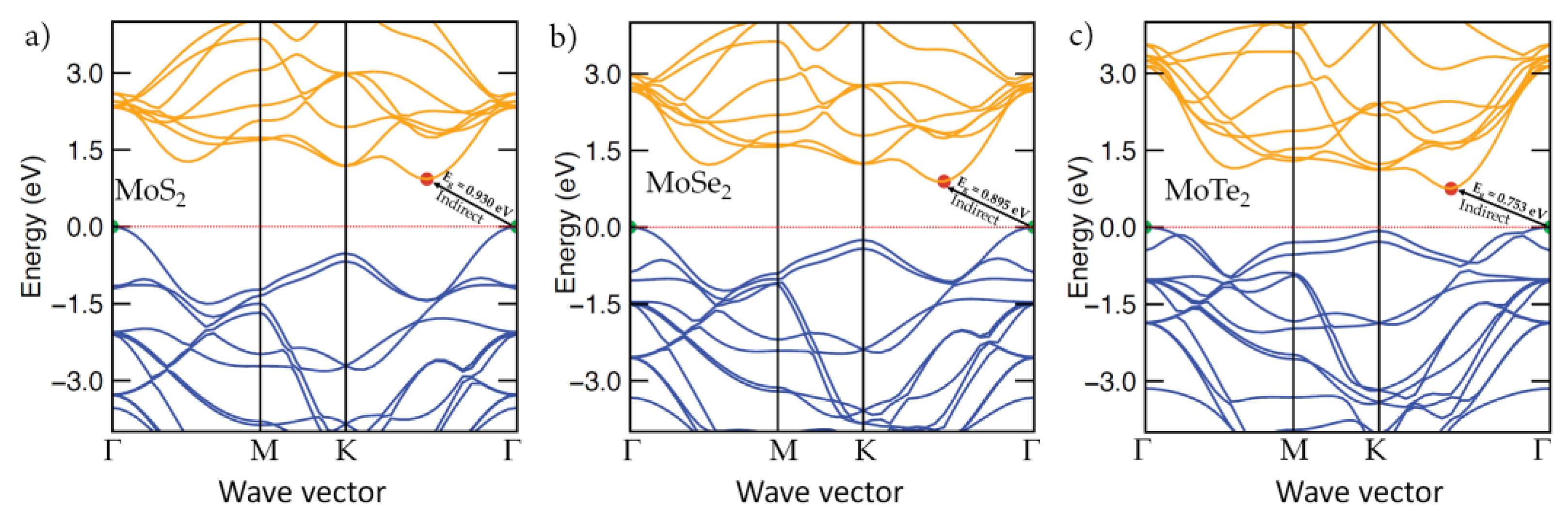
3.7. The Electronic Band Structures of Bulk MoCh2(Ch = S, Se, Te)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choi, W.; Choudhary, N.; Han, G.H.; Park, J.; Akinwande, D.; Lee, Y.H. Recent development of two-dimensional transition metal dichalcogenides and their applications. Mater. Today 2017, 20, 116–130. [Google Scholar] [CrossRef]
- Jana, M.K.; Rao, C.N.R. Two-dimensional inorganic analogues of graphene: Transition metal dichalcogenides. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Colen, J.; Liu, J.; Nguyen, M.C.; Chern, G.-W.; Louca, D. Elastic and electronic tuning of magnetoresistance in MoTe 2. Sci. Adv. 2017, 3, eaao4949. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-R.; Cheng, W.-H.; Richter, M.H.; DuChene, J.S.; Peterson, E.A.; Went, C.M.; Al Balushi, Z.Y.; Jariwala, D.; Neaton, J.B.; Chen, L.-C.; et al. Band Edge Tailoring in Few-Layer Two-Dimensional Molybdenum Sulfide/Selenide Alloys. J. Phys. Chem. C 2020, 124, 22893–22902. [Google Scholar] [CrossRef]
- Sokolikova, M.S.; Sherrell, P.C.; Palczynski, P.; Bemmer, V.L.; Mattevi, C. Direct solution-phase synthesis of 1T’ WSe2 nanosheets. Nat. Commun. 2019, 10, 712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Pan, J.; Fang, Y.; Che, X.; Wang, D.; Bu, K.; Huang, F. Metastable MoS2: Crystal Structure, Electronic Band Structure, Synthetic Approach and Intriguing Physical Properties. Chem. Eur. J. 2018, 24, 15942–15954. [Google Scholar] [CrossRef] [PubMed]
- Duerloo, K.-A.N.; Li, Y.; Reed, E.J. Structural phase transitions in two-dimensional Mo- and W-dichalcogenide monolayers. Nat. Commun. 2014, 5, 4214. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhu, H. Two-dimensional MoS2: Properties, preparation, and applications. J. Materiomics 2015, 1, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Pai, W.W.; Chan, Y.H.; Sun, W.L.; Xu, C.Z.; Lin, D.S.; Chou, M.Y.; Fedorov, A.V.; Chiang, T.C. Large quantum-spin-Hall gap in single-layer 1T′ WSe2. Nat. Commun. 2018, 9, 2003. [Google Scholar] [CrossRef]
- Li, Y.; Duerloo, K.-A.N.; Wauson, K.; Reed, E.J. Structural semiconductor-to-semimetal phase transition in two-dimensional materials induced by electrostatic gating. Nat. Commun. 2016, 7, 10671. [Google Scholar] [CrossRef]
- Qian, Z.; Jiao, L.; Xie, L. Phase Engineering of Two-Dimensional Transition Metal Dichalcogenides. Chin. J. Chem. 2020, 38, 753–760. [Google Scholar] [CrossRef]
- Yu, P.; Lin, J.; Sun, L.; Le, Q.L.; Yu, X.; Gao, G.; Hsu, C.-H.; Wu, D.; Chang, T.-R.; Zeng, Q.; et al. Metal–Semiconductor Phase-Transition in WSe2(1-x)Te2x Monolayer. Adv. Mater. 2017, 29, 1603991. [Google Scholar] [CrossRef]
- Morales-Durán, N.; MacDonald, A.H.; Potasz, P. Metal-insulator transition in transition metal dichalcogenide heterobilayer moiré superlattices. Phys. Rev. B 2021, 103, L241110. [Google Scholar] [CrossRef]
- Shimazu, Y.; Arai, K.; Iwabuchi, T. Metal–insulator transition in a transition metal dichalcogenide: Dependence on metal contacts. J. Phys. Conf. Ser. 2018, 969, 012105. [Google Scholar] [CrossRef]
- Choe, D.-H.; Sung, H.-J.; Chang, K.J. Understanding topological phase transition in monolayer transition metal dichalcogenides. Phys. Rev. B 2016, 93, 125109. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.-R.; Lin, H.; Jeng, H.-T.; Bansil, A. Thickness dependence of spin polarization and electronic structure of ultra-thin films of MoS2 and related transition-metal dichalcogenides. Sci. Rep. 2014, 4, 6270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, H.L.; Hennig, R.G. Theoretical perspective of photocatalytic properties of single-layer SnS2. Phys. Rev. B 2013, 88, 115314. [Google Scholar] [CrossRef]
- Singh, N.; Jabbour, G.; Schwingenschlögl, U. Optical and photocatalytic properties of two-dimensional MoS2. Eur. Phys. J. B 2012, 85, 392. [Google Scholar] [CrossRef]
- Kang, J.; Tongay, S.; Zhou, J.; Li, J.; Wu, J. Band offsets and heterostructures of two-dimensional semiconductors. Appl. Phys. Lett. 2013, 102, 012111. [Google Scholar] [CrossRef] [Green Version]
- Fiori, G.; Bonaccorso, F.; Iannaccone, G.; Palacios, T.; Neumaier, D.; Seabaugh, A.; Banerjee, S.K.; Colombo, L. Electronics based on two-dimensional materials. Nat. Nanotechnol. 2014, 9, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Bhimanapati, G.R.; Lin, Z.; Meunier, V.; Jung, Y.; Cha, J.; Das, S.; Xiao, D.; Son, Y.; Strano, M.S.; Cooper, V.R.; et al. Recent Advances in Two-Dimensional Materials beyond Graphene. ACS Nano 2015, 9, 11509–11539. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Bonaccorso, F.; Fal’ko, V.; Novoselov, K.S.; Roche, S.; Bøggild, P.; Borini, S.; Koppens, F.H.L.; Palermo, V.; Pugno, N.; et al. Science and technology roadmap for graphene, related two-dimensional crystals, and hybrid systems. Nanoscale 2015, 7, 4598–4810. [Google Scholar] [CrossRef] [Green Version]
- Lu, N.; Li, Z.; Yang, J. Electronic Structure Engineering via On-Plane Chemical Functionalization: A Comparison Study on Two-Dimensional Polysilane and Graphane. J. Phys. Chem. C 2009, 113, 16741–16746. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Zhao, Y.; Lu, N.; Kan, E.; Zeng, X.C.; Wu, X.; Yang, J. Tunable Magnetism in a Nonmetal-Substituted ZnO Monolayer: A First-Principles Study. J. Phys Chem. C 2012, 116, 11336–11342. [Google Scholar] [CrossRef]
- Dai, J.; Wu, X.; Yang, J.; Zeng, X.C. Unusual Metallic Microporous Boron Nitride Networks. J. Phys. Chem. Lett. 2013, 4, 3484–3488. [Google Scholar] [CrossRef]
- Mak, K.F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T.F. Atomically Thin MoS2: A New Direct-Gap Semiconductor. Phys. Rev. Lett. 2010, 105, 136805. [Google Scholar] [CrossRef] [Green Version]
- Splendiani, A.; Sun, L.; Zhang, Y.; Li, T.; Kim, J.; Chim, C.-Y.; Galli, G.; Wang, F. Emerging Photoluminescence in Monolayer MoS2. Nano Lett. 2010, 10, 1271–1275. [Google Scholar] [CrossRef]
- Eda, G.; Yamaguchi, H.; Voiry, D.; Fujita, T.; Chen, M.; Chhowalla, M. Photoluminescence from Chemically Exfoliated MoS2. Nano Lett. 2011, 11, 5111–5116. [Google Scholar] [CrossRef]
- Johari, P.; Shenoy, V.B. Tuning the Electronic Properties of Semiconducting Transition Metal Dichalcogenides by Applying Mechanical Strains. ACS Nano 2012, 6, 5449–5456. [Google Scholar] [CrossRef] [PubMed]
- Naik, M.H.; Jain, M. Origin of layer dependence in band structures of two-dimensional materials. Phys. Rev. B 2017, 95, 165125. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Long, M.; Li, X.; Zhang, Q.; Xu, H.; Chan, K.S. Effects of van der Waals interaction and electric field on the electronic structure of bilayer MoS2. J. Phys. Cond. Matt. 2014, 26, 405302. [Google Scholar] [CrossRef] [PubMed]
- Zahid, F.; Liu, L.; Zhu, Y.; Wang, J.; Guo, H. A generic tight-binding model for monolayer, bilayer and bulk MoS2. AIP Adv. 2013, 3, 052111. [Google Scholar] [CrossRef] [Green Version]
- Varsano, D.; Palummo, M.; Molinari, E.; Rontani, M. A monolayer transition-metal dichalcogenide as a topological excitonic insulator. Nat. Nanotechol. 2020, 15, 367–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastos, C.M.O.; Besse, R.; Da Silva, J.L.F.; Sipahi, G.M. Ab initio investigation of structural stability and exfoliation energies in transition metal dichalcogenides based on Ti-, V-, and Mo-group elements. Phys. Rev. Mater. 2019, 3, 044002. [Google Scholar] [CrossRef] [Green Version]
- Kam, K.K.; Parkinson, B.A. Detailed photocurrent spectroscopy of the semiconducting group VIB transition metal dichalcogenides. J. Phys. Chem. 1982, 86, 463–467. [Google Scholar] [CrossRef]
- Hamill, A.; Heischmidt, B.; Sohn, E.; Shaffer, D.; Tsai, K.-T.; Zhang, X.; Xi, X.; Suslov, A.; Berger, H.; Forró, L.; et al. Two-fold symmetric superconductivity in few-layer NbSe2. Nat. Phys. 2021, 17, 949–954. [Google Scholar] [CrossRef]
- Cho, C.-w.; Lyu, J.; Han, T.; Ng, C.Y.; Gao, Y.; Li, G.; Huang, M.; Wang, N.; Schmalian, J.; Lortz, R. Distinct Nodal and Nematic Superconducting Phases in the 2D Ising Superconductor NbSe2. 2020. Available online: https://arxiv.org/abs/2003.12467 (accessed on 6 November 2021).
- Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X.C. Tuning Electronic and Magnetic Properties of Early Transition-Metal Dichalcogenides via Tensile Strain. J. Phys. Chem. C 2014, 118, 7242–7249. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Qian, X.; Li, J. Phase transitions in 2D materials. Nat. Rev. Mater. 2021, 6, 829–846. [Google Scholar] [CrossRef]
- Cheng, P.; Sun, K.; Hu, Y.H. Mechanically-induced reverse phase transformation of MoS2 from stable 2H to metastable 1T and its memristive behavior. RSC Adv. 2016, 6, 65691–65697. [Google Scholar] [CrossRef]
- Xia, J.; Wang, J.; Chao, D.; Chen, Z.; Liu, Z.; Kuo, J.-L.; Yan, J.; Shen, Z.X. Phase evolution of lithium intercalation dynamics in 2H-MoS2. Nanoscale 2017, 9, 7533–7540. [Google Scholar] [CrossRef]
- Sánchez-Montejo, E.; Santana, G.; Domínguez, A.; Huerta, L.; Hamui, L.; López-López, M.; Limborço, H.; Matinaga, F.M.; da Silva, M.I.N.; de Oliveira, A.G.; et al. Phase stability in MoTe2 prepared by low temperature Mo tellurization using close space isothermal Te annealing. Mater. Chem. Phys. 2017, 198, 317–323. [Google Scholar] [CrossRef]
- Kansara, S.; Gupta, S.K.; Sonvane, Y. Effect of strain engineering on 2D dichalcogenides transition metal: A DFT study. Comput. Mater. Sci. 2018, 141, 235–242. [Google Scholar] [CrossRef]
- Roldán, R.; López-Sancho, M.P.; Guinea, F.; Cappelluti, E.; Silva-Guillén, J.A.; Ordejón, P. Momentum dependence of spin–orbit interaction effects in single-layer and multi-layer transition metal dichalcogenides. 2D Mater. 2014, 1, 034003. [Google Scholar] [CrossRef] [Green Version]
- Babar, V.; Vovusha, H.; Schwingenschlögl, U. Density Functional Theory Analysis of Gas Adsorption on Monolayer and Few Layer Transition Metal Dichalcogenides: Implications for Sensing. ACS Appl. Nano Mater. 2019, 2, 6076–6080. [Google Scholar] [CrossRef]
- García, Á.M.; del Corro, E.; Kalbac, M.; Frank, O. Tuning the electronic properties of monolayer and bilayer transition metal dichalcogenide compounds under direct out-of-plane compression. Phys. Chem. Chem. Phys. 2017, 19, 13333–13340. [Google Scholar] [CrossRef] [PubMed]
- Palencia-Ruiz, S.; Uzio, D.; Legens, C.; Laurenti, D.; Afanasiev, P. Stability and catalytic properties of 1T-MoS2 obtained via solvothermal synthesis. Appl. Catal. A Gen. 2021, 626, 118355. [Google Scholar] [CrossRef]
- Schönfeld, B.; Huang, J.J.; Moss, S.C. Anisotropic mean-square displacements (MSD) in single-crystals of 2H- and 3R-MoS2. Acta Crystallogr. Sect. B Struct. Sci. 1983, 39, 404–407. [Google Scholar] [CrossRef]
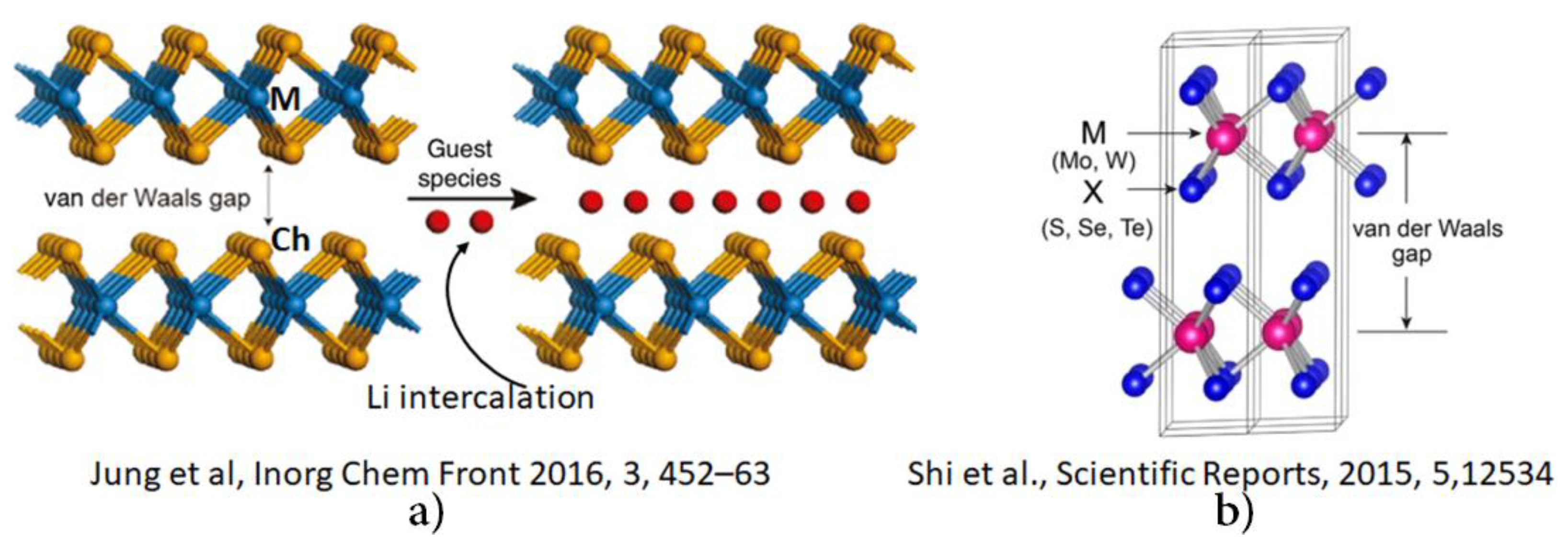
- Jung, Y.; Zhou, Y.; Cha, J.J. Intercalation in two-dimensional transition metal chalcogenides. Inorg. Chem. Front. 2016, 3, 452–463. [Google Scholar] [CrossRef]
- Jing, Y.; Liu, B.; Zhu, X.; Ouyang, F.; Sun, J.; Zhou, Y. Tunable electronic structure of two-dimensional transition metal chalcogenides for optoelectronic applications. Nanophotonics 2020, 9, 1675–1694. [Google Scholar] [CrossRef]
- Zhu, W.; Low, T.; Wang, H.; Ye, P.; Duan, X. Nanoscale electronic devices based on transition metal dichalcogenides. 2D Mater. 2019, 6, 032004. [Google Scholar] [CrossRef]
- Fukuda, M.; Zhang, J.; Lee, Y.-T.; Ozaki, T. A structure map for AB2 type 2D materials using high-throughput DFT calculations. Mater. Adv. 2021, 2, 4392–4413. [Google Scholar] [CrossRef]
- Yang, L.; Cui, X.; Zhang, J.; Wang, K.; Shen, M.; Zeng, S.; Dayeh, S.A.; Feng, L.; Xiang, B. Lattice strain effects on the optical properties of MoS2 nanosheets. Sci. Rep. 2014, 4, 5649. [Google Scholar] [CrossRef] [PubMed]
- Kulichenko, M.; Boldyrev, A.I. σ-Aromaticity in the MoS2 Monolayer. J. Phys. Chem. C 2020, 124, 6267–6273. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, X.; Liu, S.-C.; Li, Z.; Sun, Z.; Hu, C.; Xue, D.-J.; Zhang, G.; Hu, J.-S. Weak Interlayer Interaction in 2D Anisotropic GeSe2. Sci. Adv. 2019, 6, 1801810. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; MacDougall, P.J. The chalcogen bond: Can it be formed by oxygen? Phys. Chem. Chem. Phys. 2019, 21, 19969–19986. [Google Scholar] [CrossRef]
- Varadwaj, P.R. Does Oxygen Feature Chalcogen Bonding? Molecules 2019, 24, 3166. [Google Scholar] [CrossRef] [Green Version]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, R.G.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Decato, D.A.; John, E.A.; Berryman, O.B. Halogen Bonding: An Introduction. In Halogen Bonding in Solution; Huber, S., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA.: Bochum, Germany, 2021. [Google Scholar]
- Wang, W.; Zhu, H.; Feng, L.; Yu, Q.; Hao, J.; Zhu, R.; Wang, Y. Dual Chalcogen–Chalcogen Bonding Catalysis. J. Am. Chem Soc. 2020, 142, 3117–3124. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Halogen Bonding: A Halogen-Centered Noncovalent Interaction Yet to Be Understood. Inorganics 2019, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Bauzá Riera, A.; Quiñonero Santiago, D.; Deyà Serra, P.M.; Frontera Beccaria, A. Halogen bonding versus chalcogen and pnicogen bonding: A combined Cambridge structural database and theoretical study. CrystEngComm 2013, 15, 3137–3144. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metrangolo, P.; Resnati, G. Type II halogen···halogen contacts are halogen bonds. IUCrJ 2014, 1, 5–7. [Google Scholar] [CrossRef]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. A direct MP2 gradient method. Chem. Phys. Lett. 1990, 166, 275–280. [Google Scholar] [CrossRef]
- Bader, R.F. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Lu, T.; Chen, Q. Interaction Region Indicator: A Simple Real Space Function Clearly Revealing Both Chemical Bonds and Weak Interactions. Chem. Methods 2021, 1, 231–239. [Google Scholar] [CrossRef]
- Lefebvre, C.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Piquemal, J.-P.; Hénon, E. The Independent Gradient Model: A New Approach for Probing Strong and Weak Interactions in Molecules from Wave Function Calculations. ChemPhysChem 2018, 19, 724–735. [Google Scholar] [CrossRef]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Hénon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Weinhold, F.; Landis, C.R. Discovering Chemistry with Natural Bond Orbitals; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Dixit, A.; Claudot, J.; Lebègue, S.; Rocca, D. Communication: A novel implementation to compute MP2 correlation energies without basis set superposition errors and complete basis set extrapolation. J. Chem. Phys. 2017, 146, 211102. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, B.P.; Altarawy, D.; Didier, B.T.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Rev. C.01; Gaussian, Inc.: Wallinford, CT, USA, 2016. [Google Scholar]
- Noro, T.; Sekiya, M.; Koga, T. Segmented contracted basis sets for atoms H through Xe: Sapporo-(DK)-nZP sets (n = D., T., Q). Theor. Chem. Acc. 2012, 131, 1124. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Solimannejad, M. Revealing substitution effects on the strength and nature of halogen-hydride interactions: A theoretical study. J. Mol. Model. 2013, 19, 3767–3777. [Google Scholar] [CrossRef]
- Han, N.; Zeng, Y.; Li, X.; Zheng, S.; Meng, L. Enhancing Effects of Electron-Withdrawing Groups and Metallic Ions on Halogen Bonding in the YC6F4X···C2H8N2 (X = Cl, Br, I.; Y = F, CN, NO2, LiNC+, NaNC+) Complex. J. Phys. Chem. A 2013, 117, 12959–12968. [Google Scholar] [CrossRef]
- Zhang, L.; Li, D. Theoretical studies on how to tune the π-hole pnicogen bonds by substitution and cooperative effects. Int. J. Quant. Chem. 2021, 121, e26531. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. Can Counter-Intuitive Halogen Bonding Be Coulombic? ChemPhysChem 2021, 22, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, P.R.; Varadwaj, A.; Jin, B.Y. Significant evidence of C···O and C···C long-range contacts in several heterodimeric complexes of CO with CH3-X, should one refer to them as carbon and dicarbon bonds! Phys. Chem. Chem. Phys. 2014, 16, 17238–17252. [Google Scholar] [CrossRef]
- Glendening, E.E.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO (Natural Bond Orbital); Gaussian Inc.: Pittsburg, PA, USA, 2004. [Google Scholar]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comp. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Contreras-García, J.; Yang, W.; Johnson, E.R. Analysis of Hydrogen-Bond Interaction Potentials from the Electron Density: Integration of Noncovalent Interaction Regions. J. Phys. Chem. A 2011, 115, 12983–12990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otero-de-la-Roza, A.; Johnson, E.R.; Contreras-García, J. Revealing non-covalent interactions in solids: NCI plots revisited. Phys. Chem. Chem. Phys. 2012, 14, 12165–12172. [Google Scholar] [CrossRef] [PubMed]
- Boto, R.A.; Contreras-García, J.; Tierny, J.; Piquemal, J.-P. Interpretation of the reduced density gradient. Mol. Phys. 2016, 114, 1406–1414. [Google Scholar] [CrossRef] [Green Version]
- Lane, J.R.; Contreras-García, J.; Piquemal, J.-P.; Miller, B.J.; Kjaergaard, H.G. Are Bond Critical Points Really Critical for Hydrogen Bonding? J. Chem. Theory Comput. 2013, 9, 3263–3266. [Google Scholar] [CrossRef] [PubMed]
- Keith, T.A.; Gristmill Software, T.K.; Overland Park, K.S. AIMAll (Version 19.10.12); Overland Park, KS, USA. Available online: http://aim.tkgristmill.com (accessed on 6 November 2021).
- Lu, T.; Chen, F. A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD - Visual Molecular Dynamics. J. Molec. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView, 5.0.9; Semichem, Inc.: Shawnee Mission, KS, USA, 2007. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Sun, J.; Remsing, R.C.; Zhang, Y.; Sun, Z.; Ruzsinszky, A.; Peng, H.; Yang, Z.; Paul, A.; Waghmare, U.; Wu, X.; et al. Accurate first-principles structures and energies of diversely bonded systems from an efficient density functional. Nat. Chem. 2016, 8, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Yang, Z.-H.; Perdew, J.P.; Sun, J. Versatile van der Waals Density Functional Based on a Meta-Generalized Gradient Approximation. Phys. Rev. X 2016, 6, 041005. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kerker, G.P. Efficient iteration scheme for self-consistent pseudopotential calculations. Phys. Rev. B 1981, 23, 3082–3084. [Google Scholar] [CrossRef]
- Lee, C.; Hong, J.; Lee, W.R.; Kim, D.Y.; Shim, J.H. Density functional theory investigation of the electronic structure and thermoelectric properties of layered MoS2, MoSe2 and their mixed-layer compound. J. Solid State Chem. 2014, 211, 113–119. [Google Scholar] [CrossRef]
- Evans, B.L.; Hazelwood, R.A. Optical and structural properties of MoSe2. Phys. Status Solidi (A) 1971, 4, 181–192. [Google Scholar] [CrossRef]
- Knop, O.; MacDonald, R.D. Chalkogenides of the Transition elements: III. Molybdenum Ditelluride. Can. J. Chem. 1961, 39, 897–904. [Google Scholar] [CrossRef] [Green Version]
- Franconetti, A.; Quiñonero, D.; Frontera, A.; Resnati, G. Unexpected chalcogen bonds in tetravalent sulfur compounds. Phys. Chem. Chem. Phys. 2019, 21, 11313–11319. [Google Scholar] [CrossRef] [PubMed]
- Zierkiewicz, W.; Michalczyk, M.; Wysokiński, R.; Scheiner, S. On the ability of pnicogen atoms to engage in both σ and π-hole complexes. Heterodimers of ZF2C6H5 (Z = P, As, Sb, Bi) and NH3. J. Mol. Model. 2019, 25, 152. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; An, X.L.; Li, Q.Z. Se···N Chalcogen Bond and Se···X Halogen Bond Involving F2C=Se: Influence of Hybridization, Substitution, and Cooperativity. J. Phys Chem. A 2015, 119, 3518–3527. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadantsev, E.S.; Hawrylak, P. Electronic structure of a single MoS2 monolayer. Solid State Commun. 2012, 152, 909–913. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Jin, B.-Y. Halogen bonding interaction of chloromethane withseveral nitrogen donating molecules: Addressing thenature of the chlorine surface σ-hole. Phys. Chem. Chem. Phys. 2014, 16, 19573–19589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Does Chlorine in CH3Cl Behave as a Genuine Halogen Bond Donor? Crystals 2020, 10, 146. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The s-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Hobza, P. Investigations into the Nature of Halogen Bonding Including Symmetry Adapted Perturbation Theory Analyses. J. Chem. Theory Comput. 2008, 4, 232–242. [Google Scholar] [CrossRef]
- Varadwaj, A.; Marques, H.M.; Varadwaj, P.R. Is the Fluorine in Molecules Dispersive? Is Molecular Electrostatic Potential a Valid Property to Explore Fluorine-Centered Non-Covalent Interactions? Molecules 2019, 24, 379. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. Can Combined Electrostatic and Polarization Effects Alone Explain the F···F Negative-Negative Bonding in Simple Fluoro-Substituted Benzene Derivatives? A First-Principles Perspective. Computation 2018, 6, 51. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, A.; Marques, H.M.; Varadwaj, P.R. Nature of halogen-centered intermolecular interactions in crystal growth and design: Fluorine-centered interactions in dimers in crystalline hexafluoropropylene as a prototype. J. Comp. Chem. 2019, 40, 1836–1860. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Yamashita, K. Do surfaces of positive electrostatic potential on different halogen derivatives in molecules attract? like attracting like! J. Comput. Chem. 2018, 39, 343–350. [Google Scholar] [CrossRef]
- Yang, X.; Banerjee, A.; Ahuja, R. Structural Insight of the Frailty of 2D Janus NbSeTe as an Active Photocatalyst. ChemCatChem 2020, 12, 6013–6023. [Google Scholar] [CrossRef]
- Bera, A.; Singh, A.; Sorb, Y.A.; Jenjeti, R.N.; Muthu, D.V.S.; Sampath, S.; Narayana, C.; Waghmare, U.V.; Sood, A.K. Chemical ordering and pressure-induced isostructural and electronic transitions in MoSSe crystal. Phys. Rev. B 2020, 102, 014103. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. An improved and broadly accurate local approximation to the exchange–correlation density functional: The MN12-L functional for electronic structure calculations in chemistry and physics. Phys. Chem. Chem. Phys. 2012, 14, 13171–13174. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.S.; He, X.; Li, S.L.; Truhlar, D.G. MN15: A Kohn–Sham global-hybrid exchange–correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016, 7, 5032–5051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Yu, H.S.; He, X.; Truhlar, D.G. MN15-L: A New Local Exchange-Correlation Functional for Kohn–Sham Density Functional Theory with Broad Accuracy for Atoms, Molecules, and Solids. J. Chem. Theory Comp. 2016, 12, 1280–1293. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goerigk, L.; Grimme, S. Efficient and Accurate Double-Hybrid-Meta-GGA Density Functionals—Evaluation with the Extended GMTKN30 Database for General Main Group Thermochemistry, Kinetics, and Noncovalent Interactions. J. Chem. Theory Comp. 2011, 7, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Uchimaru, T. Accuracy of intermolecular interaction energies, particularly those of hetero-atom containing molecules obtained by DFT calculations with Grimme’s D2, D3 and D3BJ dispersion corrections. Phys. Chem. Chem. Phys. 2020, 22, 22508–22519. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. How Accurate Are the Minnesota Density Functionals for Noncovalent Interactions, Isomerization Energies, Thermochemistry, and Barrier Heights Involving Molecules Composed of Main-Group Elements? J. Chem. Theory Comp. 2016, 12, 4303–4325. [Google Scholar] [CrossRef] [Green Version]
- Goerigk, L.; Hansen, A.; Bauer, C.; Ehrlich, S.; Najibi, A.; Grimme, S. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. [Google Scholar] [CrossRef] [Green Version]
- Mehta, N.; Fellowes, T.; White, J.M.; Goerigk, L. CHAL336 Benchmark Set: How Well Do Quantum-Chemical Methods Describe Chalcogen-Bonding Interactions? J. Chem. Theory Comp. 2021, 17, 2783–2806. [Google Scholar] [CrossRef]
- Venkataramanan, N.S. Electronic structure, stability, and cooperativity of chalcogen bonding in sulfur dioxide and hydrated sulfur dioxide clusters: A DFT study and wave functional analysis. Struct. Chem. 2021, 1–15. [Google Scholar] [CrossRef]
- Jeffrey, G.A.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer: Berlin/Heidelberg, Germany, 1991. [Google Scholar]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond. In Structural Chemistry and Biology; Oxford University Press: Oxford, UK, 2001; Volume 19. [Google Scholar]
- Varadwaj, A.; Varadwaj, P.R.; Yamashita, K. Hybrid organic-inorganic CH3NH3PbI3perovskite building blocks: Revealing ultra-strong hydrogen bonding and mulliken inner complexes and their implications in materials design. J. Comput. Chem. 2017, 38, 2802–2818. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Very strong chalcogen bonding: Is oxygen in molecules capable of forming it? A First-Principles Perspective. Authorea 2020. [Google Scholar] [CrossRef]
- Crespo-Otero, R.; Montero, L.A.; Stohrer, W.-D.; de la Vega, J.M.G. Basis set superposition error in MP2 and density-functional theory: A case of methane-nitric oxide association. J. Chem. Phys. 2005, 123, 134107. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Kristensen, K.; Høyvik, I.-M.; Jansik, B.; Jørgensen, P.; Kjærgaard, T.; Reine, S.; Jakowski, J. MP2 energy and density for large molecular systems with internal error control using the Divide-Expand-Consolidate scheme. Phys. Chem. Chem. Phys. 2012, 14, 15706–15714. [Google Scholar] [CrossRef]
- De Azevedo Santos, L.; van der Lubbe, S.C.C.; Hamlin, T.A.; Ramalho, T.C.; Matthias Bickelhaupt, F. A Quantitative Molecular Orbital Perspective of the Chalcogen Bond. ChemistryOpen 2021, 10, 391–401. [Google Scholar] [CrossRef]
- Narth, C.; Maroun, Z.; Boto, R.A.; Chaudret, R.; Bonnet, M.-L.; Piquemal, J.-P.; Contreras-García, J. A Complete NCI Perspective: From New Bonds to Reactivity. In Applications of Topological Methods in Molecular Chemistry; Esmail, A., Remi, C., Christine, L., Bernard, S., Eds.; Springer: Cham, Switzerland, 2016; Volume 22, pp. 491–527. [Google Scholar]
- Xiong, F.; Wang, H.; Liu, X.; Sun, J.; Brongersma, M.; Pop, E.; Cui, Y. Li Intercalation in MoS2: In Situ Observation of Its Dynamics and Tuning Optical and Electrical Properties. Nano Lett. 2015, 15, 6777–6784. [Google Scholar] [CrossRef]
- Zhang, Q.; Mei, L.; Cao, X.; Tang, Y.; Zeng, Z. Intercalation and exfoliation chemistries of transition metal dichalcogenides. J. Mat. Chem. A 2020, 8, 15417–15444. [Google Scholar] [CrossRef]
- Zheng, J.; Zhang, H.; Dong, S.; Liu, Y.; Tai Nai, C.; Suk Shin, H.; Young Jeong, H.; Liu, B.; Ping Loh, K. High yield exfoliation of two-dimensional chalcogenides using sodium naphthalenide. Nat. Commun. 2014, 5, 2995. [Google Scholar] [CrossRef]
- Eremeev, S.V.; Vergniory, M.G.; Menshchikova, T.V.; Shaposhnikov, A.A.; Chulkov, E.V. The effect of van der Waal’s gap expansions on the surface electronic structure of layered topological insulators. New J. Phys. 2012, 14, 113030. [Google Scholar] [CrossRef]
- Ma, Y.; Zhao, X.; Wang, T.; Li, W.; Wang, X.; Chang, S.; Li, Y.; Zhao, M.; Dai, X. Band structure engineering in a MoS2/PbI2 van der Waals heterostructure via an external electric field. Phys. Chem. Chem. Phys. 2016, 18, 28466–28473. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W.; Carroll, M.T.; Cheeseman, J.R.; Chang, C. Properties of atoms in molecules: Atomic volumes. J. Am. Chem. Soc. 1987, 109, 7968–7979. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Resonance Delocalization Corrections. In Discovering Chemistry with Natural Bond Orbitals; Weinhold, F., Landis, C.R., Eds.; Wiley: Hoboken, NJ, USA, 2012; pp. 92–134. [Google Scholar] [CrossRef]
- Weinhold, F. Natural bond orbital analysis: A critical overview of relationships to alternative bonding perspectives. J. Comp. Chem. 2012, 33, 2363–2379. [Google Scholar] [CrossRef]
- Weinhold, F.; Glendening, E.D. NBO 6.0: Natural Bond Orbital Analysis Programs. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar]
- Banerjee, S.; Park, J.; Hwang, C.S.; Choi, J.-H.; Lee, S.-C.; Pati, S.K. Regulation of transport properties by polytypism: A computational study on bilayer MoS2. Phys. Chem. Chem. Phys. 2017, 19, 21282–21286. [Google Scholar] [CrossRef]
- Pike, N.A.; Van Troeye, B.; Dewandre, A.; Petretto, G.; Gonze, X.; Rignanese, G.-M.; Verstraete, M.J. Origin of the counterintuitive dynamic charge in the transition metal dichalcogenides. Phys. Rev. B 2017, 95, 201106. [Google Scholar] [CrossRef] [Green Version]
- Wickramaratne, D.; Zahid, F.; Lake, R.K. Electronic and thermoelectric properties of few-layer transition metal dichalcogenides. J. Chem. Phys. 2014, 140, 124710. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Wang, Z.; Li, Z.; Fu, Y.Q. Origin of Structural Transformation in Mono- and Bi-Layered Molybdenum Disulfide. Sci. Rep. 2016, 6, 26666. [Google Scholar] [CrossRef] [PubMed]
- Todorova, T.; Alexiev, V.; Prins, R.; Weber, T. Ab initio study of 2H-MoS2 using Hay and Wadt effective core pseudo-potentials for modelling the (100) surface structure. Phys. Chem. Chem. Phys. 2004, 6, 3023–3030. [Google Scholar] [CrossRef]
- Naumis, G.G. Electronic Properties of Two-Dimensional Materials. In Synthesis, Modeling, and Characterization of 2D Materials, and Their Heterostructures; Yang, E.-H., Datta, D., Ding, J., Hader, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 77–109. [Google Scholar]
- Ravichandran, L.; Banik, S. Investigation of the Failure of the MP2 Method to Describe the Out-of-Plane Bending Motions of Carbon–Carbon Double-Bonded Molecules: The Role of Atomic Orbitals. J. Phys. Chem. A 2021, 125, 9298–9317. [Google Scholar] [CrossRef] [PubMed]
- Ahlber, P. Structure and Dynamics of C9H9+ Ions: An Experimental and Theoretical Comparison. In Stable Carbocation Chemistry; Surya Prakash, G.K., Schleyer, P.V.R., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 1996. [Google Scholar]
- Smith, D.G.A.; Burns, L.A.; Patkowski, K.; Sherrill, C.D. Revised Damping Parameters for the D3 Dispersion Correction to Density Functional Theory. J. Phys. Chem. Lett. 2016, 7, 2197–2203. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K. Revealing Factors Influencing the Fluorine-Centered Non-Covalent Interactions in Some Fluorine-substituted Molecular Complexes: Insights from First-Principles Studies. ChemPhysChem 2018, 19, 1486–1499. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Jin, B.-Y. Can an entirely negative fluorine in a molecule, viz. perfluorobenzene, interact attractively with the entirely negative site (s) on another molecule (s)? Like liking like! RSC Adv. 2016, 6, 19098–19110. [Google Scholar] [CrossRef]








| Method | r(Ch···Ch) | ∠Mo1-Ch2···Ch6 | ∠Mo4-Ch6···Ch2 | μ | ΔE | ΔE(BSSE) |
|---|---|---|---|---|---|---|
| (MoTe2)···(MoTe2) | ||||||
| MP2(Full) | 3.926 | 158.8 | 158.8 | 5.8 | −3.79 | −1.98 |
| B2PLYPD3 | 3.732 | 128.7 | 128.7 | 6.1 | −2.91 | −2.44 |
| MN12-L | 3.945 | 134.3 | 134.3 | 6.7 | −1.25 | −1.05 |
| MN15 | 3.893 | 136.9 | 136.9 | 5.9 | −2.06 | −1.86 |
| M06-2X | 3.972 | 139.3 | 139.3 | 6.8 | −0.89 | −0.73 |
| MN15-L | 4.112 | 136.2 | 136.2 | 5.5 | −1.93 | −1.74 |
| ωB97XD | 4.225 | 141.6 | 141.6 | 6.5 | −1.16 | −1.05 |
| PW6B95 | 4.596 | 153.0 | 153 | 6.1 | −0.41 | −0.27 |
| (MoSe2)···(MoSe2) | ||||||
| MP2(Full) | 3.469 | 138.4 | 138.4 | 6.5 | −4.10 | −1.70 |
| B2PLYPD3 | 3.550 | 138.2 | 138.2 | 7.0 | −2.07 | −1.53 |
| MN12-L | 3.811 | 147.9 | 147.9 | 8.4 | −0.29 | 0.23 |
| MN15 | 3.825 | 147.2 | 147.2 | 7.5 | −1.05 | −0.73 |
| M06-2X | 3.852 | 147.8 | 147.8 | 8.3 | −0.25 | −0.03 |
| MN15-L | 3.982 | 149.2 | 149.2 | 7.1 | −1.02 | −0.71 |
| ωB97XD | 4.161 | 159.5 | 159.5 | 8.0 | −0.48 | −0.23 |
| PW6B95 | 4.225 | 163.3 | 163.3 | 7.3 | −0.24 | 0.02 |
| (MoS2)···(MoS2) | ||||||
| MP2(Full) | 3.331 | 136.7 | 136.7 | 6.8 | −3.09 | −2.02 |
| B2PLYPD3 | 3.487 | 143.1 | 143.1 | 7.3 | −1.42 | −1.08 |
| MN12-L | 3.850 | 158.5 | 158.5 | 9.1 | 0.56 | 0.83 |
| MN15 | 3.775 | 151.0 | 151.0 | 7.9 | −0.49 | −0.25 |
| M06-2X | 3.867 | 157.2 | 157.2 | 8.7 | 0.16 | 0.28 |
| MN15-L | 3.854 | 150.9 | 150.9 | 7.8 | −0.49 | −0.28 |
| ωB97XD | 4.196 | 161.8 | 161.8 | 8.6 | 0.09 | 0.19 |
| PW6B95 | 3.915 | 158.6 | 158.6 | 7.9 | 0.14 | 0.27 |
| Figure 3 | Dimer Type | Donor NBO(i) | Acceptor NBO(j) | E(2)/Kcal Mol−1 |
|---|---|---|---|---|
| a | (MoTe2)···(MoTe2) | BD (2)Mo4-Te6 | RY*(3)Te2 | 0.25 |
| LP (1)Te2 | BD*(3)Mo4-Te6 | 1.17 | ||
| BD (3)Mo1-Te2 | BD*(2)Mo4-Te6 | 0.32 | ||
| LP (1)Te6 | BD*(3)Mo1-Te2 | 1.17 | ||
| BD (3)Mo4-Te6 | BD*(2)Mo1-Te2 | 0.32 | ||
| BD (3)Mo4 -Te6 | RY*(3)Te2 | 0.43 | ||
| b | (MoSe2)···(MoSe2) | BD (3)Mo1 -Se2 | RY*(3)Se6 | 0.72 |
| BD (3)Mo1 -Se2 | BD*(3)Mo4-Se6 | 1.06 | ||
| LP (1)Se2 | BD*(3)Mo4-Se6 | 1.09 | ||
| BD (3)Mo4 -Se6 | RY*(3)Se2 | 0.72 | ||
| BD (3)Mo4 -Se6 | BD*(3)Mo1-Se2 | 1.06 | ||
| c | (TeMoSe)···(SeMoTe) | LP (1)Se2 | BD*(3)Mo4-Se6 | 1.15 |
| BD (3)Mo1-Se2 | RY*(3)Se6 | 0.31 | ||
| BD (3)Mo1-Se2 | BD*(3)Mo4-Se6 | 0.74 | ||
| BD (3)Mo4-Se6 | RY*(3)Se2 | 0.82 | ||
| LP (1)Se 6 | BD*(3)Mo1-Te2 | 1.03 | ||
| d | (TeMoSe)···(TeMoSe) | LP (1)Se6 | BD*(3)Mo1-Se2 | 1.09 |
| BD (3)Mo1-Se2 | RY*(3)Te6 | 0.32 | ||
| LP (1)Se2 | BD*(3)Mo4-Te6 | 1.08 | ||
| BD (3)Mo4 -Te6 | BD*(3)Mo1-Se2 | 0.38 | ||
| BD (3)Mo4 -Te6 | RY*(3)Se2 | 1.04 | ||
| e | (MoTe2) ··· (SeMoTe) | BD (2)Mo1-Te2 | RY*(3)Se6 | 0.50 |
| BD (3)Mo1-Te2 | RY*(3)Se6 | 0.76 | ||
| LP (1)Se 6 | BD*(3)Mo1-Te2 | 0.82 | ||
| BD (2)Mo4 -Se6 | RY*(3)Te2 | 0.45 | ||
| f | (MoTe2) ··· (TeMoS) | LP (1)Te2 | BD*(3)Mo4-Te6 | 0.94 |
| LP (1)Te2 | BD*(2)Mo4-Te6 | 0.63 | ||
| LP (1)Te6 | BD*(3)Mo1-Te2 | 0.73 | ||
| LP (1)Te6 | BD*(2)Mo1-Te2 | 0.56 | ||
| g | (SMoTe) ··· (SMoTe) | LP (1)Te2 | BD*(3)Mo4-S6 | 0.94 |
| LP (1)Te6 | BD*(2)Mo1-Te2 | 0.38 | ||
| LP (1) S6 | BD*(3)Mo1-Te2 | 0.89 | ||
| BD (3)Mo4 -Te6 | RY*(3)Te2 | 0.68 | ||
| h | (SeMoS) ··· (SeMoS) | BD (3)Mo1-Se2 | RY*(3)S6 | 1.11 |
| LP (1)Se2 | BD*(3)Mo4-S6 | 0.67 | ||
| LP (1) S6 | BD*(3)Mo1-Se2 | 0.92 | ||
| BD*(3)Mo4-S6 | BD*(3)Mo1 -Se2 | 1.80 | ||
| i | (MoS2) ··· (MoS2) | BD (3)Mo4-S6 | RY*(3)S2 | 0.66 |
| BD (3)Mo4-S6 | BD*(3)Mo1-S2 | 0.75 | ||
| LP (1)S6 | BD*(3)Mo1-S2 | 0.71 | ||
| BD (3)Mo1-S2 | RY*(3)S6 | 0.66 | ||
| BD (3)Mo1-S2 | BD*(3)Mo4-S6 | 0.75 | ||
| LP (1)S2 | BD*(3)Mo4-S6 | 0.71 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varadwaj, P.R.; Marques, H.M.; Varadwaj, A.; Yamashita, K. Chalcogen···Chalcogen Bonding in Molybdenum Disulfide, Molybdenum Diselenide and Molybdenum Ditelluride Dimers as Prototypes for a Basic Understanding of the Local Interfacial Chemical Bonding Environment in 2D Layered Transition Metal Dichalcogenides. Inorganics 2022, 10, 11. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics10010011
Varadwaj PR, Marques HM, Varadwaj A, Yamashita K. Chalcogen···Chalcogen Bonding in Molybdenum Disulfide, Molybdenum Diselenide and Molybdenum Ditelluride Dimers as Prototypes for a Basic Understanding of the Local Interfacial Chemical Bonding Environment in 2D Layered Transition Metal Dichalcogenides. Inorganics. 2022; 10(1):11. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics10010011
Chicago/Turabian StyleVaradwaj, Pradeep R., Helder M. Marques, Arpita Varadwaj, and Koichi Yamashita. 2022. "Chalcogen···Chalcogen Bonding in Molybdenum Disulfide, Molybdenum Diselenide and Molybdenum Ditelluride Dimers as Prototypes for a Basic Understanding of the Local Interfacial Chemical Bonding Environment in 2D Layered Transition Metal Dichalcogenides" Inorganics 10, no. 1: 11. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics10010011