
Computing Free Energies of Hydroxylated Silica Nanoclusters: Forcefield versus Density Functional Calculations



Abstract
:1. Introduction
2. Methodology
3. Results and Discussion
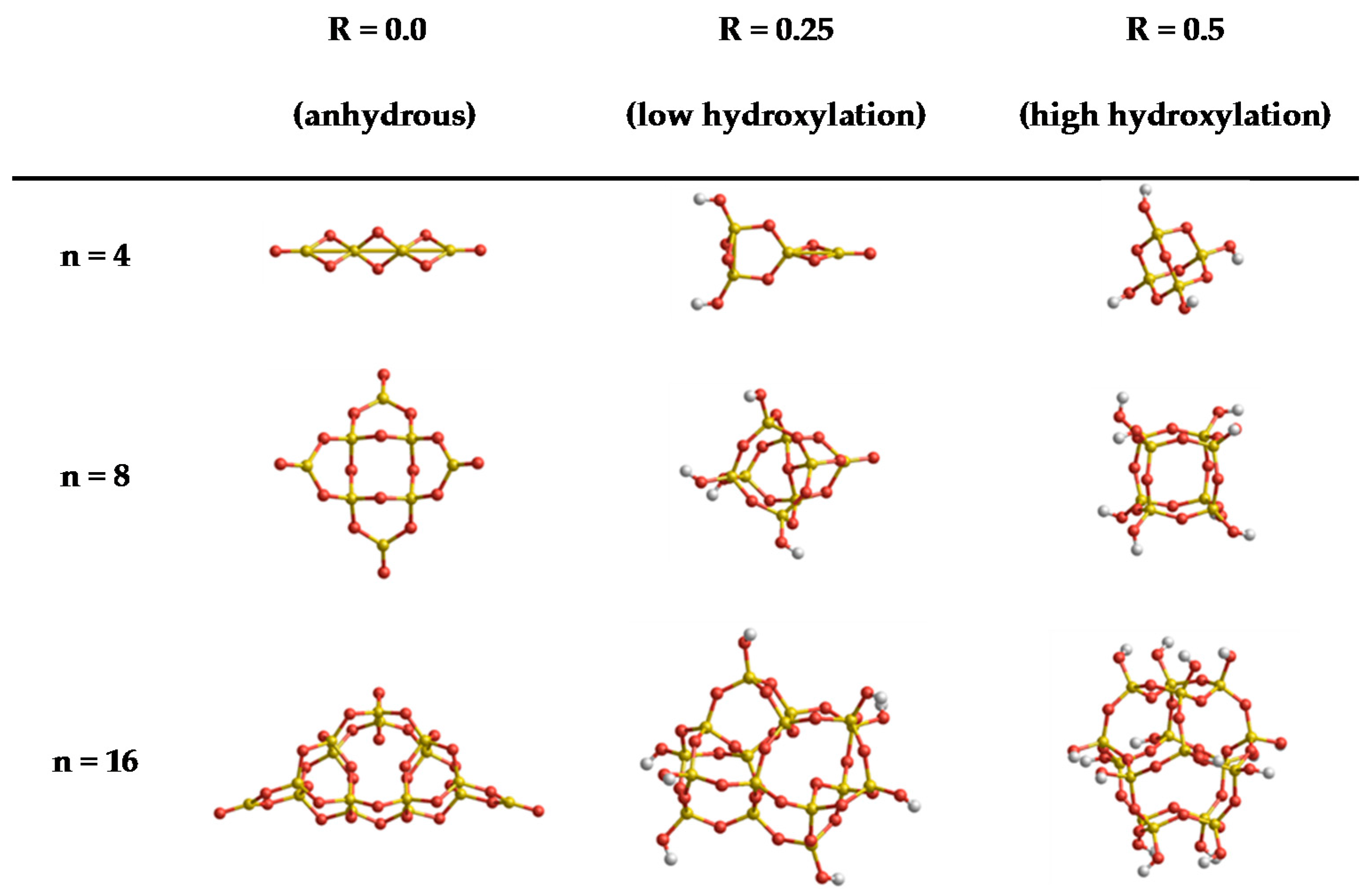
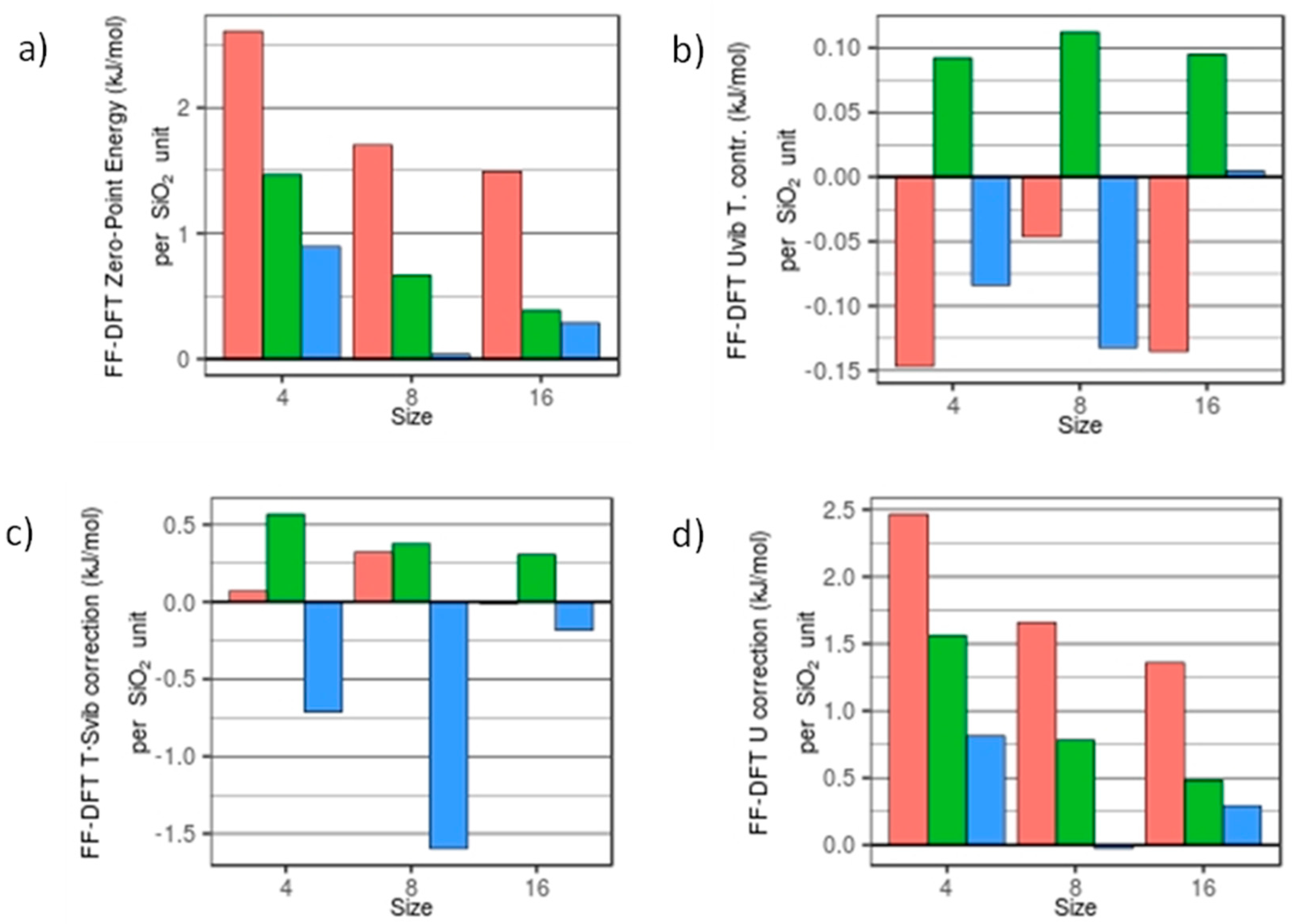
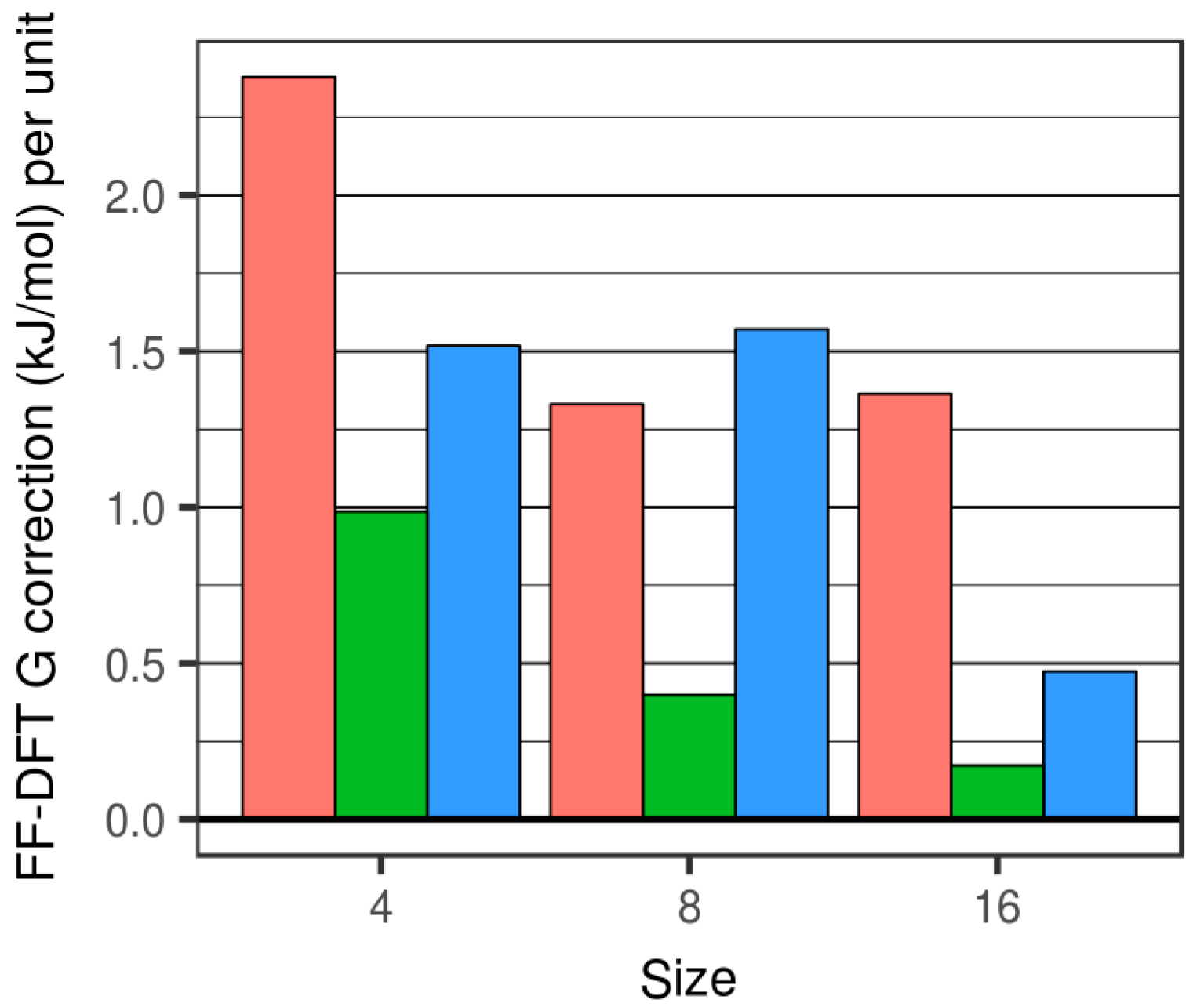
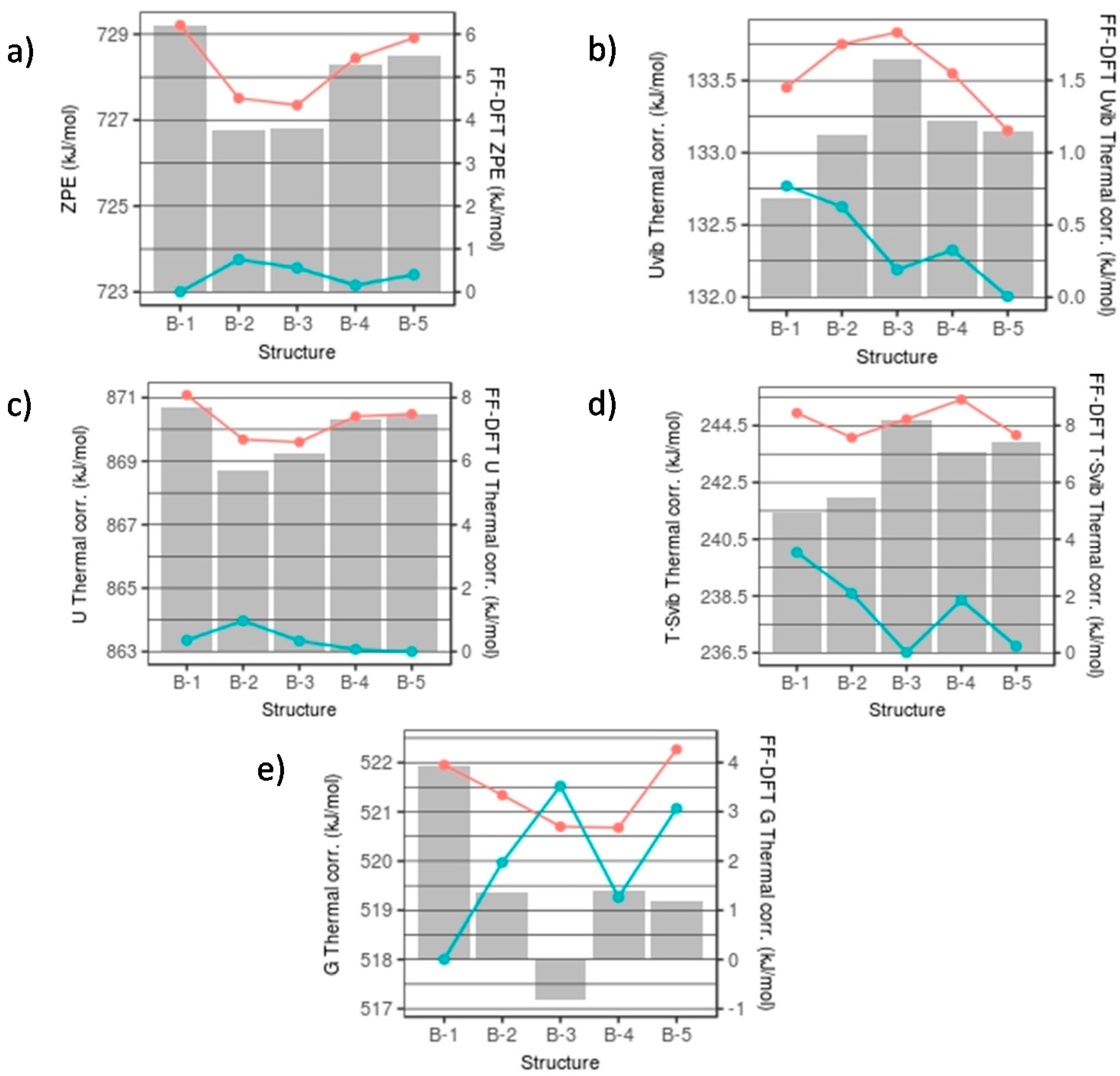
3.1. Thermodynamic Data with Respect to Size and Hydroxylation
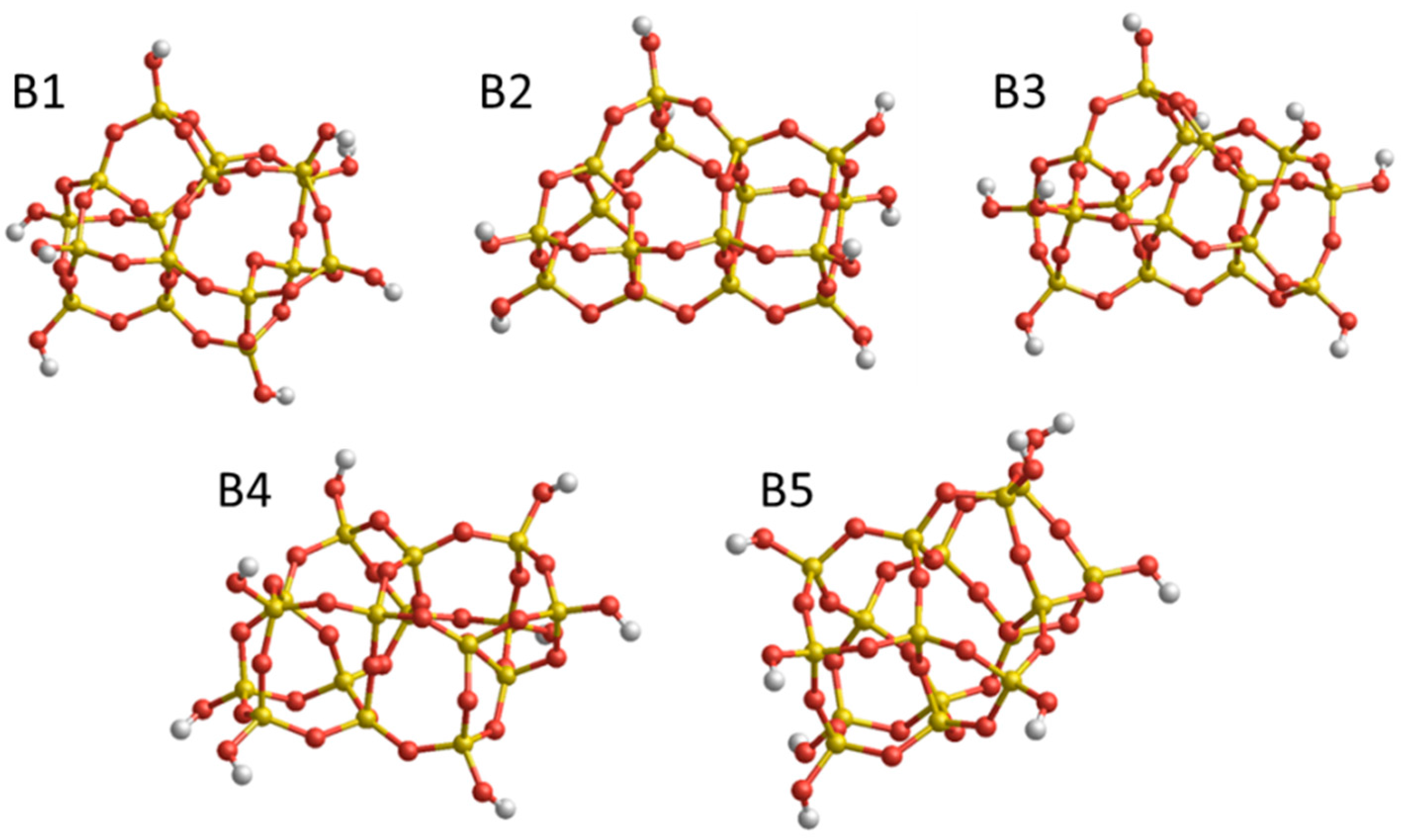
3.2. Free Energy in a Set of Isomers
3.3. Free Energy of Hydroxylation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kollman, P. Free energy calculations: Applications to chemical and biochemical phenomena. Chem. Rev. 1993, 93, 2395–2417. [Google Scholar] [CrossRef]
- Van Gunsteren, W.F.; Dolenc, J. Thirty-five years of biomolecular simulation: Development of methodology, force fields and software. Mol. Simul. 2012, 38, 1271–2181. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Tavazza, F.; Trautt, Z.T.; de Macedo, R.A.B. Considerations for choosing and using force fields and interatomic potentials in materials science and engineering. Curr. Opin. Solid State Mater. Sci. 2013, 17, 277–283. [Google Scholar] [CrossRef]
- Goumans, T.P.M.; Bromley, S.T. Efficient nucleation of stardust silicates via heteromolecular homogeneous condensation. Mon. Not. R. Astron. Soc. 2012, 420, 3344–3349. [Google Scholar] [CrossRef]
- Bromley, S.T.; Martin, J.C.G.; Plane, J.M.C. Under what conditions does (SiO)N nucleation occur? A bottom-up kinetic modelling evaluation. Phys. Chem. Chem. Phys. 2016, 18, 26913–26922. [Google Scholar] [CrossRef] [PubMed]
- Zachariah, M.R.; Tsang, W. Application of ab initio molecular orbital and reaction rate theories to nucleation kinetics. Aerosol Sci. Tech. 1993, 19, 499–513. [Google Scholar] [CrossRef]
- Köhler, T.M.; Gail, H.-P.; Sedlmayr, E. MgO dust nucleation in M-Stars: Calculation of cluster properties and nucleation rates. Astron. Astrophys. 1997, 320, 553–567. [Google Scholar]
- Bhattacharya, S.; Levchenko, S.V.; Ghiringhelli, L.M.; Scheffler, M. Efficient ab initio schemes for finding thermodynamically stable and metastable atomic structures: Benchmark of cascade genetic algorithms. New J. Phys. 2014, 16. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Levchenko, S.V.; Ghiringhelli, L.M.; Scheffler, M. Stability and Metastability of clusters in a Reactive Atmosphere: Theoretical Evidence for Unexpected Stoichiometries of MgMOx. Phys. Rev. Lett. 2013, 111. [Google Scholar] [CrossRef] [PubMed]
- Lepeshkin, S.; Baturin, V.; Tikhonov, E.; Matsko, N.; Uspenskii, Y.; Naumova, A.; Feya, O.; Schoonene, M.A.; Oganov, A.R. Super-oxidation of silicon nanoclusters: Magnetism and reactive oxygen species at the surface. Nanoscale 2016, 8, 18616–18620. [Google Scholar] [CrossRef] [PubMed]
- Mora-Fonz, M.J.; Catlow, C.R.A.; Lewis, D.W. Oligomerization and cyclization processes in the nucleation of microporous silicas. Angew. Chem. Int. Ed. 2005, 44, 3082–3086. [Google Scholar] [CrossRef] [PubMed]
- Trinh, T.T.; Jansen, A.P.J.; van Santen, R.A. Mechanism of oligomerization reactions of silica. J. Phys. Chem. B 2006, 110, 23099–23106. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, C.L.; Thomson, K.T. Density functional theory investigation into structure and reactivity of prenucleation silica species. J. Phys. Chem. C 2008, 112, 12653–12662. [Google Scholar] [CrossRef]
- Jelfs, K.; Flikkema, E.; Bromley, S.T. Evidence for atomic mixing via multiple intermediates during the dynamic interconversion of silicate oligomers in solution. Chem. Commun. 2012, 48. [Google Scholar] [CrossRef] [PubMed]
- Flikkema, E.; Bromley, S.T. A new interatomic potential for nanoscale silica. Chem. Phys. Lett. 2003, 378, 622–629. [Google Scholar] [CrossRef]
- Aguado, A.; Madden, P.A. Fully transferable interatomic potentials for large-scale computer simulations of simple metal oxides: Application to MgO. Phys. Rev. B 2004, 70. [Google Scholar] [CrossRef]
- Raiteri, P.; Demichelis, R.; Gale, J.D. Thermodynamically consistent force field for molecular dynamics simulations of alkaline-earth carbonates and their aqueous speciation. J. Phys. Chem. C 2015, 119, 24447–24458. [Google Scholar] [CrossRef]
- Raiteri, P.; Gale, J.D.; Quigley, D.; Rodger, P.M. Derivation of an accurate force-field for simulating the growth of calcium carbonate from aqueous solution: A new model for the calcite-water interface. J. Phys. Chem. C 2010, 114, 5997–6010. [Google Scholar] [CrossRef]
- Demichelis, R.; Raiteri, P.; Gale, J.D.; Quigley, D.; Gebauer, D. Stable prenucleation mineral clusters are liquid-like ionic polymers. Nat. Commun. 2011, 2, 590. [Google Scholar] [CrossRef] [PubMed]
- Pedone, A.; Malavasi, G.; Menziani, M.C.; Segre, U.; Musso, F.; Corno, M.; Civalleri, B.; Ugliengo, P. FFSiOH: A new force field for silica polymorphs and their hydroxylated surfaces based on periodic B3LYP calculations. Chem. Mater. 2008, 20, 2522–2531. [Google Scholar] [CrossRef]
- Macia, A.; Ugliengo, P.; Bromley, S.T. Modeling hydroxylated nanosilica: Testing the performance of ReaxFF and FFSiOH forcefields. J. Chem. Phys. 2017, 146. [Google Scholar] [CrossRef]
- Iler, R.K. The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties; Wiley: New York, NY, USA, 1979. [Google Scholar]
- Dove, P.M.; Rimstidt, J.D. Silica: Physical Behaviour, Geochemistry and Materials Applications; Heany, P.J., Pewitt, C.T., Gibbs, G.V., Eds.; Mineralogical Society of America: Washington, DC, USA, 1994; Volume 29, pp. 259–301. [Google Scholar]
- McQuarrie, D.A.; Simons, J.D. Molecular Thermodynamics; University Science Book: Sausalito, CA, USA, 1999. [Google Scholar]
- Jelfs, K.E.; Flikkema, E.; Bromley, S.T. Hydroxylation of silica nanoclusters (SiO2)M(H2O)N, M = 4, 8, 16, 24: Stability and structural trends. Phys. Chem. Chem. Phys. 2013, 15, 20438–20443. [Google Scholar] [CrossRef] [PubMed]
- Flikkema, E.; Jelfs, K.E.; Bromley, S.T. Structure and energetics of hydroxylated silica clusters (SiO2)M(H2O)N M = 8, 16 and N = 1–4: A global optimisation study. Chem. Phys. Lett. 2012, 554, 117–122. [Google Scholar] [CrossRef]
- Flikkema, E.; Bromley, S.T. Dedicated global optimization search for ground state silica nanoclusters: (SiO2)M (N = 6–12). J. Phys. Chem. B 2004, 108, 9638–9645. [Google Scholar] [CrossRef]
- Bromley, S.T.; Flikkema, E. Columnar-to-disk structural transition in nanoscale (SiO2)N clusters. Phys. Rev. Lett. 2005, 95, 185505. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Ugliengo, P.; Sodupe, M.; Musso, F.; Bush, I.J.; Orlando, R.; Dovesi, R. Realistic Models of Hydroxylated Amorphous Silica Surfaces and MCM-41 Mesoporous Material Simulated by Large-scale Periodic B3LYP calculations. Adv. Mater. 2008, 20, 1–5. [Google Scholar] [CrossRef]
- Schlegel, H.B. Optimization of Equilibrium Geometries and Transition Structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Gale, J.D.; Rohl, A.L. The General Utility Lattice Program (GULP). Mol. Simul. 2003, 29, 291–341. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | ΔGreact (DFT) | ΔGreact (FFSiOH) | ΔΔGreact (DFT-FFSiOH) |
|---|---|---|---|
| Si4O8 + H2O → H2Si4O9 | −323.0 | −328.5 | 5.5 |
| H2Si4O9 + H2O → H4Si4O10 | −461.8 | −459.7 | −2.1 |
| Si8O16 + 2H2O → H4Si8O18 | −392.7 | −396.4 | 3.7 |
| H4Si8O18 + 2H2O → H8Si8O20 | −244.3 | −239.6 | −4.7 |
| Si16O32 + 4H2O → H8Si16O36 | −343.2 | −348.0 | 4.8 |
| H8Si16O36 + 4H2O → H16Si16O40 | −47.9 | −46.6 | −1.3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macià Escatllar, A.; Ugliengo, P.; Bromley, S.T. Computing Free Energies of Hydroxylated Silica Nanoclusters: Forcefield versus Density Functional Calculations. Inorganics 2017, 5, 41. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics5030041
Macià Escatllar A, Ugliengo P, Bromley ST. Computing Free Energies of Hydroxylated Silica Nanoclusters: Forcefield versus Density Functional Calculations. Inorganics. 2017; 5(3):41. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics5030041
Chicago/Turabian StyleMacià Escatllar, Antoni, Piero Ugliengo, and Stefan T. Bromley. 2017. "Computing Free Energies of Hydroxylated Silica Nanoclusters: Forcefield versus Density Functional Calculations" Inorganics 5, no. 3: 41. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics5030041