
Advances in Engineered Hemoproteins that Promote Biocatalysis



Abstract
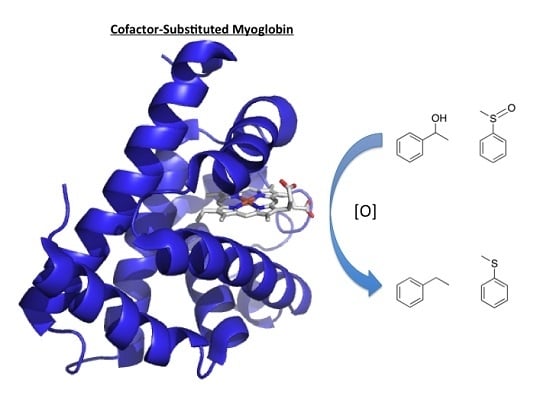
:
1. Introduction
2. Metal-Substituted Heme Proteins
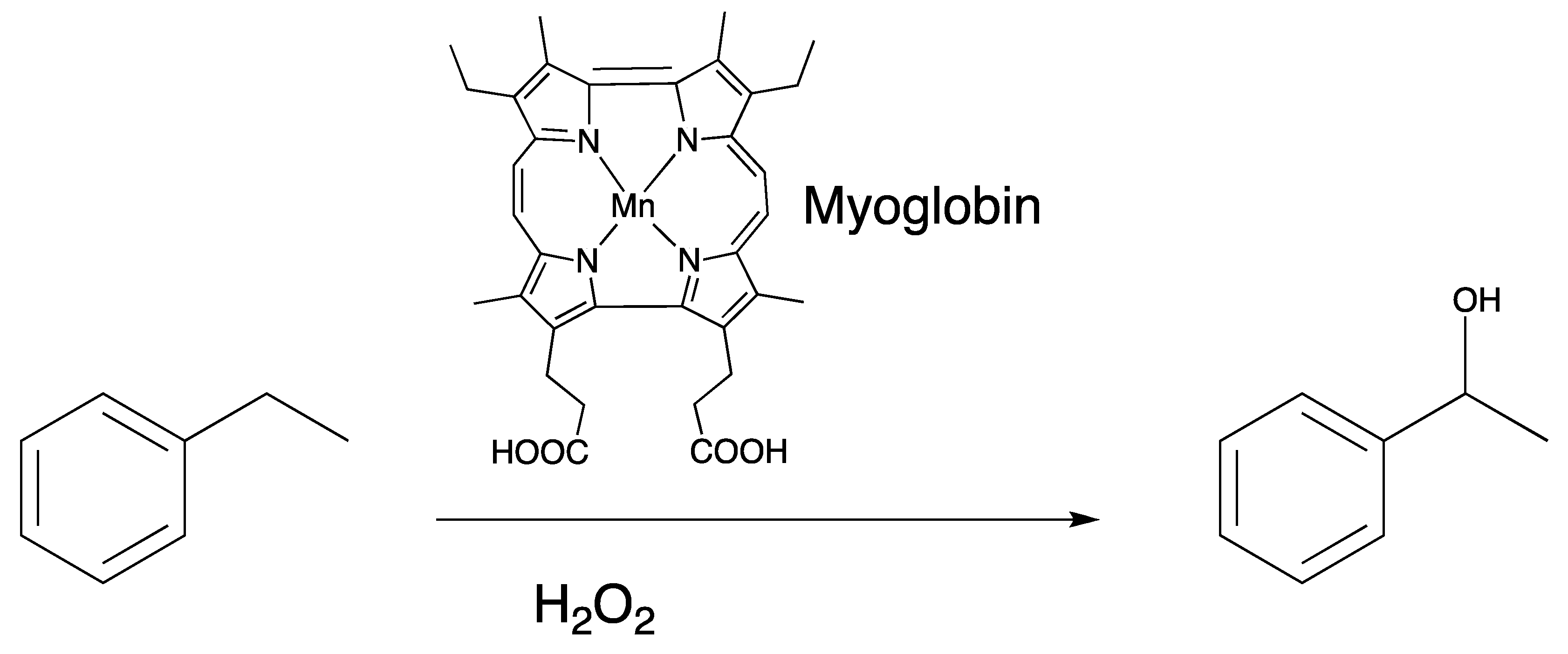
2.1. Manganese
2.2. Cobalt
2.3. Chromium
3. Rational Design of Heme Proteins that Promotes New Function
3.1. Myoglobin
3.1.1. Peroxygenase and Peroxidase Activity
3.1.2. Dehalogenase Activity
3.1.3. Oxidase Activity
3.1.4. Some Recent Examples of the Versatility of Myoglobin Mutants
3.2. Horseradish Peroxidase
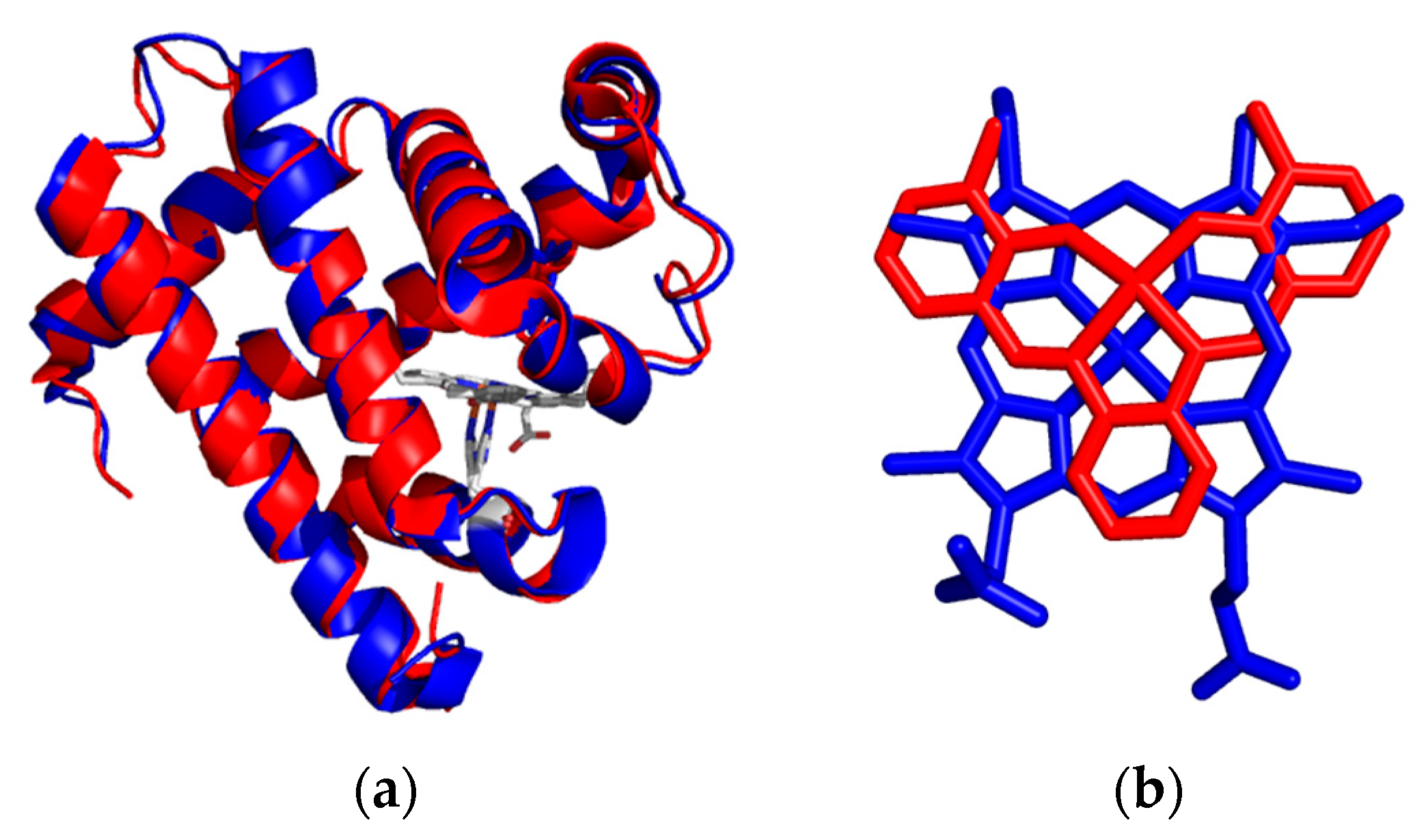
4. Insertion of Schiff-Base Cofactors into Myoglobin
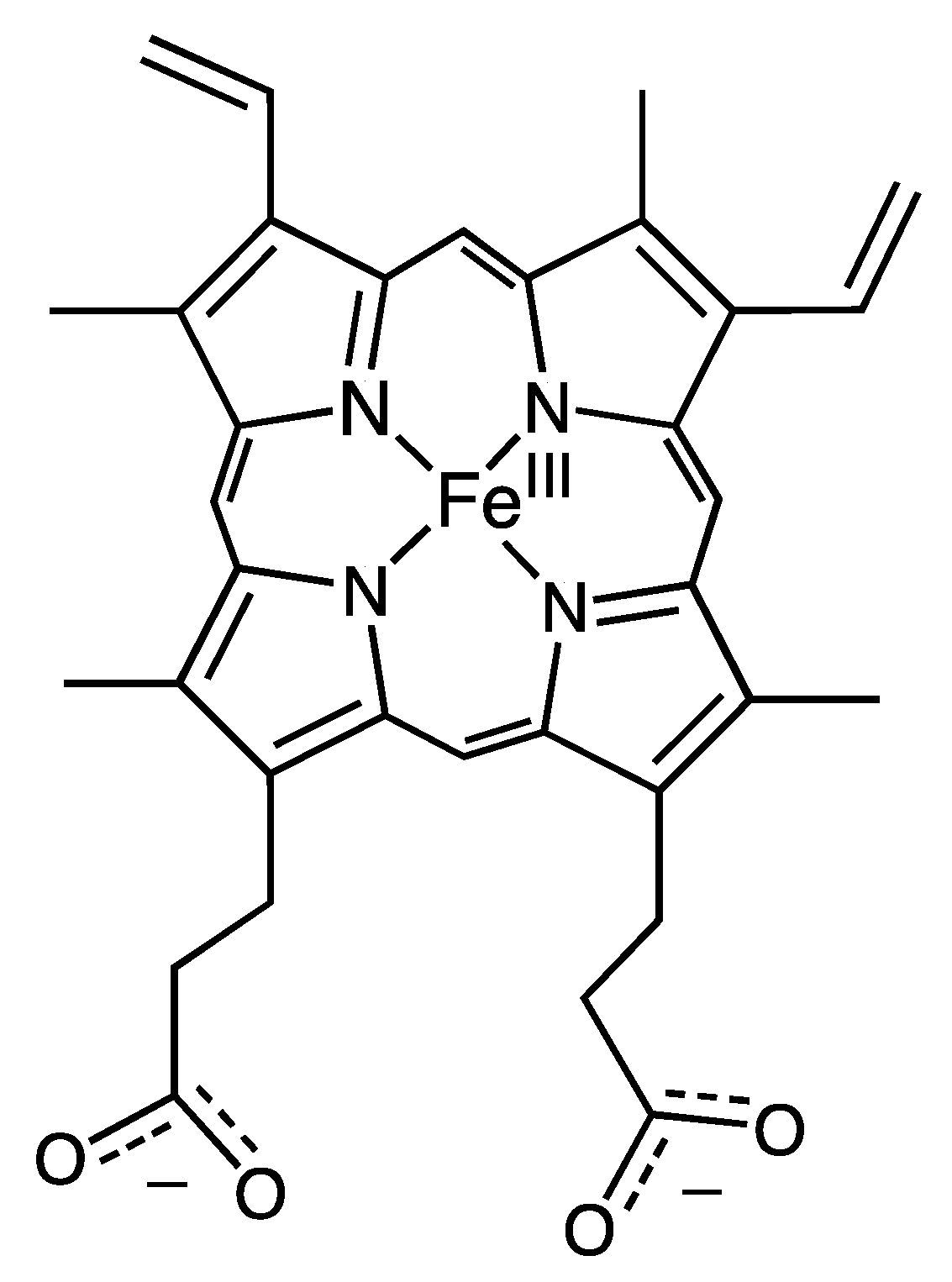
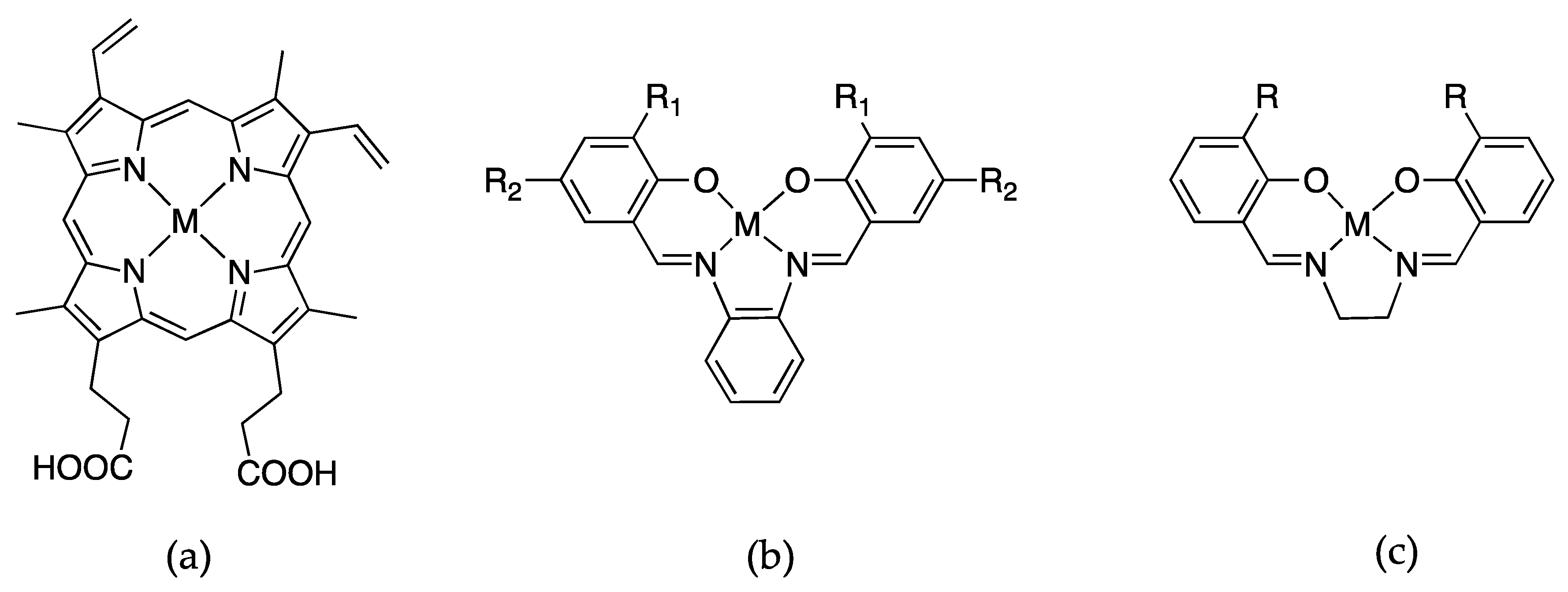
5. Porphyrin-Derivative Substitution
5.1. Chlorin
5.2. Corrole
5.3. Azahemins and Phthalocyanines
5.4. Porphycene
6. New Catalytic Functions Identified
6.1. Peroxidase Activity
6.2. C–H Bond Cleavage Activity
6.3. Hydroxylase Activity
7. Outlook and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Teale, F.W.J. Cleavage of the haem-protein link by acid methylethylketone. Biochim. Biophys. Acta 1959, 35. [Google Scholar] [CrossRef]
- Dickinson, L.C. Metal replaced hemoproteins. J. Chem. Educ. 1976, 53, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Roncone, R.; Monzani, E.; Labo, S.; Sanangelantoni, A.M.; Casella, L. Catalytic activity, stability, unfolding, and degradation pathways of engineered and reconstituted myoglobin. J. Biol. Inorg. Chem. 2005, 10, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Roncone, R.; Monzani, E.; Murtas, M.; Battaini, G.; Pennati, A.; Sanangelantoni, A.M.; Zuccotti, S.; Bolognesi, M.; Casella, L. Engineering peroxidase activity in myoglobin: The haem cavity structure and peroxide activation in the T67R/S92D mutant and its derivative reconstituted with protohaemin-l-histidine. Biochem. J. 2005, 377, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M.; Ward, T.R. Design of artificial metalloenzymes. Appl. Organomet. Chem. 2005, 19, 35–39. [Google Scholar] [CrossRef]
- Ueno, T.; Koshiyama, T.; Ohashi, M.; Kondo, K.; Kono, M.; Suzuki, A.; Yamane, T.; Watanabe, Y. Coordinated design of cofactor and active site structures in development of new protein catalysts. J. Am. Chem. Soc. 2005, 127, 6556–6562. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, N.; Ueno, T.; Unno, M.; Matsui, T.; Ikeda-Saito, M.; Watanabe, Y. Ligand design for the improvement of stability of metal complex: Protein hybrids. Chem. Commun. 2008, 229–231. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yeung, N.; Sieracki, N.; Marshall, N.M. Design of functional metalloproteins. Nature 2009, 460, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Fukumoto, K.; Watanabe, T.; Hayashi, T. Precise design of artificial cofactors for enhancing peroxidase activity of myoglobin: Myoglobin mutant H64D reconstituted with a “single-winged cofactor” is equivalent to native horseradish peroxidase in oxidation activity. Chemistry 2011, 6, 2491–2499. [Google Scholar]
- Hayashi, T.; Sano, Y.; Onoda, A. Generation of new artificial metalloproteins by cofactor modification of native hemoproteins. Isr. J. Chem. 2015, 55, 76–84. [Google Scholar] [CrossRef]
- Yonetani, T.; Asakura, T. Studies on cytochrome c peroxidase: XV. Comparison of manganese porphyrin-containing cytochrome c peroxidase, horseradish peroxidase, and myoglobin. J. Biol. Chem. 1969, 244, 4580–4588. [Google Scholar] [PubMed]
- Waterman, M.R.; Yonetani, T. Studies on modified hemoglobins: I. Properties of hybrid hemoglobins containing manganese protoporphyrin IX. J. Biol. Chem. 1970, 245, 5847–5852. [Google Scholar] [CrossRef]
- Yamamoto, H.; Kayne, F.J.; Yonetani, T. Studies on cobalt myoglobins and hemoglobins, II. Kinetic studies of reversible oxygenation of cobalt myoglobins and hemoglobins by the temperature jump relaxation method. J. Biol. Chem. 1974, 249, 691–698. [Google Scholar] [PubMed]
- Dickinson, L.C.; Chien, J.C.W. Comparative biological chemistry of cobalt hemoglobin. J. Biol. Chem. 1973, 248, 5005–5011. [Google Scholar] [PubMed]
- Gupta, R.K.; Mildvan, A.S.; Yonetani, T.; Srivastava, T.S. EPR study of 17O nuclear hyperfine interaction in cobalt-oxyhemoglobin: Conformation of bound oxygen. Biochem. Biophys. Res. Commun. 1975, 67, 1005–1012. [Google Scholar] [CrossRef]
- Woodruff, W.H.; Adams, D.H.; Spiro, T.G.; Yonetani, T. Resonance raman spectra of cobalt myoglobins and cobalt porphyrins. Evaluation of protein effects on porphyrin structure. J. Am. Chem. Soc. 1975, 97, 1695–1698. [Google Scholar] [CrossRef] [PubMed]
- Ikeda-Saito, M.; Yamamoto, H.; Imai, K.; Kayne, F.J.; Yonetani, T. Studies of cobalt myoglobins and hemoglobins. Preparation of isolated chains containg cobaltous protoporphrin IX and characterization of their equilibrium and kinetic properties of oxygenation and EPR spectra. J. Biol. Chem. 1977, 252, 620–624. [Google Scholar] [PubMed]
- Hori, H.; Ikeda-Saito, M.; Yonetani, T. Electron paramagnetic resonance and spectrophotometeric studies of the peroxide compounds of manganese-substituted horseradish peroxidase, cytochrome-c peroxidase and manganese-porphyrin model complexes. Biochim. Biophys. Acta 1987, 912, 74–81. [Google Scholar] [CrossRef]
- Nick, R.J.; Ray, G.B.; Fish, K.M.; Spiro, T.G.; Groves, J.T. Evidence for a weak Mn=O bond and a non-porphyrin radical in manganese-substituted horseradish peroxidase compound I. J. Am. Chem. Soc. 1991, 113, 1838–1840. [Google Scholar] [CrossRef]
- Mondal, M.S.; Mazumdar, S.; Mitra, S. Binding of cyanide and thiocyanate to manganese reconstituted myoglobin and formation of peroxide compound: Optical spectral, multinuclear NMR, and kinetic studies. Inorg. Chem. 1993, 32, 5362–5367. [Google Scholar] [CrossRef]
- Mondal, M.S.; Mitra, S. Kinetic studies of the two-step reactions of H2O2 with manganese-reconstituted myoglobin. Biochim. Biophys. Acta 1996, 1296, 174–180. [Google Scholar] [CrossRef]
- Ueno, T.; Ohashi, M.; Kono, M.; Kondo, K.; Suzuki, A.; Yamane, T.; Watanabe, Y. Crystal structures of artificial metalloproteins: Tight binding of FeIII(schiff-base) by mutation of Ala71 to Gly in apo-myoglobin. Inorg. Chem. 2004, 43, 2852–2858. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Hayashi, T.; Ando, T.; Hisaeda, Y.; Ueno, T.; Watanabe, Y. Hybridization of modified-heme reconstitution and distal histidine mutation to functionalize sperm whale myoglobin. J. Am. Chem. Soc. 2004, 126, 436–437. [Google Scholar] [CrossRef] [PubMed]
- Carey, J.R.; Ma, S.K.; Pfister, T.D.; Garner, D.K.; Kim, H.K.; Abramite, J.A.; Wang, Z.; Guo, Z.; Lu, Y. A site-selective dual anchoring strategy for artificial metalloprotein design. J. Am. Chem. Soc. 2004, 126, 10812–10813. [Google Scholar] [CrossRef] [PubMed]
- Garner, D.K.; Liang, L.; Barrios, D.A.; Zhang, J.-L.; Lu, Y. The important role of covalent anchor positions in tuning catalytic properties of a rationally designed mnsalen-containing metalloenzyme. ACS Catal. 2011, 1, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-L.; Garner, D.K.; Liang, L.; Barrios, D.A.; Lu, Y. Noncovalent modulation of pH-dependent reactivity of a Mn-salen cofactor in myoglobin with hydrogen peroxide. Chemistry 2009, 15, 7481–7489. [Google Scholar] [CrossRef] [PubMed]
- Paulson, D.R.; Addison, A.W.; Dolphin, D.; James, B.R. Preparation of ruthenium(II) and ruthenium(III) myoglobin and the reaction of dioxygen, and carbon monoxide, with ruthenium(II) myoglobin. J. Biol. Chem. 1979, 254, 7002–7006. [Google Scholar] [PubMed]
- Aoyama, Y.; Aoyagi, K.; Toi, H.; Ogoshi, H. Reconstituted myoglobins with rhodium(III) complexes of mesoporphyrin and deuteroporphryin IX. Inorg. Chem. 1983, 22, 3046–3050. [Google Scholar] [CrossRef]
- Stynes, D.V.; Liu, S.; Marcus, H. Distal histidine coordination to iron in phthalocyanine-reconstituted myoglobin. Inorg. Chem. 1985, 24, 4335–4338. [Google Scholar] [CrossRef]
- Neya, S.; Kaku, T.; Funasaki, N.; Shiro, Y.; Iizuka, T.; Imai, K.; Hori, H. Novel ligand binding properties of the myoglobin substituted with monoazahemin. J. Biol. Chem. 1995, 270, 13118–13123. [Google Scholar] [CrossRef] [PubMed]
- Neya, S.; Hori, H.; Imai, K.; Kawamura-Konishi, Y.; Suzuki, H.; Shiro, Y.; Iizuka, T.; Funasaki, N. Remarkable functional aspects of myoglobin induced by diazaheme prosthetic group. J. Biochem. 1997, 121, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Dejima, H.; Matsuo, T.; Sato, H.; Murata, D.; Hisaeda, Y. Blue myoglobin reconstituted with an iron porphycene shows extremely high oxygen affinity. J. Am. Chem. Soc. 2002, 124, 11226–11227. [Google Scholar] [CrossRef] [PubMed]
- Neya, S.; Imai, K.; Hori, H.; Ishikawa, H.; Ishimori, K.; Okuno, D.; Nagatomo, S.; Hoshino, T.; Hata, M.; Funasaki, N. Iron hemiporphycene as a functional prosthetic group for myoglobin. Inorg. Chem. 2003, 42, 1456–1461. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Dejima, H.; Hirota, S.; Murata, D.; Sato, H.; Ikegami, T.; Hori, H.; Hisaeda, Y.; Hayashi, T. Ligand binding properties of myoglobin reconstituted with iron porphycene: Unusual O2 binding selectivity against CO binding. J. Am. Chem. Soc. 2004, 126, 16007–16017. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Murata, D.; Makino, M.; Sugimoto, H.; Matsuo, T.; Sato, H.; Shiro, Y.; Hisaeda, Y. Crystal structure and peroxidase activity of myoglobin reconstituted with iron porphycene. Inorg. Chem. 2006, 45, 10530–10536. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-z.; Taniguchi, I.; Mulchandani, A. Redox properties of engineered ruthenium myoglobin. Bioelectrochemistry 2009, 75, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Heinecke, J.L.; Yi, J.; Pereira, J.C.M.; Richter-Addo, G.B.; Ford, P.C. Nitrite reduction by CoII and MnII substituted myoglobins. Towards understanding necessary components of Mb nitrite reductase activity. J. Inorg. Biochem. 2012, 107, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Oohora, K.; Kihira, Y.; Mizohata, E.; Inoue, T.; Hayashi, T. C(sp3)–H bond hydroxylation catalyzed by myoglobin reconstituted with manganese porphycene. J. Am. Chem. Soc. 2013, 135, 17282–17285. [Google Scholar] [CrossRef] [PubMed]
- Stone, K.L.; Hua, J.; Choudhry, H. Manganese-substituted myoglobin: Characterization and reactivity of an oxidizing intermediate towards a weak C–H bond. Inorganics 2015, 3, 219–229. [Google Scholar] [CrossRef]
- Dolphin, D. The Porphyrins: Structure and Synthesis; Academic Press: New York, NY, USA, 1978; Volume 2. [Google Scholar]
- Bordeaux, M.; Singh, R.; Fasan, R. Intramolecular C(sp3)–H amination of arylsulfonyl azides with engineered and artificial myoglobin-based catalysts. Bioorg. Med. Chem. 2014, 22, 5697–5704. [Google Scholar] [CrossRef] [PubMed]
- Hori, H. Light absorption, electron paramagnetic resonance and resonance raman characteristics of nitridochromium(V) protoporphyrin-IX and its reconstituted hemoproteins. Biochim. Biophys. Acta 1991, 1077, 392–399. [Google Scholar] [CrossRef]
- Zahran, Z.N.; Chooback, L.; Copeland, D.M.; West, A.H.; Richter-Addo, G.B. Crystal structures of manganese- and cobalt-substituted myoglobin in complex with NO and nitrite reveal unusual ligand conformations. J. Inorg. Biochem. 2008, 102, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, S.-I.; Matsui, T.; Watanabe, Y. Conversion of myoglobin into a highly stereo-specific peroxygenase by the L29H/H64L mutation. J. Am. Chem. Soc. 1996, 118, 9784–9785. [Google Scholar] [CrossRef]
- Matsui, T.; Ozaki, S.-I.; Watanabe, Y. On the formation and reactivity of compund I of the His-64 myoglobin mutants. J. Biol. Chem. 1997, 272, 32735–32738. [Google Scholar] [CrossRef] [PubMed]
- Hara, I.; Ueno, T.; Shin-ichi, O.; Itoh, S.; Lee, K.; Ueyama, N.; Watanabe, Y. Oxidative modification of tryptophan 43 in the heme vicinity of the F43W/H64L myoglobin mutant. J. Biol. Chem. 2001, 276, 36067–36070. [Google Scholar] [CrossRef] [PubMed]
- Pfister, T.D.; Ohki, T.; Ueno, T.; Hara, I.; Adachi, S.; Makino, Y.; Ueyama, N.; Lu, Y.; Watanabe, Y. Monooxygenation of an aromatic ring by F43W/H64D/V68I myoglobin mutant and hydrogen peroxide. J. Biol. Chem. 2005, 280, 12858–12866. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Nakajima, H.; Ueno, T. Reactivities of oxo and peroxo intermediates studied by hemoproteins mutants. Acc. Chem. Res. 2007, 40, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Urayama, P.; Phillips, G.N., Jr.; Gruner, S.M. Probing substates in sperm whale myoglobin using high-pressure crystallography. Structure 2002, 10, 51–60. [Google Scholar] [CrossRef]
- Du, J.; Huang, X.; Sun, S.; Wang, C.; Lebioda, L.; Dawson, J.H. Amphitrite ornata dehaloperoxidase (DHP): Investigations of structural factors that influence the mechanism of halophenol dehalogenation using “peroxidase-like” myoglobin mutants and “myoglobin-like” DHP mutants. Biochemistry 2011, 50, 8172–8180. [Google Scholar] [CrossRef] [PubMed]
- Sigman, J.A.; Kwok, B.C.; Lu, Y. From myoglobin to heme-copper oxidase: Design and engineering of a CuB center into sperm whale myoglobin. J. Am. Chem. Soc. 2000, 122, 8192–8196. [Google Scholar] [CrossRef]
- Zhao, X.; Yeung, N.; Wang, Z.; Guo, Z.; Lu, Y. Effects of metal ions in the CuB center on the redox properties of heme in heme-copper oxidases: Spectroelectrochemical studies of an engineered heme-copper center in myoglobin. Biochemistry 2005, 44, 1210–1214. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yeung, N.; Russell, B.S.; Garner, D.K.; Lu, Y. Catalytic reduction of NO to N2O by a designed heme copper center in myoglobin: Implications for the role of metal ions. J. Am. Chem. Soc. 2006, 128, 6766–6767. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Cui, C.; Liu, X.; Petrik, I.D.; Wang, J.; Lu, Y. A designed metalloenzyme achieving the catalytic rate of a native enzyme. J. Am. Chem. Soc. 2015, 137, 11570–11573. [Google Scholar] [CrossRef] [PubMed]
- Bordeaux, M.; Tyagi, V.; Fasan, R. Highly diastereoselective and enantioselective olefin cyclopropanation using engineered myoglobin-based catalysts. Angew. Chem. Int. Ed. 2015, 54, 1744–1748. [Google Scholar] [CrossRef] [PubMed]
- Giovani, S.; Singh, R.; Fasan, R. Efficient conversion of primary azides to aldehydes catalyzed by active site variants of myoglobin. Chem. Sci. 2016, 7, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Sreenilayam, G.; Fasan, R. Myoglobin-catalyzed intermolecular carbene N–H insertion with arylamine substrates. Chem. Commun. 2015, 51, 1532–1534. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, V.; Bonn, R.B.; Fasan, R. Intermolecular carbene S–H insertion catalysed by engineered myoglobin-based catalysts. Chem. Sci. 2015, 6, 2488–2494. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, V.; Fasan, R. Myoglobin-catalyzed olefination of aldehydes. Angew. Chem. Int. Ed. 2016, 55, 2512–2516. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, S.-I.; Ortiz de Montellano, P.R. Molecular engineering of horseradish peroxidase: Thioether sulfoxidation and styrene epoxidation by Phe-141 leucine and threonine mutants. J. Am. Chem. Soc. 1995, 117, 7056–7064. [Google Scholar] [CrossRef]
- Gupta, K.C.; Sutar, A.K. Catalytic activities of Schiff base transition metal complexes. Coord. Chem. Rev. 2008, 252, 1420–1450. [Google Scholar] [CrossRef]
- Cozzi, P.G. Metal-salen Schiff base complexes in catalysis: Practical aspects. Chem. Soc. Rev. 2004, 33, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Bois, J.D.; Tomooka, C.S.; Hong, J.; Carreira, E.M. Nitridomanganese(V) complexes: Design, preparation, and use as nitrogen atom-transfer reagents. Acc. Chem. Res. 1997, 30, 364–372. [Google Scholar] [CrossRef]
- Sun, W.; Herdtweck, E.; Kuhn, F.E. Catalytic aziridinations with copper(I) salen complexes. New J. Chem. 2005, 29, 1577–1580. [Google Scholar] [CrossRef]
- Wang, C.; Kurahashi, T.; Inomata, K.; Hada, M.; Fujii, H. Oxygen-atom transfer from iodosylarene adducts of a manganese(IV) salen complex: Effect of arenes and anions on I(III) of the coordinated iodosylarene. Inorg. Chem. 2013, 52, 9557–9566. [Google Scholar] [CrossRef] [PubMed]
- Coulter, E.D.; Sono, M.; Chang, C.K.; Lopez, O.; Dawson, J.H. Electron paramagnetic resonance spectroscopy as a probe of coordination structure in green heme systems: Iron chlorins and iron formylporphyrins reconstituted into myoglobin. Inorg. Chim. Acta 1995, 240, 603–608. [Google Scholar] [CrossRef]
- Sotiriou-Leventis, C.; Chang, C.K. Kinetic study of CO and O2 binding to horse heart myoglobin reconstituted with synthetic iron chlorin green hemes. Inorg. Chim. Acta 2000, 311, 113–118. [Google Scholar] [CrossRef]
- Matsuo, T.; Hayashi, A.; Abe, M.; Matsuda, T.; Hisaeda, Y.; Hayashi, T. Meso-unsubstituted iron corrole in hemoproteins: Remarkable differences in effects on peroxidase activities between myoglobin and horseradish peroxidase. J. Am. Chem. Soc. 2009, 131, 15124–15125. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.H.; Busch, D.H. Complexes derived from strong field ligands. XIX. Magnetic properties of transition metal derivatives of 4,4′,4″,4′′′-tetrasulfophthalocyanine. Inorg. Chem. 1965, 4, 469–471. [Google Scholar] [CrossRef]
- Dolotova, O.; Yuzhakova, O.; Solovyova, L.; Shevchenko, E.; Negrimovsky, V.; Lukyanets, E.; Kaliya, O. Water-soluble manganese phthalocyanines. J. Porphyr. Phthalocya. 2013, 17, 881–888. [Google Scholar] [CrossRef]
- Matsuo, T.; Tsuruta, T.; Maehara, K.; Sato, H.; Hisaeda, Y.; Hayashi, T. Preparation and O2 binding study of myoglobin having a cobalt porphycene. Inorg. Chem. 2005, 44, 9391–9396. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. Cytochrome P450: Structure, Mechanism, and Biochemistry; Springer: New York, NY, USA, 1995; Volume 2. [Google Scholar]
- Davydov, R.; Makris, T.M.; Kofman, V.; Werst, D.E.; Sligar, S.G.; Hoffman, B.M. Hydroxylation of camphor by reduced oxy-cytochrome P450cam: Mechanistic implications of EPR and ENDOR studies of catalytic intermediates in native and mutant enzymes. J. Am. Chem. Soc. 2001, 123, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Kumar, D.; Visser, S.P.D.; Altun, A.; Thiel, W. Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef] [PubMed]
- Gelb, M.H.; William, A.; Toscano, J.; Sligar, S.G. Chemical mechanisms for cytochrome P-450 oxidation: Spectral and catalytic properties of a manganese-substituted protein. Proc. Natl. Acad. Sci. USA 1982, 79, 5758–5762. [Google Scholar] [CrossRef] [PubMed]
- Schlichting, I.; Berendzen, J.; Chu, K.; Stock, A.M.; Maves, S.A.; Benson, D.E.; Sweet, R.M.; Ringe, D.; Petsko, G.A.; Sligar, S.G. The catalytic pathway of cytochrome P450cam at atomic resolution. Science 2000, 287, 1615–1622. [Google Scholar] [CrossRef] [PubMed]
- Baylon, J.L.; Lenov, I.L.; Sligar, S.G.; Tajkhorshid, E. Characterizing the membrane-bound state of cytochrome P450 3A4: Structure, depth of insertion, and orientation. J. Am. Chem. Soc. 2013, 135, 8542–8551. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
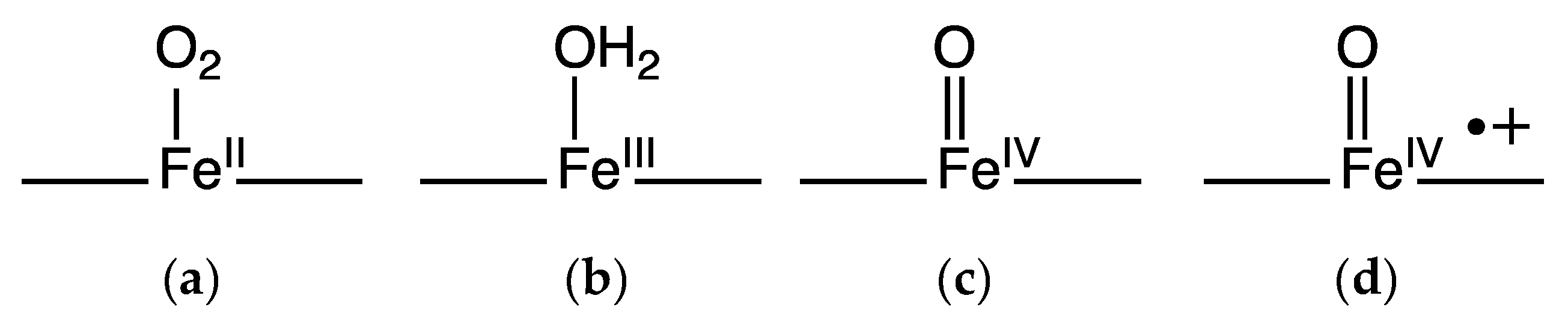{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hemoprotein | Metal | Comment | References |
|---|---|---|---|
| Hemoglobin | Co | Co-substituted hemoglobin binds O2 with high affinity but less than the iron version. | [14,15,16,18] |
| Mn | Mn-substituted hemoglobin does not bind O2 and was used to study cooperativity of the subunits upon O2 binding by selectively incorporating Mn-heme into the subunits. | [13] | |
| Myoglobin | Co | Co-substituted myoglobin binds O2 with high affinity and there have been several reports of ligand binding. | [17,18,38,41] |
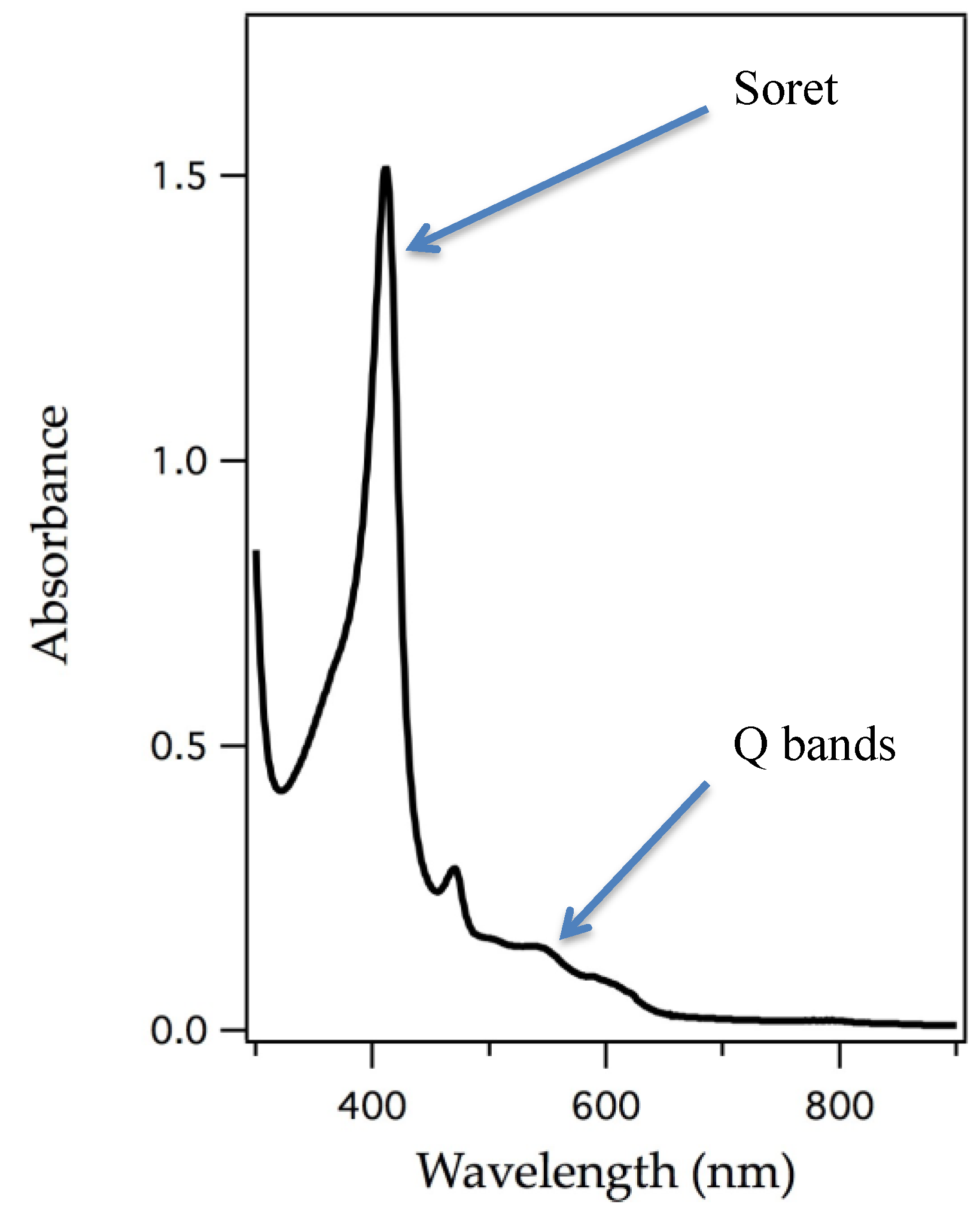
| Mn | Mn-substituted myoglobin reacts with hydroperoxides giving rise to Soret shifts and EPR signals showing Mn(IV) character and there have been several reports of ligand binding. | [19,21,22,40] | |
| Cr | Cr(V)nitride-substituted myoglobin was prepared by azide photolysis of the Cr(III)-N3 protoporphyrin IX and subsequent incorporation into myoglobin. | [42] | |
| Ru | Ru-substituted myoglobin binds CO. | [28] | |
| Rh | A Rh-CH3 moiety was observed in Rh-substituted myoglobin. | [29] | |
| Cytochrome c Peroxidase | Mn | Mn-substituted cytochrome c peroxidase reacts with hydroperoxides giving rise to Soret shifts and EPR signals showing Mn(IV) character. | [19] |
| Cr | Cr(V)nitride-substituted cytochrome c peroxidase was prepared by azide photolysis of the Cr(III)–N3 protoporphyrin IX and subsequent incorporation into cytochrome c peroxidase. | [42] | |
| Horseradish Peroxidase | Mn | Mn-substituted horseradish peroxidase reacts with hydroperoxides giving rise to Soret shifts and EPR signals showing Mn(IV) character. | [19,20] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stone, K.L.; Ahmed, S.M. Advances in Engineered Hemoproteins that Promote Biocatalysis. Inorganics 2016, 4, 12. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics4020012
Stone KL, Ahmed SM. Advances in Engineered Hemoproteins that Promote Biocatalysis. Inorganics. 2016; 4(2):12. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics4020012
Chicago/Turabian StyleStone, Kari L., and Syeda M. Ahmed. 2016. "Advances in Engineered Hemoproteins that Promote Biocatalysis" Inorganics 4, no. 2: 12. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics4020012