
Binuclear Heteroleptic Ru(III) Dithiocarbamate Complexes: A Step towards Tunable Antiproliferative Agents


Abstract
:1. Introduction
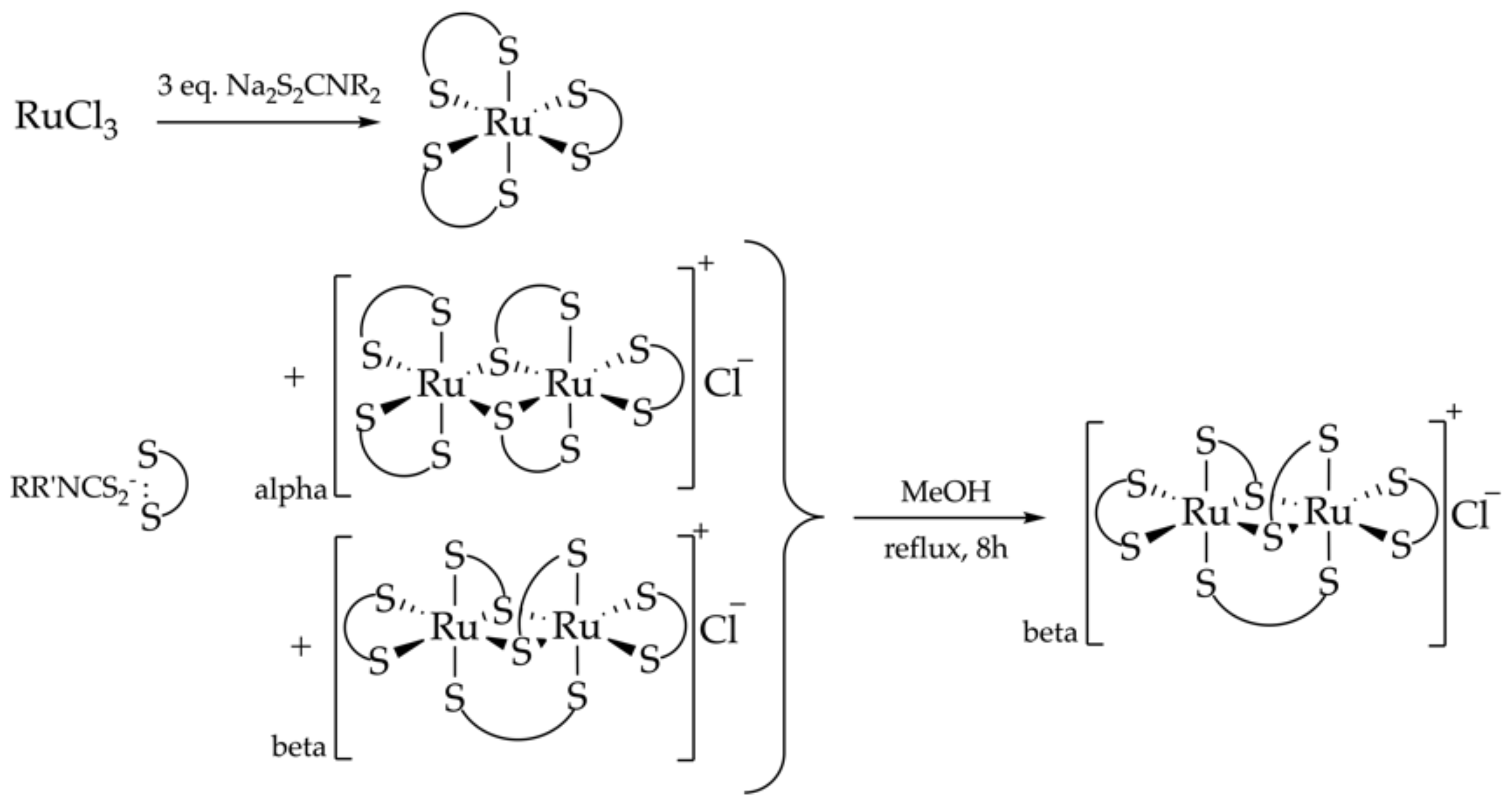
Synthesis of Binuclear Homoleptic Complexes of Ru(III)
2. Results
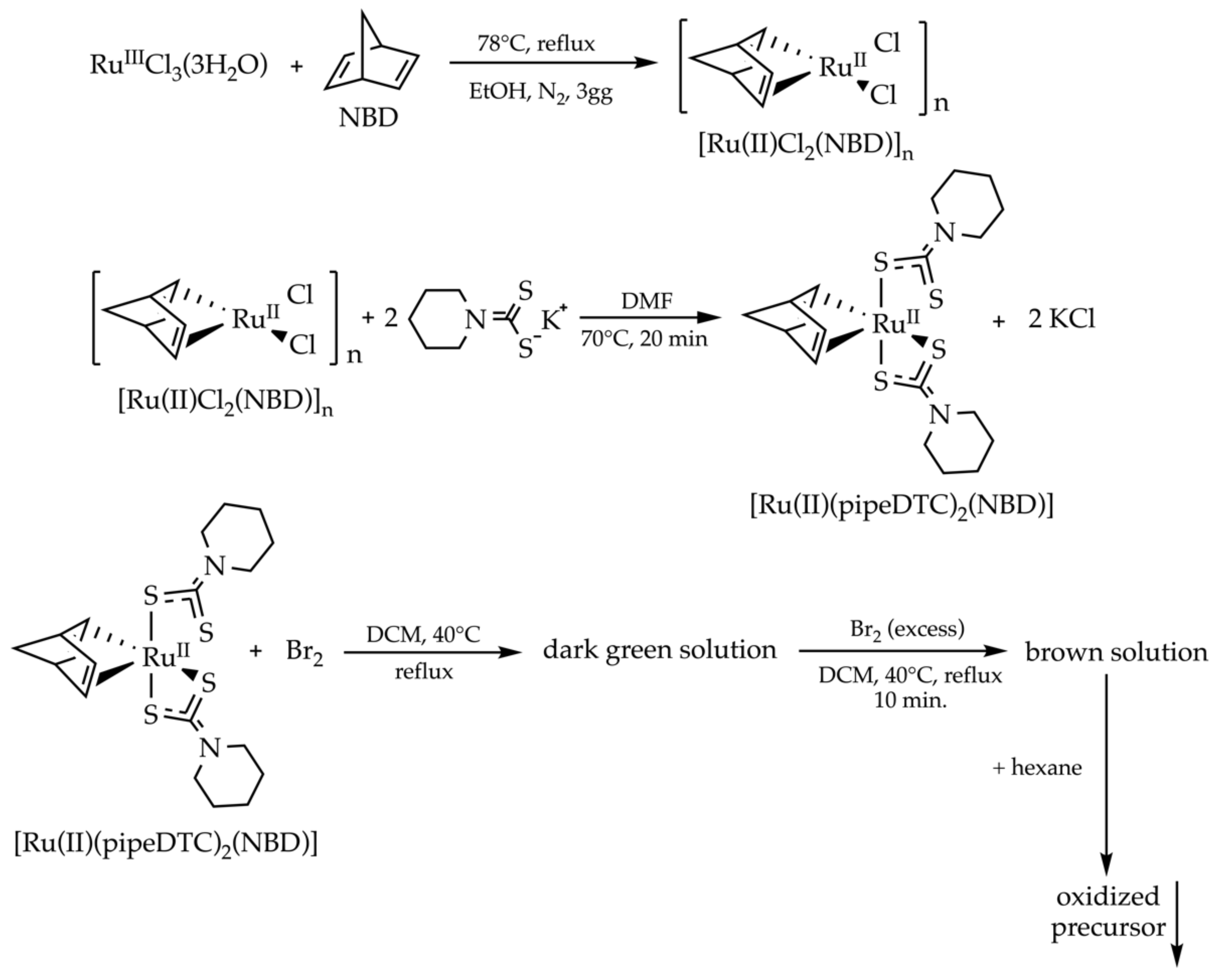
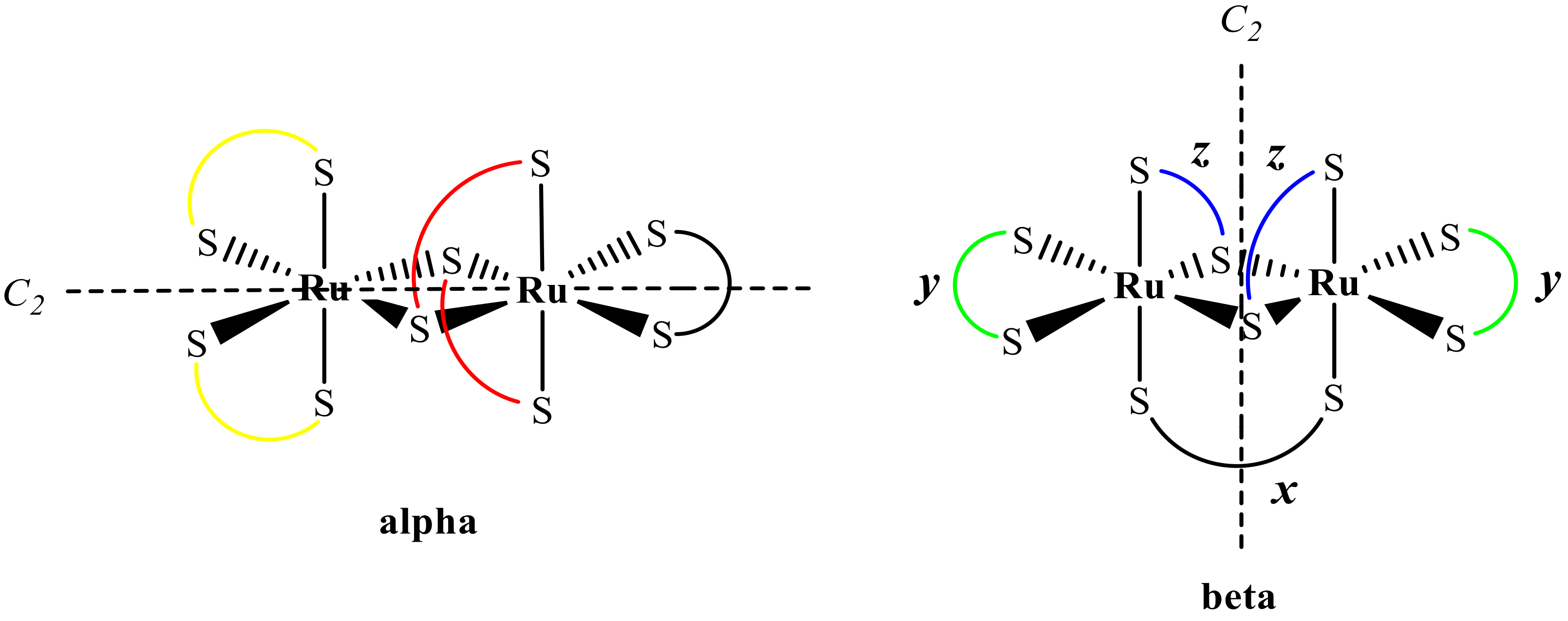
2.1. A Synthetic Strategy for the Synthesis of Binuclear Heteroleptic Complexes of Ru(III)
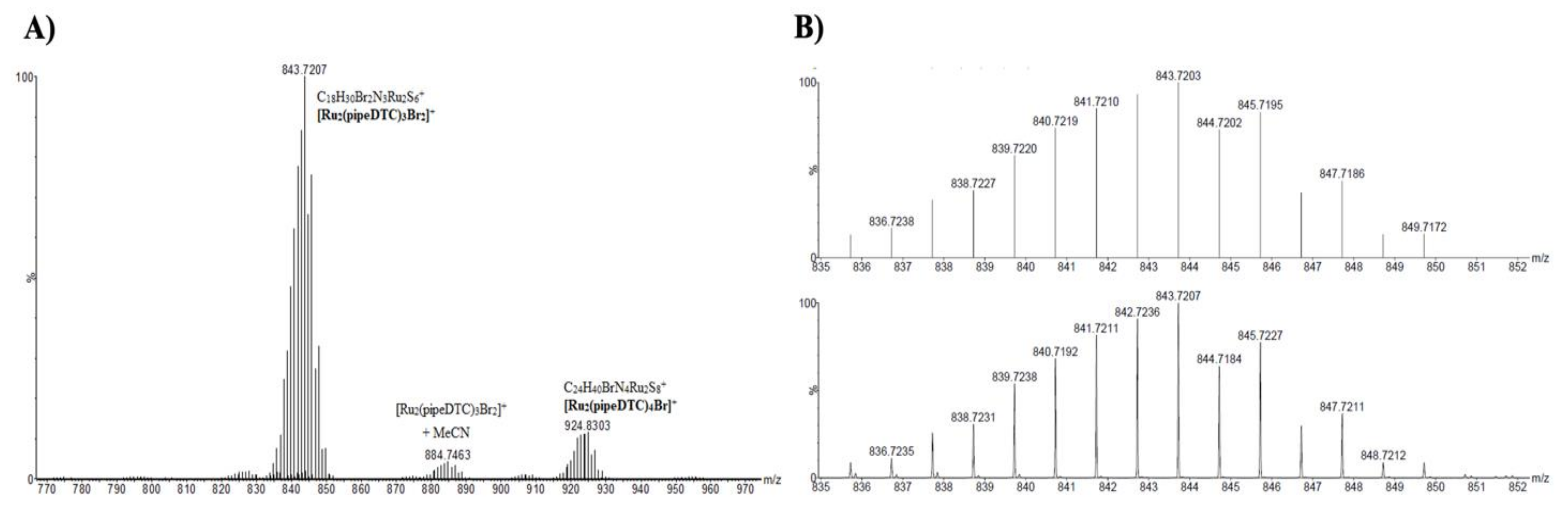
2.2. ESI-MS Characterization
2.3. FT-IR Spectroscopy
2.4. UV-Vis Spectroscopy
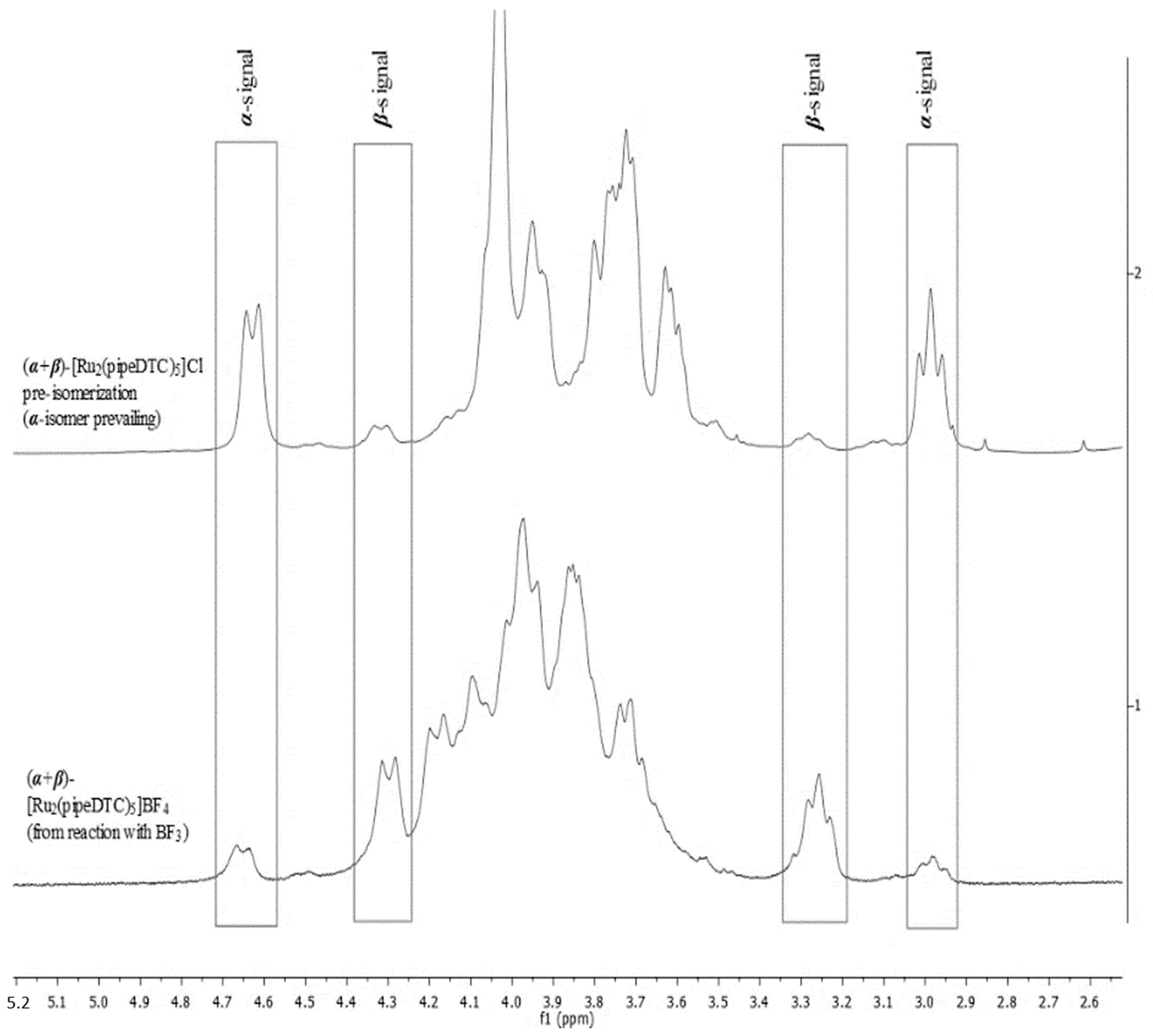
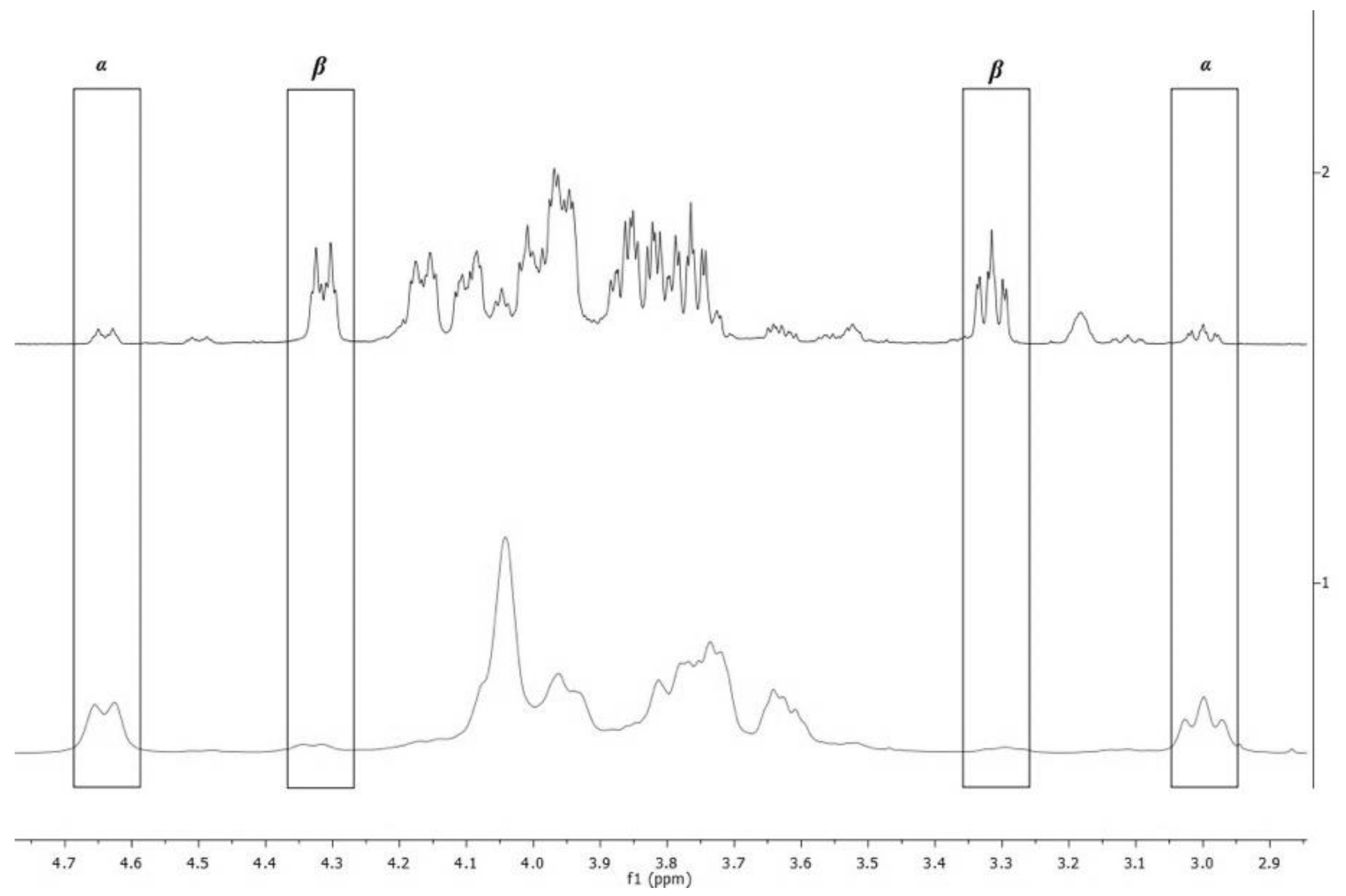
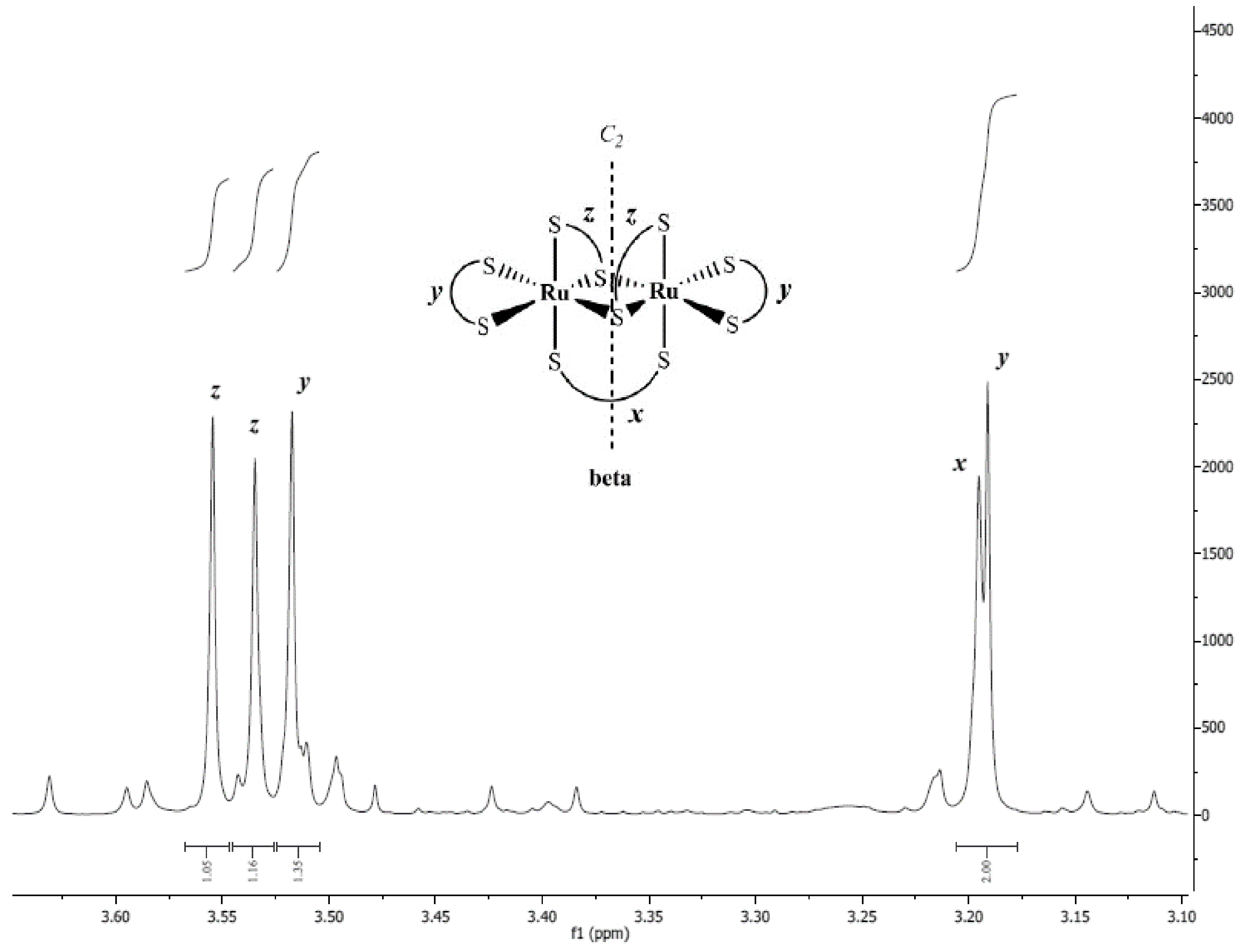
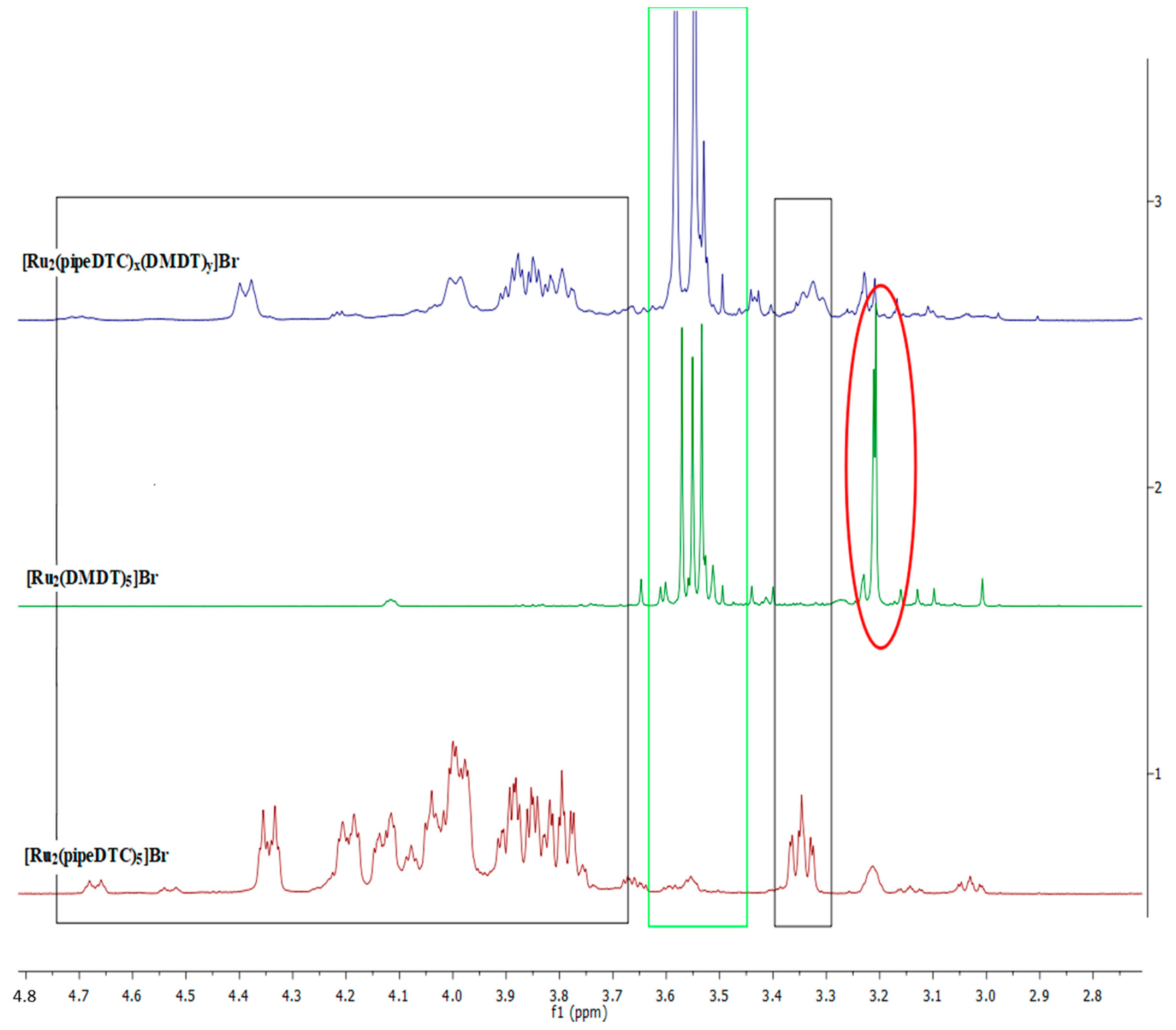
2.5. 1H-NMR Spectroscopy
2.6. Antiproliferative Activity
2.6.1. Cell Viability
Effects of the Treatments on Cell Growth of Breast Adenocarcinoma Line MDA-MB-231
Effects of the Treatments on Cell Growth of the PC3 Prostate Cancer Line
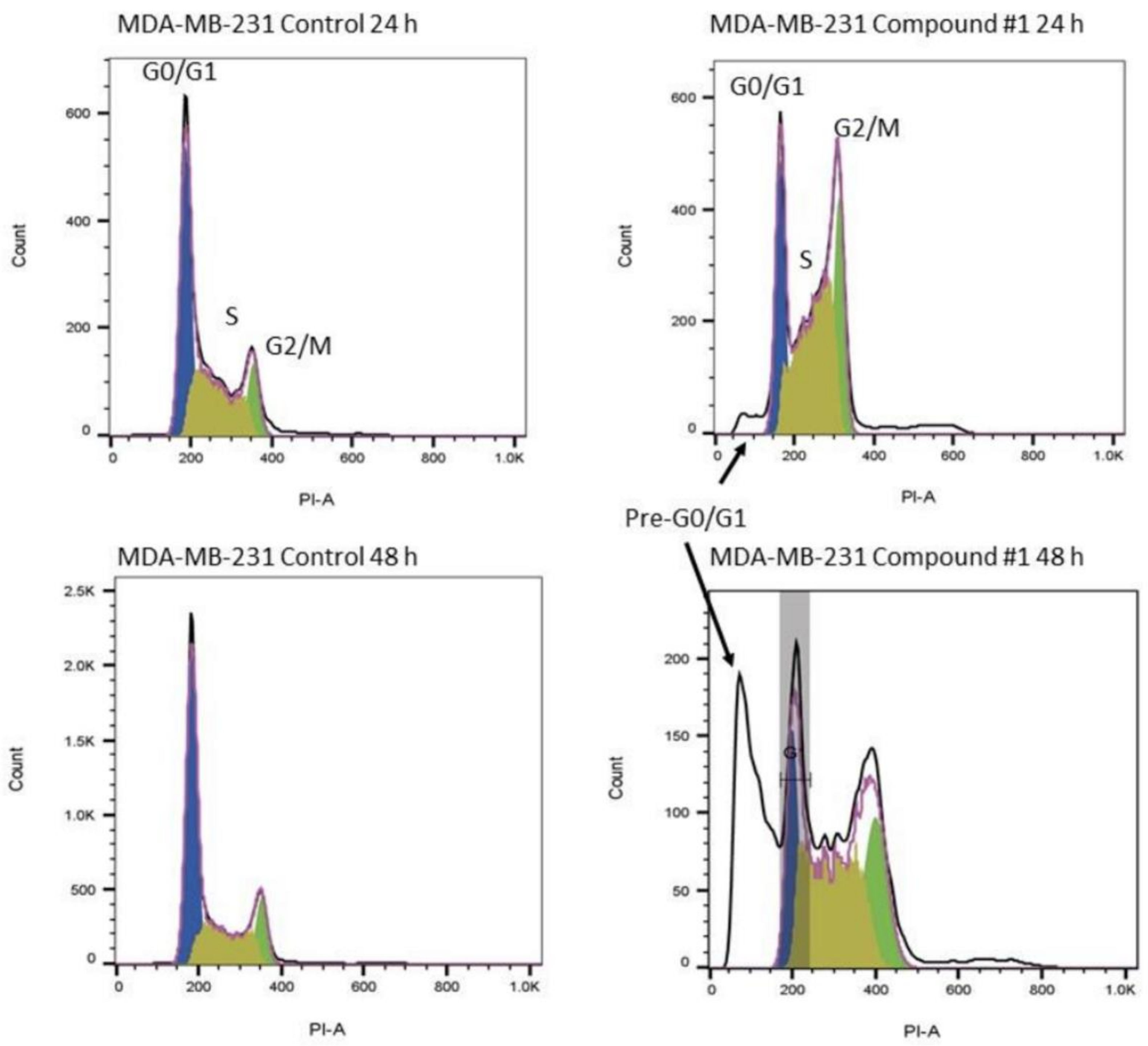
2.6.2. Cell Cycle Studies
Effects of Treatment on the Cell Cycle of the Breast Adenocarcinoma Line MDA-MB-231
Effects of Treatment on the Cell Cycle of the PC3 Prostate Cancer Line
3. Discussion
4. Materials and Methods
4.1. Materials
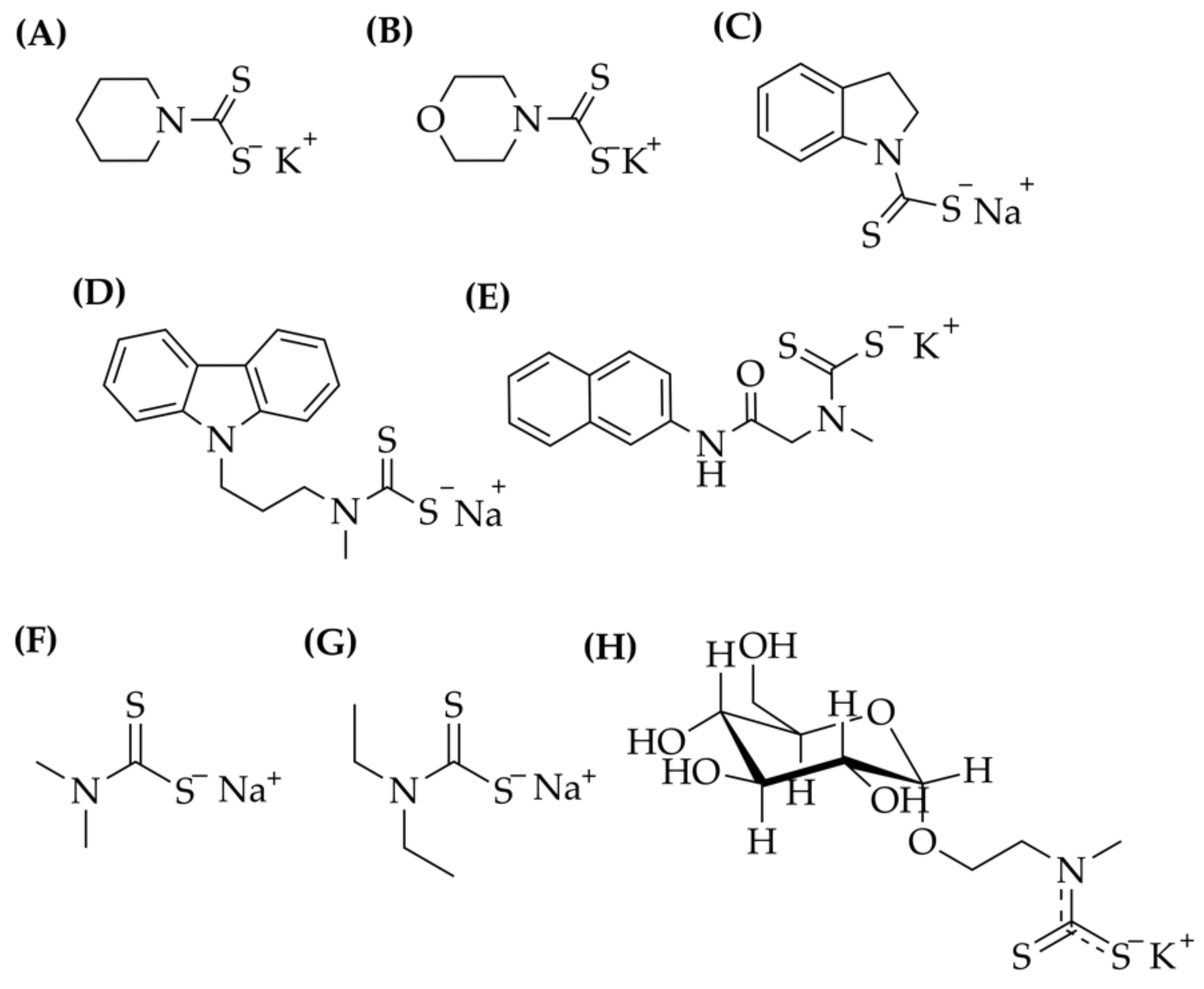
4.2. Synthesis of the Ligands
4.2.1. Piperidine Dithiocarbamate Potassium Salt
4.2.2. Morpholine Dithiocarbamate Potassium Salt
4.2.3. Indoline Dithiocarbamate Sodium Salt (Na(indolineDTC))
4.2.4. (3-(9H-Carbazol-9-yl)propyl)(methyl)dithiocarbamate Sodium Salt: Na(Carbz-pr-N(Me)-DTC)
Amine Precursor Synthesis: 3-(9H-Carbazol-9-yl)-N-methylpropan-1-amine(•HCl): Carbz-pr-NH(Me)(•HCl)
Dithiocarbamate Synthesis: Na(Carbz-pr-N(Me)-DTC)
(N.B.: This Procedure Leads to a Product Comprehensive of 1 Equivalent of NaCl)
Methyl(2-(naphtalen-2-ylamino)-2-oxoethyl)dithiocarbamate Potassium Salt: K(β-Napht-Sar-DTC)
Coupling (Z-SarOH)-(β-naphthylamine)
Hydrogenation of the Coupling Product: Z-Protecting Group Cleavage
Dithiocarbamate Synthesis: K(β-Napht-Sar-DTC)
(N.B.: This Procedure Leads to a Product Comprehensive of 1 Equivalent of KCl)
4.2.5. β-D-Glucoside-Conjugated Dithiocarbamate (gluc-MAE-DTC)
4.3. Synthesis of the Bimetallic Ruthenium Precursor
4.3.1. [Ru(II)Cl2(NBD)]n
4.3.2. [Ru(II)(pipeDTC)2(NBD)]
4.4. Synthesis of Binuclear Heteroleptic Complexes of Ru(III)
4.4.1. [Ru2(pipeDTC)5]Br
4.4.2. [Ru2(pipeDTC)3(morphDTC)2]Br
4.4.3. [Ru2(pipeDTC)x(indolineDTC)y]Br
4.4.4. [Ru2(pipeDTC)x(Carbz-pr-N(Me)-DTC)y]Br
4.4.5. [Ru2(pipeDTC)x(β-Napht-Sar-DTC)y]Br
4.4.6. [Ru2(pipeDTC)x(DMDT)y]Br
4.4.7. [Ru2(pipeDTC)x(DEDT)y]Br
4.4.8. [Ru2(pipeDTC)3(gluc-MAE-DTC)2]Br
4.5. Viability Studies
4.6. Cell Cycle Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef] [PubMed]
- van Rijt, S.H.; Sadler, P.J. Current applications and future potential for bioinorganic chemistry in the development of anticancer drugs. Drug Discov. Today 2009, 14, 1089–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mjos, K.D.; Orvig, C. Metallodrugs in Medicinal Inorganic Chemistry. Chem. Rev. 2014, 114, 4540–4563. [Google Scholar] [CrossRef]
- Cocchietto, M.; Zorzet, S.; Sorc, A.; Sava, G. Primary tumor, lung and kidney retention and antimetastasis effect of NAMI-A following different routes of administration. Investig. New Drugs 2003, 21, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, a new redox-active anticancer agent--preclinical development and results of a clinical phase I study in tumor patients. Chem. Biodivers. 2008, 5, 2140–2155. [Google Scholar] [CrossRef] [PubMed]
- Betanzos-Lara, S.; Novakova, O.; Deeth, R.J.; Pizarro, A.M.; Clarkson, G.J.; Liskova, B.; Brabec, V.; Sadler, P.J.; Habtemariam, A. Bipyrimidine ruthenium(II) arene complexes: Structure, reactivity and cytotoxicity. J. Biol. Inorg. Chem. 2012, 17, 1033–1051. [Google Scholar] [CrossRef]
- Weiss, A.; Berndsen, R.H.; Dubois, M.; Müller, C.; Schibli, R.; Griffioen, A.W.; Dyson, P.J.; Nowak-Sliwinska, P. In vivo anti-tumor activity of the organometallic ruthenium(ii)-arene complex [Ru(η6-p-cymene)Cl2(pta)] (RAPTA-C) in human ovarian and colorectal carcinomas. Chem. Sci. 2014, 5, 4742–4748. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.J. Ruthenium metallopharmaceuticals. Coord. Chem. Rev. 2002, 232, 69–93. [Google Scholar] [CrossRef]
- Abid, M.; Shamsi, F.; Azam, A. Ruthenium Complexes: An Emerging Ground to the Development of Metallopharmaceuticals for Cancer Therapy. Mini-Rev. Med. Chem. 2016, 16, 772–786. [Google Scholar] [CrossRef]
- Borch, R.F.; Pleasants, M.E. Inhibition of cis-platinum nephrotoxicity by diethyldithiocarbamate rescue in a rat model. Proc. Natl. Acad. Sci. USA 1979, 76, 6611–6614. [Google Scholar] [CrossRef] [Green Version]
- Fuertes, M.A.; Alonso, C.; Pérez, J.M. Biochemical Modulation of Cisplatin Mechanisms of Action: Enhancement of Antitumor Activity and Circumvention of Drug Resistance. Chem. Rev. 2003, 103, 645–662. [Google Scholar] [CrossRef] [PubMed]
- Hogarth, G. Metal-dithiocarbamate complexes: Chemistry and biological activity. Mini-Rev. Med. Chem. 2012, 12, 1202–1215. [Google Scholar] [CrossRef] [PubMed]
- Nagy, E.M.; Pettenuzzo, A.; Boscutti, G.; Marchio, L.; Dalla Via, L.; Fregona, D. Ruthenium(II/III)-based compounds with encouraging antiproliferative activity against non-small-cell lung cancer. Chemistry 2012, 18, 14464–14472. [Google Scholar] [CrossRef]
- Scintilla, S.; Brustolin, L.; Gambalunga, A.; Chiara, F.; Trevisan, A.; Nardon, C.; Fregona, D. Ru(III) anticancer agents with aromatic and non-aromatic dithiocarbamates as ligands: Loading into nanocarriers and preliminary biological studies. J. Inorg. Biochem. 2016, 165, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Nardon, C.; Brustolin, L.; Fregona, D. Is matching ruthenium with dithiocarbamato ligands a potent chemotherapeutic weapon in oncology? Future Med. Chem. 2016, 8, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Brustolin, L.; Nardon, C.; Pettenuzzo, N.; Zuin Fantoni, N.; Quarta, S.; Chiara, F.; Gambalunga, A.; Trevisan, A.; Marchio, L.; Pontisso, P.; et al. Synthesis, chemical characterization and cancer cell growth-inhibitory activities of Cu(ii) and Ru(iii) aliphatic and aromatic dithiocarbamato complexes. Dalton Trans. 2018, 47, 15477–15486. [Google Scholar] [CrossRef] [PubMed]
- Fregona, D.; Nardon, C.; Brustolin, L.; Pettenuzzo, N. Coordination Compounds, Syntheses, Nanoformulation and Use Thereof in Oncology. PCT/IB2017/057594. 2 December 2017. Available online: https://patentscope2.wipo.int/search/en/detail.jsf?docId=WO2018100562 (accessed on 19 February 2022).
- Brustolin, L.; Pettenuzzo, N.; Nardon, C.; Quarta, S.; Montagner, I.; Pontisso, P.; Rosato, A.; Conte, P.; Merigliano, S.; Fregona, D. Labelled micelles for the delivery of cytotoxic Cu(II) and Ru(III) compounds in the treatment of aggressive orphan cancers: Design and biological in vitro data. J. Inorg. Biochem. 2020, 213, 111259. [Google Scholar] [CrossRef]
- Wheeler, S.H.; Mattson, B.M.; Miessler, G.L. Electrochemical and chemical properties of dithiocarbamato complexes of ruthenium(II), ruthenium(III), and ruthenium(IV). Inorg. Chem. 1978, 17, 340–350. [Google Scholar] [CrossRef]
- Mattson, B.M.; Heiman, J.R.; Pignolet, L.H. Oxidation of tris(N,N-disubstituted-dithiocarbamato) complexes of ruthenium(III). X-ray structure determination of bis(N,N-diethyldithiocarbamato)-.mu.-tris(N,N-diethyldithiocarbamato)-diruthenium(III) tetrafluoroborate, [Ru2(Et2dtc)5]BF4. Inorg. Chem. 1976, 15, 564–571. [Google Scholar] [CrossRef]
- Mason, R.; Rae, A.I.M. The crystal structure of ruthenium carbonyl, Ru3(CO)12. J. Chem. Soc. A Inorg. Phys. Theor. 1968, 0, 778–779. [Google Scholar] [CrossRef]
- Lippard, S.J.; Coucouvanis, D.; Zubieta, J.A. Crystal and molecular structure of bis(ethyl thioxanthato)-.mu.-bis(ethyl thioxanthato)-.mu.’-bis(ethylthio)diiron(III). Inorg. Chem. 1970, 9, 2775–2781. [Google Scholar] [CrossRef]
- Malatesta, L. Sui ditiocarbammati di rutenio, rodio e palladio. Gazz. Chim. Ital. 1938, 68, 195. [Google Scholar]
- Hendrickson, A.R.; Hope, J.M.; Martin, R.L. Tris- and pentakis-dialkyldithiocarbamates of ruthenium, [Ru(S2CNR2)3] and [Ru2(S2CNR2)5] (n = +1, 0, and –1): Chemical and electrochemical interrelations. J. Chem. Soc. Dalton Trans. 1976, 20, 2032–2039. [Google Scholar] [CrossRef]
- Pasek, E.A.; Straub, D.K. Tris(N,N-disubstituted dithiocarbamato)iron(IV) tetrafluoroborates. Inorg. Chem. 1972, 11, 259–263. [Google Scholar] [CrossRef]
- Martin, R.L.; Rohde, N.M.; Robertson, G.B.; Taylor, D. Structural characterization of tris(pyrrolidyldithiocarbamato)iron(IV) perchlorate. Iron sulfide (FeS6) complex of unusually high oxidation state. J. Am. Chem. Soc. 1974, 96, 3647–3649. [Google Scholar] [CrossRef]
- Brown, K.L.; Golding, R.M.; Healy, P.C.; Jessop, K.J.; Tennant, W.C. Manganese(IV) dithiocarbamato complexes. Aust. J. Chem. 1974, 27, 2075–2081. [Google Scholar] [CrossRef]
- Hendrickson, A.R.; Martin, R.L.; Taylor, D. Synthesis and properties of dimeric cobalt(III) dithiocarbamate complexes [Co2(R2dtc)5]+: X-ray structural analysis of pentakis(diethyldithiocarbamato)dicobalt(III) tetrafluoroborate. J. Chem. Soc. Dalton Trans. 1975, 21, 2182–2188. [Google Scholar] [CrossRef]
- Hendrickson, A.R.; Martin, R.L.; Taylor, D. A binuclear rhodium(III) dithiocarbamate complex, [Rh2(Me2NCS2)5] BF4. Aust. J. Chem. 1976, 29, 269–273. [Google Scholar] [CrossRef]
- Given, K.W.; Mattson, B.M.; Pignolet, L.H. Synthesis, properties, and x-ray structural characterization of novel seven-coordinate halogenotris(dithiocarbamato) complex of ruthenium(IV). Inorg. Chem. 1976, 15, 3152–3156. [Google Scholar] [CrossRef]
- Bradley, D.C.; Gitlitz, M.H. Preparation and properties of NN-dialkyldithiocarbamates of early transition elements. J. Chem. Soc. A: Inorg. Phys. Theor. 1969, 0, 1152–1156. [Google Scholar] [CrossRef]
- Brown, D.A.; Glass, W.K.; Burke, M.A. The general use of i.r. spectral criteria in discussions of the bonding and structure of metal dithiocarbamates. Spectrochim. Acta Part A Mol. Spectrosc. 1976, 32, 137–143. [Google Scholar] [CrossRef]
- Kellner, R.; St. Nikolov, G. Far IR spectra of dithiocarbamate complexes correlations with structure parameters. J. Inorg. Nucl. Chem. 1981, 43, 1183–1188. [Google Scholar] [CrossRef]
- Mohammad, T.M. Aslam Comparative study on the IR spectra of some transition metal dithiocarbamates. Biosci. Biotechnol. Res. Asia 2016, 5, 833–836. [Google Scholar] [CrossRef]
- Ronconi, L.; Giovagnini, L.; Marzano, C.; Bettio, F.; Graziani, R.; Pilloni, G.; Fregona, D. Gold dithiocarbamate derivatives as potential antineoplastic agents: Design, spectroscopic properties, and in vitro antitumor activity. Inorg. Chem. 2005, 44, 1867–1881. [Google Scholar] [CrossRef]
- Manoussakis, G.E.; Bolos, C.A. Synthesis and characterization of a series of new mixed ligand complexes of manganese(III), iron(III), nickel(II), copper(II) and zinc(II) with schiff bases of N, N-diethylamino-dithiocarbamate as ligands. Inorg. Chim. Acta 1985, 108, 215–220. [Google Scholar] [CrossRef]
- Jørgensen, C.K. Absorption spectra of transition group complexes of sulphur-containing ligands. J. Inorg. Nucl. Chem. 1962, 24, 1571–1585. [Google Scholar] [CrossRef]
- Meyer, T.J. Optical and thermal electron transfer in metal complexes. Acc. Chem. Res. 2002, 11, 94–100. [Google Scholar] [CrossRef]
- Kassam, F.; Enright, K.; Dent, R.; Dranitsaris, G.; Myers, J.; Flynn, C.; Fralick, M.; Kumar, R.; Clemons, M. Survival outcomes for patients with metastatic triple-negative breast cancer: Implications for clinical practice and trial design. Clin. Breast Cancer 2009, 9, 29–33. [Google Scholar] [CrossRef]
- Yang, L.; Wu, X.; Wang, Y.; Zhang, K.; Wu, J.; Yuan, Y.C.; Deng, X.; Chen, L.; Kim, C.C.; Lau, S.; et al. FZD7 has a critical role in cell proliferation in triple negative breast cancer. Oncogene 2011, 30, 4437–4446. [Google Scholar] [CrossRef]
- Stavridi, F.; Karapanagiotou, E.M.; Syrigos, K.N. Targeted therapeutic approaches for hormone-refractory prostate cancer. Cancer Treat. Rev. 2010, 36, 122–130. [Google Scholar] [CrossRef]
- Sun, M.; Zhu, L.; Kan, W.; Wei, Y.; Ma, D.; Fan, X.; Huang, W.; Xu, H. Ternary donor–acceptor phosphine oxide hosts with peculiar high energy gap for efficient blue electroluminescence. J. Mater. Chem. C 2015, 3, 9469–9478. [Google Scholar] [CrossRef]
- Pettenuzzo, N.; Brustolin, L.; Coltri, E.; Gambalunga, A.; Chiara, F.; Trevisan, A.; Biondi, B.; Nardon, C.; Fregona, D. Cu(II) and Au(III) Complexes with Glycoconjugated Dithiocarbamato Ligands for Potential Applications in Targeted Chemotherapy. ChemMedChem 2019, 14, 1162–1172. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binuclear Complex | Detected Binuclear Complex | Empirical Formula | m/z |
|---|---|---|---|
| [Ru2(pipeDTC)5]Br | [Ru2(pipeDTC)5]+ | C30H50N5Ru2S10+ | (1003.9365) |
| 1003.9354 | |||
| [Ru2(pipeDTC)x(morphDTC)y]Br | [Ru2(pipeDTC)3(morphDTC)2]+ | C28H46N5O2Ru2S10+ | (1007.8950) |
| 1007.9023 | |||
| [Ru2(pipeDTC)x(indolineDTC)y]Br | [Ru2(pipeDTC)4(indolineDTC)]+ | C33H48N5Ru2S10+ | (1037.9202) |
| 1037.9205 | |||
| [Ru2(pipeDTC)3(indolineDTC)2]+ | C36H46N5Ru2S10+ | (1071.9054) | |
| 1071.9031 | |||
| [Ru2(pipeDTC)2(indolineDTC)3]+ | C39H44N5Ru2S10+ | (1105.8899) | |
| 1105.8823 | |||
| [Ru2(pipeDTC)x(Carbz-pr-N(Me)-DTC)y]Br | [Ru2(pipeDTC)4(Carbz-pr-N(Me)-DTC)]+ | C41H57N6Ru2S10+ | (1156.9948) |
| 1156.9969 | |||
| [Ru2(pipeDTC)3(Carbz-pr-N(Me)-DTC)2]+ | C52H64N7Ru2S10+ | (1310.053) | |
| 1310.0536 | |||
| [Ru2(pipeDTC)x(β-Napht-Sar-DTC)y]Br | Ru2(pipeDTC)4(β-Napht-Sar-DTC)]Br | C38H53N6ORu2S10+ | (1132.9569) |
| 1132.9583 | |||
| Ru2(pipeDTC)3(β-Napht-Sar-DTC)2]Br | C46H56N7O2Ru2S10+ | (1261.9784) | |
| 1261.9814 | |||
| [Ru2(pipeDTC)x(DMDT)y]Br | [Ru2(pipeDTC)2(DMDT)3]+ | C21H38N5Ru2S10+ | (883.8422) |
| 883.8406 | |||
| [Ru2(pipeDTC)3(DMDT)2]+ | C24H42N5Ru2S10+ | (923.8736) | |
| 923.8732 | |||
| [Ru2(pipeDTC)4(DMDT)]+ | C27H46N5Ru2S10+ | (963.905) | |
| 963.9039 | |||
| [Ru2(pipeDTC)x(DEDT)y]Br | [Ru2(pipeDTC)3(DEDT)2]+ | C28H50N5Ru2S10+ | (979.9364) |
| 979.9362 | |||
| [Ru2(pipeDTC)4(DEDT)]+ | C29H50N5Ru2S10+ | (991.9365) | |
| 991.9379 | |||
| [Ru2(pipeDTC)x(gluc-MAE-DTC)y]Br | [Ru2(pipeDTC)3(gluc-MAE-DTC)2]+ | C38H66N5O12Ru2S10+ | (1308.0011) |
| 1307.9977 | |||
| [Ru2(pipeDTC)4(gluc-MAE-DTC)]+ | C34H58N5O6Ru2S10+ | (1155.9688) | |
| 1155.9681 |
| Compounds | Vibrational Modes and Relative Frequencies (cm−1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ν(C=O) | νa(C-O) | ν(C=C) | ν(C-H)ar. | ν(C-H) | ρ(C-H) | τ(C-H) | ν(N-CSS) | νa(C-SS) | |
| [Ru2(pipeDTC)5]Br | - | - | - | - | 2928, 2851 | 850 | - | 1504, 1440 | 1000 |
| [Ru2(pipeDTC)x(DMDT)y]Br | - | - | - | - | 2926, 2851 | 883, 851 | - | 1532, 1441 | 1000, 944 |
| [Ru2(pipeDTC)x(DEDT)y]Br | - | - | - | - | 2971, 2928, 2851 | 849 | - | 1510, 1438 | 1000, 946 |
| [Ru2(pipeDTC)x(morphDTC)y]Br | - | 1107 | - | - | 2933, 2853 | 878, 849 | - | 1535, 1500, 1438 | 1000, 946 |
| [Ru2(pipeDTC)x(indolineDTC)y]Br | - | - | 1601 | - | 2930, 2851 | 851 | 748 | 1532, 1482, 1429 | 1000, 937 |
| [Ru2(pipeDTC)x(Carbz-pr-N(Me)-DTC)y]Br | - | - | 1626, 1594 | 3047 | 2933, 2849 | 885, 849 | 750, 722 | 1503, 1483, 1441 | 1000, 998 |
| [Ru2(pipeDTC)x(β-Napht-Sar-DTC)y]Br | 1691 (NC=O) | - | 1632, 1608, 1585 | 3014 | 2935, 2851 | 886, 851, 819 | 744, 723 | 1529, 1503, 1441 | 1001 |
| [Ru2(pipeDTC)3(Gluc-MAE-DTC)2]Br | - | 1132 (C-O-C) | - | - | 2933, 2858 | 891, 850, 798 | 723 | 1526, 1442 | 1003 |
| [Ru2(pipeDTC)4(Gluc-MAE-DTC)]Br | - | 1132 (C-O-C) | - | - | 2935, 2858 | 889, 858, 798 | 718 | 1524, 1507, 1443 | 1003 |
| Vibrational Modes and Relative Frequencies (cm−1) | |||
|---|---|---|---|
| Compounds | νs(C-SS) | νa(Ru-S) | νs(Ru-S) |
| [Ru2(pipeDTC)5]Br | 544 or 508 | 406 | 326 |
| [Ru2(pipeDTC)x(DMDT)y]Br | 578, 546, 508 | 406, 365 | 325 |
| [Ru2(pipeDTC)x(DEDT)y]Br | 573, 550, 508 | 405, 366 | 321 |
| [Ru2(pipeDTC)x(morphDTC)y]Br | 547, 508 | 406 | 300 |
| [Ru2(pipeDTC)x(indolineDTC)y]Br | 552, 508 | 420, 406 | 353 |
| [Ru2(pipeDTC)x(Carbz-pr-N(Me)-DTC)y]Br | 563, 547, 508 | 437, 407 | 424 |
| [Ru2(pipeDTC)x(β-Napht-Sar-DTC)y]Br | 547, 566, 508 | 474, 406 | 366 |
| [Ru2(pipeDTC)3(Gluc-MAE-DTC)2]Br | 549, 515, 507 | 424, 406 | 367 |
| [Ru2(pipeDTC)4(Gluc-MAE-DTC)]Br | n.a. | n.a. | n.a. |
| λ (nm) | |||
|---|---|---|---|
| Compounds | Region I | Region II | Region III |
| [Ru2(pipeDTC)5]Br | 209, 245 (sh) | 272 (sh), 285 | 335 (sh), 450 |
| [Ru2(pipeDTC)3(morphDTC)2]Br | 211, 242 | 267 (sh), 289 | 339 (sh), 466 |
| [Ru2(pipeDTC)x(indolineDTC)y]Br | 209, 244 (sh) | 266, 290 | 373 (sh), 462 |
| [Ru2(pipeDTC)x(Carbz-pr-N(Me)-DTC)y]Br | 228 (sh), 235 | 260, 283, 292 | 328 (sh), 342 (sh), 445 |
| [Ru2(pipeDTC)x(β-Napht-Sar-DTC)y]Br | 211 (sh), 220 (sh), 244 | 284 | 338 (sh), 472 |
| [Ru2(pipeDTC)x(DMDT)y]Br | 211, 223 (sh), 246 (sh) | 274 | 338 (sh), 446 |
| [Ru2(pipeDTC)x(DEDT)y]Br | 209, 222 (sh), 248 (sh) | 267 (sh), 284 | 336 (sh), 460 |
| [Ru2(pipeDTC)3(gluc-MAE-DTC)2]Br | 217, 242 (sh) | 287 | 338 (sh), 472 |
| [Ru2(pipeDTC)4(gluc-MAE-DTC)]Br | 214, 244 | 265, 286 (sh) | 339 (sh), 467 |
| IC50 (μM) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Viability Test | Trypan Blue | WST-1 | ||||||
| Cell Line | MDA-MB-231 | PC3 | MDA-MB-231 | PC3 | ||||
| Treatment Time | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h |
| Compound #1 | 0.9 ± 0.3 | 1.0 ± 0.1 | 1.05 ± 0.12 | 0.66 ± 0.05 | 0.6 ± 0.1 | 0.6 ± 0.1 | 1.1 ± 0.1 | 0.4 ± 0.2 |
| Compound #2 | 1.3 ± 0.2 | 0.25 ± 0.1 | 1.17 ± 0.13 | 0.27 ± 0.09 | 0.39 ± 0.1 | 0.25 ± 0.3 | 1.47 ± 0.3 | 1.08 ± 0.2 |
| Compound #3 | N.A | ~1.6 | N.A. | N.A. | 0.01 ± 0.02 | 0.12 ± 0.05 | N.A | N.A. |
| % of the Cell Cycle Stages in MDA-MB-231 | ||||||
|---|---|---|---|---|---|---|
| G0/G1 | S | G2/M | ||||
| Time 0 | 44.5 ± 4.6 | 39.8 ± 0 | 12.05 ± 0.3 | |||
| Treatment time | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h |
| Control | 48.0 ± 1.5 | 59.1 ± 0.7 | 39.1 ± 0.1 | 32.2 ± 1.1 | 11.6 ± 1.0 | 13.05 ± 0.1 |
| Compound #1 | 21.9 ± 1.3 | 13.0 ± 1.0 | 43.5 ± 3.8 | 31.3 ± 1.7 | 25.0 ± 4.5 | 17.95 ± 0.9 |
| Compound #2 | 22.4 ± 1.2 | 17.7 ± 1.4 | 51.2 ± 0.3 | 32.9 ± 0.5 | 20.7 ± 0.2 | 15.15 ± 1.2 |
| Compound #3 | 47.8 ± 5.4 | 63.7 ± 3.8 | 27.7 ± 6.8 | 25.1 ± 0.2 | 19.8 ± 4.3 | 16.35 ± 0.2 |
| % of the Cell Cycle Stages in PC3 | ||||||
|---|---|---|---|---|---|---|
| G0/G1 | S | G2/M | ||||
| Time 0 | 39.3 ± 5.3 | 33.4 ± 2.5 | 18.2 ± 3.0 | |||
| Treatment time | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h |
| Control | 24.3 ± 12.3 | 28.1 ± 2.3 | 46.4 ± 7.5 | 43.2 ± 1.5 | 19.1 ± 3.0 | 19.8 ± 1.0 |
| Compound #1 | 40.1 ± 2.9 | 32.3 ± 0.6 | 31.4 ± 1.0 | 27.0 ± 0.1 | 16.7 ± 0.1 | 14.2 ± 0.6 |
| Compound #2 | 42.2 ± 1.3 | 32.7 ± 2.5 | 37.4 ± 4.0 | 26.4 ± 0.9 | 7.3 ± 6.2 | 10.7 ± 1.0 |
| Compound #3 | 31.9 ± 2.7 | 28.7 ± 2.2 | 37.6 ± 2.7 | 36.9 ± 1.5 | 20.0 ± 1.0 | 20.1 ± 2.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallio, A.E.; Brustolin, L.; Pettenuzzo, N.; Fregona, D. Binuclear Heteroleptic Ru(III) Dithiocarbamate Complexes: A Step towards Tunable Antiproliferative Agents. Inorganics 2022, 10, 37. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics10030037
Gallio AE, Brustolin L, Pettenuzzo N, Fregona D. Binuclear Heteroleptic Ru(III) Dithiocarbamate Complexes: A Step towards Tunable Antiproliferative Agents. Inorganics. 2022; 10(3):37. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics10030037
Chicago/Turabian StyleGallio, Andrea E., Leonardo Brustolin, Nicolò Pettenuzzo, and Dolores Fregona. 2022. "Binuclear Heteroleptic Ru(III) Dithiocarbamate Complexes: A Step towards Tunable Antiproliferative Agents" Inorganics 10, no. 3: 37. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics10030037