Various Oxygen-Centered Phosphanegold(I) Cluster Cations Formed by Polyoxometalate (POM)-Mediated Clusterization: Effects of POMs and Phosphanes


Abstract
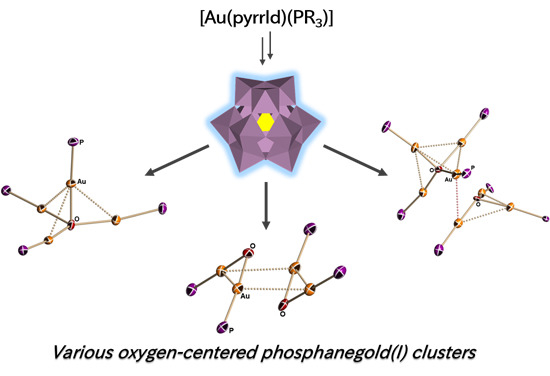
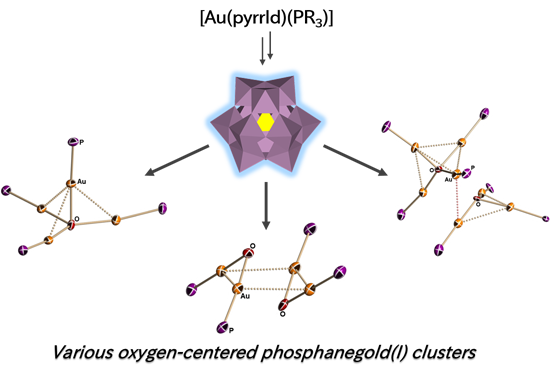
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Compositional Characterization

{kind=link}
{kind=link}
{kind=link}
| Parameters | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Formula | C216H146Au12F36-Mo24O84P14 | C144H104Au8F24-Mo12O46P8Si | C168H168Au8O46-P8SiW12 | C168H168Au8Mo12-O46P8Si | C147H147Au7O42-P8W12 |
| Formula weight | 9869.07 | 6029.13 | 6980.81 | 5925.89 | 6418.37 |
| Color, shape | Orange-yellow, rod | Yellow, block | Pale-yellow, block | Yellow, block | Colorless, plate |
| Crystal system | Triclinic | Triclinic | Rhombohedral | Rhombohedral | Triclinic |
| Space group | P-1 | P-1 | R-3 | R-3 | P-1 |
| T/K | 100 | 120 | 120 | 100 | 100 |
| a/Å | 16.191(2) | 16.296(3) | 20.478(3) | 20.3813(7) | 17.7243(11) |
| b/Å | 29.165(4) | 16.357(3) | 17.8341(10) | ||
| c/Å | 35.365(4) | 16.854(3) | 36.726(7) | 36.679(2) | 31.7570(18) |
| α/° | 99.801(2) | 77.37(3) | 104.8260(10) | ||
| β/° | 99.153(2) | 75.89(3) | 92.640(2) | ||
| γ/° | 104.963(2) | 80.58(3) | 120 | 120 | 109.6850(10) |
| V/Å3 | 15533(3) | 4222.3(15) | 13337(4) | 13195.2(11) | 9040.0(9) |
| Z | 2 | 1 | 3 | 3 | 2 |
| Dcalc/g cm−3 | 2.110 | 2.371 | 2.607 | 2.237 | 2.358 |
| F000 | 9208 | 2822 | 9594 | 8442 | 5852 |
| GOF | 1.027 | 1.066 | 1.311 | 1.074 | 1.373 |
| R1 (I > 2.00 σ(I)) | 0.0723 | 0.0915 | 0.0547 | 0.0376 | 0.1466 |
| R (all data) | 0.1077 | 0.1042 | 0.0558 | 0.0390 | 0.2417 |
| wR2 (all data) | 0.1753 | 0.2502 | 0.1217 | 0.0862 | 0.4614 |
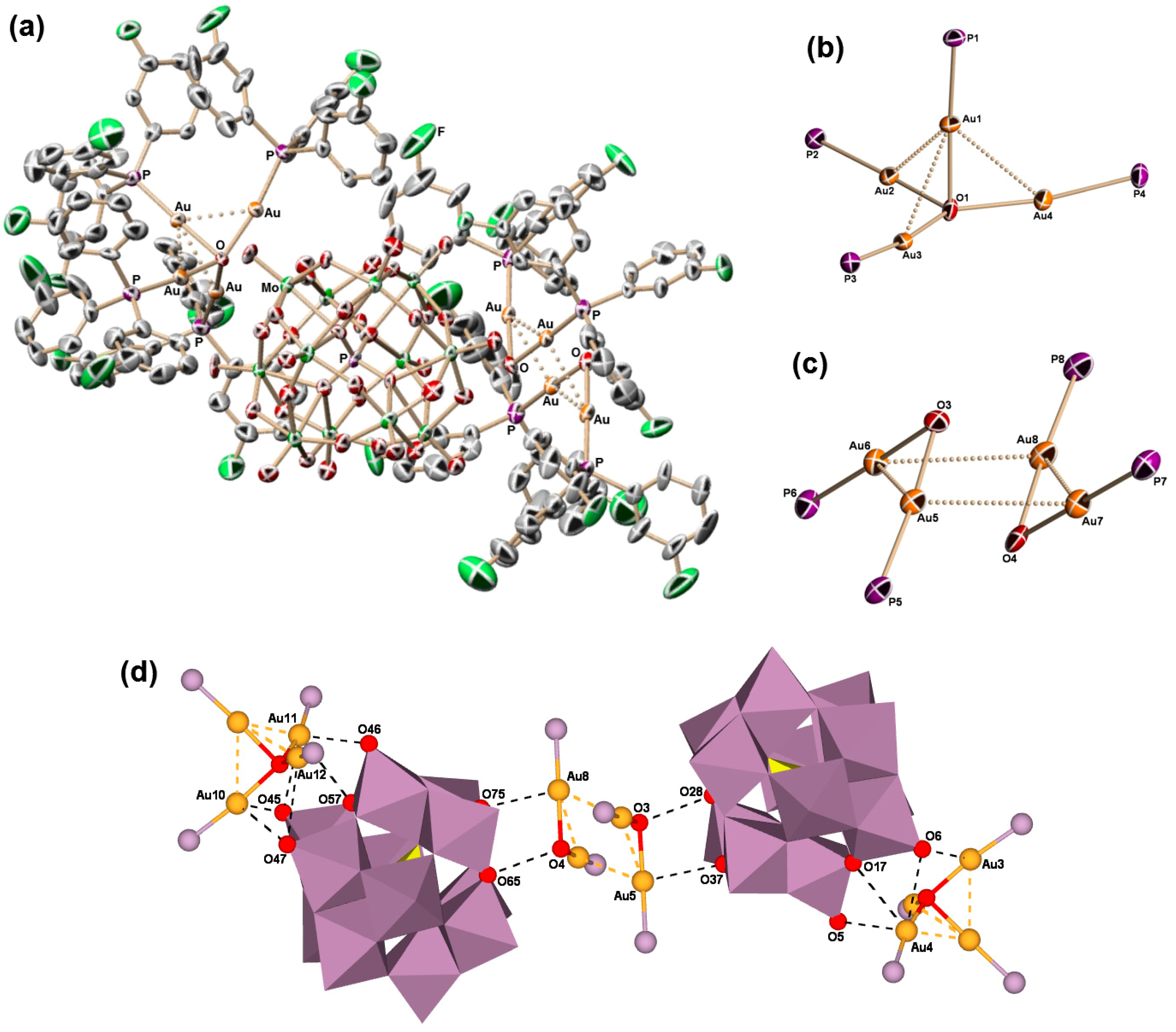
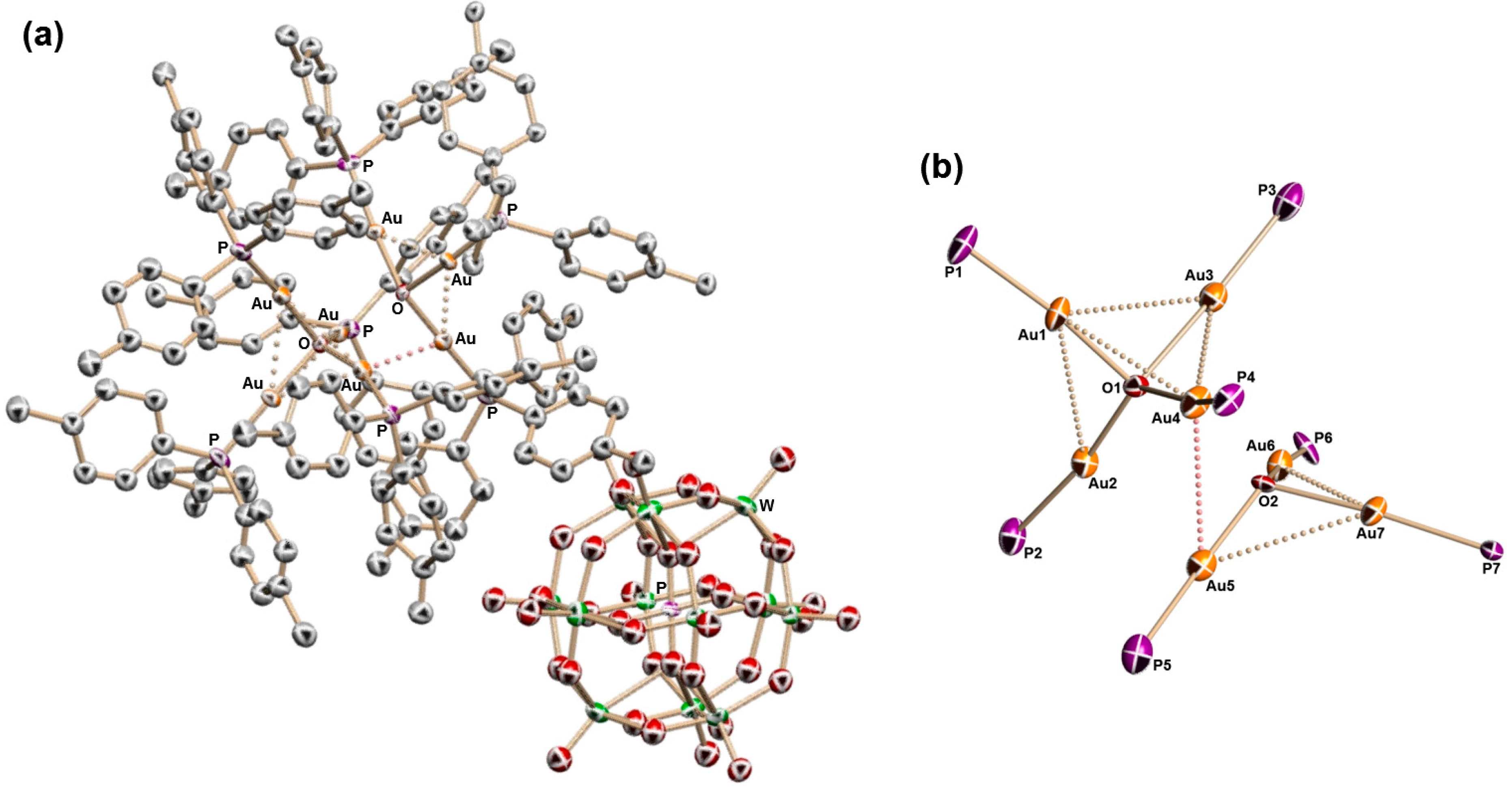
2.2. Molecular Structures of 1–5


2.3. Solid-State CPMAS 31P and Solution 31P{1H} NMR
| Compound | Solid-State CPMAS 31P | Solution 31P{1H} |
|---|---|---|
| 1 | −3.4, 24.4 | −3.23, 26.31 (main), −0.40, 43.57 (minor) |
| 2 | 19.6, 24.4 | 25.67 (main), 43.38 (minor) |
| 3 | 18.3, 28.5 | insoluble |
| 4 | 17.4, 27.7 | insoluble |
| 5 | −14.6, 23.1 | −14.88, 22.39 |
3. Experimental Section
3.1. Materials
3.2. Instrumentation and Analytical Procedures
3.3. Syntheses
3.4. X-ray Crystallography
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pope, M.T.; Müller, A. Polyoxometalate Chemistry: An Old Field with New Dimensions in Several Disciplines. Angew. Chem. Int. Ed. Engl. 1991, 30, 34–48. [Google Scholar] [CrossRef]
- Hill, C.L.; Prosser-McCartha, C.M. Homogeneous catalysis by transition metal oxygen anion clusters. Coord. Chem. Rev. 1995, 143, 407–455. [Google Scholar] [CrossRef]
- Okuhara, T.; Mizuno, N.; Misono, M. Catalytic Chemistry of Heteropoly Compounds. Adv. Catal. 1996, 41, 113–252. [Google Scholar]
- Proust, A.; Thouvenot, R.; Gouzerh, P. Functionalization of polyoxometalates: Towards advanced applications in catalysis and materials science. Chem. Commun. 2008, 1837–1852. [Google Scholar]
- Long, D.-L.; Tsunashima, R.; Cronin, L. Polyoxometalates: Building Blocks for Functional Nanoscale Systems. Angew. Chem. Int. Ed. 2010, 49, 1736–1758. [Google Scholar] [CrossRef]
- Nomiya, K.; Sakai, Y.; Matsunaga, S. Chemistry of Group IV Metal Ion-Containing Polyoxometalates. Eur. J. Inorg. Chem. 2011, 2011, 179–196. [Google Scholar] [CrossRef]
- Schulz-Dobrick, M.; Jansen, M. Supramolecular Intercluster Compounds Consisting of Gold Clusters and Keggin Anions. Eur. J. Inorg. Chem. 2006, 2006, 4498–4502. [Google Scholar] [CrossRef]
- Schulz-Dobrick, M.; Jansen, M. Synthesis and Characterization of Intercluster Compounds Consisting of Various Gold Clusters and Differently Charged Keggin Anions. Z. Anorg. Allg. Chem. 2008, 634, 2880–2884. [Google Scholar] [CrossRef]
- Noguchi, R.; Hara, A.; Sugie, A.; Nomiya, K. Synthesis of novel gold(I) complexes derived by AgCl-elimination between [AuCl(PPh3)] and silver(I) heterocyclic carboxylates, and their antimicrobial activities. Molecular structure of [Au(R,S-Hpyrrld)(PPh3)] (H2pyrrld = 2-pyrrolidone-5-carboxylic acid. Inorg. Chem. Commun. 2006, 9, 355–359. [Google Scholar] [CrossRef]
- Nomiya, K.; Yoshida, T.; Sakai, Y.; Nanba, A.; Tsuruta, S. Intercluster Compound between a Tetrakis{triphenylphosphinegold(I)}oxonium Cation and a Keggin Polyoxometalate (POM): Formation during the Course of Carboxylate Elimination of a Monomeric Triphenylphosphinegold(I) Carboxylate in the Presence of POMs. Inorg. Chem. 2010, 49, 8247–8254. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Nomiya, K.; Matsunaga, S. Novel intercluster compound between a heptakis{triphenylphosphinegold(I)}dioxonium cation and an α-Keggin polyoxometalate anion. Dalton Trans. 2012, 41, 10085–10090. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Matsunaga, S.; Nomiya, K. Two types of tetranuclear phosphanegold(I) cations as dimers of dinuclear units, [{(Au{P(p-RPh)3})2(μ-OH)}2]2+ (R = Me, F), synthesized by polyoxometalate-mediated clusterization. Dalton Trans. 2013, 42, 11418–11425. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Matsunaga, S.; Nomiya, K. Novel Intercluster Compounds Composed of a Tetra{phosphanegold(I)}oxonium Cation and an α-Keggin Polyoxometalate Anion Linked by Three Monomeric Phosphanegold(I) Units. Chem. Lett. 2013, 42, 1487–1489. [Google Scholar] [CrossRef]
- Yoshida, T.; Yasuda, Y.; Nagashima, E.; Arai, H.; Matsunaga, S.; Nomiya, K. Template Effects of Polyoxometalate (POM) in the Formation of Dimer-of-Dinuclear Phosphanegold(I) Clusters by POM-Mediated Clusterization. Kanagawa university: Hiratsuka, Japan, Unpublished work. 2014. [Google Scholar]
- Schmidbaur, H. Ludwig Mond Lecture. High-carat gold compounds. Chem. Soc. Rev. 1995, 24, 391–400. [Google Scholar] [CrossRef]
- Schmidbaur, H.; Schier, A. A briefing on aurophilicity. Chem. Soc. Rev. 2008, 37, 1931–1951. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, M.C.; Laguna, A. Chalcogenide centred gold complexes. Chem. Soc. Rev. 2008, 37, 1951–1966. [Google Scholar] [CrossRef]
- Nesmeyanov, A.N.; Perevalova, E.G.; Struchkov, Y.T.; Antipin, M.Y.; Grandberg, K.I.; Dyadchenko, V.P. Tris{triphenylphosphinegold}oxonium salts. J. Organomet. Chem. 1980, 201, 343–349. [Google Scholar] [CrossRef]
- Yang, Y.; Ramamoorthy, V.; Sharp, P.R. Late transition metal oxo and imido complexes. 11. Gold(I) oxo complexes. Inorg. Chem. 1993, 32, 1946–1950. [Google Scholar] [CrossRef]
- Angermaier, K.; Schmidbaur, H. A New Structural Motif of Gold Clustering at Oxide Centers in the Dication [Au6O2(PMe3)6]2+. Inorg. Chem. 1994, 33, 2069–2070. [Google Scholar] [CrossRef]
- Chung, S.-C.; Krüger, S.; Schmidbaur, H.; Rösch, N. A Density Functional Study of Trigold Oxonium Complexes and of Their Dimerization. Inorg. Chem. 1996, 35, 5387–5392. [Google Scholar] [CrossRef]
- Rocchiccioli-Deltcheff, C.; Fournier, M.; Franck, R.; Thouvenot, R. Vibrational Investigations of Polyoxometalates. 2. Evidence for Anion–Anion Interactions in Molybdenum(VI) and Tungsten(VI) Compounds Related to the Keggin Structure. Inorg. Chem. 1983, 22, 207–216. [Google Scholar] [CrossRef]
- Wells, A.F. Structural Inorganic Chemistry, 4th ed.; Clarendon Press: Oxford, UK, 1975; p. 1020. [Google Scholar]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- North, E.O.; Haney, W. Silicomolybdic Acid. Inorg. Synth. 1939, 1, 127–129. [Google Scholar]
- Tézé, A.; Hervé, G. α-, β-, and γ-Dodecatungstosilicic Acids: Isomers and Related Lacunary Compounds. Inorg. Synth. 1990, 27, 85–96. [Google Scholar]
- Aoki, S.; Kurashina, T.; Kasahara, Y.; Nishijima, T.; Nomiya, K. Polyoxometalate (POM)-based, multi-functional, inorganic-organi, hybrid compounds: Syntheses and molecular structures of silanol- and/or siloxane bond-containing species grafted on mono- and tri-lacunary Keggin POMs. Dalton Trans. 2011, 40, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS, Program for Area Detector Adsorption Correction; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Spek, A.L. PLATON, an Integrated Tool for the Analysis of the Results of a Single Crystal Structure Determination. Acta Crystallogr. 1990, 46, c34. [Google Scholar]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshida, T.; Yasuda, Y.; Nagashima, E.; Arai, H.; Matsunaga, S.; Nomiya, K. Various Oxygen-Centered Phosphanegold(I) Cluster Cations Formed by Polyoxometalate (POM)-Mediated Clusterization: Effects of POMs and Phosphanes. Inorganics 2014, 2, 660-673. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics2040660
Yoshida T, Yasuda Y, Nagashima E, Arai H, Matsunaga S, Nomiya K. Various Oxygen-Centered Phosphanegold(I) Cluster Cations Formed by Polyoxometalate (POM)-Mediated Clusterization: Effects of POMs and Phosphanes. Inorganics. 2014; 2(4):660-673. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics2040660
Chicago/Turabian StyleYoshida, Takuya, Yuta Yasuda, Eri Nagashima, Hidekazu Arai, Satoshi Matsunaga, and Kenji Nomiya. 2014. "Various Oxygen-Centered Phosphanegold(I) Cluster Cations Formed by Polyoxometalate (POM)-Mediated Clusterization: Effects of POMs and Phosphanes" Inorganics 2, no. 4: 660-673. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics2040660