
Synthesis and Reactivity of a Cerium(III) Scorpionate Complex Containing a Redox Non-Innocent 2,2′-Bipyridine Ligand

Abstract
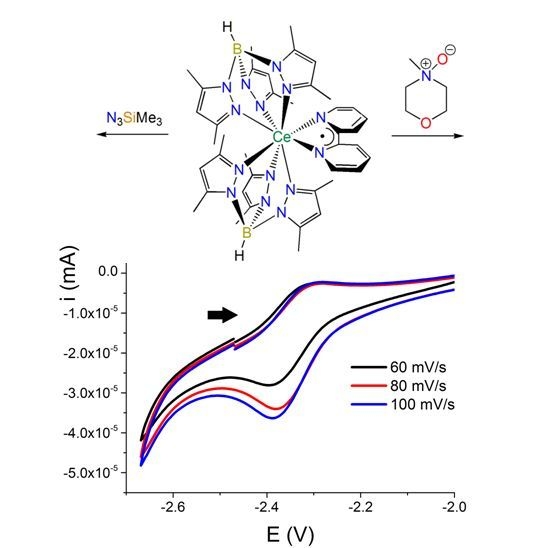
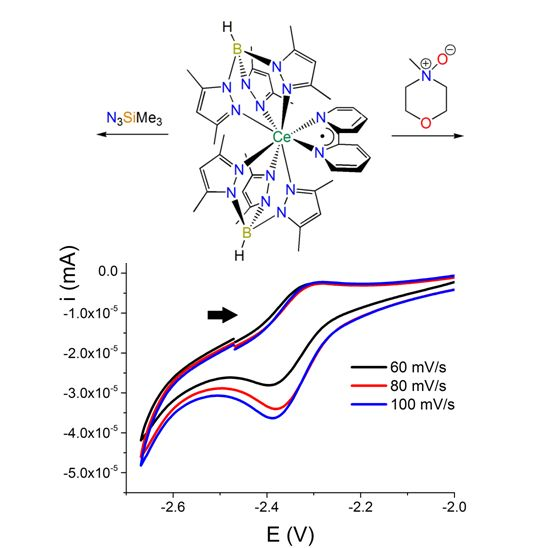
:
1. Introduction
2. Results and Discussion
2.1. Preparation of a Ce(II) Synthon
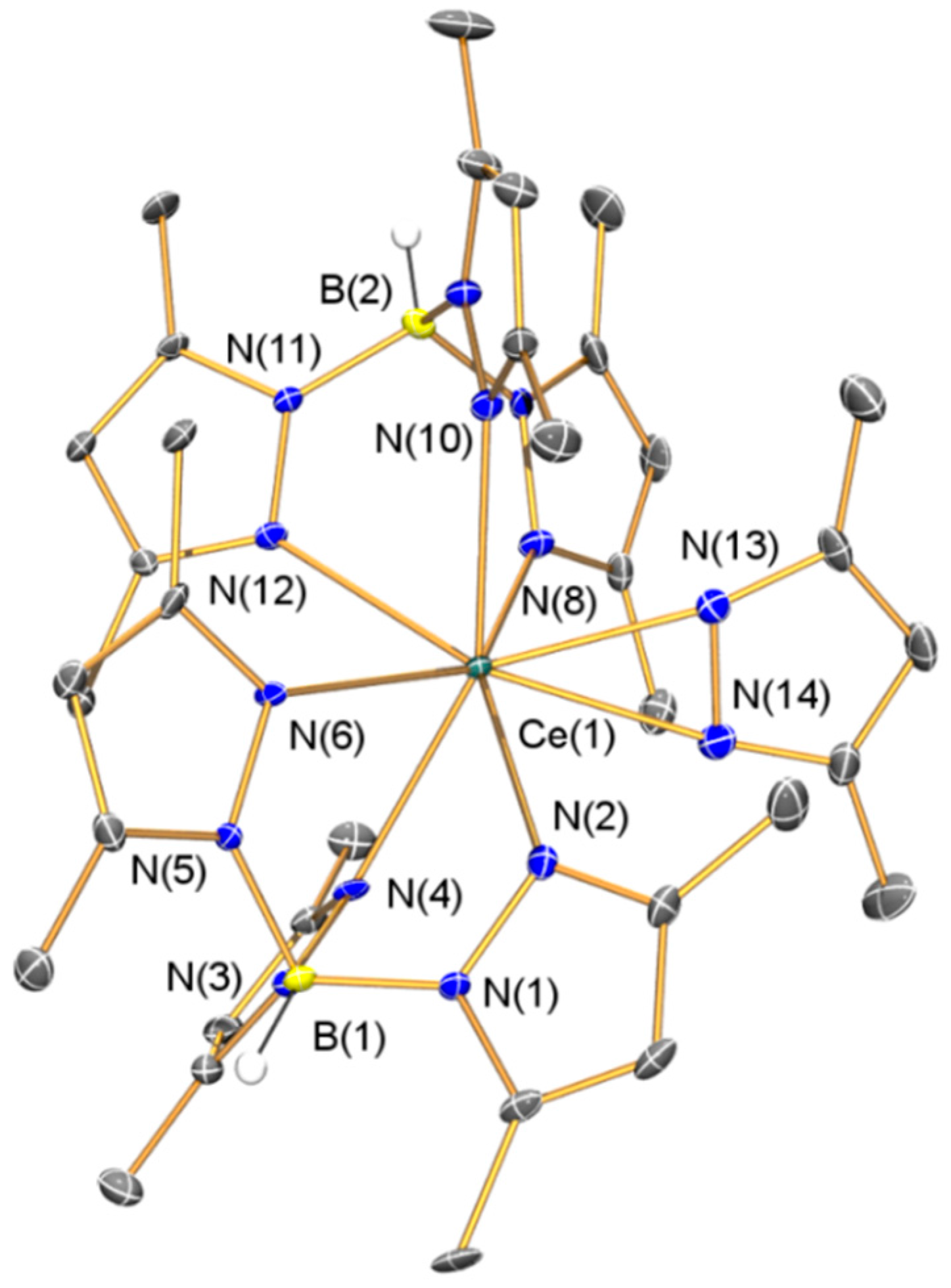
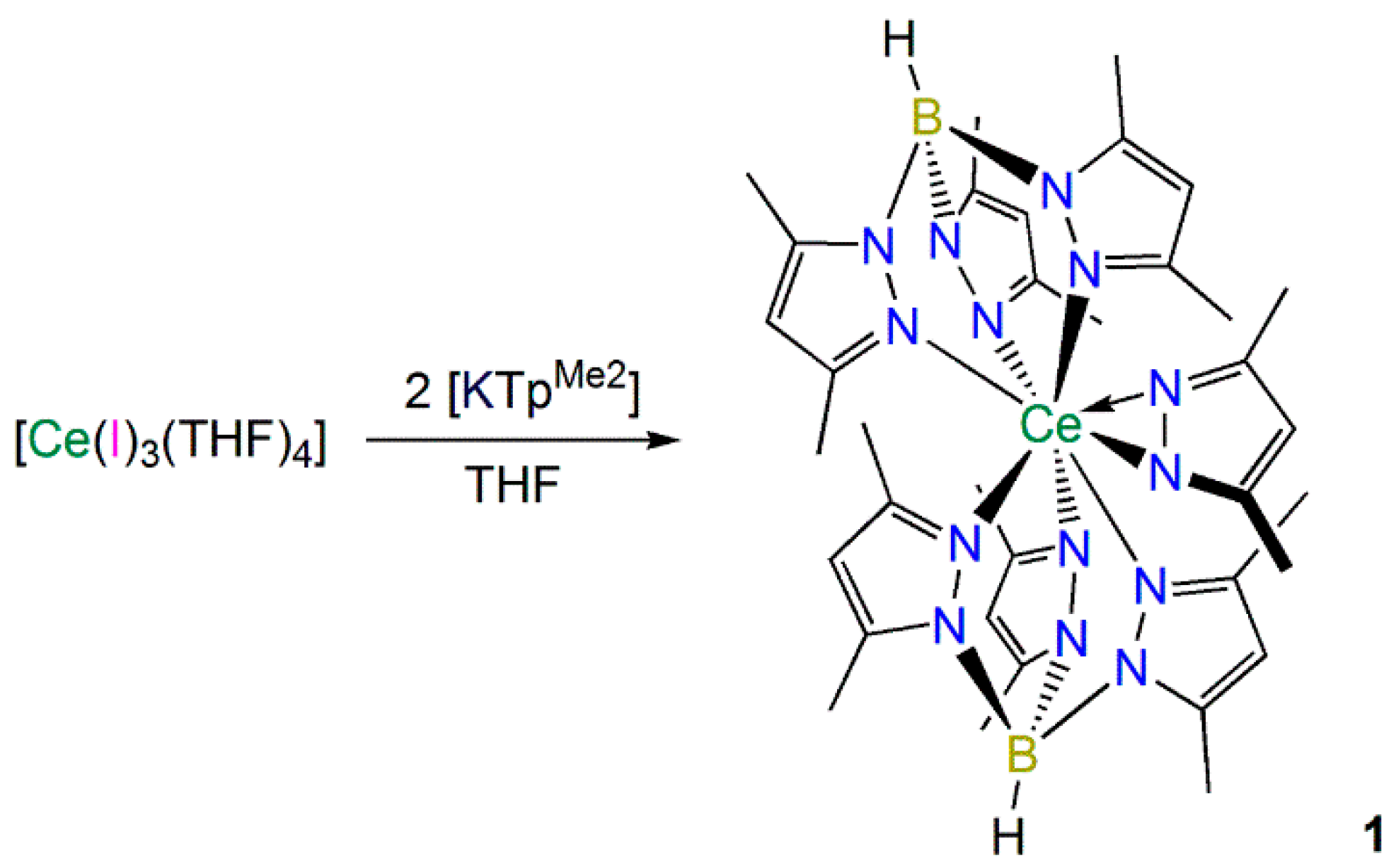
2.1.1. Synthesis and Structural Characterization of [Ce(TpMe2)2(κ2-dmpz)] (1)


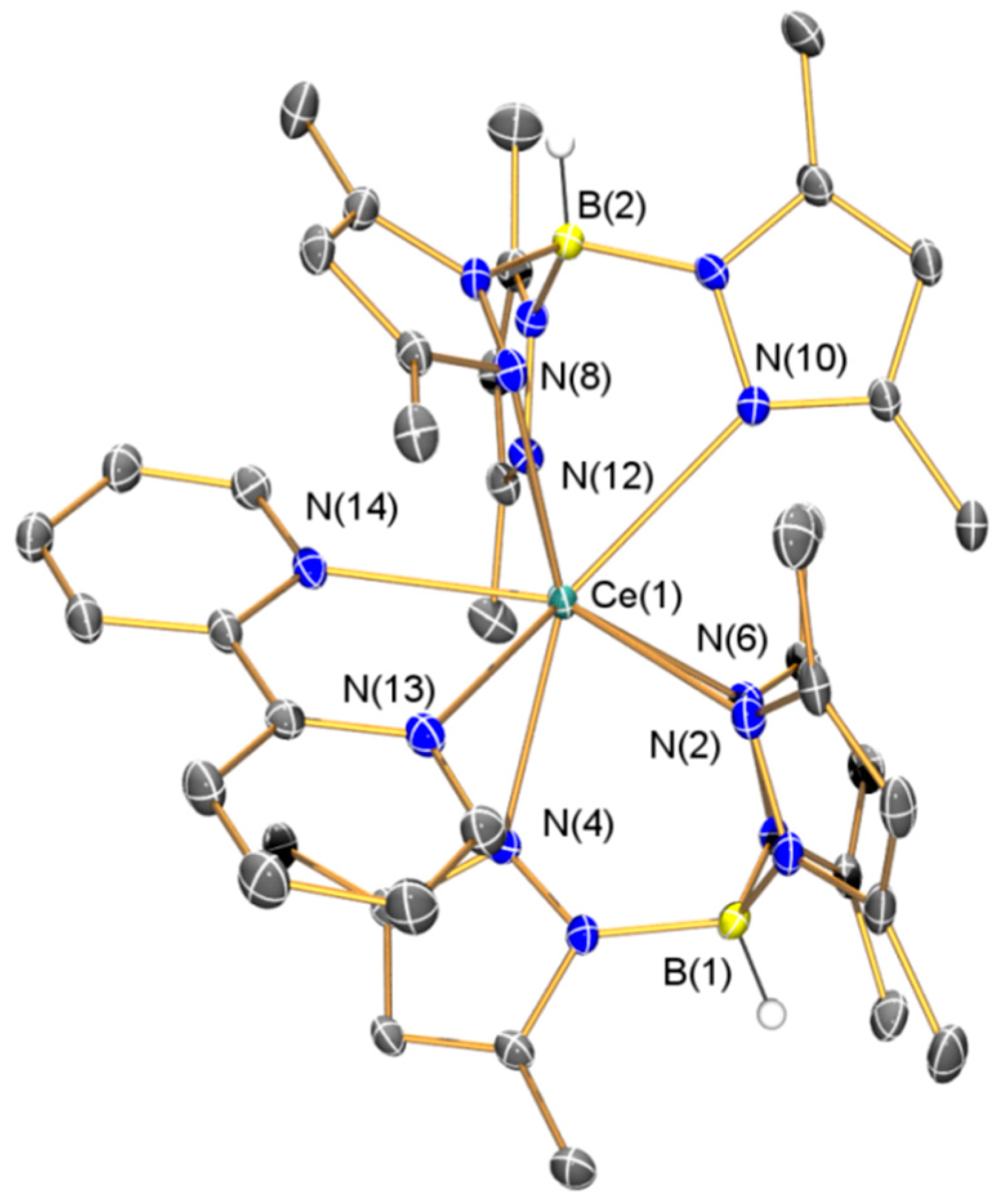
2.1.2. Synthesis and Structural Characterization of [Ce(TpMe2)(bipy)] (2)


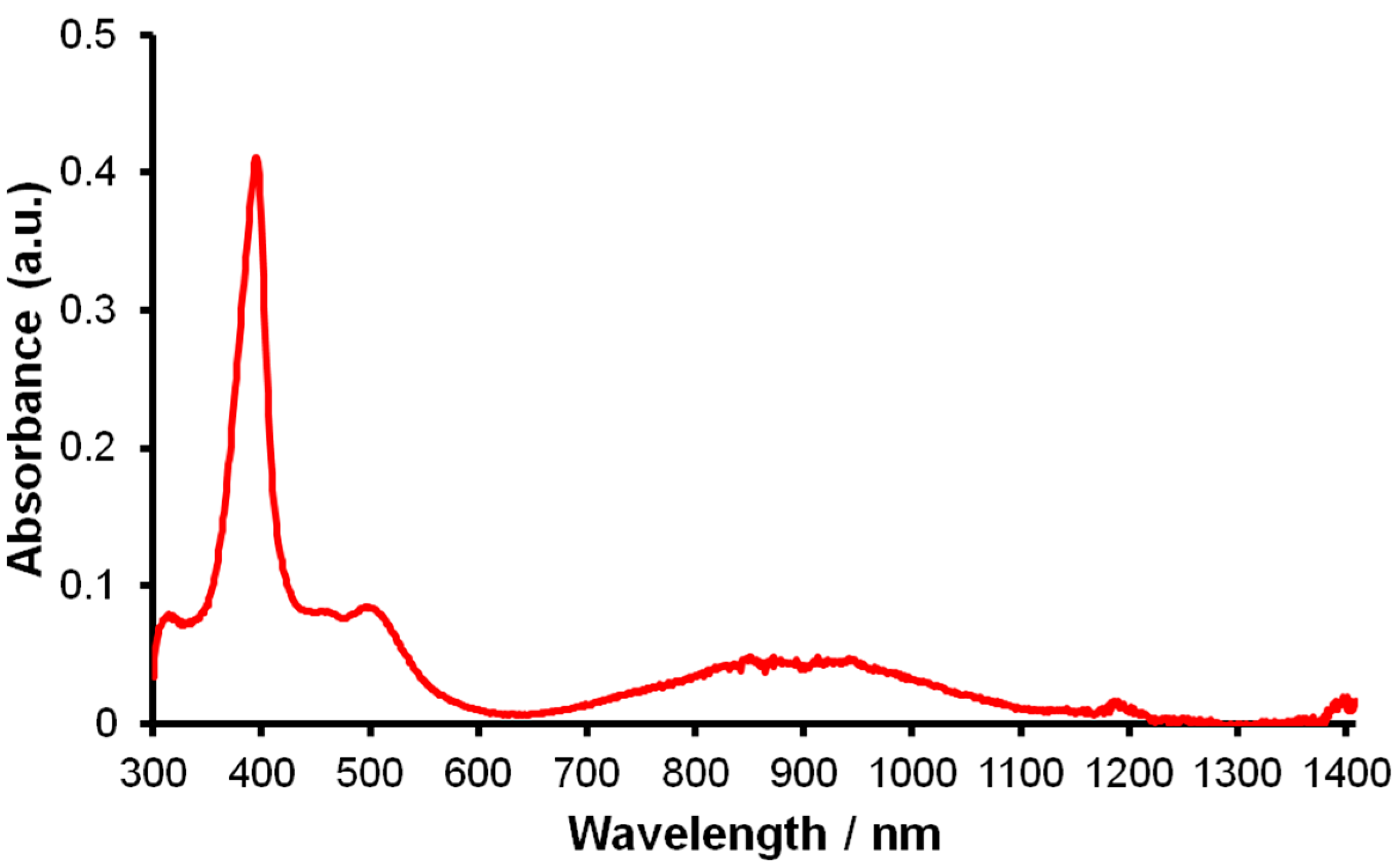
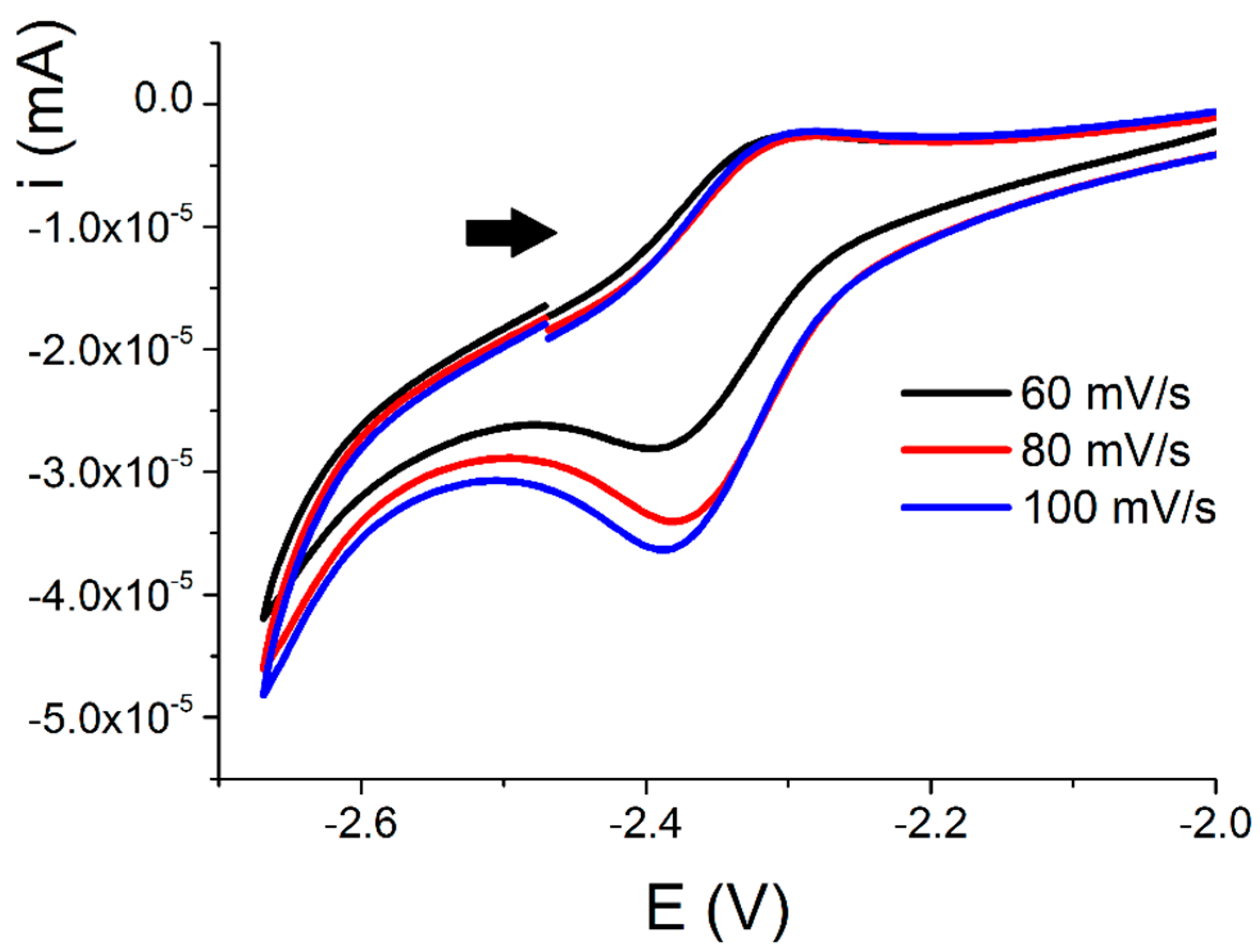
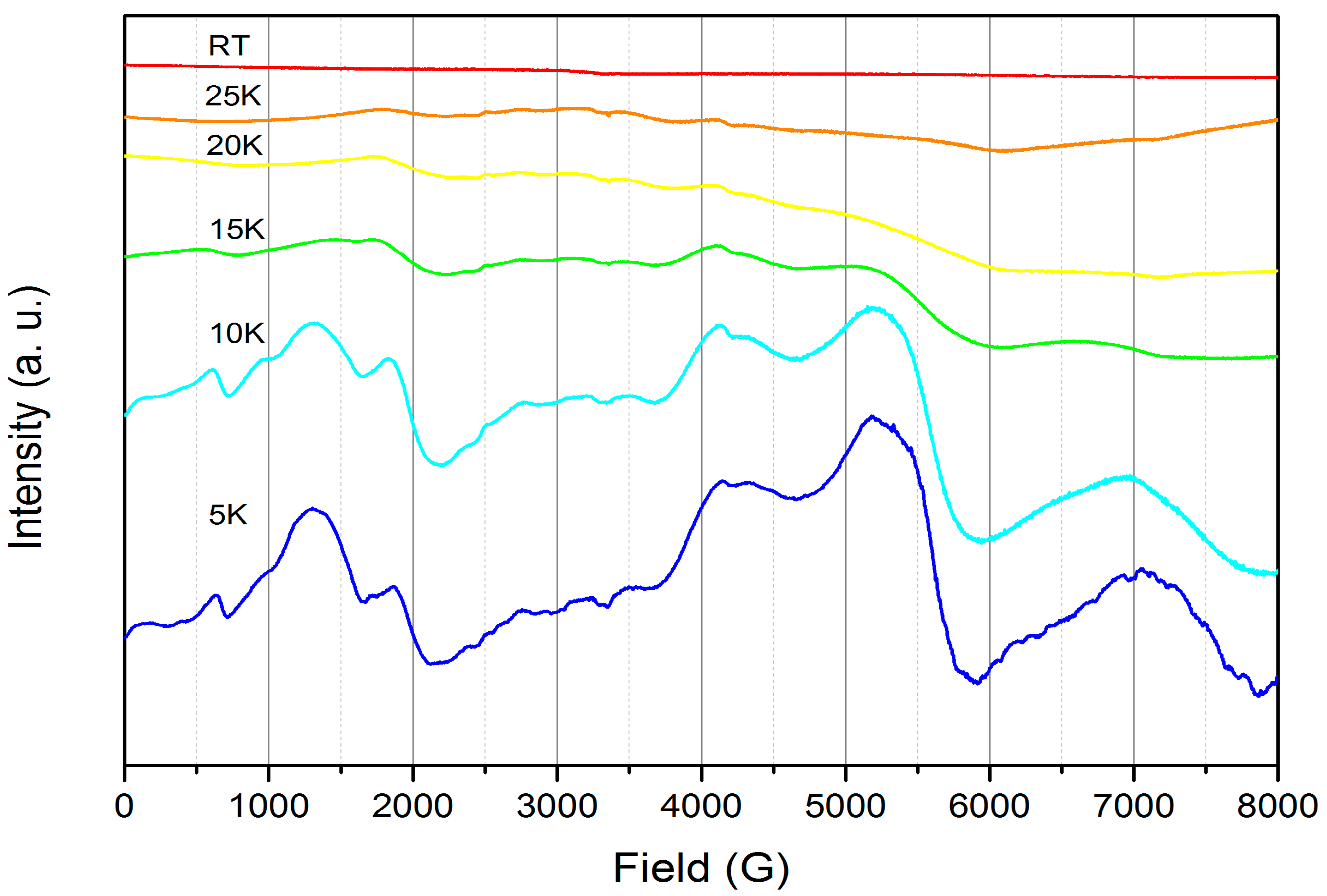
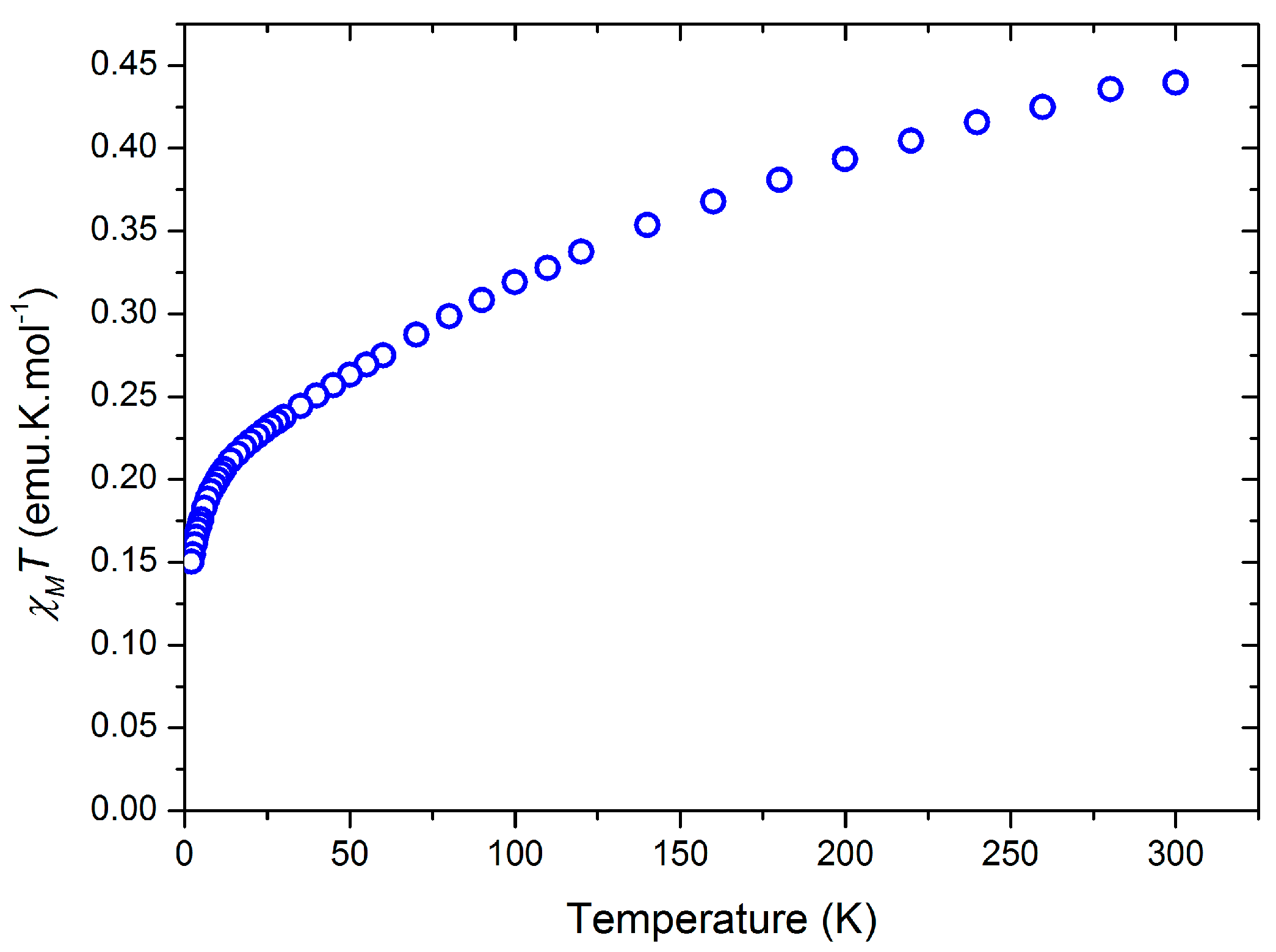
2.1.3. Further Characterization of [Ce(TpMe2)(bipy)] (2)





2.2. Reactivity of a Ce(II) Synthon
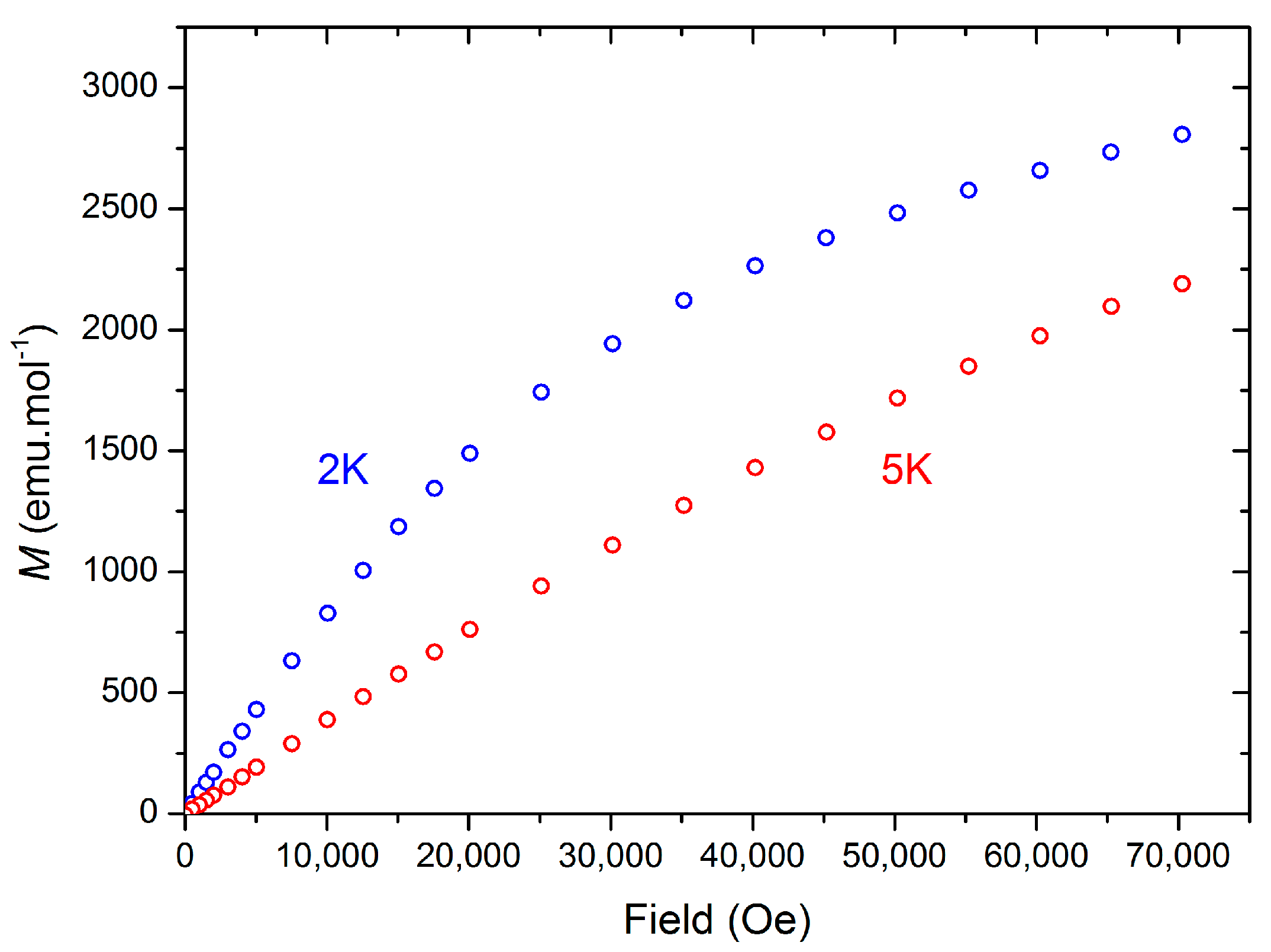
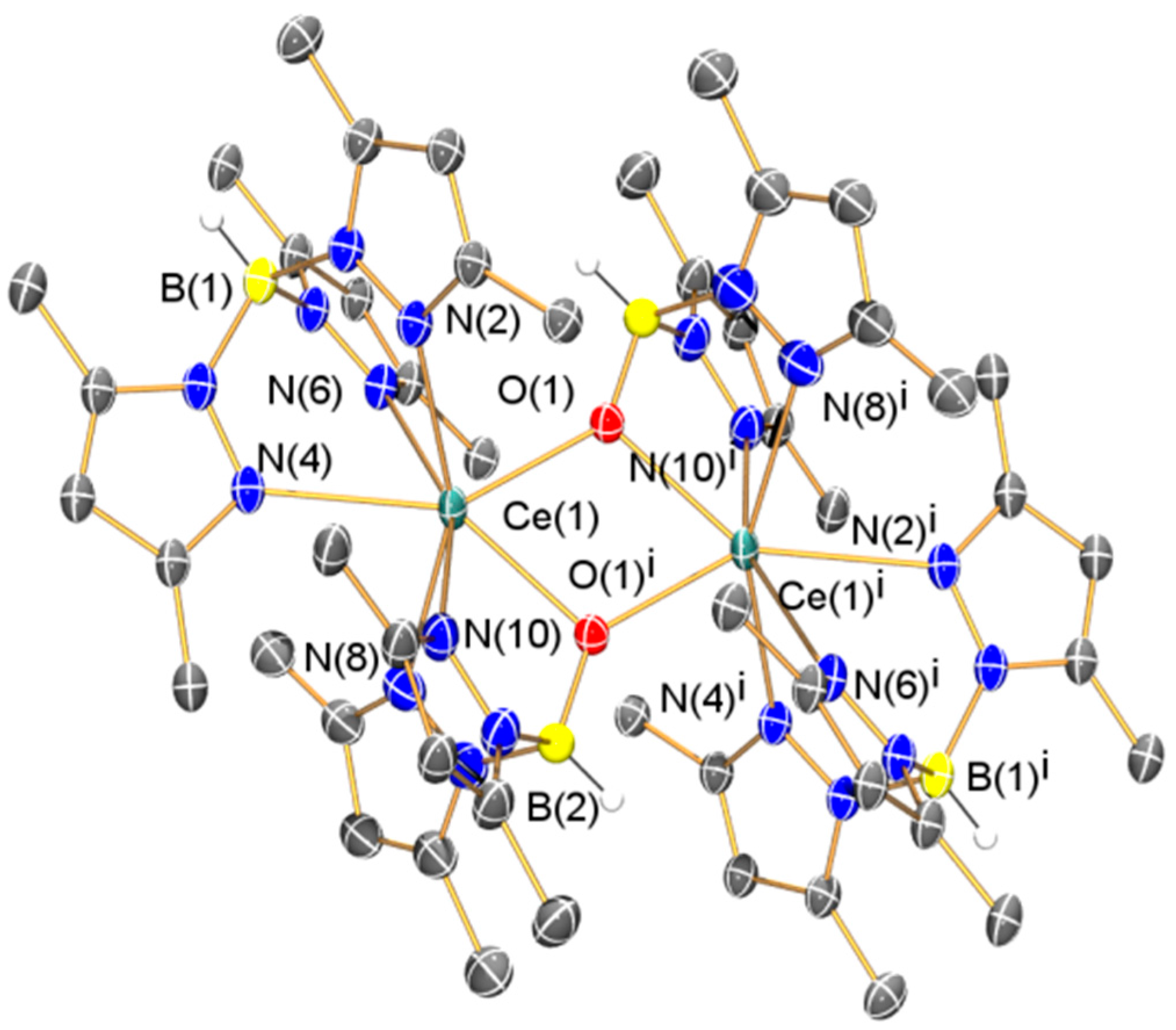
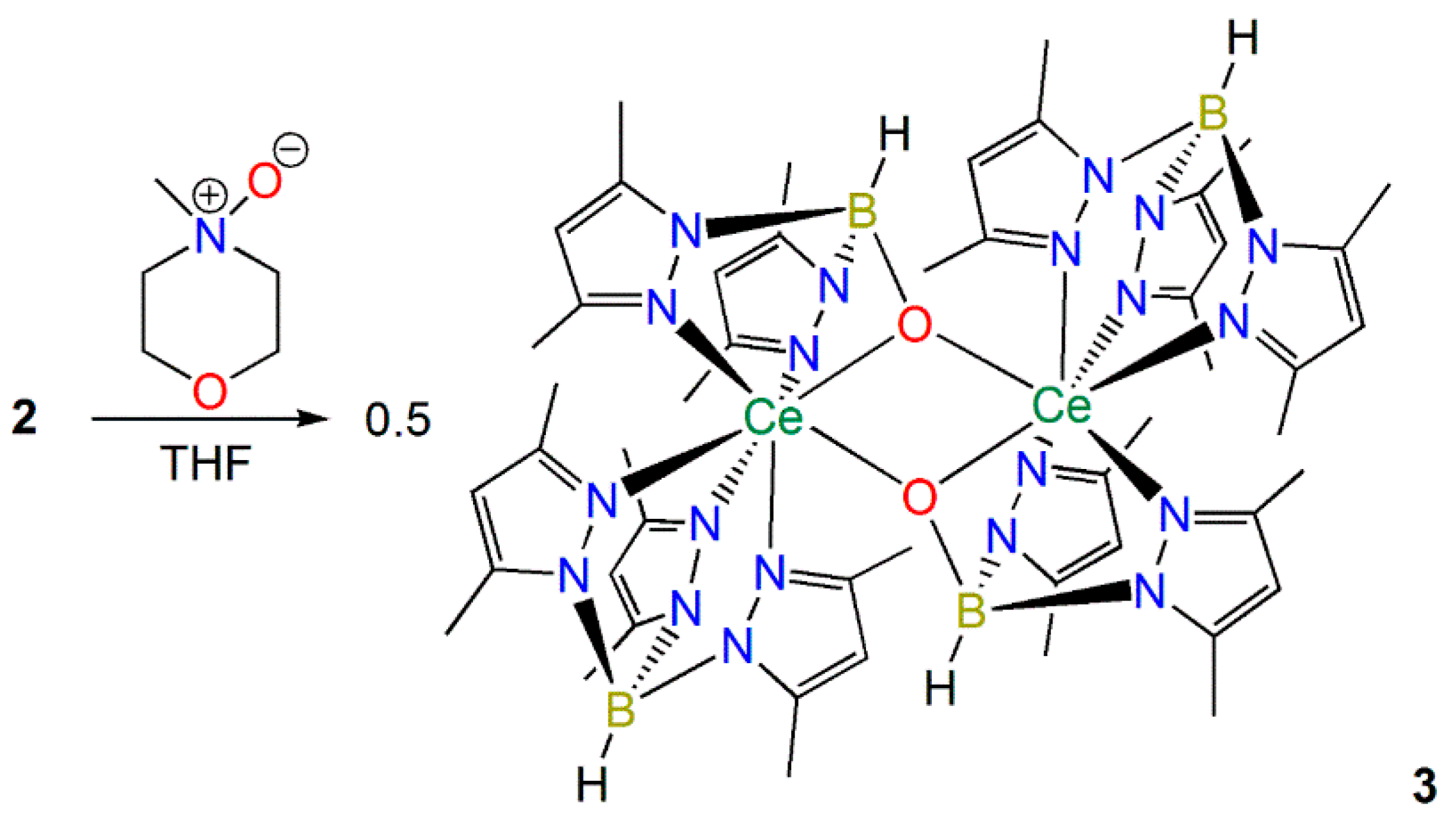
2.2.1. Synthesis and Structural Characterization of [Ce(TpMe2)(μ-BOpMe2)]2 (3)



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1·C7H8 | |||
| Ce(1)–N(2) | 2.653(5) | Ce(1)–N(4) | 2.582(4) |
| Ce(1)–N(6) | 2.631(5) | Ce(1)–N(8) | 2.624(5) |
| Ce(1)–N(10) | 2.546(4) | Ce(1)–N(12) | 2.667(4) |
| Ce(1)–N(13) | 2.385(5) | Ce(1)–N(14) | 2.505(5) |
| N(13)–N(14) | 1.368(6) | Ce(1)–N(13)–N(14) | 78.7(3) |
| Ce(1)–N(14)–N(13) | 69.0(3) | N(13)–Ce(1)–N(14) | 32.38(14) |
| 2·(C4H8O)2 | |||
| Ce(1)–N(2) | 2.591(4) | Ce(1)–N(4) | 2.650(3) |
| Ce(1)–N(6) | 2.711(4) | Ce(1)–N(8) | 2.648(4) |
| Ce(1)–N(10) | 2.719(3) | Ce(1)–N(12) | 2.577(4) |
| Ce(1)–N(13) | 2.592(4) | Ce(1)–N(14) | 2.612(4) |
| C(35)–C(36) | 1.431(6) | C(35)–N(13) | 1.373(6) |
| C(36)–N(14) | 1.379(5) | N(13)–Ce(1)–N(14) | 62.40(12) |
| Ce(1)–N(13)–C(35) | 122.1(3) | Ce(1)–N(14)–C(36) | 120.7(3) |
| N(13)–C(35)–C(36) | 117.0(4) | N(14)–C(36)–C(35) | 117.8(4) |
| 3·(C4H8O)2 | |||
| Ce(1)–N(2) | 2.668(4) | Ce(1)–N(4) | 2.584(4) |
| Ce(1)–N(6) | 2.561(4) | Ce(1)–N(7) | 2.591(4) |
| Ce(1)–N(9) | 2.578(4) | Ce(1)–O(1) | 2.292(3) |
| Ce(1)–O(1i) | 2.378(3) | B(1)–O(1) | 1.407(6) |
| Ce(1)···Ce(1i) | 3.7987(5) | Ce(1)–O(1)–Ce(1i) | 108.84(12) |
| O(1)–Ce(1)–O(1i) | 71.16(12) | ||
| 4·C7H8 | |||
| Ce(1)–N(2) | 2.656(5) | Ce(1)–N(4) | 2.655(5) |
| Ce(1)–N(6) | 2.565(4) | Ce(1)–N(8) | 2.665(5) |
| Ce(1)–N(10) | 2.539(5) | Ce(1)–N(12) | 2.646(5) |
| Ce(1)–N(13) | 2.340(6) | N(13)–N(14) | 1.188(8) |
| N(14)–N(15) | 1.165(8) | Ce(1)–N(13)–N(14) | 165.5(5) |
| N(13)–N(14)–N(15) | 178.4(8) | ||
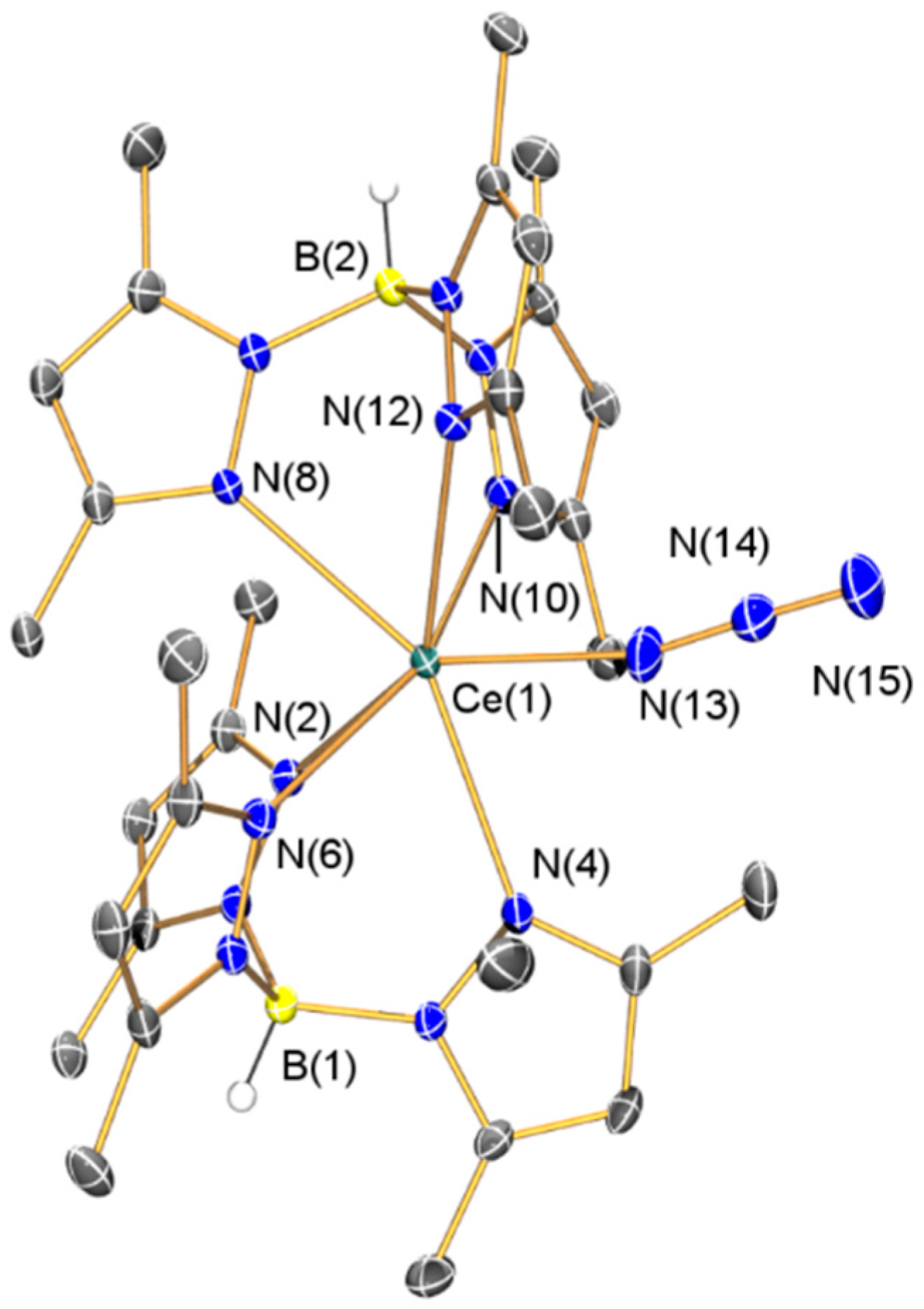
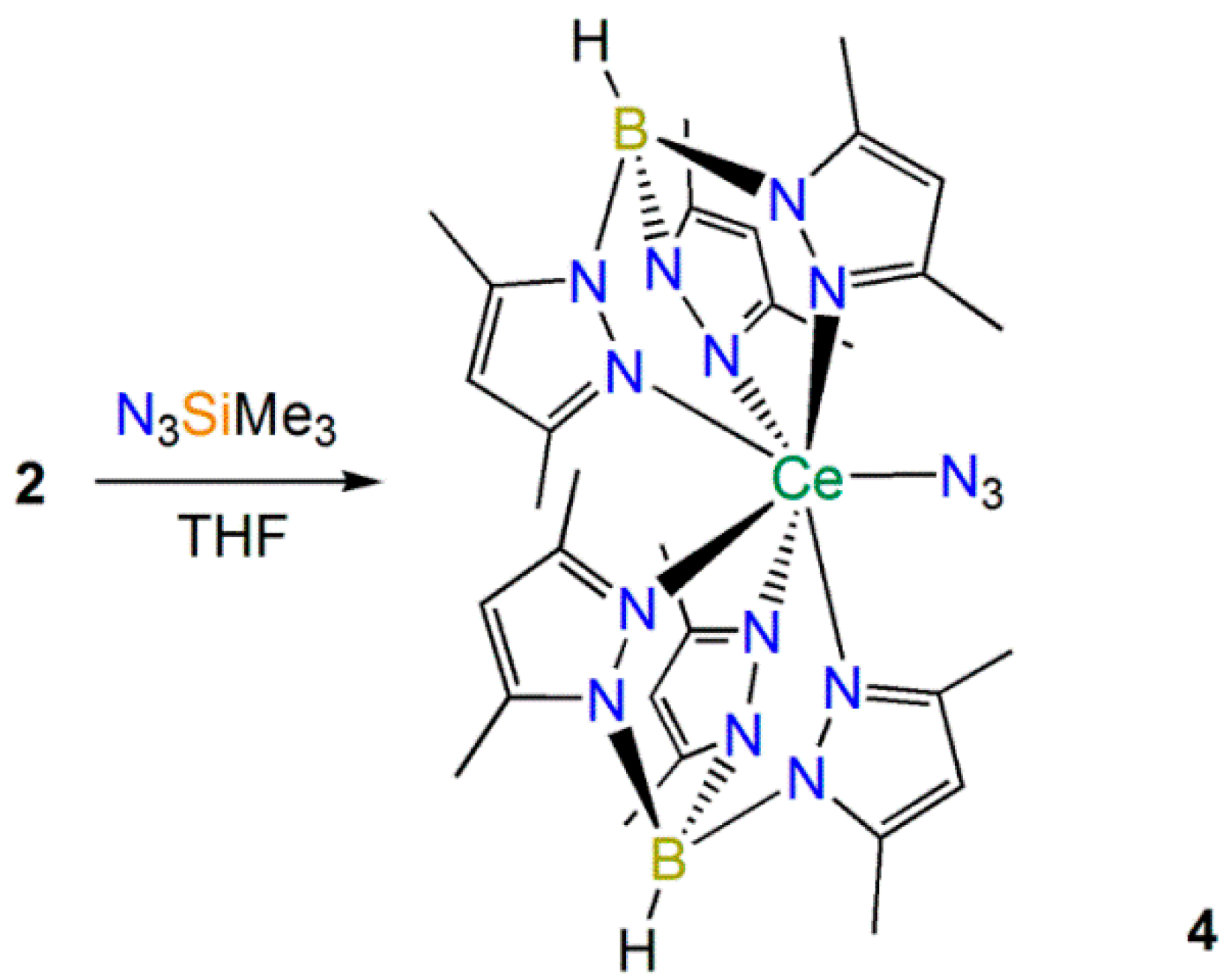
2.2.2. Synthesis and Structural Characterization of [Ce(TpMe2)2(N3)] (4)


3. Experimental Section
3.1. General Procedures
3.2. Synthesis
3.3. X-ray Crystallography
| Parameter | 1·C7H8 | 2·(C4H8O)2 |
|---|---|---|
| Formula | C42H59B2CeN14 | C48H68B2CeN14O2 |
| Fw | 921.77 | 1034.90 |
| cryst size, mm | 0.171 × 0.152 × 0.095 | 0.116 × 0.110 × 0.095 |
| cryst syst | Triclinic | Monoclinic |
| space group | P–1 | P21/c |
| a, Å | 11.1262(9) | 14.2034(4) |
| b, Å | 14.6920(14) | 20.6250(7) |
| c, Å | 14.7874(14) | 17.1106(6) |
| α, ° | 70.037(9) | 90 |
| β, ° | 85.302(8) | 95.066(3) |
| γ, ° | 81.302(7) | 90 |
| V, Å3 | 2244.7(4) | 4992.9(3) |
| Z | 2 | 4 |
| ρcalcd, g cm−3 | 1.364 | 1.377 |
| μ, mm−1 | 1.061 | 0.965 |
| no. of reflections measd | 15541 | 22174 |
| no. of unique reflns, Rint | 8201, 0.0954 | 9125, 0.0630 |
| no. of reflns with F2 > 2σ(F2) | 6502 | 7004 |
| transmn coeff range | 0.806–1.000 | 0.965–1.000 |
| R, Rw a (F2 > 2σ(F2)) | 0.0642, 0.1312 | 0.0523, 0.1058 |
| R, Rw a (all data) | 0.0845, 0.1414 | 0.0746, 0.1163 |
| S a | 1.046 | 1.029 |
| Number of parameters | 547 | 708 |
| max., min. diff map, e Å−3 | 3.59, −0.17 | 1.55, −1.61 |
| Parameter | 3·(C4H8O)2 | 4·C7H8 |
|---|---|---|
| Formula | C62H90B4Ce2N20O2 | C37H52B2CeN15 |
| Fw | 1447.62 | 868.67 |
| cryst size, mm | 0.320 × 0.260 × 0.164 | 0.279 × 0.204 × 0.129 |
| cryst syst | Triclinic | Triclinic |
| space group | P–1 | P–1 |
| a, Å | 10.0579(4) | 11.2609(3) |
| b, Å | 13.3606(6) | 12.5209(3) |
| c, Å | 13.6905(7) | 16.7464(4) |
| α, ° | 87.438(4) | 105.035(2) |
| β, ° | 73.681(4) | 96.314(2) |
| γ, ° | 71.869(4) | 112.313(3) |
| V, Å3 | 1676.10(14) | 2051.38(10) |
| Z | 1 | 2 |
| ρcalcd, g cm−3 | 1.291 | 1.406 |
| μ, mm−1 | 10.730 | 1.157 |
| no. of reflections measd | 13,249 | 28,373 |
| no. of unique reflns, Rint | 5891, 0.0581 | 28,373, 0.060 |
| no. of reflns with F2 > 2σ(F2) | 5342 | 22,381 |
| transmn coeff range | 0.197–1.000 | 0.687–1.000 |
| R, Rw a (F2 > 2σ(F2)) | 0.0448, 0.1119 | 0.0515, 0.1456 |
| R, Rw a (all data) | 0.0496, 0.1153 | 0.0654, 0.0515 |
| S a | 1.015 | 1.097 |
| Number of parameters | 368 | 510 |
| max., min. diff map, e Å−3 | 1.53, −2.10 | 1.63, −1.08 |
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nugent, W.A.; Mayer, J.M. Metal-Ligand Multiple Bonds; Wiley-Interscience: New York, NY, USA, 1988. [Google Scholar]
- Giesbrecht, G.R.; Gordon, J.C. Lanthanide alkylidene and imido complexes. Dalton Trans. 2004, 2387–2393. [Google Scholar] [CrossRef] [PubMed]
- Summerscales, O.T.; Gordon, J.C. Complexes containing multiple bonding interactions between lanthanoid elements and main-group fragments. RSC Adv. 2013, 3, 6682–6692. [Google Scholar] [CrossRef]
- Gregson, M.; Lu, E.; McMaster, J.; Lewis, W.; Blake, A.J.; Liddle, S.T. A cerium(IV)–carbon multiple bond. Angew. Chem. Int. Ed. 2013, 52, 13016–13019. [Google Scholar] [CrossRef] [PubMed]
- Schädle, D.; Meermann-Zimmerman, M.; Schädle, C.; Maichle-Mössmer, C.; Anwander, R. Rare-earth metal complexes with terminal imido ligands. Eur. J. Inorg. Chem. 2015, 2015, 1334–1339. [Google Scholar]
- So, Y.-M.; Wang, G.-C.; Li, Y.; Sung, H.H.-Y.; Williams, I.D.; Lin, Z.; Leung, W.-H. A tetravalent cerium complex containing a Ce=O bond. Angew. Chem. Int. Ed. 2014, 53, 1626–1629. [Google Scholar] [CrossRef] [PubMed]
- Lu, E.; Li, Y.; Chen, Y. A scandium terminal imido complex: Synthesis, structure and DFT studies. Chem. Commun. 2010, 46, 4469–4471. [Google Scholar] [CrossRef] [PubMed]
- Hayton, T.W. Metal-ligand multiple bonding in uranium: Structure and reactivity. Dalton Trans. 2010, 39, 1129–1404. [Google Scholar] [CrossRef] [PubMed]
- Hayton, T.W. Recent developments in actinide-ligand multiple bonding. Chem. Commun. 2013, 49, 2956–2973. [Google Scholar] [CrossRef] [PubMed]
- Zi, G.; Jia, L.; Werkema, E.L.; Walter, M.D.; Gottfriedsen, J.P.; Andersen, R.A. Preparation and reactions of base-free bis(1,2,4-tri-tert-butylcyclopentadienyl)uranium oxide, Cp'2UO. Organometallics 2005, 24, 4251–4264. [Google Scholar] [CrossRef]
- Kraft, S.J.; Walensky, J.; Fanwick, P.E.; Hall, M.B.; Bart, S.C. Crystallographic evidence of a base-free uranium(IV) terminal oxo species. Inorg. Chem. 2010, 49, 7620–7622. [Google Scholar] [CrossRef] [PubMed]
- Matson, E.M.; Kiemicki, J.J.; Anderson, N.H.; Fanwick, P.E.; Bart, S.C. Isolation of a uranium(III) benzophenone ketyl radical that displays redox-active ligand behavior. Dalton Trans. 2014, 43, 17885–17888. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.H.; Odoh, S.O.; Yao, Y.; Williams, U.J.; Schaefer, B.A.; Kiernicki, J.J.; Lewis, A.J.; Goshert, M.D.; Fanwick, P.E.; Schelter, E.J.; et al. Harnessing redox activity for the formation of uranium tris(imido) compounds. Nat. Chem. 2014, 6, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Zi, G.; Walter, M.D. Synthesis, structure and reactivity of a thorium metallocene containing a 2,2ʹ-bipyridyl ligand. Organometallics 2012, 31, 672–679. [Google Scholar] [CrossRef]
- Nugent, L.J.; Baybarz, R.D.; Burnett, J.L.; Ryan, J.L. Electron-transfer and f-d absorption bands of some lanthanide and actinide complexes and the standard (II–III) oxidation potential for each member of the lanthanide and actinide series. J. Phys. Chem. 1973, 77, 1528–1539. [Google Scholar] [CrossRef]
- Kraft, S.J.; Fanwick, P.E.; Bart, S.C. Synthesis and characterization of a uranium(III) complex containing a redox-active 2,2ʹ-bipyridine ligand. Inorg. Chem. 2010, 49, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Roitershtein, D.; Domingos, A.; Pereira, L.C.J.; Ascenso, J.R.; Marques, N. Coordination of 2,2ʹ-bipyridyl and 1,10-phenanthroline to yttrium and lanthanum complexes based on a scorpionate ligand. Inorg. Chem. 2003, 42, 7666–7673. [Google Scholar] [CrossRef] [PubMed]
- Marques, N.; Sella, A.; Takats, J. Chemistry of the lanthanides using pyrazolylborate ligands. Chem. Rev. 2002, 102, 2137–2159. [Google Scholar] [CrossRef] [PubMed]
- Booth, C.H.; Walter, M.D.; Kazhdan, D.; Hu, Y.-J.; Lukens, W.W.; Bauer, E.D.; Maron, L.; Eisenstein, O.; Andersen, R.A. Decamethylytterbocene complexes of bipyridines and diazabutadienes: Multiconfigurational ground states and open-shell singlet formation. J. Am. Chem. Soc. 2009, 131, 6480–6491. [Google Scholar] [CrossRef] [PubMed]
- Nocton, G.; Booth, C.H.; Maron, L.; Andersen, R.A. Influence of the torsion angle in 3,3ʹ-dimethyl-2,2ʹ-bipyridine on the intermediate valence of Yb in (C5Me5)2Yb(3,3ʹ-Me2-bipy). Organometallics 2013, 32, 5305–5312. [Google Scholar] [CrossRef]
- Izod, K.; Liddle, S.T.; Clegg, W. A convenient route to lanthanide triiodide THF solvates. Crystal structures of LnI3(THF)4 [Ln = Pr] and LnI3(THF)3.5 [Ln = Nd, Gd, Y]. Inorg. Chem. 2004, 43, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Trofimenko, S. Boron-pyrazole chemistry. IV. Carbon- and boron-substituted poly(1-pyrazolyl)borates. J. Am. Chem. Soc. 1967, 89, 6288–6294. [Google Scholar] [CrossRef]
- Kunrath, F.A.; Casagrande, O.L., Jr.; Toupet, L.; Carpentier, J.-F. Synthesis and reactivity in salt metathesis reactions of trivalent [La(TpMe2)2X] (X = Cl, I) complexes: Crystal structures of [La(TpMe2)2Cl] and [La(TpMe2)2(κ2-pzMe2)]. Polyhedron 2004, 23, 2437–2445. [Google Scholar] [CrossRef]
- Galler, J.L.; Goodchild, S.; Gould, J.; McDonald, R.; Sella, A. Tris-pyrazolylborate complexes of redox inactive lanthanides—The structures of [(TpMe2)2NdX] (X = Cl, NPh2, dpm, H2BEt2). Polyhedron 2004, 23, 253–262. [Google Scholar] [CrossRef]
- Hillier, A.C.; Zhang, X.; Maunder, G.H.; Liu, S.Y.; Eberspacher, T.A.; Metz, M.V.; McDonald, R.; Domingos, A.; Marques, N.; Day, V.W.; et al. Synthesis and structural comparison of a series of divalent Ln(TpR,Rʹ)2 (Ln = Sm, Eu, Yb) and trivalent Sm(TpMe2)2X (X = F, Cl, I, BPh4) complexes. Inorg. Chem. 2001, 40, 5106–5116. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-Y.; Maunder, G.H.; Sella, A.; Stevenson, M.; Tocher, D.A. Synthesis and molecular structures of hydrotris(dimethylpyrazolyl)borate complexes of the lanthanides. Inorg. Chem. 1996, 35, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Amberger, H.-D.; Edelmann, F.T.; Gottfriedsen, J.; Herbst-Irmer, R.; Jank, S.; Kilimann, M.; Reddmann, H.; Schäfer, M. Synthesis, molecular, and electronic structure of (η8-C8H8)Ln(scorpionate) half-sandwich complexes: An experimental key to a better understanding of f-element-cyclooctatetraenyl bonding. Inorg. Chem. 2009, 48, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.F. The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J. Chem. Soc. 1959, 2003–2005. [Google Scholar] [CrossRef]
- Van Vleck, J.H. Theory of Electric and Magnetic Susceptibilities; Oxford University Press: Oxford, UK, 1932. [Google Scholar]
- Akita, M.; Otha, K.; Takahashi, Y.; Hikichi, S.; Moro-oka, Y. Synthesis and structure determination of Rh–diene complexes with the hydridotris(3,5-diisopropylpyrazolyl)borate ligand, TpiPrRh(diene) (diene = cod, nbd): Dependence of the v(B–H) values on the hapticity of the TpiPr ligand (κ2 vs. κ3). Organometallics 1997, 16, 4121–4128. [Google Scholar] [CrossRef]
- Lopes, I.; Lin, G.Y.; Domingos, A.; Marques, N.; Takats, J. Unprecedented transformation of a hydrotris(pyrazolyl)borate ligand at a metal center: Synthesis and rearrangement of the first mixed Tp/Cp lanthanide complex, Sm(TpMe2)2(Cp). J. Am. Chem. Soc. 1999, 121, 8110–8111. [Google Scholar] [CrossRef]
- Shannon, R.D.; Prewitt, C.T. Revised values of effective ionic radii. Acta Crystallogr. Sect. B 1970, 26, 1046–1048. [Google Scholar] [CrossRef]
- Bergbreiter, D.E.; Killough, J.M. Reactions of potassium-graphite. J. Am. Chem. Soc. 1978, 100, 2126–2134. [Google Scholar] [CrossRef]
- Mehdoui, T.; Berthet, J.-C.; Thuéry, P.; Salmon, L.; Rivière, E.; Ephritikhine, M. Lanthanide(III)/actinide(III) differentiation in the cerium and uranium complexes [M(C5Me5)2(L)]0,+ (L = 2,2ʹ-bipyridine, 2,2ʹ:6ʹ,2ʹʹ-terpyridine): Structural magnetic and reactivity studies. Chem. Eur. J. 2005, 11, 6994–7006. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, C.V.; Creutz, C.; Schwarz, H.A.; Sutin, N. Reduction potentials for 2,2ʹ-bipyridine and 1,10ʹ-phenanthroline couples in aqueous solutions. J. Am. Chem. Soc. 1983, 105, 5617–5623. [Google Scholar] [CrossRef]
- Schelter, E.J. Cerium under the lens. Nat. Chem. 2013, 5, 348. [Google Scholar] [CrossRef] [PubMed]
- Domingos, A.; Elsegood, M.R.J.; Hillier, A.C.; Lin, G.; Liu, S.Y.; Lopes, I.; Marques, N.; Maunder, G.H.; McDonald, R.; Sella, A.; et al. Facile pyrazolylborate ligand degradation at lanthanide centers: X-ray crystal structures of pyrazolylborinate-bridged bimetallics. Inorg. Chem. 2002, 41, 6761–6768. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, A.; Cladis, D.P.; Forrest, W.P.; Fanwick, P.E.; Bart, S.C. Reductive heterocoupling mediated by Cp*2U(2,2ʹ-bpy). Chem. Commun. 2012, 48, 1671–1673. [Google Scholar] [CrossRef] [PubMed]
- Dröse, P.; Gottfriedsen, J.; Hrib, C.G.; Jones, P.G.; Hilfert, L.; Edelmann, F.T. The first cationic complex of tetravalent cerium. Z. Anorg. Allg. Chem. 2011, 637, 369–373. [Google Scholar] [CrossRef]
- CrysAlis PRO, version 37; Agilent Technologies: Yarnton, UK, 2010.
- Sheldrick, G.M. Crystal structure refinement with SHELX. Acta Cryst. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- POVRAY, version 3.7.0. Persistence of Vision Pty. Ltd.: Williamstown, Australia, 2004. Available online: http://www.povray.org/ (Accessed on 16 August 2015).
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortu, F.; Zhu, H.; Boulon, M.-E.; Mills, D.P. Synthesis and Reactivity of a Cerium(III) Scorpionate Complex Containing a Redox Non-Innocent 2,2′-Bipyridine Ligand. Inorganics 2015, 3, 534-553. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics3040534
Ortu F, Zhu H, Boulon M-E, Mills DP. Synthesis and Reactivity of a Cerium(III) Scorpionate Complex Containing a Redox Non-Innocent 2,2′-Bipyridine Ligand. Inorganics. 2015; 3(4):534-553. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics3040534
Chicago/Turabian StyleOrtu, Fabrizio, Hao Zhu, Marie-Emmanuelle Boulon, and David P. Mills. 2015. "Synthesis and Reactivity of a Cerium(III) Scorpionate Complex Containing a Redox Non-Innocent 2,2′-Bipyridine Ligand" Inorganics 3, no. 4: 534-553. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics3040534