
Water Oxidation by Ru-Polyoxometalate Catalysts: Overpotential Dependency on the Number and Charge of the Metal Centers

Abstract
:1. Introduction
2. Computational Methods
2.1. Electronic Structure Calculations
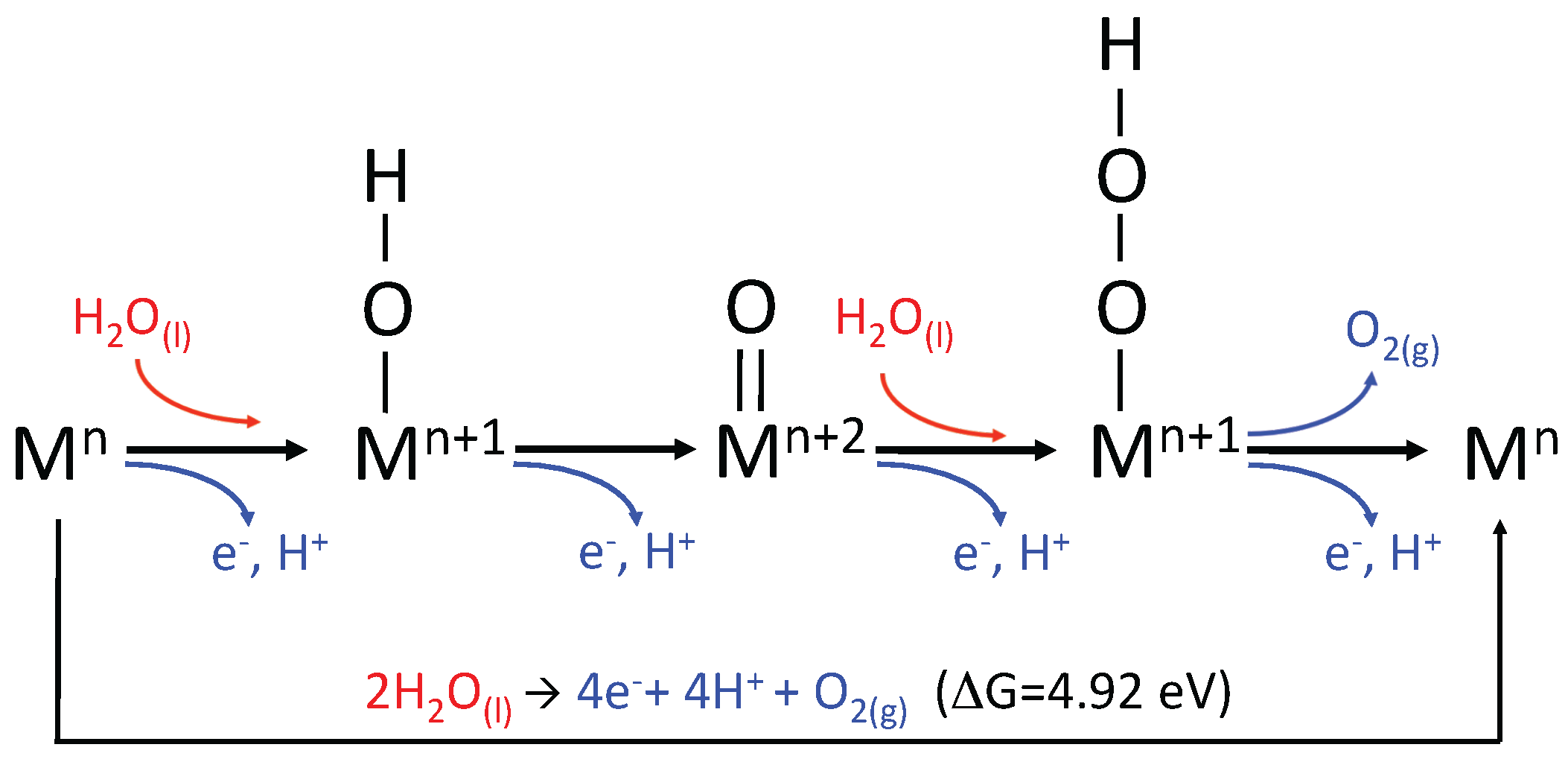
2.2. The Water Oxidation Cycle

2.3. Reaction Thermodynamics
3. Results and Discussion
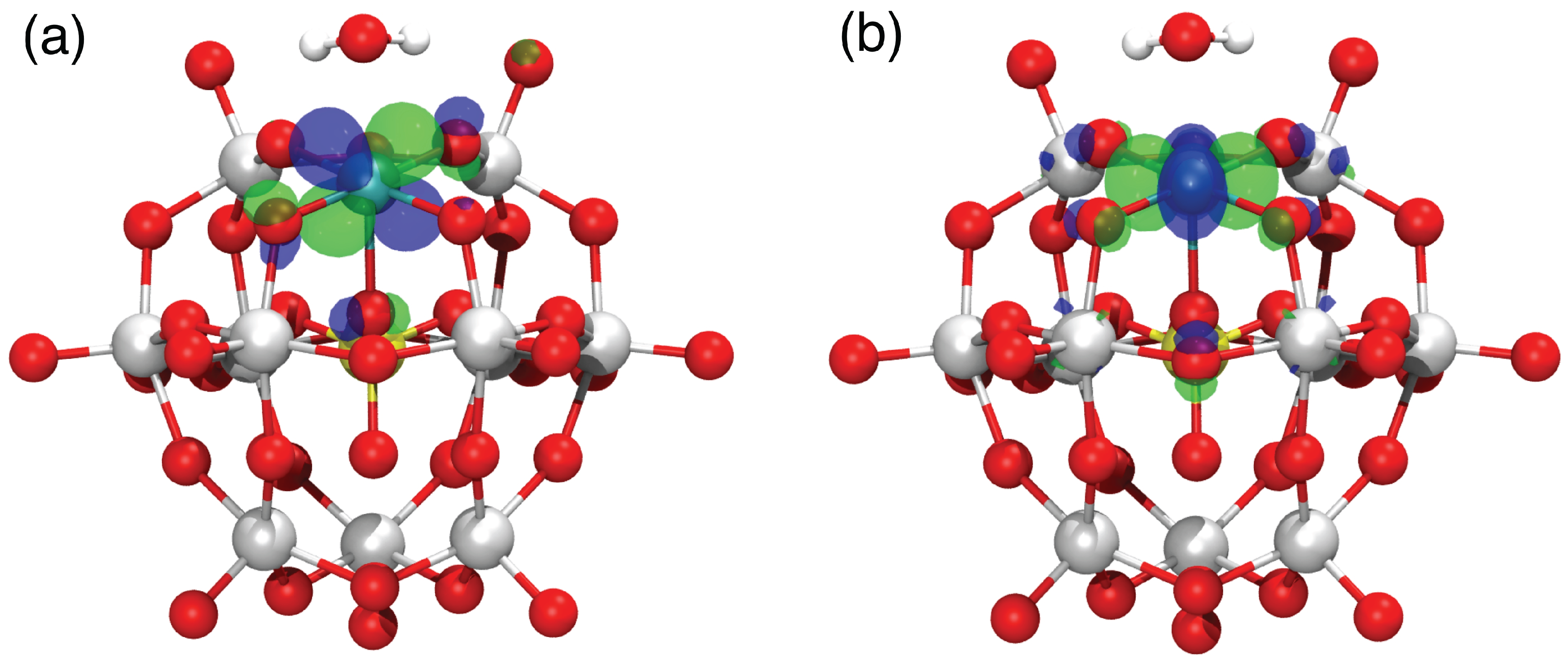
3.1. Electronic Structure of Ru-POM: Dependency on the Ru Oxidation State


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OS | Conf. | Q | 2S + 1 | (PBE) | μ(B3LYP) |
|---|---|---|---|---|---|
| Ru(III) | −5 | 2 | 0.74 | 0.81 | |
| Ru(IV) | −4 | 3 | 1.31 | 1.48 | |
| Ru(V) | −3 | 2 | 0.76 | 0.67 |
3.2. Thermodynamics of the Water Oxidation Cycle
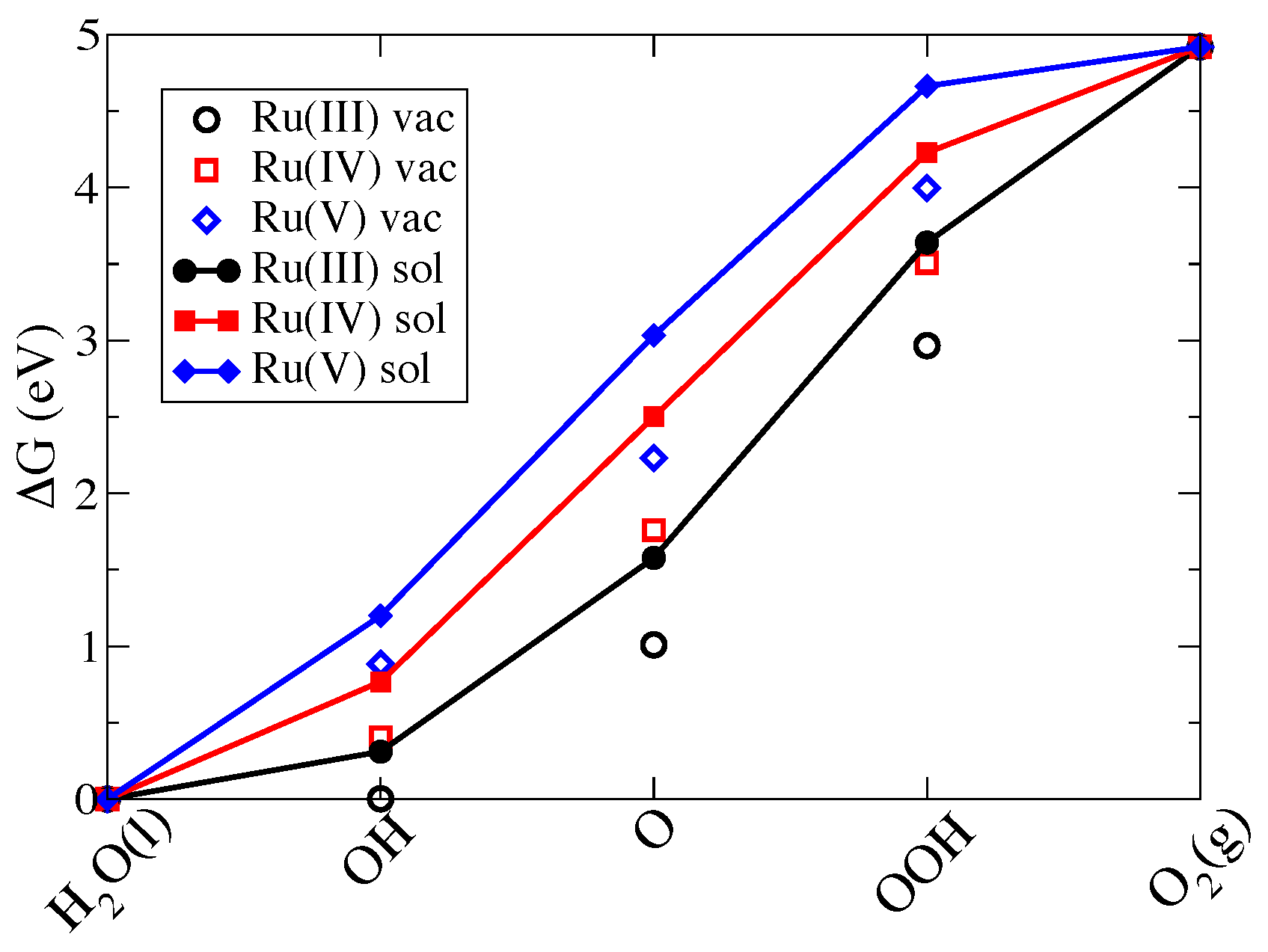
3.2.1. Ru-POM and the Effect of Ru Oxidation State

| Intermediate | In Vacuum | In Solution | ||||||
|---|---|---|---|---|---|---|---|---|
| Ru(III) | Ru(IV) | Ru(V) | Ru(IV) | Ru(III) | Ru(IV) | Ru(V) | Ru(IV) | |
| HO(l) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| *OH | 0.00 | 0.40 | 0.89 | 0.78 | 0.31 | 0.77 | 1.20 | 0.72 |
| *O | 1.01 | 1.76 | 2.23 | 2.50 | 1.58 | 2.50 | 3.03 | 2.65 |
| *OOH | 2.97 | 3.01 | 4.00 | 4.20 | 3.64 | 4.28 | 4.66 | 4.35 |
| O(g) | 4.92 | 4.92 | 4.92 | 4.92 | 4.92 | 4.92 | 4.92 | 4.92 |
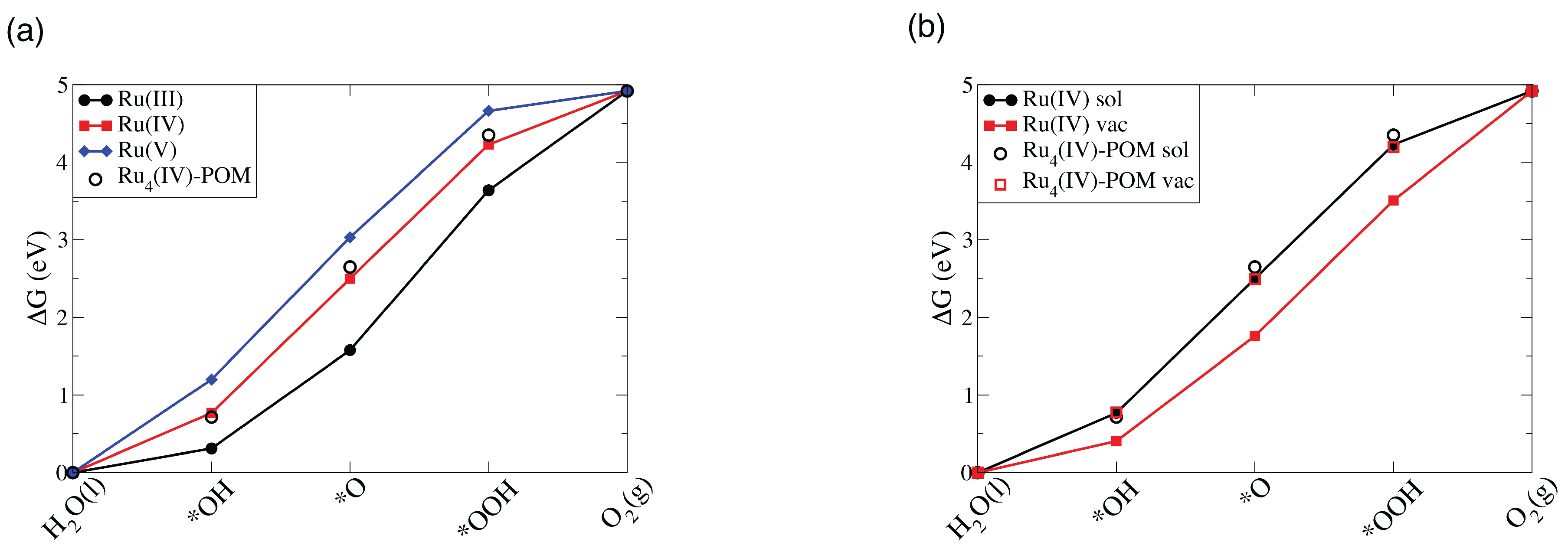
3.2.2. Ru-POM vs. Ru-POM: The Effect of the Number of Ru Centers

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix
A. Estimating the Error Bar in the Computed Thermodynamics
| Intermediate | CP2K | Gaussian |
|---|---|---|
| HO(l) | 0.00 | 0.00 |
| *OH | −0.05 | 0.00 |
| *O | 1.17 | 1.01 |
| *OOH | 2.98 | 2.97 |
| O(g) | 4.92 | 4.92 |
References
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef] [PubMed]
- Balzani, V.; Credi, A.; Venturi, M. Photochemical conversion of solar energy. ChemSusChem 2008, 1, 26–58. [Google Scholar] [CrossRef] [PubMed]
- Suga, M.; Akita, F.; Hirata, K.; Ueno, G.; Murakami, H.; Nakajima, Y.; Shimizu, T.; Yamashita, K.; Yamamoto, M.; Ago, H.; et al. Native structure of photosystem II at 1.95 A resolution viewed by femtosecond X-ray pulses. Nature 2015, 517, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Kanan, M.W.; Nocera, D.G. In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+. Science 2008, 321, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Karkas, M.D.; Verho, O.; Johnston, E.V.; Pkermark, B. Artificial Photosynthesis: Molecular Systems for Catalytic Water Oxidation. Chem. Rev. 2014, 114, 11863–12001. [Google Scholar] [CrossRef] [PubMed]
- Gersten, S.W.; Samuels, G.J.; Meyer, T.J. Catalytic oxidation of water by an oxo-bridged ruthenium dimer. J. Am. Chem. Soc. 1982, 104, 4029–4030. [Google Scholar] [CrossRef]
- Dau, H.; Limberg, C.; Reier, T.; Risch, M.; Roggan, S.; Strasser, P. The Mechanism of Water Oxidation: From Electrolysis via Homogeneous to Biological Catalysis. ChemCatChem 2010, 2, 724–761. [Google Scholar] [CrossRef]
- Limburg, B.; Bouwman, E.; Bonnet, S. Molecular water oxidation catalysts based on transition metals and their decomposition pathways. Coord. Chem. Rev. 2012, 256, 1451–1467. [Google Scholar] [CrossRef]
- Sartorel, A.; Carraro, M.; Scorrano, G.; Zorzi, R.D.; Geremia, S.; McDaniel, N.; Bernhard, S.; Bonchio, M. Polyoxometalate embedding of a tetraruthenium(IV)-oxo-core by template-directed metalation of [γ-SiW10O36]8-: a totally inorganic oxygen-evolving catalyst. J. Am. Chem. Soc. 2008, 130, 5006–5007. [Google Scholar] [CrossRef] [PubMed]
- Geletii, Y.V.; Botar, B.; Kögerler, P.; Hillesheim, D.A.; Musaev, D.G.; Hill, C.G. An all-inorganic, stable, and highly active tetraruthenium homogeneous catalyst for water oxidation. Agew. Chem. Int. Ed. 2008, 47, 3896–3899. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Tan, J.M.; Besson, C.; Geletii, Y.V.; Musaev, D.G.; Kuznetsov, A.E.; Luo, Z.; Hardcastle, K.I.; Hill, C.L. A Fast Soluble Carbon-Free Molecular Water Oxidation Catalyst Based on Abundant Metals. Science 2010, 328, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Al-Oweini, R.; Sartorel, A.; Bassil, B.S.; Natali, M.; Berardi, S.; Scandola, F.; Kortz, U.; Bonchio, M. Photocatalytic Water Oxidation by a Mixed-Valent MnIII3MnIVO3 Manganese Oxo Core that Mimics the Natural Oxygen-Evolving Center. Agew. Chem. Int. Ed. 2014, 53, 11182–11185. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, N.D.; Coughlin, F.J.; Tinker, L.L.; Bernhard, S. Cyclometalated iridium(III) Aquo complexes: efficient and tunable catalysts for the homogeneous oxidation of water. J. Am. Chem. Soc. 2008, 130, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Wasylenko, D.J.; Palmer, R.D.; Berlinguette, C.P. Homogeneous water oxidation catalysts containing a single metal site. Chem. Commun. 2013, 49, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Hong, D.; Suenobu, T.; Yamaguchi, S.; Ogura, T.; Fukuzumi, S. Catalytic mechanism of water oxidation with single-site ruthenium-heteropolytungstate complexes. J. Am. Chem. Soc. 2011, 133, 11605–11613. [Google Scholar] [CrossRef] [PubMed]
- Lang, Z.L.; Yang, G.C.; Ma, N.N.; Wen, S.Z.; Yan, L.K.; Guan, W.; Su, Z.M. DFT characterization on the mechanism of water splitting catalyzed by single-Ru-substituted polyoxometalates. Dalton Trans. 2013, 42, 10617–10625. [Google Scholar] [CrossRef] [PubMed]
- Piccinin, S.; Sartorel, A.; Bonchio, M.; Fabris, S. Water oxidation surface mechanisms replicated by a totally inorganic tetraruthenium-oxo molecular complex. Proc. Natl. Acad. Sci. USA 2013, 110, 4917–4922. [Google Scholar] [CrossRef] [PubMed]
- Romain, S.; Vigara, L.; Llobet, A. Oxygen-Oxygen Bond Formation Pathways Promoted by Ruthenium Complexes. Acc. Chem. Res. 2009, 42, 1944–1953. [Google Scholar] [CrossRef] [PubMed]
- Piccinin, S.; Fabris, S. First principles study of water oxidation catalyzed by a tetraruthenium-oxo core embedded in polyoxometalate ligands. Phys. Chem. Chem. Phys. 2011, 13, 7666–7674. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Densityfunctional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Stephens, P.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623. [Google Scholar] [CrossRef]
- Kang, R.; Yao, J.; Chen, H. Are DFT Methods Accurate in Mononuclear Ruthenium-Catalyzed Water Oxidation? An ab Initio Assessment. J. Chem. Theory Comput. 2013, 9, 1872–1879. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982. [Google Scholar] [CrossRef]
- Kwapien, K.; Piccinin, S.; Fabris, S. Energetics of Water Oxidation Catalyzed by Cobalt Oxide Nanoparticles: Assessing the Accuracy of DFT and DFT+U Approaches against Coupled Cluster Methods. J. Phys. Chem. Lett. 2013, 4, 4223–4230. [Google Scholar] [CrossRef] [PubMed]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270. [Google Scholar] [CrossRef]
- Andrae, D.; Haussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chem. Acc. 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the first row transition elements. J. Chem. Phys 1987, 86, 866. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comp. Phys. Comm. 2005, 167, 103–128. [Google Scholar] [CrossRef] [Green Version]
- Klamt, A.; Schuurmann, G. COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 799–805. [Google Scholar] [CrossRef]
- Valiev, M.; Bylaska, E.; Govind, N.; Kowalski, K.; Straatsma, T.; Dam, H.V.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.; et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comp. Phys. Comm. 2010, 181, 1477–1489. [Google Scholar] [CrossRef]
- Liu, F.; Concepcion, J.J.; Jurss, J.W.; Cardolaccia, T.; Templeton, J.L.; Meyer, T.J. Mechanisms of water oxidation from the blue dimer to photosystem II. Inorg. Chem. 2008, 47, 1727–1752. [Google Scholar] [CrossRef] [PubMed]
- Rossmeisl, J.; Qu, Z.W.; Zhu, H.; Kroes, G.J.; Nørskov, J.K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 2007, 607, 83–89. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jonsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B. 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Logadottir, A.; Nørskov, J.K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Nørskov, J.K.; Taylor, C.D.; Janik, M.J.; Neurock, M. Calculated phase diagrams for the electrochemical oxidation and reduction of water over Pt(111). J. Phys. Chem. B 2006, 110, 21833–21839. [Google Scholar] [CrossRef] [PubMed]
- Stull, D.; Prophet, H. JANAF Thermochemical Tables, 2nd ed.; U.S. National Bureau of Standards: Washington, DC, USA, 1971. [Google Scholar]
- Cheng, J.; Liu, X.; Kattirtzi, J.A.; VandeVondele, J.; Sprik, M. Aligning Electronic and Protonic Energy Levels of Proton-Coupled Electron Transfer in Water Oxidation on Aqueous TiO2. Angew. Chem. 2014, 126, 12242–12246. [Google Scholar] [CrossRef]
- Man, I.C.; Su, H.Y.; Calle-Vallejo, F.; Hansen, H.A.; Martinez, J.I.; Inoglu, N.G.; Kitchin, J.; Jaramillo, T.F.; Norskov, J.K.; Rossmeisl, J. Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces. ChemCatChem 2011, 3, 1159–1165. [Google Scholar] [CrossRef]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piccinin, S.; Fabris, S. Water Oxidation by Ru-Polyoxometalate Catalysts: Overpotential Dependency on the Number and Charge of the Metal Centers. Inorganics 2015, 3, 374-387. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics3030374
Piccinin S, Fabris S. Water Oxidation by Ru-Polyoxometalate Catalysts: Overpotential Dependency on the Number and Charge of the Metal Centers. Inorganics. 2015; 3(3):374-387. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics3030374
Chicago/Turabian StylePiccinin, Simone, and Stefano Fabris. 2015. "Water Oxidation by Ru-Polyoxometalate Catalysts: Overpotential Dependency on the Number and Charge of the Metal Centers" Inorganics 3, no. 3: 374-387. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics3030374