
Magnetic and Photo-Physical Properties of Lanthanide Dinuclear Complexes Involving the 4,5-Bis(2-Pyridyl-N-Oxidemethylthio)-4′,5′-Dicarboxylic Acid-Tetrathiafulvalene-, Dimethyl Ester Ligand

 ,
,  , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis

2.2. Crystal Structure of [Dy2(hfac)6(L)]·(CH2Cl2)·(C6H14)0.5 (1)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | [Dy2(hfac)6(L)2]·(CH2Cl2)·0.5C6H14 (1) |
|---|---|
| Formula | C78H51Cl2Dy2F36N4O24S12 |
| M/g·mol−1 | 2892.84 |
| Crystal system | Orthorhombic |
| Space group | Pcab (No.61) |
| Cell parameters | a = 17.3005(5) Å |
| b = 34.2288(10) Å | |
| c = 37.6768(10) Å | |
| Volume/Å3 | 22,311.3(11) |
| Z | 8 |
| T/K | 150 (2) |
| 2θ range/° | 2.16 ≤ 2θ ≤ 54.96 |
| ρcalc/g·cm−3 | 1.722 |
| μ/mm−1 | 1.728 |
| Number of reflections | 103,297 |
| Independent reflections | 25,452 |
| Rint | 0.0815 |
| Fo2 > 2σ(Fo)2 | 13,844 |
| Number of variables | 1396 |
| R1, wR2 | 0.0805, 0.2175 |
| Compounds | 1 |
|---|---|
| Dy1–O9 | 2.263(8) |
| Dy1–O10 | 2.324(6) |
| Dy1–O11 | 2.376(7) |
| Dy1–O12 | 2.347(7) |
| Dy1–O13 | 2.359(7) |
| Dy1–O14 | 2.366(7) |
| Dy1–O15 | 2.381(7) |
| Dy1–O16 | 2.380(8) |
| Dy2–O17 | 2.344(7) |
| Dy2–O18 | 2.264(7) |
| Dy2–O19 | 2.359(7) |
| Dy2–O20 | 2.361(8) |
| Dy2–O21 | 2.352(8) |
| Dy2–O22 | 2.326(8) |
| Dy2–O23 | 2.382(7) |
| Dy2–O24 | 2.394(8) |


2.3. Electrochemical Properties
| E11/2/V | E21/2/V | E31/2/V | |||||
|---|---|---|---|---|---|---|---|
| OxE11/2 | redE11/2 | OxE21/2 | redE21/2 | OxE31/2 | redE31/2 | ||
| L | 0.79 | 0.70 | 1.17 | 1.09 | / | / | |
| 1 | 0.76 | 0.67 | 1.15 | 1.02 | 1.28 | 1.20 | |
| 2 | 0.76 | 0.66 | 1.13 | 0.99 | 1.28 | 1.20 | |
2.4. Photo-Physical Properties
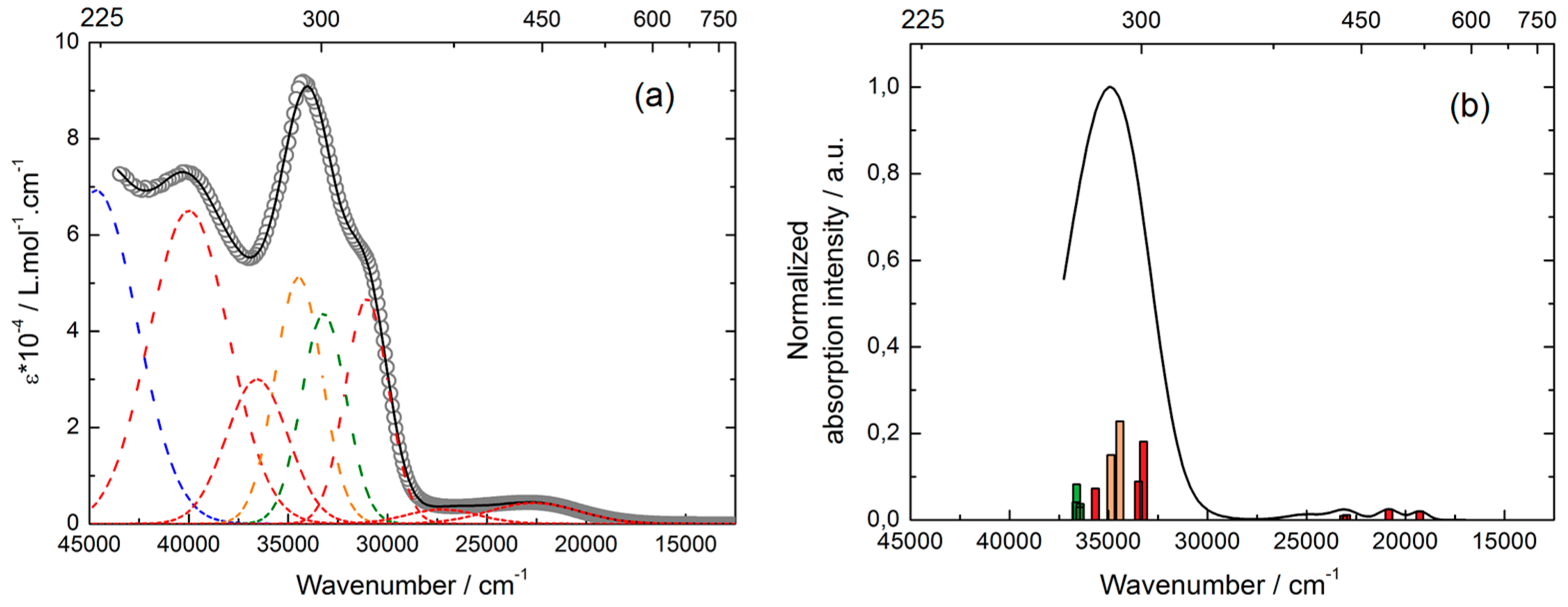
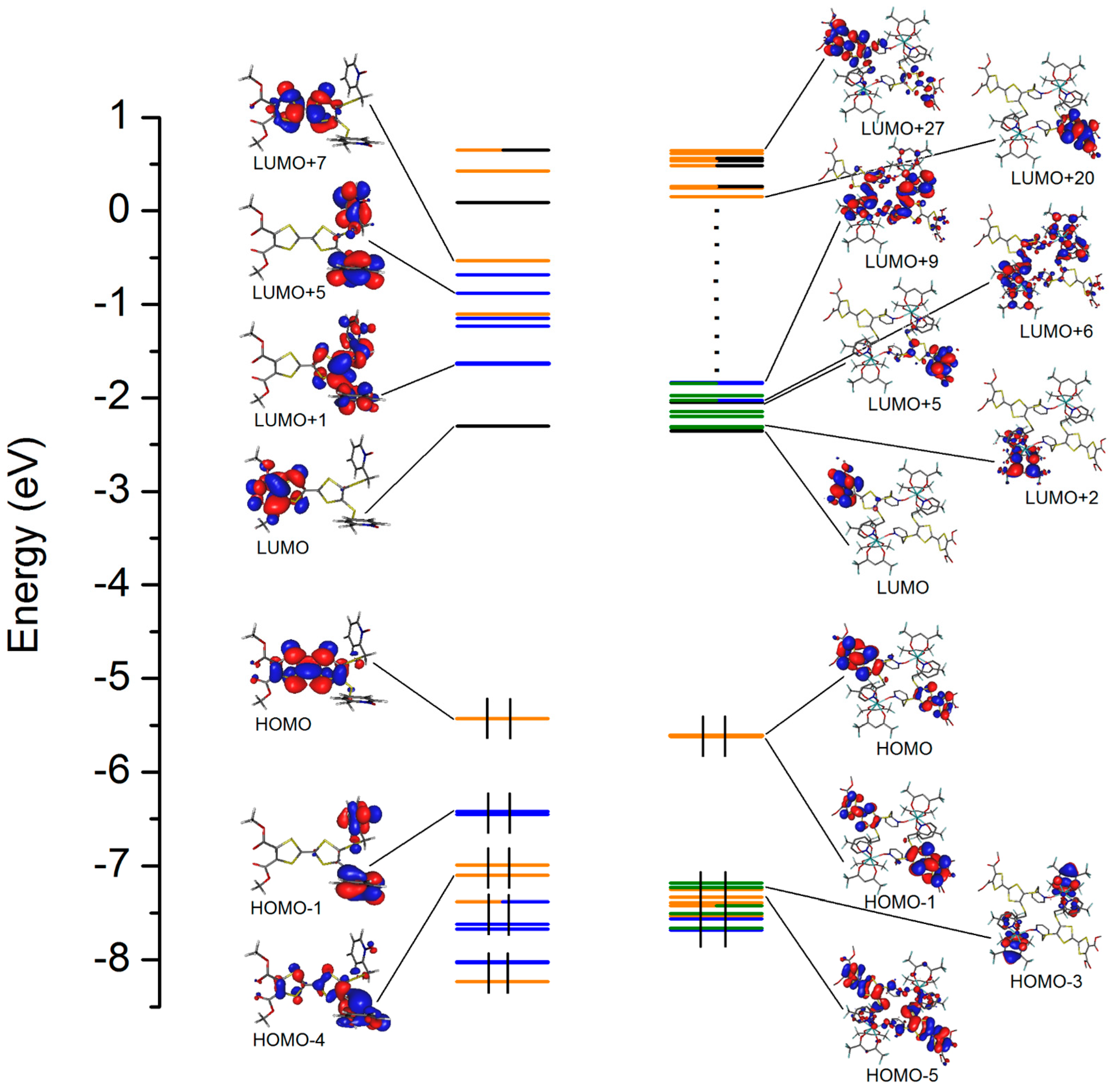
2.4.1. Absorption Properties



| Energy Exp. (cm−1) | Energy Th. (cm−1) | Osc. | Type | Assignment | Transition | |
|---|---|---|---|---|---|---|
| L | 22,600 | 18,810 | 0.03 | ILCT | πTTF→π*Ester | H→L (99%) |
| 27,600 | 24,463 | 0.03 | ILCT | πTTF→π*PyNO | H→L + 1 (88%) | |
| 30,600 | 32,414 | 0.06 | IA | πPyNO→π*PyNO | H-1→L + 1 (74%) | |
| 32,857 | 0.07 | H-2→L + 1 (70%) | ||||
| 32,200 | 33,652 34,426 34,640 | 0.18 0.22 0.11 | ID + ILCT | πTTF→π*TTF + πTTF→π*PyNO | H→L + 7 (60%) H-3/-4→L + 1 (43/28%) | |
| 34,000 | 36,605 | 0.11 | ID + IA | πTTF→π*TTF + | H→L + 7/ + 9 (10/44%) | |
| 36,788 | 0.14 | πPyNO→π*PyNO | H-1→L + 2/ + 5/ + 6 (9/27/10%) | |||
| 38,200 | 39,635 40,437 40,773 41,331 41,698 | 0.08 0.05 0.07 0.06 0.04 | ID + ILCT | πTTF→π*TTF + πTTF→π*PyNO | H-3/-4→L + 4 (38/40%) H-5→L + 1 (38%) H-3→L + 2 (25%) H-10→L (41%) | |
| 44,400 | / | / | IA | πPyNO→π*PyNO | / | |
| Y | 22,600 | 19,282 | 0.03 | ILCT | πTTF→π*Ester | H/-1→L (76/22%) |
| 20,840 | 0.04 | H/-1→L + 5 (22/72%) | ||||
| 27,300 | 22,985 | 0.02 | ILCT | πTTF→π*PyNO | H→L + 6/ + 8/ + 9 (20/19/21%) | |
| 23,132 | 0.02 | H-1→L + 6/ + 9 (17/33%) | ||||
| 31,000 | 33,234 33,485 | 0.30 0.15 | ID + ILCT | πTTF→π*TTF + πTTF→π*Ester | H/-1→L + 20 (9/30%) H→L + 21 (15%) H-4/-5→L (21/29%) | |
| 33,200 | 36,440 36,455 36,618 36,639 | 0.27 0.10 | Ihfac | πhfac→π*hfac | H→L + 10 (53%) + H-3→L (65%) | |
| 34,500 | 34,432 34,886 | 0.38 | ID | πTTF→π*TTF | H/-1→L + 21 (12/41%) H/-1→L + 22 (9/30%) | |
| 36,600 | 35,662 | 0.12 | ILCT | πTTF→π*Ester | H-4/-5→L + 5 (13/7%) | |
| 40,000 | / | / | ILCT | πTTF→π*PyNO | / | |
| 44,700 | / | / | IA | πL2→π*PyImPy | / |
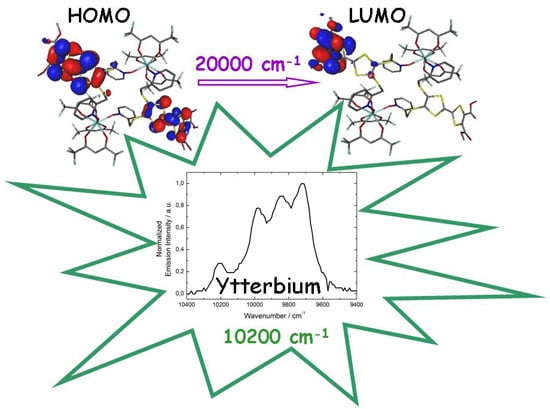
2.4.2. Emission Properties

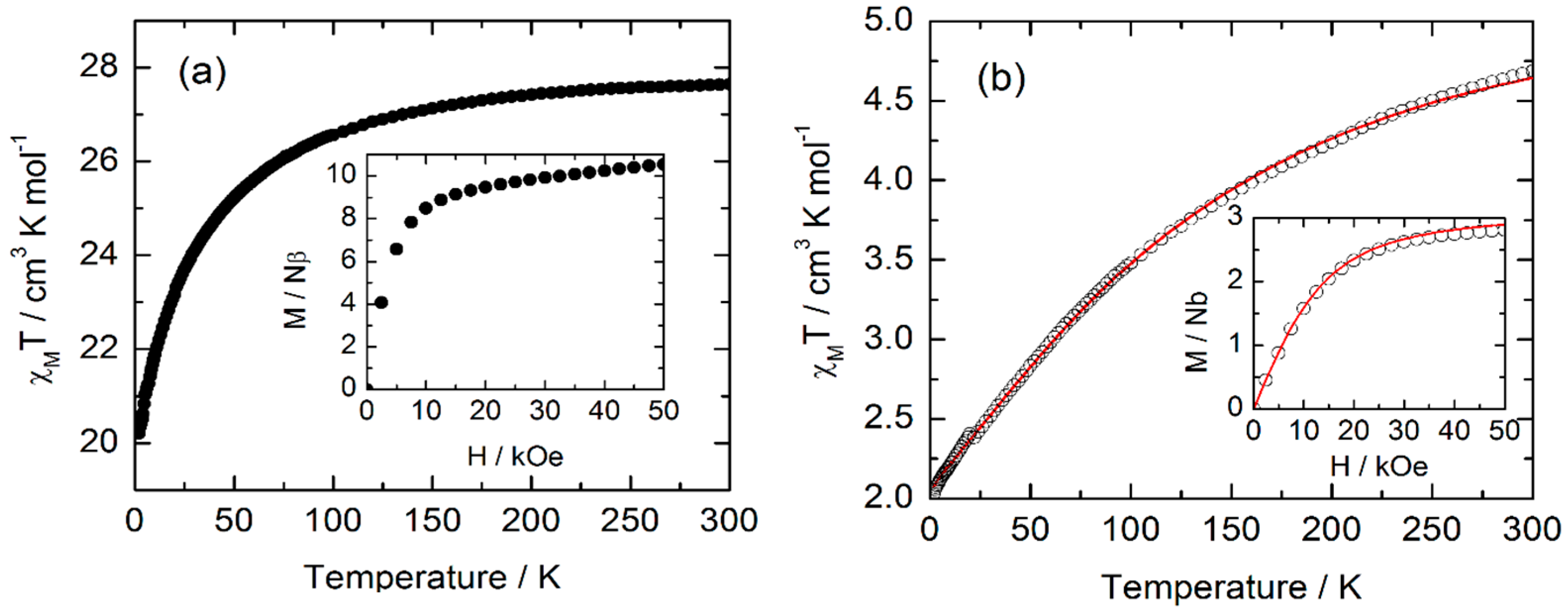
2.5. Magnetic Properties

2.6. Correlation Magnetism-Luminescence
3. Experimental Section
3.1. Synthesis
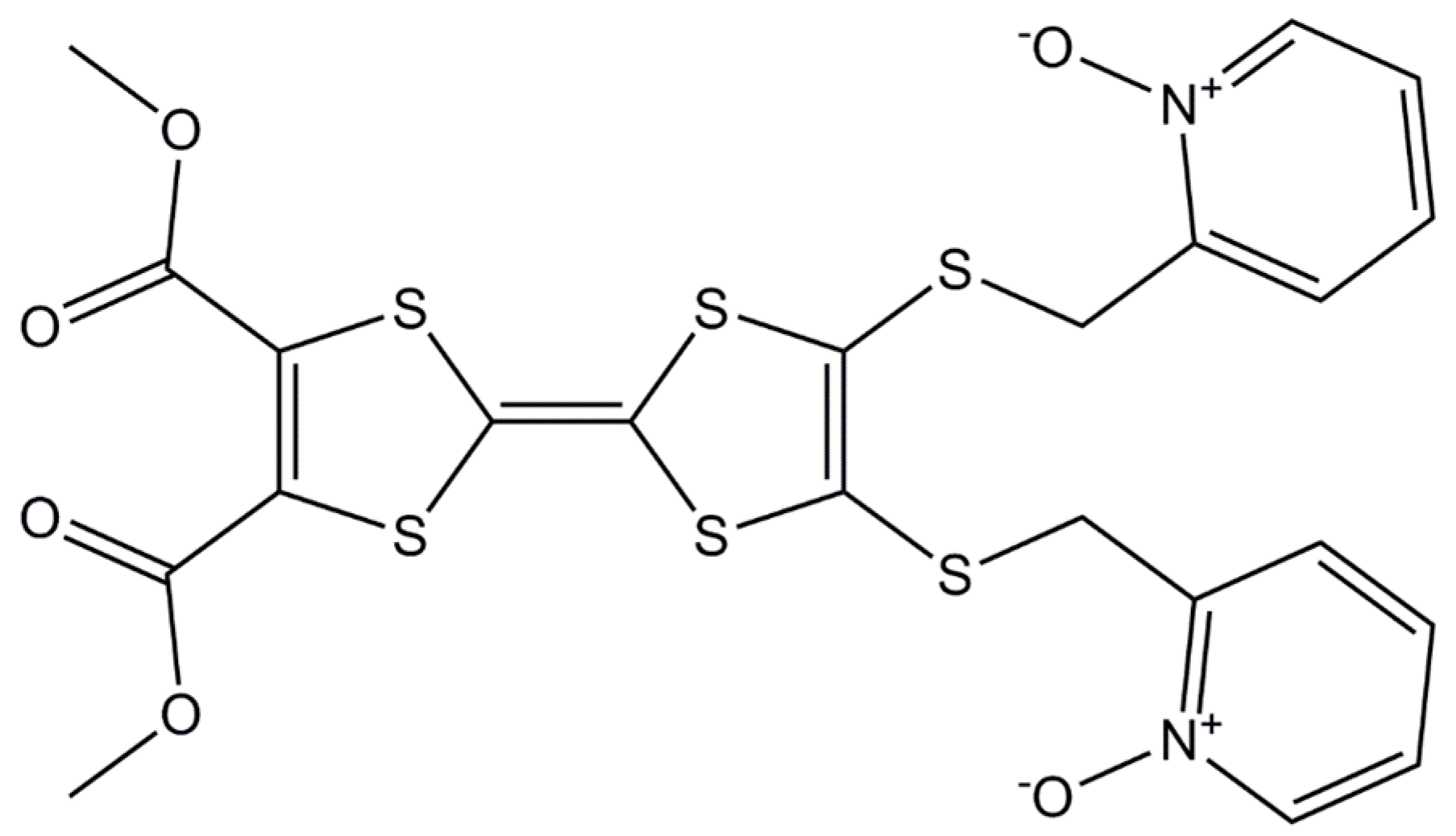
3.2. Synthesis of 4,5-Bis(2-Pyridyl-N-Oxidemethylthio)-4′,5′-Dicarboxylic Acid-Tetrathiafulvalene-, Dimethyl Ester (L)
3.3. Synthesis of Complexes 1 and 2
3.4. Crystallography
3.5. Physical Measurements
3.6. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ishikawa, N.; Sugita, M.; Ishikawa, T.; Koshihara, S.; Kaizu, Y. Lanthanide double-decker complexes functioning as magnets at the single-molecular level. J. Am. Chem. Soc. 2003, 125, 8694–8695. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, D.N.; Winpenny, R.E.P.; Layfield, R.A. Lanthanide single-molecule magnets. Chem. Rev. 2013, 113, 5110–5148. [Google Scholar] [CrossRef] [PubMed]
- Luzon, J.; Sessoli, R. Lanthanides in molecular magnetism: So fascinating so challenging. Dalton Trans. 2012, 41, 13556–13567. [Google Scholar] [CrossRef] [PubMed]
- Sessoli, R.; Powell, A.K. Strategies towards single molecule magnets based on lanthanide ions. Coord. Chem. Rev. 2009, 253, 2328–2341. [Google Scholar] [CrossRef]
- Layfield, R.A. Organometallic single-molecule magnets. Organometallics 2014, 33, 1084–1099. [Google Scholar] [CrossRef]
- Feltham, H.L.C.; Brooker, S. Review of purely 4f and mixed-metal nd-4f single-molecule magnets containing only lanthanide ion. Coord. Chem. Rev. 2014, 276, 1–33. [Google Scholar] [CrossRef]
- Benelli, C.; Gatteschi, D. Magnetism of lanthanides in molecular materials with transition-metal ions and organic radicals. Chem. Rev. 2002, 102, 2369–2387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Guo, Y.-N.; Tang, J. Recent advances in dysprosium-based single molecule magnets: Structural overview and synthetic strategies. Coord. Chem. Rev. 2013, 257, 1728–1763. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, L.; Tang, J. Lanthanide single molecule magnets: Progress and perspective. Dalton Trans. 2015, 44, 3923–3929. [Google Scholar] [CrossRef] [PubMed]
- Ungur, L.; Lin, S.-Y.; Tang, J.; Chibotaru, L.F. Single-molecule toroics in Ising-type lanthanide molecular clusters. Chem. Soc. Rev. 2014, 43, 6894–6905. [Google Scholar] [CrossRef] [PubMed]
- Liddle, S.T.; van Slageren, J. Improving f-element single molecule magnets. Chem. Soc. Rev. 2015, 44, 6655–6669. [Google Scholar] [CrossRef] [PubMed]
- Bünzli, J.C.G.; Piguet, C. Lanthanide-containing molecular and supramolecular polymetallic functional assemblies. Chem. Rev. 2002, 102, 1897–1928. [Google Scholar] [CrossRef] [PubMed]
- Moro, F.; Mills, D.P.; Liddle, S.T.; van Slageren, J. The inherent single-molecule magnet character of trivalent uranium. Angew. Chem. Int. Ed. 2013, 52, 3430–3433. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, J.T.; Antunes, M.A.; Pereira, L.C.J.; Marçalo, J.; Almeida, M. Zero-field slow magnetic relaxation in a uranium(III) complex with a radical ligand. Chem. Commun. 2014, 50, 10262–10264. [Google Scholar] [CrossRef] [PubMed]
- King, D.M.; Tuna, F.; McMaster, J.; Lewis, W.; Blake, A.J.; McInnes, E.J.L.; Liddle, S.T. Single-molecule magnetism in a single-ion triamidoamine uranium(V) terminal mono-oxo complex. Angew. Chem. Int. Ed. 2013, 52, 4921–4924. [Google Scholar] [CrossRef] [PubMed]
- Mougel, V.; Chatelain, L.; Pécaut, J.; Caciuffo, R.; Colineau, E.; Griveau, J.-C.; Mazzanti, M. Uranium and manganese assembled in a wheel-shaped nanoscale single-molecule magnet with high spin-reversal barrier. Nat. Chem. 2012, 4, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Magnani, N.; Colineau, E.; Griveau, J.-C.; Apostolidis, C.; Walter, O.; Caciuffo, R. A plutonium-based single-molecule magnet. Chem. Commun. 2014, 50, 8171–8173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meihaus, K.R.; Minasian, S.G.; Lukens, W.W., Jr.; Kozimor, S.A.; Shuh, D.K.; Tyliszczak, T.; Long, J.R. Influence of pyrazolate vs. N-heterocyclic carbene ligands on the slow magnetic relaxation of homoleptic trischelate lanthanide(III) and uranium(III) complexes. J. Am. Chem. Soc. 2014, 136, 6056–6068. [Google Scholar] [CrossRef] [PubMed]
- Le Roy, J.J.; Gorelsky, S.I.; Korobkov, I.; Murugesu, M. Slow magnetic relaxation in uranium(III) and neodymium(III) cyclooctatetraenyl complexes. Organometallics 2015, 34, 1415–1418. [Google Scholar] [CrossRef]
- Dei, A.; Gatteschi, D. Molecular (Nano) magnet as test grounds of quantum mechanics. Angew. Chem. Int. Ed. 2011, 50, 11852–11858. [Google Scholar] [CrossRef] [PubMed]
- Leuenberger, M.N.; Loss, D. Quantum computing in molecular magnets. Nature 2001, 410, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.; Edwards, R.S.; Aliaga-Alcalde, N.; Christou, G. Quantum coherence in an exchange-coupled dimer of single-molecule magnets. Science 2003, 302, 1015–1018. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.W.; Rebic, S.; Sparkes, B.M.; Twamley, J.; Buchler, B.C.; Lam, P.K. Memory-enhanced noiseless cross-phase modulation. Light Sci. Appl. 2012, 1, e40. [Google Scholar] [CrossRef]
- Sessoli, R.; Tsai, H.L.; Schake, A.R.; Wang, S.; Vincent, J.B.; Folting, K.; Gatteschi, D.; Christou, G.; Hendrickson, D.N. High-spin molecules: [Mn12O12(O2CR)16(H2O)4]. J. Am. Chem. Soc. 1993, 115, 1804–1816. [Google Scholar] [CrossRef]
- Mannini, M.; Pineider, F.; Sainctavit, P.; Danieli, C.; Otero, E.; Sciancalepore, C.; Talarico, A.M.; Arrio, M.-A.; Cornia, A.; Gatteschi, D.; et al. Magnetic memory of a single-molecule quantum magnet wired to a gold surface. Nat. Mater. 2009, 8, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Li, X.; Cao, Y. Optical storage arrays: A perspective for future big data storage. Light Sci. Appl. 2014, 3, e177. [Google Scholar] [CrossRef]
- Papasimakis, N.; Thongrattanasiri, S.; Zheludev, N.I.; de Abajo, F.J. The magnetic response of grapheme split-ring metamaterials. Light Sci. Appl. 2013, 2, e78. [Google Scholar] [CrossRef]
- Sanvito, S. Molecular Spintronics. Chem. Soc. Rev. 2011, 40, 3336–3355. [Google Scholar] [CrossRef] [PubMed]
- Bogani, L.; Wernsdorfer, W. Molecular spintronics using single-molecule magnets. Nat. Mater. 2008, 7, 179–186. [Google Scholar] [CrossRef] [PubMed]
- De Bettancourt-Dias, A. Luminescence of Lanthanide Ions in Coordination Compounds and Nanomaterials; Wiley: Hoboken, NJ, USA, 2014. [Google Scholar]
- Weissman, S.I. Intramolecular energy transfer, the fluorescence of complexes of europium. J. Chem. Phys. 1942, 10, 214–217. [Google Scholar] [CrossRef]
- Crosby, G.A.; Kasha, M. Intramolecular energy transfer in ytterbium organic chelates. Spectrochim. Acta 1958, 10, 377–382. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Wang, C.; Zhao, L.; Wua, J.; Tang, J. Chiral mononuclear lanthanide complexes and field-induced single-ion magnet behaviour of Dy analogue. Dalton Trans. 2015, 44, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Rouquette, J.; Thibaud, J.-M.; Ferreira, R.A.S.; Carlos, L.D.; Donnadieu, B.; Vieru, V.; Chibotaru, L.F.; Konczewicz, L.; Haines, J.; et al. A high-temperature molecular ferroelectric Zn/Dy complex exhibiting single-ion-magnet behavior and lanthanide luminescence. Angew. Chem. Int. Ed. 2015, 54, 2236–2240. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-L.; Chen, C.-L.; Xiao, H.-P.; Wang, A.-L.; Liu, C.-M.; Zheng, X.; Gao, L.-J.; Yanga, X.-G.; Fang, S.-M. Luminescent, magnetic and ferroelectric properties of noncentrosymmetric chain-like complexes composed of nine-coordinate lanthanide ions. Dalton Trans. 2013, 42, 15317–15325. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Kobayashi, H.; Kobayashi, A.; Cassoux, P. Superconductivity, antiferromagnetism, and phase diagram of a series of organic conductors: λ-(BETS)2FexGa1−xBryCl4−y. Adv. Mater. 2000, 12, 1685–1689. [Google Scholar] [CrossRef]
- Uji, S.; Shinagawa, H.; Terashima, T.; Terakura, C.; Yakabe, T.; Terai, Y.; Tokumoto, M.; Kobayashi, A.; Tanaka, H.; Kobayashi, H. Magnetic-field-induced superconductivity in a two-dimensional organic conductor. Nature 2001, 410, 908–910. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Fujiwara, E.; Kobayashi, H. Single-component molecular metals with extended-TTF dithiolate ligands. Chem. Rev. 2004, 104, 5243–5264. [Google Scholar] [CrossRef] [PubMed]
- Enoki, T.; Miyasaki, A. Magnetic TTF-based charge-transfer complexes. Chem. Rev. 2004, 104, 5449–5478. [Google Scholar] [CrossRef] [PubMed]
- Coronado, E.; Day, P. Magnetic molecular conductors. Chem. Rev. 2004, 104, 5419–5448. [Google Scholar] [CrossRef] [PubMed]
- Ouahab, L.; Enoki, T. Multiproperty molecular materials: TTF-based conductiong and magnetic molecular materials. Eur. J. Inorg. Chem. 2004, 2004, 933–941. [Google Scholar] [CrossRef]
- Fujiwara, H.; Wada, K.; Hiraoka, T.; Hayashi, T.; Sugimoto, T.; Nakazumi, H.; Yokogawa, K.; Teramura, M.; Yasuzuka, S.; Murata, K.; et al. Stable metallic behavior and antiferromagnetic ordering of Fe(III) d spins in (EDO-TTFVO)2·FeCl4. J. Am. Chem. Soc. 2005, 127, 14166–14167. [Google Scholar] [CrossRef] [PubMed]
- Lorcy, D.; Bellec, N.; Fourmigué, M.; Avarvari, N. Tetrathiafulvalene-based group XV ligands: Synthesis, coordination chemistry and radical cation salts. Coord. Chem. Rev. 2009, 253, 1398–1438. [Google Scholar] [CrossRef] [Green Version]
- Pointillart, F.; Golhen, S.; Cador, O.; Ouahab, L. Paramagnetic 3D coordination complexes involving redox-active tetrathiafulvalene derivatives: An efficient approach to elaborate multi-properties materials. Dalton Trans. 2013, 42, 1949–1960. [Google Scholar] [CrossRef] [PubMed]
- Cosquer, G.; Pointillart, F.; Golhen, S.; Cador, O.; Ouahab, L. Slow magnetic relaxation in condensed versus dispersed dysprosium(III) mononuclear complexes. Chem. Eur. J. 2013, 19, 7895–7903. [Google Scholar] [CrossRef] [PubMed]
- Da Cunha, T.T.; Jung, J.; Boulon, M.-E.; Campo, G.; Pointillart, F.; Pereira, C.L.M.; Le Guennic, B.; Cador, O.; Bernot, K.; Pineider, F.; et al. Magnetic poles determinations and robustness of memory effect upon solubilization in a Dy-III-based single ion magnet. J. Am. Chem. Soc. 2013, 135, 16332–16335. [Google Scholar] [CrossRef] [PubMed]
- Pointillart, F.; Golhen, S.; Cador, O.; Ouahab, L. Slow magnetic relaxation in a redox-active tetrathiafulvalene-based ferromagnetic dysprosium complex. Eur. J. Inorg. Chem. 2014, 2014, 4558–4563. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, X.-M.; Cui, L.; Deng, K.; Zeng, Q.-D.; Zuo, J.-L. Tetrathiafulvalene-supported triple-decker phthalocyaninato dysprosium(III) complex: Synthesis, properties and surface assembly. Sci. Rep. 2014, 4, 5928. [Google Scholar] [CrossRef] [PubMed]
- Pointillart, F.; Bernot, K.; Golhen, S.; le Guennic, B.; Guizouarn, T.; Ouahab, L.; Cador, O. Magnetic memory in an isotopically enriched and magnetically isolated mononuclear dysprosium complex. Angew. Chem. Int. Ed. 2015, 54, 1504–1507. [Google Scholar] [CrossRef] [PubMed]
- Pointillart, F.; Jung, J.; Berraud-Pache, R.; le Guennic, B.; Dorcet, V.; Golhen, S.; Cador, O.; Maury, O.; Guyot, Y.; Decurtins, S.; et al. Luminescence and single-molecule magnet behavior in lanthanide complexes involving a tetrathiafulvalene-fused dipyridophenazine ligand. Inorg. Chem. 2015, 54, 5384–5397. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Pointillart, F.; Lefeuvre, B.; Dorcet, V.; Golhen, S.; Cador, O.; Ouahab, L. Multiple single-molecule magnet behaviors in dysprosium dinuclear complexes involving a multiple functionalized tetrathiafulvalene-based ligand. Inorg. Chem. 2015, 54, 4021–4028. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, S.; Burton-Pye, B.P.; Khan, T.; Martin, L.R.; Wray, S.D.; Skabara, P.J. Interaction between tetrathiafulvalene carboxylic acid and ytterbium DO3A: Solution state self-assembly of a ternary complex which is luminescent in the near IR. Chem. Commun. 2002, 1668–1669. [Google Scholar] [CrossRef]
- Pope, S.J.A.; Burton-Pye, B.P.; Berridge, R.; Khan, T.; Skabara, P.; Faulkner, S. Self-assembly of luminescent ternary complexes between seven-coordinate lanthanide(III) complexes and chromophore bearing carboxylates and phosphonates. Dalton Trans. 2006, 2907–2912. [Google Scholar] [CrossRef] [PubMed]
- Pointillart, F.; le Guennic, B.; Golhen, S.; Cador, O.; Maury, O.; Ouahab, L. High nuclearity complexes of lanthanide involving tetrathiafulvalene ligands: Structural, magnetic, and photophysical properties. Inorg. Chem. 2013, 52, 1610–1620. [Google Scholar] [CrossRef] [PubMed]
- Ran, Y.-F.; Steinmann, M.; Sigrist, M.; Liu, S.-X.; Hauser, J.; Decurtins, S. Tetrathiafulvalene-based lanthanide coordination complexes: Synthesis, crystal structure, optical and electrochemical characterization. C. R. Chim. 2012, 15, 838–844. [Google Scholar] [CrossRef]
- Feng, M.; Pointillart, F.; le Guennic, B.; Lefeuvre, B.; Golhen, S.; Cador, O.; Maury, O.; Ouahab, L. Unprecedented sensitization of visible and near-infrared lanthanide luminescence by using a tetrathiafulvalene-based chromophore. Chem. Asian J. 2014, 9, 2814–2825. [Google Scholar] [CrossRef] [PubMed]
- Pointillart, F.; le Guennic, B.; Cauchy, T.; Golhen, S.; Cador, O.; Maury, O.; Ouahab, L. A series of tetrathiafulvalene-based lanthanide complexes displaying either single molecule magnet or luminescence-direct magnetic and photo-physical correlations in the ytterbium analogue. Inorg. Chem. 2013, 52, 5978–5990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soussi, K.; Jung, J.; Pointillart, F.; le Guennic, B.; Lefeuvre, B.; Golhen, S.; Cador, O.; Guyot, Y.; Maury, O.; Ouahab, L. Magnetic and photo-physical investigations into DyIII and YbIII complexes involving tetrathiafulvalene ligand. Inorg. Chem. Front. 2015, 2, 1105–1117. [Google Scholar] [CrossRef]
- Simonsen, B.; Svenstrup, N.; Lau, J.; Simonsen, O.; Mork, P.; Kristensen, G.J.; Becher, J. Sequential functionalisation of bis-protected tetrathiafulvalene-dithiolates. Synthesis 1996, 3, 407–418. [Google Scholar] [CrossRef]
- Lin, H.-H.; Yan, Z.-M.; Dai, J.; Zhang, D.-Q.; Zuo, J.-L.; Zhu, Q.-Y.; Jia, D.-X. A water-soluble derivative of tetrathiafulvalene exhibiting pH sensitive redox properties. New J. Chem. 2005, 29, 509–513. [Google Scholar] [CrossRef]
- Llunell, M.; Casanova, D.; Cirera, J.; Bofill, J.M.; Alemany, P.; Alvarez, S. SHAPE; Version 2.1; Universitat de Barcelona: Barcelona, Spain, 2013. [Google Scholar]
- Cosquer, G.; Pointillart, F.; le Guennic, B.; le Gal, Y.; Golhen, S.; Cador, O.; Ouahab, L. 3d4f heterobimetallic dinuclear and tetranuclear complexes involving tetrathiafulvalene as ligands: X-ray structures and magnetic and photophysical investigations. Inorg. Chem. 2012, 51, 8488–8501. [Google Scholar] [CrossRef] [PubMed]
- Pointillart, F.; Cauchy, T.; Maury, O.; le Gal, Y.; Golhen, S.; Cador, O.; Ouahab, L. Tetrathiafulvalene-amido-2-pyridine-N-oxide as efficient charge-transfer antenna ligand for the sensitization of YbIII luminescence in a series of lanthanide paramagnetic coordination complexes. Chem. Eur. J. 2010, 16, 11926–11941. [Google Scholar] [CrossRef] [PubMed]
- Pointillart, F.; le Guennic, B.; Golhen, S.; Cador, O.; Maury, O.; Ouahab, L. A redox-active luminescent ytterbium based single molecule magnet. Chem. Commun. 2013, 49, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves Silva, F.R.; Malta, O.L.; Reinhard, C.; Güdel, H.U.; Piguet, C.; Moser, J.; Bünzli, J.C.G. Visible and near-infrared luminescence of lanthnaide-containing dimetallic triple-stranded helicates: Energy transfer mechanisms in the SmIII and YbIII molecular edifices. J. Phys. Chem. A 2002, 106, 1670–1677. [Google Scholar] [CrossRef]
- Lapadula, G.; Bourdolle, A.; Allouche, F.; Conley, M.; del Rosa, I.; Maron, L.; Lukens, W.W.; Guyot, Y.; Andraud, C.; Brasselet, S.; et al. Near-IR two photon microscopy imaging of silica nanoparticles functionalized with isolated sensitized Yb(III) centers. Chem. Mater. 2014, 26, 1062–1073. [Google Scholar] [CrossRef]
- Kahn, O. Molecular Magnetism; VCH: Weinhem, Germany, 1993. [Google Scholar]
- Orbach, R. Spin-lattice relaxation in rare-earth salts. Proc. R. Soc. Lond. A 1961, 264, 458–484. [Google Scholar] [CrossRef]
- Rudowicz, C. Transformation relations for the conventional Okq and normalised O′kq Stevens operator equivalents with k = 1 to 6 and −k ≤ q ≤ k. J. Phys. C Solid State Phys. 1985, 18, 1415–1430. [Google Scholar] [CrossRef]
- Görlder-Walrand, C.; Binnemans, K. Rationalization of Crystal-Field Parametrization. In Hanbook on the Physics and Chemistry of Rare Earths; Elsevier: Philadelphia, PA, USA, 1996; Volume 23, pp. 121–283. [Google Scholar]
- Richardson, M.F.; Wagner, W.F.; Sands, D.E. Rare-earth trishexafluoroacetylacetonates and related compounds. J. Inorg. Nucl. Chem. 1968, 30, 1275–1289. [Google Scholar] [CrossRef]
- Binet, L.; Fabre, J.M.; Montginoul, C.; Simonsen, K.B.; Becher, J. Preparation and chemistry of new unsymmetrically substituted tetrachalcogenofulvalenes bearing CN(CH2)2X and HO(CH2)2X groups (X = S or Se). J. Chem. Soc. Perkin Trans. 1 1996, 783–788. [Google Scholar] [CrossRef]
- Polasek, M.; Sedinova, M.; Kotek, J.; Vander Elst, L.; Muller, N.R.; Hermann, P.; Lukes, I. Pyridine-N-oxide analogues of DOTA and their gadolinium(III) complexes endowed with a fast water exchange on the square-antiprismatic isomer. Inorg. Chem. 2009, 48, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELX97—Programs for Crystal Structure Analysis (Release 97-2); Tammanstrasse 4, D-3400; Institüt für Anorganische Chemie der Universität: Göttingen, Germany, 1998. [Google Scholar]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Cryst. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Dolg, M.; Stoll, H.; Preuss, H. A combination of quasirelativistic pseudopotential and ligand field calculations for lanthanoid compounds. Theor. Chim. Acta 1993, 85, 441–450. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Cossi, M.; Barone, V. Time-dependent density functional theory for molecules in liquid solutions. J. Chem. Phys. 2001, 115, 4708–4717. [Google Scholar] [CrossRef]
- Improta, R.; Barone, V.; Scalmani, G.; Frisch, M.J. A state-specific polarizable continuum model time dependent density functional theory method for excited state calculations in solution. J. Chem. Phys. 2006, 125, 054103. [Google Scholar] [CrossRef] [PubMed]
- Allouche, A.-R. Gabedit—A graphical user interface for computational chemistry softwares. J. Comput. Chem. 2011, 32, 174–182. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pointillart, F.; Speed, S.; Lefeuvre, B.; Riobé, F.; Golhen, S.; Le Guennic, B.; Cador, O.; Maury, O.; Ouahab, L. Magnetic and Photo-Physical Properties of Lanthanide Dinuclear Complexes Involving the 4,5-Bis(2-Pyridyl-N-Oxidemethylthio)-4′,5′-Dicarboxylic Acid-Tetrathiafulvalene-, Dimethyl Ester Ligand. Inorganics 2015, 3, 554-572. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics3040554
Pointillart F, Speed S, Lefeuvre B, Riobé F, Golhen S, Le Guennic B, Cador O, Maury O, Ouahab L. Magnetic and Photo-Physical Properties of Lanthanide Dinuclear Complexes Involving the 4,5-Bis(2-Pyridyl-N-Oxidemethylthio)-4′,5′-Dicarboxylic Acid-Tetrathiafulvalene-, Dimethyl Ester Ligand. Inorganics. 2015; 3(4):554-572. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics3040554
Chicago/Turabian StylePointillart, Fabrice, Saskia Speed, Bertrand Lefeuvre, François Riobé, Stéphane Golhen, Boris Le Guennic, Olivier Cador, Olivier Maury, and Lahcène Ouahab. 2015. "Magnetic and Photo-Physical Properties of Lanthanide Dinuclear Complexes Involving the 4,5-Bis(2-Pyridyl-N-Oxidemethylthio)-4′,5′-Dicarboxylic Acid-Tetrathiafulvalene-, Dimethyl Ester Ligand" Inorganics 3, no. 4: 554-572. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics3040554