
Synthesis and Characterisation of the Europium (III) Dimolybdo-Enneatungsto-Silicate Dimer, [Eu(α-SiW9Mo2O39)2]13−

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of K13[Eu(α-SiW9Mo2O39)2] 21H2O
2.2. Elemental Analysis, TGA and IR Spectroscopy
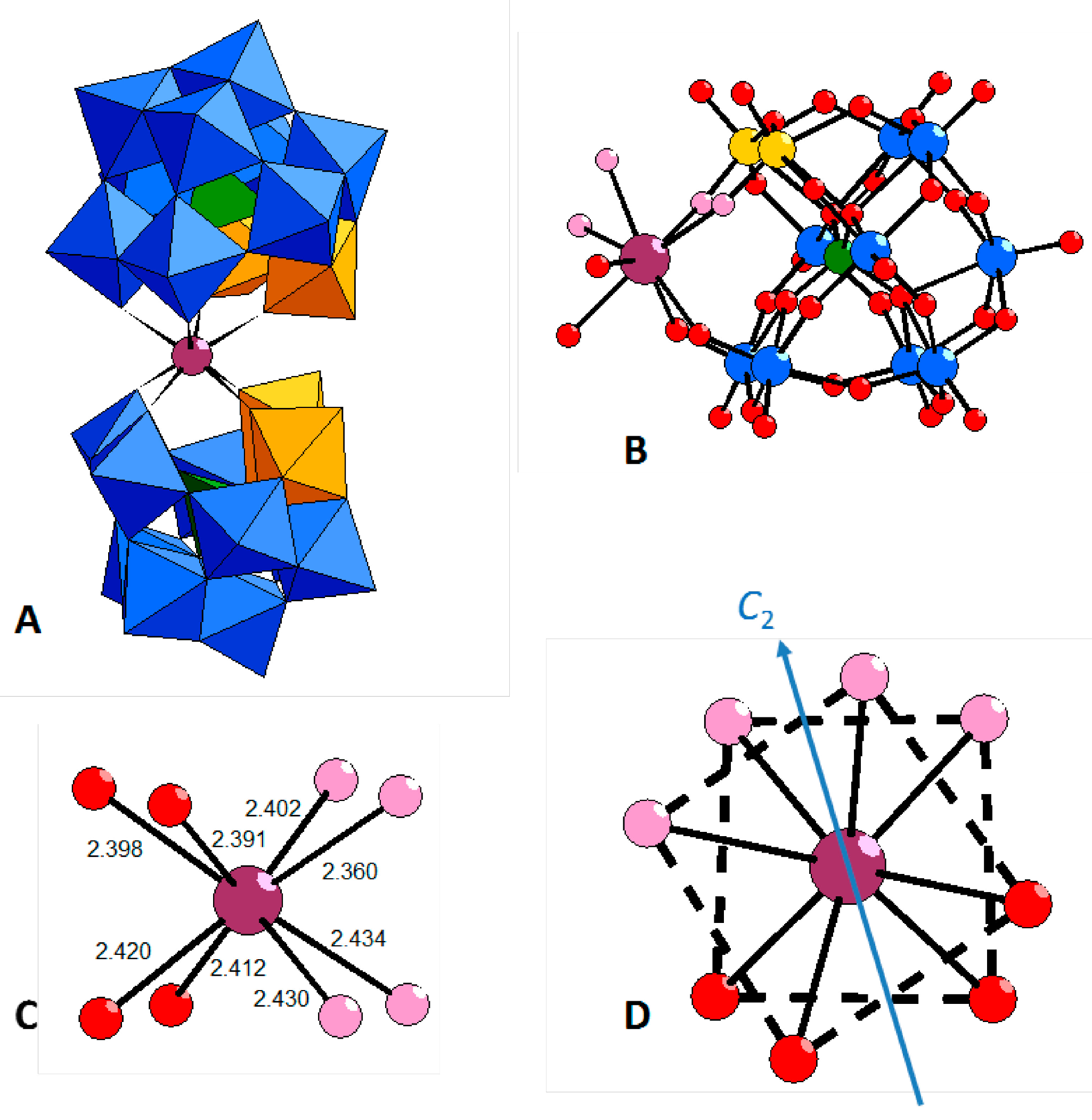
2.3. Structure

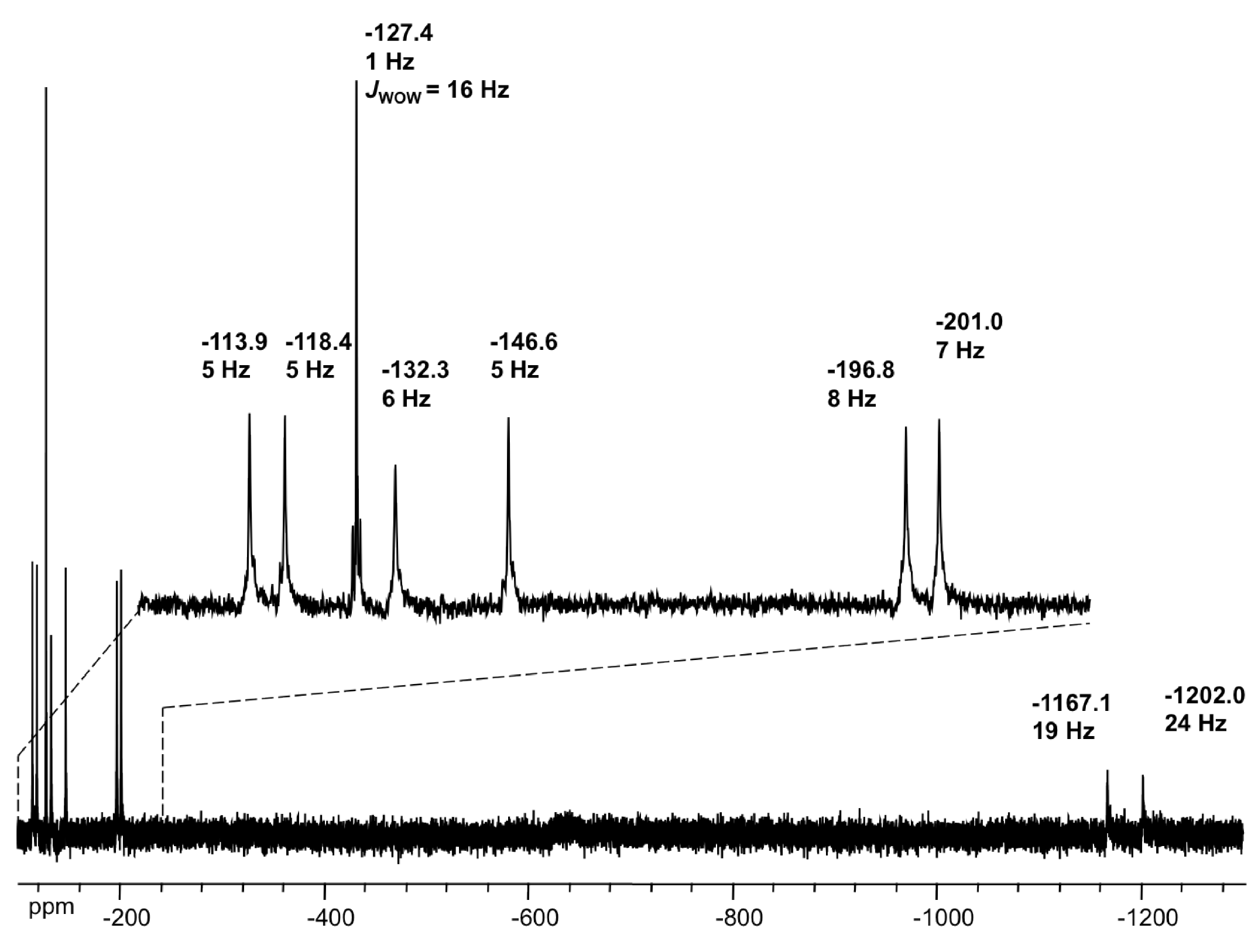
2.4. NMR Spectroscopy

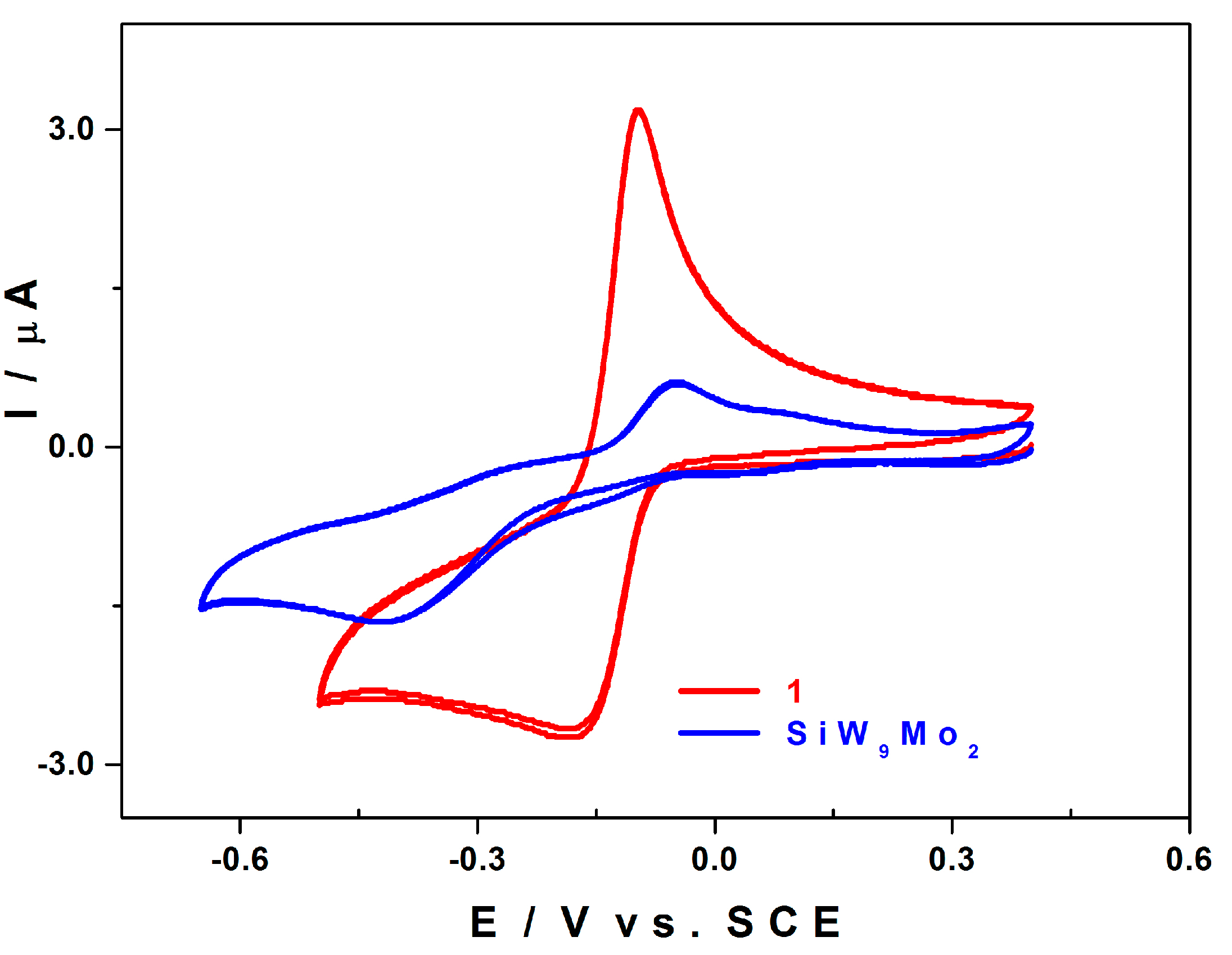
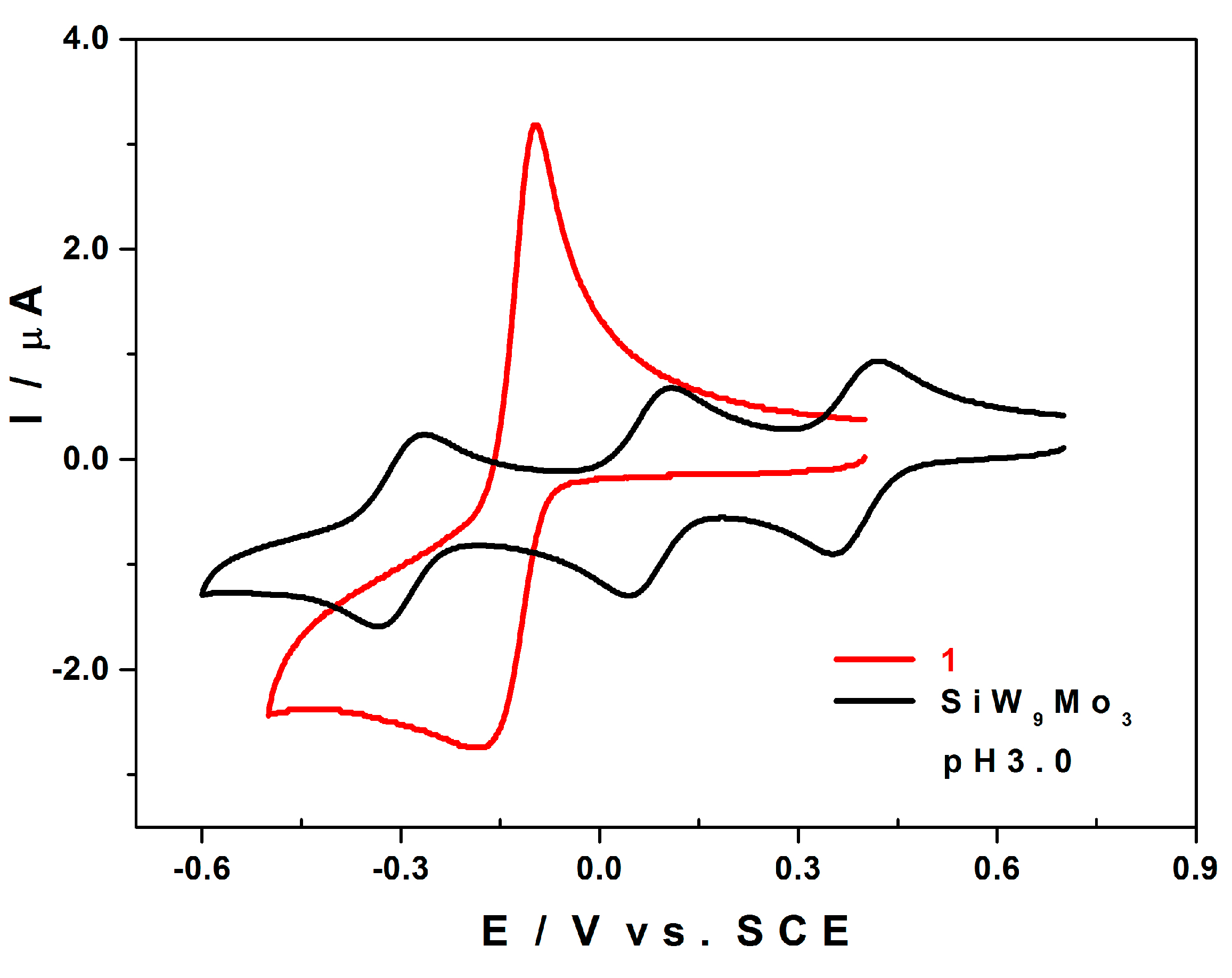
2.5. Electrochemistry

2.5.1. Compared Redox Behaviour of 1 and of [SiW9Mo3O40]4− at pH 3.0

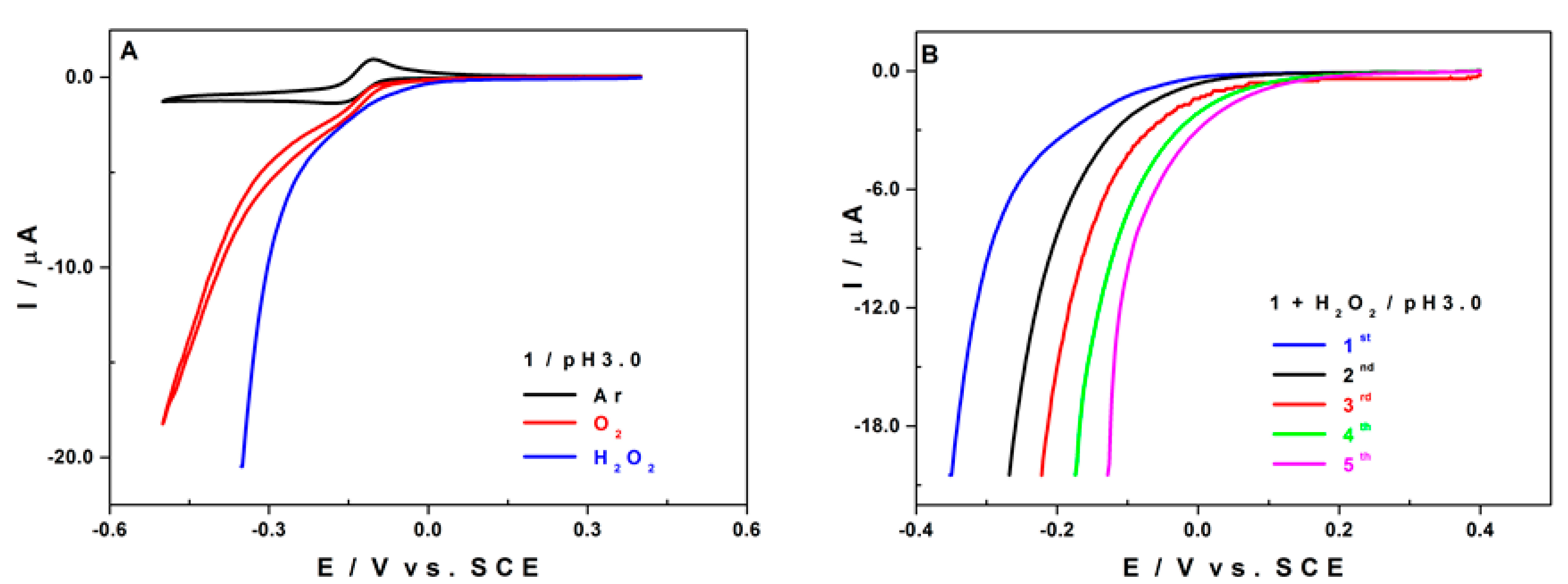
2.5.2. Electro-Catalytic Activity of 1 towards O2 and H2O2 Reduction

3. Experimental Section
3.1. Synthesis of K13[Eu(α-SiW9Mo2O39)2] 21H2O
3.2. TGA, and IR Spectroscopy
3.3. X-ray Crystallography

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | 1 |
|---|---|
| Formula | EuK13Mo4O94Si2W18 |
| Formula weight, g | 5913.50 |
| Crystal system | triclinic |
| Space group | P-1 |
| a/Å | 12.2447(10) |
| b/Å | 12.7803(10) |
| c/Å | 33.762(3) |
| α/° | 83.799(2) |
| β/° | 85.106(2) |
| γ/° | 66.214(2) |
| V/Å3 | 4801.4(6) |
| Z | 2 |
| Dcalc/g cm−3 | 4.090 |
| μ/mm−1 | 23.307 |
| Data/parameters | 37169/26808 |
| Rint | 0.0560 |
| GOF | 0.946 |
| R1 (I > 2σ(I)) | 0.0720 |
| wR2 | 0.1786 |
3.4. NMR Spectroscopy
3.5. Electrochemistry
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contribution
Conflicts of Interest
References
- Berzelius, J.J. Beitrag zur näheren Kenntniss des Molybdäns. Ann. Phys. 1826, 82, 369–392. [Google Scholar] [CrossRef]
- Keggin, J.F. Structure of the Molecule of 12-Phosphotungstic. Nature 1933, 131, 908–909. [Google Scholar] [CrossRef]
- Rhule, J.T.; Hill, C.L.; Judd, D.A.; Schinazi, R.F. Polyoxometalates in Medicine. Chem. Rev. 1998, 98, 327–358. [Google Scholar] [CrossRef] [PubMed]
- Prudent, R.; Moucadel, V.; Laudet, B.; Barette, C.; Lafanechère, L.; Hasenknopf, B.; Li, J.; Bareyt, S.; Lacôte, E.; Thorimbert, S.; Malacria, M.; Gouzerh, P.; Cochet, C. Identification of polyoxometalates as nanomolar noncompetitive inhibitors of protein kinase CK2. Chem. Biol. 2008, 15, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Geletii, Y.V.; Zhao, C.; Vickers, J.W.; Zhu, G.; Luo, Z.; Song, J.; Lian, T.; Musaev, D.G.; Hill, C.L. Polyoxometalate water oxidation catalysts and the production of green fuel. Chem. Soc. Rev. 2012, 41, 7572–7589. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, N.; Kamata, K. Catalytic oxidation of hydrocarbons with hydrogen peroxide by vanadium-based polyoxometalates. Coord. Chem. Rev. 2011, 255, 2358–2370. [Google Scholar] [CrossRef]
- Mitchell, S.G.; de la Fuente, J.M. The synergistic behavior of polyoxometalates and metal nanoparticles: from synthetic approaches to functional nanohybrid materials. J. Mater. Chem. 2012, 22, 18091–18100. [Google Scholar] [CrossRef]
- Mondloch, J.E.; Bayram, E.; Finke, R.G. A review of the kinetics and mechanisms of formation of supported-nanoparticle heterogeneous catalysts. J. Mol. Catal. A 2012, 355, 1–38. [Google Scholar] [CrossRef]
- Oms, O.; Dolbecq, A.; Mialane, P. Diversity in structures and properties of 3d-incorporating polyoxotungstates. Chem. Soc. Rev. 2012, 41, 7497–7536. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.-T.; Yang, G.-Y. Recent advances in paramagnetic-TM-substituted polyoxometalates (TM = Mn, Fe, Co, Ni, Cu). Chem. Soc. Rev. 2012, 41, 7623–7646. [Google Scholar] [CrossRef] [PubMed]
- Bünzli, J.-C.G.; Piguet, C. Taking advantage of luminescent lanthanide ions. Chem. Soc. Rev. 2005, 34, 1048–1077. [Google Scholar] [CrossRef] [PubMed]
- Binnemans, K. Lanthanide-based luminescent hybrid materials. Chem. Rev. 2009, 109, 4283–4374. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, G.A. The position of cerium in the periodic system and the complex molybdates of tetravalent cerium, Atti della Accademia Nazionale dei Lincei, Classe di Scienze Fisiche. Mat. Nat. Rend. 1914, 23, 805–812. (In Italian) [Google Scholar]
- Peacock, R.D.; Weakley, T.J.R. Heteropolytungstate complexes of the lanthanide elements. Part I. Preparation and reactions. J. Chem. Soc. A 1971, 1836–1839. [Google Scholar] [CrossRef]
- Granadeiro, C.M.; de Castro, B.; Balula, S.S.; Cunha-Silva, L. Lanthanopolyoxometalates: From the structure of polyanions to the design of functional materials. Polyhedron 2013, 52, 10–24. [Google Scholar] [CrossRef]
- Sadakane, M.; Dickman, M.; Pope, M. Controlled Assembly of Polyoxometalate Chains from Lacunary Building Blocks and Lanthanide-Cation Linkers. Angew. Chem. Int. Ed. 2000, 39, 2914–2916. [Google Scholar] [CrossRef]
- Luo, Q.; Howell, R.C.; Bartis, J.; Dankova, M.; Horrocks, W.D.; Rheingold, A.L.; Francesconi, L.C. Lanthanide Complexes of [α-2-P2W17O61]10− : Solid State and Solution Studies. Inorg. Chem. 2002, 41, 6112–6117. [Google Scholar] [CrossRef] [PubMed]
- Gaunt, A.J.; May, I.; Sarsfield, M.J.; Collison, D.; Helliwell, M.; Denniss, I.S. A rare structural characterisation of the phosphomolybdate lacunary anion, [PMo11O39]7?. Crystal structures of the Ln(III) complexes, (NH4)11[Ln(PMo11O39)2] 16H2O (Ln = CeIII, SmIII, DyIII or LuIII) Electronic supplementary information (ESI) available: IR. Dalton Trans. 2003, 2767–2771. [Google Scholar] [CrossRef]
- Bassil, B.S.; Dickman, M.H.; von der Kammer, B.; Kortz, U. The monolanthanide-containing silicotungstates [Ln(β2-SiW11O39)2]13− (Ln = La, Ce, Sm, Eu, Gd, Tb, Yb, Lu): a synthetic and structural investigation. Inorg. Chem. 2007, 46, 2452–2458. [Google Scholar] [CrossRef] [PubMed]
- Ostuni, A.; Bachman, R.E.; Pope, M.T. Multiple Diastereomers of [Mn+ (αm-P2W17O61)2](20−n)− (M = UIV , ThIV , CeIII ; m = 1, 2). Syn- and Anti-Conformations of the Polytungstate Ligands in α1α1, α1α2 and α2α2 Complexes. J. Clust. Sci. 2003, 14, 431–446. [Google Scholar] [CrossRef]
- Niu, J.; Wang, K.; Chen, H.; Zhao, J.; Ma, P.; Wang, J.; Li, M.; Bai, Y.; Dang, D. Assembly Chemistry between Lanthanide Cations and Monovacant Keggin Polyoxotungstates: Two Types of Lanthanide Substituted Phosphotungstates [{(α-PW11O39H)Ln(H2O)3}2]6− and [{(α-PW11O39)Ln(H2O)(η2,μ-1,1)-CH3COO}2]10−. Cryst. Growth Des. 2009, 9, 4362–4372. [Google Scholar] [CrossRef]
- Sadakane, M.; Ostuni, A.; Pope, M.T. Formation of 1:1 and 2:2 complexes of Ce(III) with the heteropolytungstate anion α2-[P2W17O61]10−, and their interaction with proline. The structure of [Ce2(P2W17O61)2(H2O)8]14−. Dalt. Trans. 2002, 63–67. [Google Scholar] [CrossRef]
- Zhang, C.; Bensaid, L.; McGregor, D.; Fang, X.; Howell, R.C.; Burton-Pye, B.; Luo, Q.; Todaro, L.; Francesconi, L.C. Influence of the Lanthanide Ion and Solution Conditions on Formation of Lanthanide Wells–Dawson Polyoxotungstates. J. Clust. Sci. 2006, 17, 389–425. [Google Scholar] [CrossRef]
- Kortz, U. Rare-Earth Substituted Polyoxoanions: [{La(CH3COO)(H2O)2 (2-P2W17O61)}2]16− and [{Nd(H2O)3(α2-P2W17O61)}2]14−. J. Clust. Sci. 2003, 14, 205–214. [Google Scholar] [CrossRef]
- Mialane, P.; Dolbecq, A.; Marrot, J.; Sécheresse, F. Oligomerization of Yb(III)-substituted Dawson polyoxotungstates by oxalato ligands. Inorg. Chem. Commun. 2005, 8, 740–742. [Google Scholar] [CrossRef]
- Dickman, M.H.; Gama, G.J.; Kim, K.; Pope, M.T. The structures of europium(III)- and uranium(IV) derivatives of [P5W30O110]15−: Evidence for “cryptohydration”. J. Clust. Sci. 1996, 7, 567–583. [Google Scholar] [CrossRef]
- Naruke, H.; Yamase, T. Crystal structure of K18.5H1.5[Ce3(CO3)(SbW9O33)(W5O18)3] 14H2O. J. Alloys Compd. 1998, 268, 100–106. [Google Scholar] [CrossRef]
- Müller, A.; Beugholt, C.; Bögge, H.; Schmidtmann, M. Influencing the Size of Giant Rings by Manipulating Their Curvatures: Na6[Mo120O366(H2O)48H12{Pr(H2O)5}6] (∼200H2O) with Open Shell Metal Centers at the Cluster Surface. Inorg. Chem. 2000, 39, 3112–3113. [Google Scholar] [CrossRef] [PubMed]
- Julião, D.; Fernandes, D.M.; Cunha-Silva, L.; Ananias, D.; Balula, S.S.; Freire, C. Sandwich lanthano-silicotungstates: Structure, electrochemistry and photoluminescence properties. Polyhedron 2013, 52, 308–314. [Google Scholar] [CrossRef]
- VanPelt, C.E.; Crooks, W.J.; Choppin, G.R. Stability constant determination and characterization of the complexation of trivalent lanthanides with polyoxometalates. Inorg. Chim. Acta 2003, 346, 215–222. [Google Scholar] [CrossRef]
- Yamase, T.; Ozeki, T.; Ueda, K. Structure of NaSr4[EuW10O36] 34.5H2O. Acta Cryst. 1993, C49, 1572–1574. [Google Scholar] [CrossRef]
- Bartis, J.; Sukal, S.; Dankova, M.; Kraft, E.; Kronzon, R.; Blumenstein, M.; Francesconi, L.C. Lanthanide complexes of polyoxometalates: characterization by tungsten-183 and phosphorus-31 nuclear magnetic resonance spectroscopy. Dalton Trans. 1997, 1937–1944. [Google Scholar] [CrossRef]
- Klymenko, O.V.; Svir, I.; Amatore, C. Molecular electrochemistry and electrocatalysis: a dynamic view. Mol. Phys. 2014, 112, 1273–1283. [Google Scholar] [CrossRef]
- Cadot, E.; Thouvenot, R.; Teze, A.; Herve, G. Syntheses and multinuclear NMR characterizations of alpha-[SiMo2W9O39]8− and alpha-[SiMo3−xVxW9O40](4+x)− (x = 1, 2) heteropolyoxometalates. Inorg. Chem. 1992, 31, 4128–4133. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS, program for scaling and correction of area detector data; University of Göttingen: Germany, 1997. [Google Scholar]
- Blessing, R.H. An empirical correction for absorption anisotropy. Acta Cryst. 1995, C51, 33–38. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELX-TL, version 5.03, Software Package for the Crystal Structure Determination; Siemens Analytical X-ray Instrument Division: Madison, WI, USA, 1994.
- Vilà, N.; Aparicio, P.A.; Sécheresse, F.; Poblet, J.M.; López, X.; Mbomekallé, I.M. Electrochemical behavior of α1/α2-[Fe(H2O)P2W17O61]7− isomers in solution: experimental and DFT studies. Inorg. Chem. 2012, 51, 6129–6138. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parent, L.; De Oliveira, P.; Teillout, A.-L.; Dolbecq, A.; Haouas, M.; Cadot, E.; Mbomekallé, I.M. Synthesis and Characterisation of the Europium (III) Dimolybdo-Enneatungsto-Silicate Dimer, [Eu(α-SiW9Mo2O39)2]13−. Inorganics 2015, 3, 341-354. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics3030341
Parent L, De Oliveira P, Teillout A-L, Dolbecq A, Haouas M, Cadot E, Mbomekallé IM. Synthesis and Characterisation of the Europium (III) Dimolybdo-Enneatungsto-Silicate Dimer, [Eu(α-SiW9Mo2O39)2]13−. Inorganics. 2015; 3(3):341-354. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics3030341
Chicago/Turabian StyleParent, Loïc, Pedro De Oliveira, Anne-Lucie Teillout, Anne Dolbecq, Mohamed Haouas, Emmanuel Cadot, and Israël M. Mbomekallé. 2015. "Synthesis and Characterisation of the Europium (III) Dimolybdo-Enneatungsto-Silicate Dimer, [Eu(α-SiW9Mo2O39)2]13−" Inorganics 3, no. 3: 341-354. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics3030341