
A DFT Study on the O2 Adsorption Properties of Supported PtNi Clusters


1
Department of Physics, King’s College London, WC2R2LS London, UK
2
Instituto de Física, Universidad Nacional Autónoma de Mexico, Apartado Postal 20-364, Mexico DF 01000, Mexico
*
Author to whom correspondence should be addressed.
Inorganics 2017, 5(3), 43; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics5030043
Submission received: 17 May 2017
/
Revised: 26 June 2017
/
Accepted: 26 June 2017
/
Published: 4 July 2017
(This article belongs to the Special Issue Inorganic Nanoclusters: Advances in Understanding Structure and Properties)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:We present a systematic study on the adsorption properties of molecular oxygen on Pt, Ni and PtNi clusters previously deposited on MgO(100) by means of density functional theory calculations. We map the different adsorption sites for a variety of cluster geometries, including icosahedra, decahedra, truncated octahedra and cuboctahedra, in the size range between 25–58 atoms. The average adsorption energy depends on the chemical composition, varying from 2 eV for pure Ni, 1.07 for pure Pt and 1.09 for a PtNi nanoalloy. To correlate the adsorption map to the adsorption properties, we opt for a geometrical descriptor based on the metallic coordination up to the second coordination shell. We find an almost linear relationship between the second coordination shell and adsorption energy, with low coordination sites, such as those located at the (111)/(111) and (111)/(100) cluster edges-displaying adsorption energies above 1 eV, while higher coordination sites such as (111) cluster facets have an interaction of 0.4 eV or lower. The inclusion of van der Waals corrections leads to an overall increase of the O adsorption energy without an alteration of the general adsorption trends.
1. Introduction
The development of novel mobility technologies for cleaner vehicle emissions is nowadays essential in order to mitigate the current high levels of pollution seen on internal combustion engines. One foreseeable technology involves the electrochemical conversion of energy via efficient fuel cells (FCs), namely using hydrogen as its primarily source of fuel [1]. All FCs consist of an anode, cathode, as well as of a proton-conductive electrolyte. FCs are generally classified according to the type of electrolyte used, and excellent technology reviews exist on the literature, ranging from proton-conducting metal–organic frameworks [2], crystalline porous-materials [3], as well as carbon- and nitrogen-based porous solids [4]. Among all of the existing fuel cells, the proton exchange membrane fuel cell (PEMFC) has been actively developed for use in vehicles [5,6]. PEMFCs currently provide the necessary power needed to travel even long-distances. However, its mass commercialization is currently hindered both by the slow oxygen reduction reaction (ORR) at the cathode and the extremely high cost of platinum (Pt). This has triggered an active search for cheaper Pt-based alloys and the miniaturization of the metal catalyst towards the nanoscale. One proposed solution is to combine it with other late transition metals such as nickel (Ni), cobalt (Co), chromium (Cr), copper (Cu) and iron (Fe) [7,8,9]. The goal of the alloyed material is to improve the catalytic activity towards the oxygen reduction reaction (ORRs), offered by current commercial Pt cathode catalysts, without the scaling costs.
In this regard, recent experimental studies have shown that a five-fold decrease in the amount of Pt used in PEMFC stacks is needed in order to reach levels for mass-production and commercialisation for use in light-duty vehicles [10]. A promising alloy corresponds to PtNi. Work performed by Stamenkovic et al. showed an exceptional catalytic activity (10-fold) towards ORR of an extended PtNi(111) surface compared to a monometallic Pt(111) surface; and up to 90 times greater than the carbon supported Pt catalysts used in PEMFC [11]. Their results were rationalized due to an unusual shift in the calculated PtNi d-band centre, as well as due to having a peculiar atomic segregation pattern involving a Pt-rich outer layer. Further experimental work has focused on the preparation of small PtNi nanoparticles (NPs) ≤ 5 nm, with varying Pt-loading. These NPs have well-controlled octahedral geometries (FCC-type atomic arrangements) displaying extensive (111) facets, aiming for similar catalytic properties as the extended surface [12,13,14,15,16,17,18,19,20,21]. Thus, the maximisation of large surface areas offered by bimetallic NPs makes them ideal as catalysts for novel fuel cell applications.
Previous computational results based on Monte Carlo (MC) simulations have addressed the structural stability of cuboctahedral PtNi NPs, ranging from 2.5 to 5 nm [22]. Using a many-body potential to describe the interactions between metal atoms, MC simulations at 600 K showed that PtNi tends to form surface-sandwich structures, with a segregation pattern in which Pt atoms are enriched at the outermost and third shells, while the Ni atoms are enriched in the second shell. These results suggest an economical catalyst design, where Pt atoms are located at the outermost layers with Ni occupying core positions within the bimetallic NP. Similar segregation patterns were reported for smaller-gas-phase-PtNi clusters of less than 20 atoms, using a combined genetic algorithm global optimization approach and density functional theory (DFT) calculations [23]. In terms of cluster reactivity, recent work has shown that sub-nanometre gas-phase PtNi clusters (up to 55 atoms in size) can bind O above 1 eV, due to a substantial geometrical reconstruction of the metal-metal bridge underneath the adsorbed molecule [24]. More recent DFT calculations reported fully-dealloyed PtNi particle (∼8 nm) surfaces exhibiting triangulated surface arrangements, as a regular Pt(111) surface, while reducing the rate-determining ORR step significantly [25].
In this work, we performed a systematic density functional theory (DFT) study on O adsorption on a variety of PtNi clusters supported on MgO(100), as well as on their monometallic (Pt, Ni) counterparts, ranging between 25–58 atoms. Using the calculated adsorption energies (), we construct an O adsorption map for supported PtNi, Pt and Ni clusters presenting a variety of cluster geometries, ranging from truncated octahedra (TO), cuboctahedra (CO), icosahedra (Ih) and decahedra (Dh). Clusters are either in contact with their (100) and (111) facets to the oxide substrate. For supported PtNi clusters, we try to preserve a PtNi bulk-like composition, implying that Ni doping occurs at the bimetallic cluster core sites (i.e., core–shell clusters). However, due to cluster sizes and geometries, Ni atoms are in some cases located inevitably at cluster surface sites (e.g., TO, Dh). For all cases considered, we calculated the general coordination number (GCN), a quantity which establishes a link between geometry, adsorption and activity [26,27,28,29]. The GCN can distinguish symmetrically equivalent sites, and it has been recently used to successfully describe O adsorption trends over large (∼2 nm) supported PtNi clusters [30]. The reader must bear in mind that a fuel cell works under complex electrochemical processes [31]. In particular, a net electric field at the electrode-electrolyte can alter the overall ORR performance. Thus, our work aims at providing a fundamental understanding on molecular O adsorption using a variety of adsorption scenarios on supported PtNi clusters.
2. Methodology
Periodic density functional theory (DFT) calculations are performed using the Quantum Espresso (QE) plane-wave implementation code [32]. The Perdew-Burke-Ernzerhof (PBE) exchange-correlation (xc) functional was used along with a combination of ultrasoft pseudopotentials to describe Pt (5d6s), Ni (3d4s), Mg (2p3s3p) and O (2s2p) atoms [33,34,35]. A kinetic energy for wave-functions and charge density cutoff of 45 and 360 Ry, respectively, is used. To improve SCF convergence, a Marzari–Vanderbilt smearing value of 0.001 Ry is used. All DFT calculations have been performed spin-polarised. The pristine MgO(100) surface is modelled using a (6 × 6) three atomic layer slab, using the calculated PBE bulk lattice constant of 4.238 Å. Although our value is slightly larger than the experimental value of 4.21 Å, it is in close agreement with calculated PBE values (4.26 Å and 4.30 Å) and hybrid (PBE0) functionals (4.21 Å) [36,37]. The MgO(100) substrate has been considered on the basis that it is a often used in experiments, and it is a well-characterized and rigid substrate, which strongly interacts with a metallic cluster. Its checkerboard (100) surface allows us to analyse any effects between flat and corrugated cluster/oxide interfaces. The size of the supercell considered (17.9803 Å × 17.9803 Å × 29.2380 Å) provides a sufficiently large vacuum region necessary to avoid any spurious interactions between neighbouring periodic images. Due to the large size of the oxide slab and overall supercell, the Brillouin zone was sampled at the -point only. The inclusion of van der Waals effects is accounted for via an empirical dispersion correction on the PBE functional (DFT+D), which includes a pairwise addition C/R correction term [38]. The effect of London dispersion forces is checked via DFT+D single-point calculations on the supported clusters’ relaxed configurations.
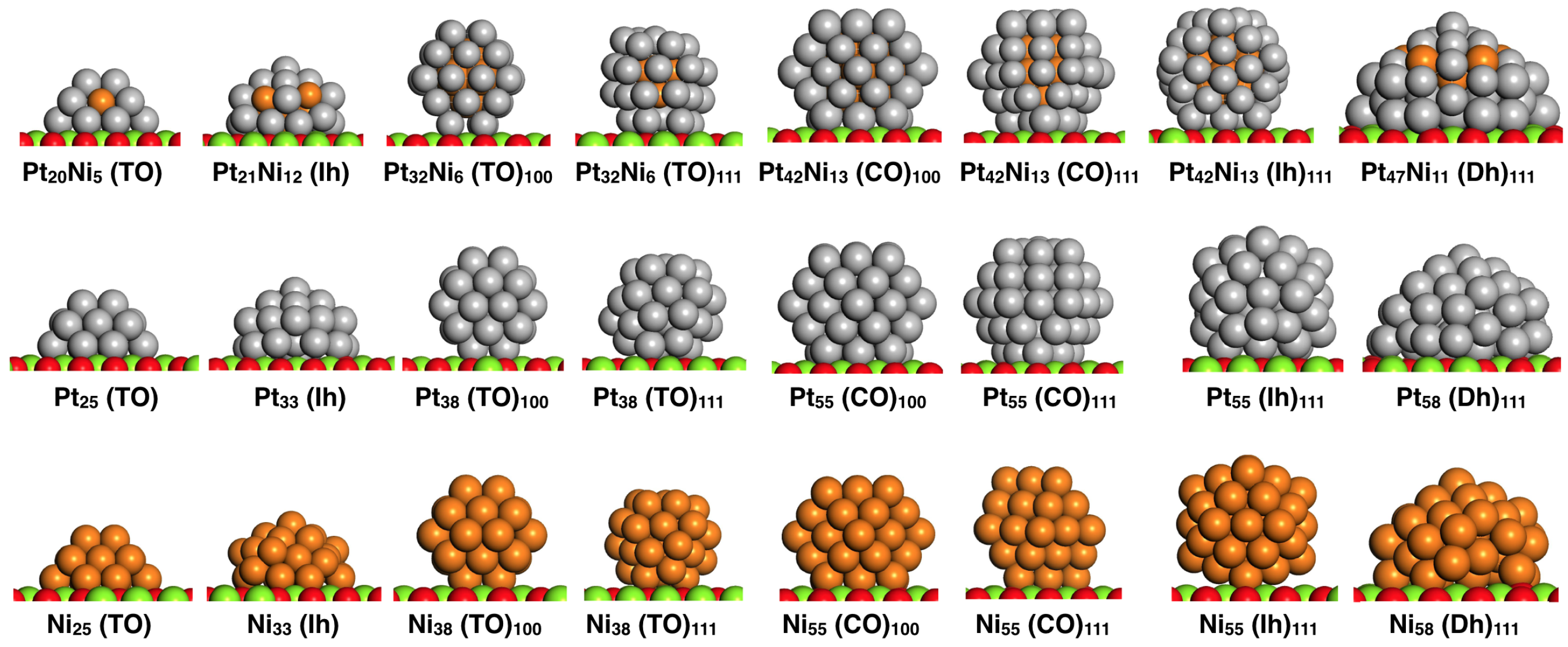
O adsorption is studied at different sites on four different structural models, as highlighted in Figure 1: truncated octahedra (TO), cuboctahedra (CO), icosahedra (Ih) and decahedra (Dh), with supported metal cluster sizes ranging from 25 up to 58 atoms. Besides the shape and size effects, we considered clusters to be either in contact with their (100) and (111) facets to the oxide substrate, in such a way as to estimate the contribution from the strain due to a mismatch with the oxide surface. Similar structural models have been recently used in a force-field parametrisation describing the interaction between Pt clusters and the MgO(100) substrate via classical molecular dynamics (MD) simulations [39]. In our work, supported PtNi clusters display a deliberate PtNi chemical arrangement. Depending on size and geometry, some Ni atoms can occupy surface sites, such as clusters PtNi(TO), PtNi(Ih), and PtNi(Dh). After construction in the gas-phase, the metal clusters are subsequently placed over the MgO(100) substrate, maximising the number of metal–oxygen (M–O) bonds. Each structure is initially positioned in such a way that the shortest height from the substrate is at least ∼2 Å. This takes into account the calculated optimal distance for Pt and Ni atoms (1.985 Å and 1.796 Å) above one oxygen atom of the oxide surface. We must stress that the considered supported PtNi, Pt and Ni clusters—though locally DFT-relaxed—may not correspond to the lowest energy isomer as no global optimizations were carried out. However, they act as geometrical scenarios to understand O adsorption properties over a variety of distinct sites within the supported clusters.
3. Energetic Analysis
Adsorption energies () are calculated as total energy differences between the interacting configuration (), the bare (relaxed) supported cluster (), and the O molecule in the gas-phase:
where positive values indicate an exothermic O adsorption. A geometrical characterisation of the O adsorption site is done using the generalized coordination number (GCN). Introduced by Sautet and co-workers for monometallic Pt systems, the GCN is a geometrical descriptor as robust as the d-band center, which establishes a direct link between geometry, adsorption map and activity [26,27,28,29]. The GCN can be defined as the coordination number at the adsorption site weighted by the overall coordination of the neighbouring atoms:
where the sum runs over all neighbouring atoms n of the atom i of a given site. Then, each neighbour is weighted, according to its coordination number, divided by the maximum coordination number of the adsorption site. We kept CN = 18 as the O molecule is adsorbed, on the majority of cases, at a bridge site (on top of two metallic atoms), as done recently for larger supported PtNi clusters [30]. When the O molecule is adsorbed at the cluster’s FCC (face-centered cubic) and HCP (hexagonal close packing) sites, it is technically sharing a bond to three metallic atoms, both molecularly or dissociatively adsorbed, as it implies having the O molecule over a hollow site at one of the cluster’s facets. Thus, the same CN value is used in order to make all calculated GNC values comparable. The threshold distances for the monometallic cases correspond to the calculated Pt and Ni bulk lattice values (2.812 Å and 2.487 Å, respectively). For the PtNi case, we considered the PtNi phase experimental bulk value (2.718 Å) [40]. This is due to the fact that it has been experimentally reported that PtNi bulk alloy has an ordered FCC lattice. However, it has a random occupation of platinum and nickel according to the stoichiometry, thus making it difficult to calculate a DFT bulk value [41]. For all cases, a ±0.3 Å lower and upper bound range to the threshold value is considered, to take into account any bond length distortions suffered upon O adsorption by those metal atoms located at both cluster core/shell sites.
We further quantified the supported PtNi, Pt and Ni clusters’ core and shell strain. This quantity is calculated as the percentage difference between the average nearest-neighbour (NN) distance—for those atoms occupying core (d) and shell (d) positions—with respect to previously mentioned Pt, Ni and PtNi threshold distances (d). Thus, negative values in Equation (3) indicate a compressive strain felt by the cluster:
4. Results and Discussion
4.1. O Adsorption Trends on Supported PtNi Nanoparticles
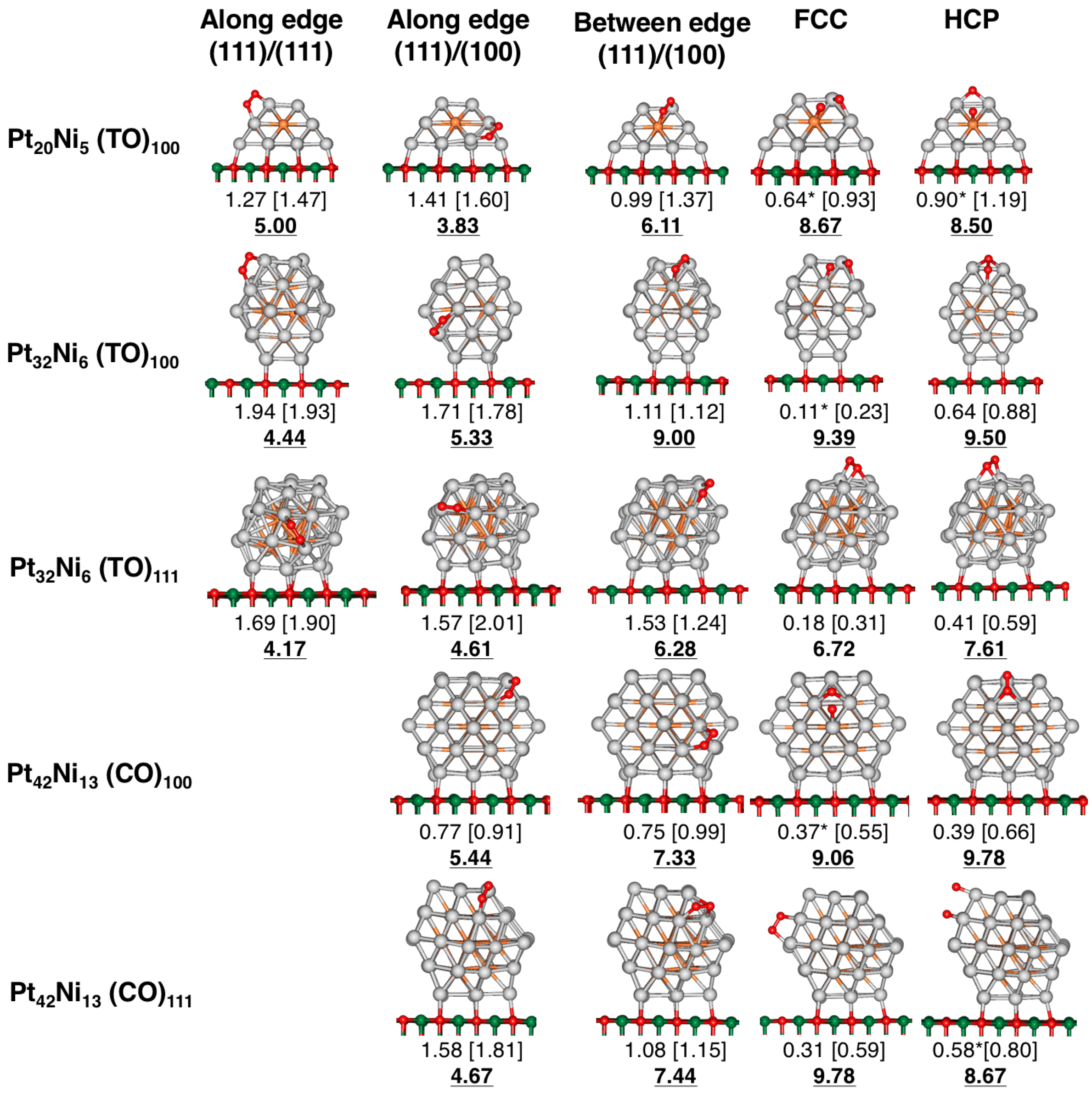
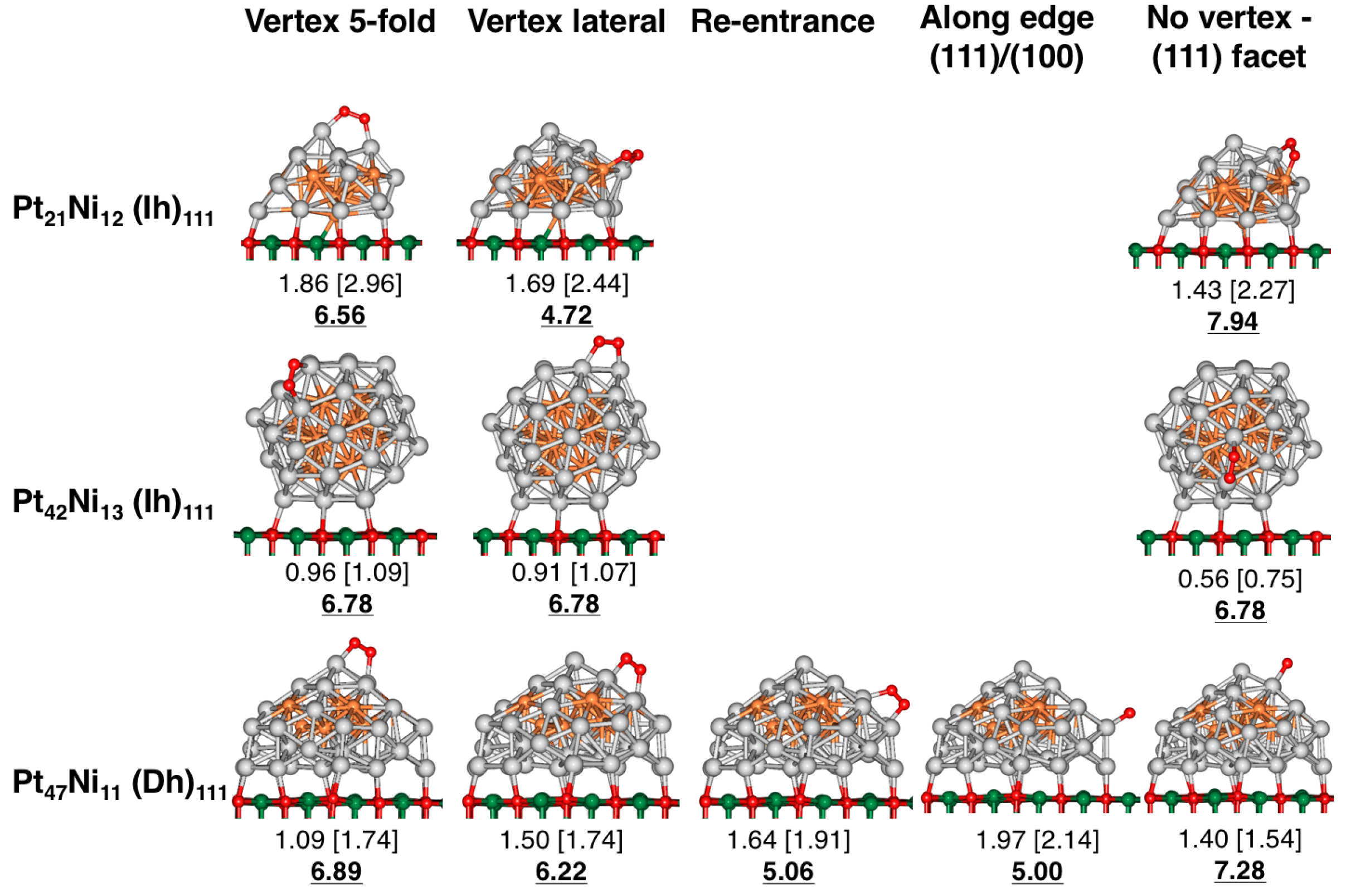
Initially, the O molecule is placed horizontally towards the cluster surface, bridging between two metal atoms, as it has been highlighted in recent theoretical studies as the preferred adsorption configuration [42,43,44]. Five different O adsorption sites are considered for those supported PtNi clusters involving TO and CO geometries: (1) “along edge(111)/(111)”; (2) “along edge(111)/(100)”; (3) “between edge (111)/(100)” as well as (4) “FCC” and (5) “HCP” sites (see Figure 2). For the Dh and Ih geometries, O adsorption sites involve 5-fold vertices (“vertex 5-fold”, “vertex lateral”), cluster re-entrances (“Re-entrace”), sites involving a cluster edge (“along edge (111)/(100)”) and those sites located at the small (111) surfaces within the cluster not involving a 5-fold vertex (“no vertex—(111) facet”), as shown in Figure 3. Similarly, O adsorption is studied on the monometallic Pt and Ni counterparts (see Figures S1–S4, in Supplementary Materials).
Our results showed that, for supported PtNi clusters, the strongest calculated E values for TO and CO configurations are located along the cluster edges. In particular, when O is adsorbed at the “along edge (111)/(111)”, “along edge (111)/(100)” and “between edge (111)/(100)” sites, calculated E values range from 0.75 eV up to 1.94 eV. Strong E values—ranging from 0.91 eV up to 1.97 eV—are also reported for the Dh and Ih configurations, for most of the adsorption sites involving vertices and edges: “vertex 5-fold”, “vertex-lateral”, “along edge (111)/(100)” and “re-entrances”. Weak E values are obtained when O is adsorbed in a small, cluster (111) facet involving a PtNi (Ih) cluster—such as in the “no vertex—(111) facet” site—at 0.56 eV. Weaker adsorption trends are found for the TO and CO clusters involving the FCC and HCP, with E values, ranging from 0.18 up to 0.64 eV for molecular O adsorption. We must point out that we also observe spontaneous O dissociation, when one of the O atoms is in contact with an Ni atom located at the cluster surface involving both FCC and HCP sites, such as in the PtNi (TO) cluster (see Figure 2). Other structures involving a dissociated O molecule are: PtNi (TO) (FCC site), PtNi (CO) (FCC site), as well as PtNi (CO) (HCP site).
For monometallic Pt supported clusters, similar adsorption trends are observed. For the TO and CO geometries, E values range from 0.85 up to 1.59 eV for those O sites involving cluster edges. Again, FCC and HCP sites have the weakest E values (0.42 to 0.86 eV) for O molecular adsorption, where spontaneous O adsorption is also observed on Pt and Pt clusters. For the Ih and Dh geometries, E values on edges and vertices range from 0.50 up to 1.71 eV, while adsorption at the (111) facets clearly involves weaker adsorption energies (0.34–1.21 eV) (see Figures S1 and S2, in Supplementary Materials). However, molecular O interaction with supported Ni clusters edges is stronger, compared to both supported Pt and PtNi clusters, with overall values of the order of 1.52 up to 2.51 eV. The strong adsorption energy values can be attributed to a larger electronegativity difference between the O and Ni atoms, compared to Pt ones (O = 3.44, Pt = 2.28, Ni =1.91, Mg =1.31 in the Pauling scale, respectively). Interestingly, the O molecule is also found to spontaneously dissociate on both FCC and HCP sites of both TO and CO geometries. Regarding Ih and Dh configurations, vertex sites are found to have the strongest O adsorption values (1.88 up to 2.51 eV), while the weakest are found on the supported Ni (111) facets (1.54 up to 1.83 eV), as shown in Figures S3 and S4, in Supplementary Materials.
An O adsorption map is constructed for all the different adsorption sites for supported PtNi, Pt and Ni clusters (see Figure 4). Figure 4 includes calculated (PBE) reference values for the O adsorption on the flat Pt(111) surface (0.46–0.86 eV) [43,45,46,47], including experimental values (0.4–0.5 eV) [48,49] as well as for those for the stepped Pt(321) surface (1.23 to 1.56 eV) [50]. Values for PtNi gas-phase clusters O adsorption (0.5–2.5 eV) [24] are also highlighted by dashed green lines. We identify four adsorption sites having values resembling those of the Pt(111) surface: FCC, HCP, “no vertex—(111) facet”, as well as “vertex lateral”. From Figure 4, it is clear that weak O values, lower than 0.5 eV, are found namely for FCC and HCP sites, involving mostly CO and TO structures’ (111) facets.
The relevance of (111) facets on metal clusters as candidates for low-barrier O dissociation has been recently discussed for pure Pt truncated octahedral (TO) gas-phase clusters at atom sizes 38, 79 and 116 by Jennings and co-workers [43,51]. In their work, sites involving the edge-bridge site between the (111) and (100) facets are reported as the most stable for O adsorption on both pure Pt and bimetallic PtTi clusters (2.01 and 1.76 eV, respectively). Moreover, O adsorption energy values are also competitive on FCC and HCP sites over pure Pt (1.79 and 1.84 eV). These values are stronger compared to the same FCC/HCP sites on the PtTi, core-shell bimetallic cluster (0.38 and 0.74 eV). Although it is expected that oxygen dissociation will occur preferentially at sites with the strongest E values—where O is most likely to adsorb on the cluster—they reported near barrier-free dissociation at the (111) facet of the pure Pt cluster, namely involving the hollow FCC/HCP (0.00 and 0.04 eV) sites. Larger barriers where found for the bimetallic case involving those same FCC/HCP adsorption sites (0.62 and 0.34 eV). This behaviour was explained due to an easily distorted Pt(111) facet-namely via the central Pt atom-facilitating O dissociation, while an increase in the rigidity of the (111) facet in the bimetallic case was due to Ti-alloying at core lead to larger dissociation barriers. For larger TO particles [43], it was reported that only those hollow sites close to the edges of the (111) facet facilitate O dissociation, with similar adsorption and dissociation trends observed for the larger 116-atom truncated octahedron clusters [51]. Overall, it was reported that the rigidity of the small (111) cluster surface tends to decrease for 3d metals from the 4–8 group (Ti to Fe). Similar distortions as of the pure Pt cluster were reported for groups 10–12 (Ni, Cu and Zn) [52], our calculations being in good agreement with the previous adsorption trends. We must also highlight that recent experimental and theoretical work by Huang and co-workers have showed a promising catalytic activity and durability by Mo-doped supported PtNi octahedra nanoparticles towards the ORR [53]. In their work, theoretical calculations indicate that Mo-doping may increase the oxygen binding energies at sites closer to the center of the (111) facet of the nanoparticle, which bind oxygen too weakly. This type of nanoparticle surface engineering could lead to further improvements to the catalytic activity of the PtNi system.
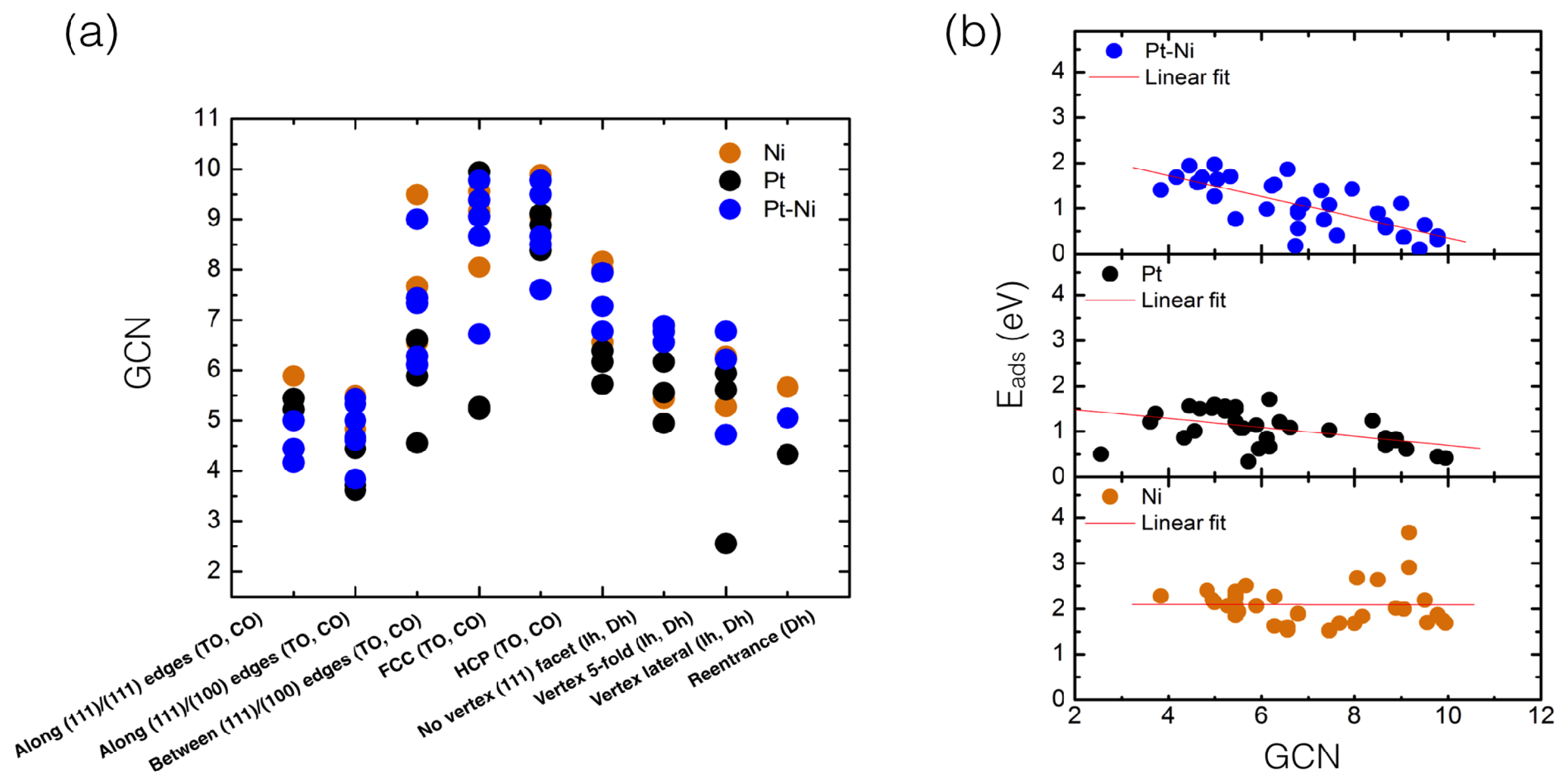
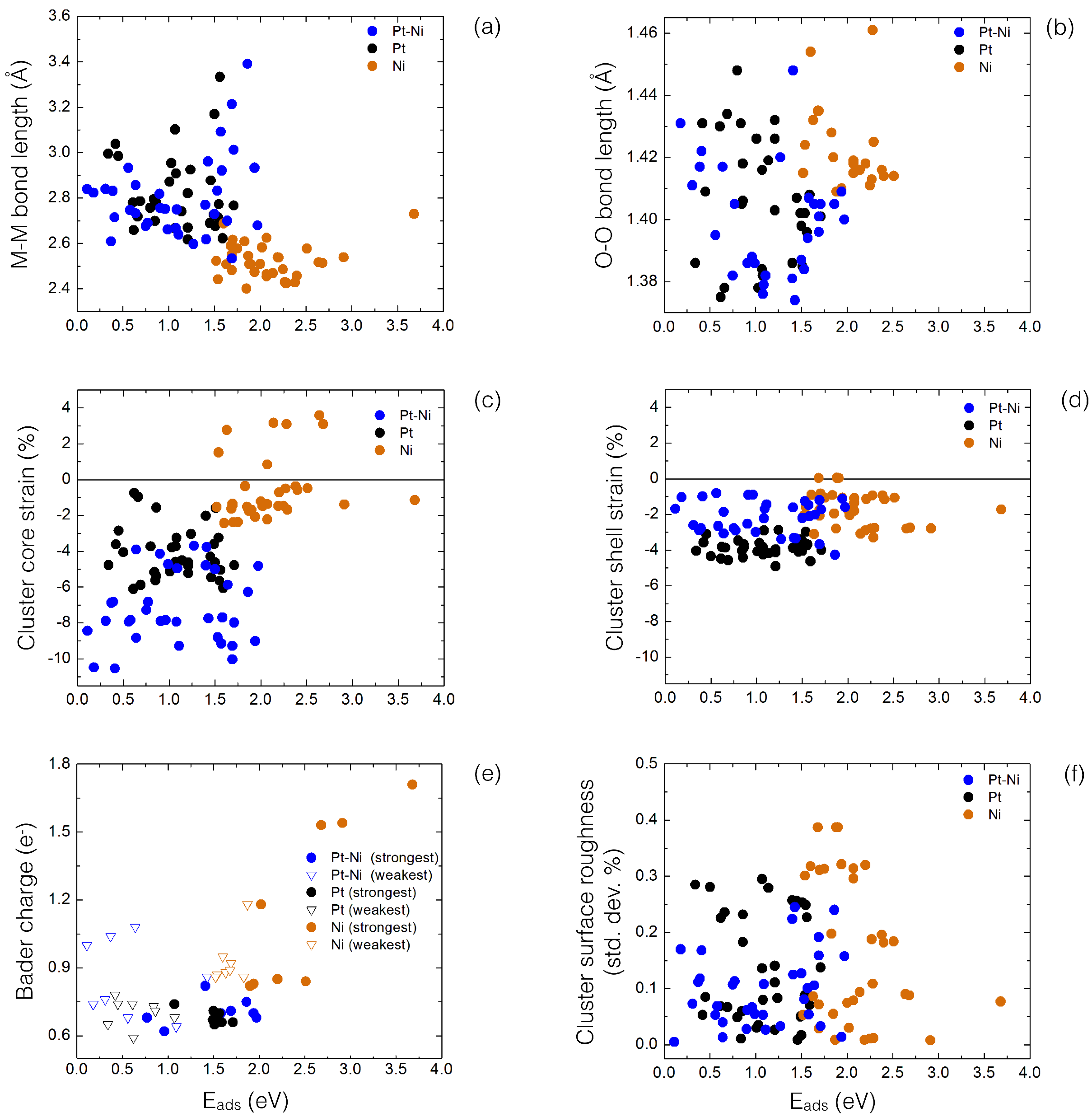
Based on our calculated values, molecular O adsorption trends for supported PtNi, Pt and Ni clusters can be further understood in terms of their surface coordination chemistry. Thus, each adsorption site was assigned a GCN value (see Figure 5a). GCN values establish a direct link between geometry and the adsorption map, despite the size, shape and composition of the supported clusters [26,27,28,29,30]. From Figure 5a, it is clear that those adsorption sites having the largest GCN value correspond to namely the FCC and HCP sites (>6), while smaller GCN values (<5) are calculated to sites involving clusters’ edges and vertices (see calculated GCN values in Tables S1–S3, in Supplementary Materials). We have also plotted the corresponding GCN values as a function of the calculated , as shown in Figure 5b. Despite the ubiquitous differences in cluster sizes and geometries, calculated GCN values show for PtNi supported clusters an overall linear fit relationship. Thus, as the GCN value increases, a reduction on the values is seen, particularly above a GCN > 8, where most of the calculated values involve FCC and HCP sites. Concomitantly, the strongest values (>0.64 eV and above) can be found at cluster edge sites, such as along edges (111)/(111) sites, where GCNs between 4 and 5 are calculated. For supported Pt clusters, a similar trend on calculated GCN values is observed (Figure 5b). However, the slope of the linear fit is less pronounced, compared to supported PtNi clusters, due to elemental differences between the mono- and bi-metallic system. Figure 5a,b value dispersion can be understood due to drastic changes in the Pt–Pt bond lengths at the adsorption site, as both PtNi and Pt clusters try to accommodate the incoming O molecule, thus directly effecting the GCN at the adsorption site (see Figure 6a). On the other hand, supported Ni clusters strongly interact with the adsorbed O molecule in the GCN value range of 4 to 6, with spontaneous O dissociation seen at those FCC/HCP sites located on the Ni cluster (111) facets, having GCN values > 8. Nonetheless, no clear linear trend for Ni clusters is observed from Figure 5a at the cluster sizes considered. Regarding the O molecule, an O-O bond length value clustering around ∼1.42 Å can be observed for Ni values—not including those dissociated configurations—compared to more dispersed PtNi and Pt values (Figure 6b) and our calculated DFT gas-phase value (1.23 Å).
Figure 6c,d shows both the calculated core and shell supported PtNi, Pt and Ni cluster strain, as it has been recently shown a direct link between the NP reactivity and its compressive strain, particularly in bimetallic cases [8,44,54]. The most dramatic effects are observed for supported PtNi as they are characterised by a large core compressive strain (between −4% and −10%). This is mostly due to the fact that the outer Pt atom layer needs to form chemical bonds with internal Ni atoms. Supported monometallic Pt clusters have a milder compression percentage, between −2% and −6%. However, supported Ni clusters may show a tensile strain, such as the TO(100) structure. The supported TO(100) cluster has 12 Ni atoms at the cluster/oxide interface, who need to match 12 O surface sites. Having the latter an average interface bond lengths of 2.52 Å, compared to the average Ni–Ni cluster bond length of 2.45 Å, it thus enlarges the Ni–Ni cluster core distances, thus leading to overall positive core strain values. Regarding shell strain, we observe that the calculated values for supported PtNi, Pt and Ni clusters are rather small, no larger than −4%, with the outer layer not greatly affected by the internal cluster strain.
A charge transfer Bader analysis is performed for supported PtNi, Pt and Ni clusters at the “strongest” and “weakest” calculated O adsorption sites (see Figure 6e). In particular, Figure 6e displays the amount of charge on the O molecule. Overall, the O molecule withdraws an small amount of charge (∼0.35 e) per O atom in contact with PtNi and Pt sites. A slightly larger amount of charge is withdrawn for Ni (∼0.5 e), in line with the larger E values calculated for Ni clusters. This excess of charge on the O molecule creates a metal–superoxo (M–O) type intermediate (one electron transferred) [42]. Charge transfer is in line with both O (3.44), Pt (2.28), Ni (1.91), and Mg (1.31) Pauling electronegativities. As a general trend, our calculations indicate that the amount of charge transferred from the support to the supported clusters varies as a function of interfacial metal atoms (number of contact O surface sites), with less charge being transferred from the substrate to Ni clusters, compared to Pt and PtNi clusters.
At the cluster/oxide interface, we observe a modest tendency of supported PtNi clusters to reduce the overall roughness of the (100) and (111) interface layers (see Figure 6f). The Pt metal–metal (M–M) bond lengths—of all those interfacial PtNi cluster layers in direct contact with the oxide surface—are slightly larger (∼2.76 Å), compared to the average M–M bond lengths of all PtNi clusters (∼2.65 Å). This is in line with those elongations observed for the M–M distances at the interface of monometallic Pt and Ni clusters (∼2.77 Å and ∼2.56 Å, respectively), compared to the overall cluster bond length average (Pt ∼2.72 Å and Ni ∼2.45 Å). These elongations can be understood as the metal atoms located at the cluster/oxide interface try to match those O atoms (average O–O distance ∼2.99 Å). Even though weaker E values are calculated for TO and CO clusters in contact with the oxide through their large (100) facets at low roughness (less than 0.1%), the dispersion of the overall values is too wide to provide any conclusive analysis on a strong MgO oxide substrate effect on the calculated E values. This suggests a rather “inert” nature of the MgO(100) substrate, with the metal cluster chemistry at the adsorption site ruling O values. Our calculations seem to indicate that the main role of the MgO(100) support is thus to provide an “anchor” point for the cluster, on both (100) and (111) interfaces, and, to a lesser extent, to facilitate charge being transferred to the supported cluster (which eventually is redistributed among the cluster and O molecule). One could envisage different cluster/oxide interfaces that could promote the controlled preparation of supported PtNi clusters grow in size, where tailored O adsorption (and eventual dissociation) can take place [30].
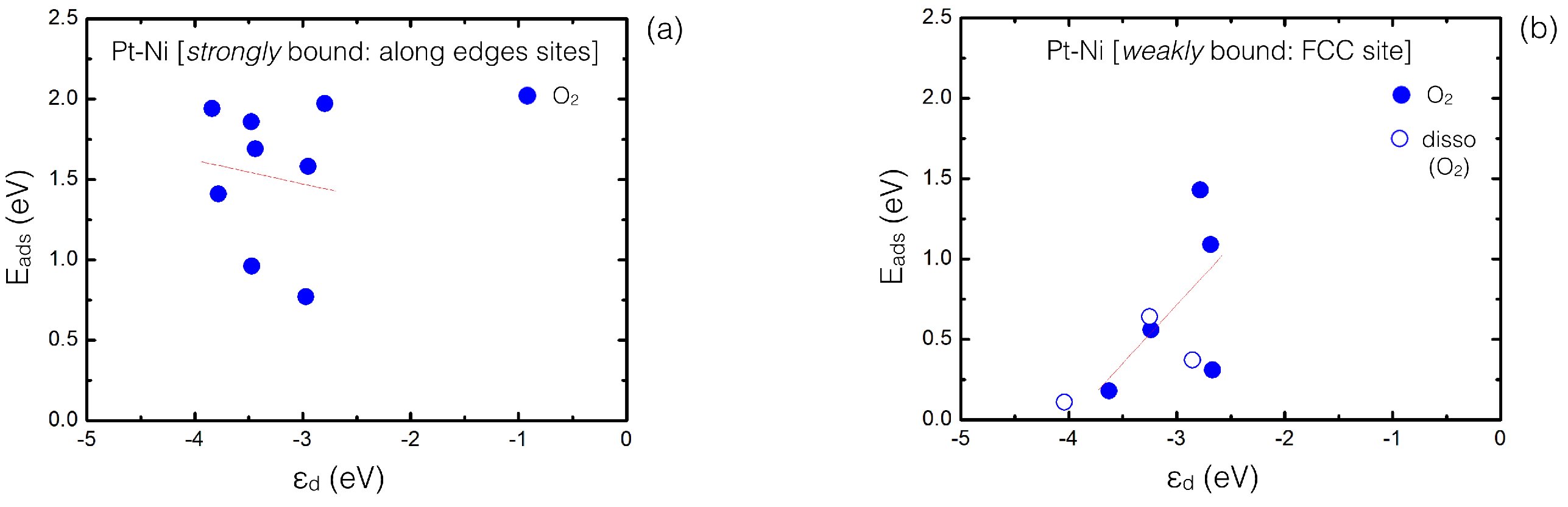
From an electronic point of view, the d-band center model can be also used to analyze O adsorption. With positioned lower (higher) with respect to the Fermi energy describing weaker (stronger) interactions, Figure 7 shows the calculated plotted against the calculated values for those strongly/weakly bound adsorption sites (see calculated values in Tables S4, in Supplementary Materials). Overall, we observe that the values from the strongest adsorption sites are positioned on an energy window between −2.80 eV and −3.84 eV, while are slightly shifted to lower values for the weakest configurations (−2.67 to −4.04 eV). For the weakest sites, the lowest values (position ∼−4 eV) are caused primarily by the Pt atoms located at the central (111) facets of TO structures and the least coordinated Pt atoms for CO configurations. While the d-band center model adsorption trend is followed for the weakest sites—despite involving some configurations with a dissociated O molecule—this is not the case for the strongest sites, where the trend is not so linear anymore. Comparisons between different PtNi clusters are remarkably difficult due to the size range considered and the variety of cluster geometries, including the wide range of their compressive strain. Thus, our calculations indicate that it is not straightforward to adopt the d-band center model for supported bimetallic cluster systems—with a few tens of atoms—where interatomic distortions are induced by alloying play a significant role.
4.2. Inclusion of van der Waals Corrections
Finally, we would like to comment on how London dispersion forces (DFT-D) may affect the overall O adsorption picture. The inclusion of the DFT-D semi-empirical dispersion correction on the calculated PBE values via single-point (SCF) calculations have, in most cases, modestly increased the calculated E values. In general, predicted PBE energetic ordering between adsorption sites was preserved, with only a minor energetic rearrangement among those adsorption sites where adsorption is the strongest. DFT+D relaxations were also performed for the different adsorption sites, only for the supported Pt, Ni and PtNi (TO) structures, confirming an overall preference for strong O adsorption at edge sites, such as (100)/(111) and (111)/(111). For supported PtNi clusters, DFT+D relaxations predict the along edge (111)/(100) site as the strongest O adsorption site (1.62 eV), closely followed by along edge (111)/(111) site (1.47 eV), just as the PBE energetic ordering. Similar gains in energy are calculated for the FCC and HCP sites. For the Pt clusters, the strongest O adsorption site is now the along edge (111)/(100), instead of the predicted PBE along edge (111)/(111) at 1.96 eV. Being now the O molecule at close proximity to the oxide surface (2.99 Å), DFT+D is able to capture a stronger overlap between the electron orbitals of the O molecule and those of the oxide surface. This results in an E increase up to 1.96 eV, from the 1.38 eV single-point (DFT+D) calculation, an increase also calculated for the between edge (111)/(100) site, from 1.67 to 1.71 eV. Analogous trends are observed for Ni clusters, where along edge (111)/(100) site remains as the one having the largest E value (2.55 eV), while marginal gains are calculated for sites along edge (111)/(111) and between edge (111)/(100) (2.25 and 1.79 eV, respectively).
5. Conclusions
Our calculations show that O adsorption on supported PtNi, Pt and Ni clusters—at the sub-nanometer 25–58 atoms size range—is a rather complex case, due to the fact that the overall electronic structure and geometrical properties are deeply intertwined, directly affecting the calculated E values.
We observe that O adsorption is regulated by two distinct groups of adsorption sites, where: (a) the strongest E values are calculated at cluster edges (above 1 eV), such as the along edge (111)/(111) and (111)/(100) as well as the between edge (111)/(100) sites, which tend to have a low GCN value (<6); and (b) the FCC and HCP sites, where the weakest E values (less than 0.64 eV) are calculated, involving GCN values >8, which resemble those O adsorption energies of the flat Pt(111) surface. Calculated d-band center positions for those Pt atoms weakly (strongly) binding with the O adsorbate-molecularly or dissociatively—are located at lower (higher) energies with respect to the Fermi level. From the supported PtNi cluster sizes considered in this work, it may be difficult to extend the d-band model to such small, and finite chemical systems, where distortions to the interatomic distances and the strain associated to them play a fundamental rule in the calculated values. Our calculations seems to indicate that the generalized coordination number (GCN) is a more robust reactivity descriptor even at the supported cluster sizes considered. Although Pt and Ni atoms have a strong mismatch, we kept the simplest GCN implementation, without any particular weighting in order to distinguish both atoms, as we have recently shown that the effect is almost negligible and none of those provide a better understanding of the physics of our systems [30]. Overall, the robustness of the GCN arises from the fact that is a purely geometrical descriptor, very sensitive to the deformation induced by metal alloying and by the environment (e.g., strain epitaxy due to the considered substrate).
Finally, our results provide—as far as we are concerned—the first O adsorption map across a wide range of bimetallic (PtNi), and monometallic (Pt) and (Ni) supported clusters. This is done over a wide size range (25–58 atoms) and considering eight different geometries, each of which involve five different adsorption sites, with two types of cluster/oxide interfaces-namely (100) and (111)—considered as they may determine different supported cluster core/shell strains. Developing clear O adsorption trends on supported metal clusters can provide a deeper understanding of potential adsorption sites to target low-barrier O dissociation, following the Sabatier principle. This will imply that the best catalysts should be the ones that bind both atoms and molecules not too weakly nor too strongly, in order to activate the reactants and to efficiently desorb the products [55]. We show that it is theoretically possible to construct supported core-shell systems, where the core of the nanoparticle (cluster) is made of a cheaper metal (Ni) compared to a more expensive metal on the shell (Pt). This not only reduces the amount of Pt-loading by alloying, but preserves the Pt chemistry at a mere 1 atomic-layer Pt-shell, where O adsorption process can occur, with calculated values of the order of those seen for a full monometallic Pt nanoparticle and bulk Pt(111) surface. Having systematically analysed the effects of O adsorption over supported over mono (Pt, Ni) and bimetallic (PtNi) clusters (<1 nm), we foresee that a potential tailoring of physico-chemical properties and a rational novel metal nanocatalysts design will ultimately depend on a synergistic combination of controlled NP preparation methods (such as size, geometric and segregation effects) and a profound knowledge of the complex cluster/oxide interface.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2304-6740/5/3/43/s1, Figure S1: O adsorption sites considered for supported Pt clusters in the size range 25–55 atoms, involving truncated octahedral (TO) and cuboctahedral (CO) geometries, Figure S2: O adsorption sites considered for supported Pt clusters in the size range 33–58 atoms, involving icosahedral (Ih) and decahedral (Dh) geometries, Figure S3: O adsorption sites considered for supported Pt clusters in the size range 25–55 atoms, involving truncated octahedral (TO) and cuboctahedral (CO) geometries, Figure S4: O adsorption sites considered for supported Pt clusters in the size range 33–58 atoms, involving icosahedral (Ih) and decahedral (Dh) geometries; Table S1: Structural properties of supported PtNi clusters upon molecular O adsorption at all the different inequivalent sites considered in this work, Table S2: Structural properties of supported Pt clusters upon molecular O adsorption at all the different inequivalent sites considered in this work, Table S3: Structural properties of supported Ni clusters upon molecular O adsorption at all the different inequivalent sites considered in this work. Table S4: Calculated average d-band center of supported PtNi clusters on MgO(100) upon O adsorption.
Acknowledgments
This work has been supported by the UK Engineering and Physical Sciences Research Council (EPSRC) under the Critical Mass “TOwards an Understanding of CAtalysis on Nanoalloys” (TOUCAN) project, grant No. EP/J010812/1. Francesca Baletto benefited from the financial support of the EPSRC, under Grant No. EP/GO03146/1 and the Royal Society grant No. RG120207. Lauro Oliver Paz-Borbón is thankful for the financial support from the Universidad Nacional Autónoma de México, Dirección General Asuntos del Personal Académico (DGAPA-UNAM), under Project IA102716. Density Functional Theory (DFT) calculations have been performed on the ARCHER (Cray XC30 system), at the UK’s National High-Performance Computing (HPC) Facility; as well as at Dirección General de Cómputo y de Tecnologías de Información y Comunicación (DGTIC-UNAM), through SC16-1-IG-78, SC15-1-IG-82 and LANCAD-UNAM-DGTIC-307 grants.
Author Contributions
Francesca Baletto conceived and designed the calculations; Lauro Oliver Paz-Borbón performed the calculations and analysed the data; Francesca Baletto and Lauro Oliver Paz-Borbón wrote the paper. Lauro Oliver Paz-Borbón drew the figures and compiled the Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Steele, B.C.H.; Heinzel, A. Materials for Fuel-Cell Technologies. Nature 2001, 414, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Sadakiyo, M.; Shigematsu, A.; Kitagawa, H. Proton-Conductive Metal–Organic Frameworks. Bull. Chem. Soc. Jpn. 2016, 89, 1–10. [Google Scholar] [CrossRef]
- Meng, X.; Wang, H.-N.; Song, S.-Y.; Zhang, H.-J. Proton-Conducting Crystalline Porous Materials. Chem. Soc. Rev. 2017, 46, 464–480. [Google Scholar] [CrossRef] [PubMed]
- Sakaushi, K.; Antonietti, M. Carbon- and Nitrogen-Based Porous Solids: A Recently Emerging Class of Materials. Bull. Chem. Soc. Jpn. 2015, 88, 386–398. [Google Scholar] [CrossRef]
- Shao, M.; Chang, Q.; Dodelet, J.-P.; Chenitz, R. Recent Advances in Electrocatalysts for Oxygen Reduction Reaction. Chem. Rev. 2016, 116, 3594–3657. [Google Scholar] [CrossRef] [PubMed]
- Debe, M.K. Electrocatalyst Approaches and Challenges for Automotive Fuel Cells. Nature 2012, 486, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Greeley, J.; Stephens, I.E.L.; Bondarenko, A.S.; Johansson, T.P.; Hansen, H.A.; Jaramillo, T.F.; Rossmeisl, J.; Chorkendorff, I.; Norskov, J.K. Alloys of Platinum and Early Transition Metals as Oxygen Reduction Electrocatalysts. Nat. Chem. 2009, 1, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Strasser, P.; Koh, S.; Anniyev, T.; Greeley, J.; More, K.; Yu, C.; Liu, Z.; Kaya, S.; Nordlund, D.; Ogasawara, H.; et al. Lattice-Strain Control of the Activity in Dealloyed Core–Shell Fuel Cell Catalysts. Nat. Chem. 2010, 2, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, V.R.; Mun, B.S.; Arenz, M.; Mayrhofer, K.J.J.; Lucas, C.A.; Wang, G.; Ross, P.N.; Marković, N.M. Trends in Electrocatalysis on Extended and Nanoscale Pt-Bimetallic Alloy Surfaces. Nat. Mater. 2007, 6, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity Benchmarks and Requirements for Pt, Pt-alloy, and non-Pt Oxygen Reduction Catalysts for PEMFCs. Appl. Catal. B Environ. 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Stamenkovic, V.R.; Fowler, B.; Mun, B.S.; Wang, G.; Ross, P.N.; Lucas, C.A.; Marković, N.M. Improved Oxygen Reduction Activity on Pt3Ni(111) via Increased Surface Site Availability. Science 2007, 315, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Strasser, P. Catalysts by Platonic Design. Science 2015, 349, 379–380. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, M.K.; Moylan, T.E.; Kukreja, R.S.; Atwan, M.H.; Tessema, M.M. Solvothermal Synthesis of Platinum Alloy Nanoparticles for Oxygen Reduction Electrocatalysis. J. Am. Chem. Soc. 2012, 134, 8535–8542. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Gan, L.; Heggen, M.; Rudi, S.; Strasser, P. Compositional Segregation in Shaped Pt Alloy Nanoparticles and Their Structural Behaviour During Electrocatalysis. Nat. Mater. 2013, 12, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-I.; Shao, M.; Lu, N.; Ruditskiy, A.; Peng, H.-C.; Park, J.; Guerrero, S.; Wang, J.; Kim, M.J.; Xia, Y. Synthesis and Characterization of Pd@Pt–Ni Core–Shell Octahedra with High Activity toward Oxygen Reduction. ACS Nano 2014, 8, 10363–10371. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-I.; Xie, S.; Shao, M.; Odell, J.H.; Lu, N.; Peng, H.-C.; Protsailo, L.; Guerrero, S.; Park, J.; Xia, X.; et al. Synthesis and Characterization of 9 nm Pt–Ni Octahedra with a Record High Activity of 3.3 A/mgPt for the Oxygen Reduction Reaction. Nano Lett. 2013, 13, 3420–3425. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Gan, L.; Li, H.-H.; Yu, S.-H.; Heggen, M.; Strasser, P. Octahedral PtNi Nanoparticle Catalysts: Exceptional Oxygen Reduction Activity by Tuning the Alloy Particle Surface Composition. Nano Lett. 2012, 12, 5885–5889. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhao, Z.; Chen, Y.; Zhu, E.; Li, M.; Duan, X.; Huang, Y. A Rational Design of Carbon-Supported Dispersive Pt-based Octahedra as Efficient Oxygen Reduction Reaction Catalysts. Energy Environ. Sci. 2014, 7, 2957–2962. [Google Scholar] [CrossRef]
- Zhang, C.; Hwang, S.Y.; Peng, Z. Size-dependent Oxygen Reduction Property of Octahedral Pt–Ni Nanoparticle Electrocatalysts. J. Mater. Chem. A 2014, 2, 19778–19787. [Google Scholar] [CrossRef]
- Zhang, C.; Hwang, S.Y.; Trout, A.; Peng, Z. Solid-State Chemistry-Enabled Scalable Production of Octahedral Pt–Ni Alloy Electrocatalyst for Oxygen Reduction Reaction. J. Am. Chem. Soc. 2014, 136, 7805–7808. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Li, J.; Jiang, L.; Shang, M.; Zhang, S.; Jiang, Z.; Wei, X.; Huang, Y.; Sun, G.; Wang, J.-Q. Structure of PtnNi Nanoparticles Electrocatalysts Investigated by X-ray Absorption Spectroscopy. J. Phys. Chem. C 2013, 117, 20584–20591. [Google Scholar] [CrossRef]
- Wang, G.; van Hove, M.A.; Ross, P.N.; Baskes, M.I. Monte Carlo Simulations of Segregation in Pt–Ni Catalyst Nanoparticles. J. Chem. Phys. 2005, 122, 024706. [Google Scholar] [CrossRef] [PubMed]
- Radillo-Díaz, A.; Coronado, Y.; Pérez, L.; Garzón, I.L. Structural and Electronic Properties of PtPd and PtNi Nanoalloys. Eur. Phys. J. D 2009, 52, 127–130. [Google Scholar] [CrossRef]
- Di Paola, C.; Baletto, F. Oxygen Adsorption on Small PtNi Nanoalloys. Phys. Chem. Chem. Phys. 2011, 13, 7701–7707. [Google Scholar] [CrossRef] [PubMed]
- Fortunelli, A.; Goddard, W.A., III; Sementa, L.; Barcaro, G.; Negreiros, F.R.; Jaramillo-Botero, A. The Atomistic Origin of the Extraordinary Oxygen Reduction Activity of Pt3Ni7 Fuel Cell Catalysts. Chem. Sci. 2015, 6, 3915–3925. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Sautet, P.; Loffreda, D. Understanding Adsorption-Induced Effects on Platinum Nanoparticles: An Energy-Decomposition Analysis. J. Phys. Chem. Lett. 2014, 5, 3120–3124. [Google Scholar] [CrossRef] [PubMed]
- Calle-Vallejo, F.; Martínez, J.I.; García-Lastra, J.M.; Sautet, P.; Loffreda, D. Fast Prediction of Adsorption Properties for Platinum Nanocatalysts with Generalized Coordination Numbers. Angew. Chem. Int. Ed. 2014, 53, 8316–8319. [Google Scholar] [CrossRef] [PubMed]
- Calle-Vallejo, F.; Loffreda, D.; Koper, M.T.M.; Sautet, P. Introducing Structural Sensitivity into Adsorption–Energy Scaling Relations by Means of Coordination Numbers. Nat. Chem. 2015, 7, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Calle-Vallejo, F.; Tymoczko, J.; Colic, V.; Vu, Q.H.; Pohl, M.D.; Morgenstern, K.; Loffreda, D.; Sautet, P.; Schuhmann, W.; Bandarenka, A.S. Finding Optimal Surface Sites on Heterogeneous Catalysts by Counting Nearest Neighbors. Science 2015, 350, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Asara, G.G.; Paz-Borbón, L.O.; Baletto, F. “Get in Touch and Keep in Contact”: Interface Effect on the Oxygen Reduction Reaction (ORR) Activity for Supported PtNi Nanoparticles. ACS Catal. 2016, 6, 4388–4393. [Google Scholar] [CrossRef]
- Litster, S.; McLean, G. PEM Fuel Cell Electrodes. J. Power Sources 2004, 130, 61–76. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, M.; et al. QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Rappe, A.M.; Rabe, K.M.; Kaxiras, E.; Joannopoulos, J.D. Optimized pseudopotentials. Phys. Rev. B 1990, 41, 1227–1230. [Google Scholar] [CrossRef]
- Garrity, K.F.; Bennett, J.W.; Rabe, K.M.; Vanderbilt, D. Pseudopotentials for High-Throughput DFT Calculations. Comput. Mater. Sci. 2014, 81, 446–452. [Google Scholar] [CrossRef]
- Broqvist, P.; Grönbeck, H.; Panas, I. Surface Properties of Alkaline Earth Metal Oxides. Surf. Sci. 2004, 554, 262–271. [Google Scholar] [CrossRef]
- Paier, J.; Marsman, M.; Hummer, K.; Kresse, G.; Gerber, I.C.; Àngyàn, J. Screened Hybrid Density Functionals Applied to Solids. J. Chem. Phys. 2006, 124, 154709. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Accurate Description of van der Waals Complexes by Density Functional Theory Including Empirical Corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Rossi, K.; Ellaby, T.; Paz-Borbón, L.O.; Atanasov, I.; Pavan, L.; Baletto, F. Melting of Large Pt@MgO(100) Icosahedra. J. Phys. Condens. Matter 2017, 29, 145402. [Google Scholar] [CrossRef] [PubMed]
- Engelke, M.J. Local Atomic Arrangements in Ni–Pt: The Bulk and Near-Surface Regimes. Ph.D. Thesis, ETH Zürich, Swiss Federal Institute of Technology, Zurich, Switzerland, 2010. [Google Scholar]
- Darlington, M.J.D. Stability of Single Crystal Pt and Pt3Ni Surfaces During Electro-Oxidation. Ph.D. Thesis, The University of Liverpool, Liverpool, UK, 2014. [Google Scholar]
- Li, T.; Balbuena, P.B. Computational Studies of the Interactions of Oxygen with Platinum Clusters. J. Phys. Chem. B 2001, 105, 9943–9952. [Google Scholar] [CrossRef]
- Jennings, P.C.; Aleksandrov, H.A.; Neyman, K.M.; Johnston, R.L. A DFT Study of Oxygen Dissociation on Platinum Based Nanoparticles. Nanoscale 2014, 6, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Peles, A.; Shoemaker, K. Electrocatalysis on Platinum Nanoparticles: Particle Size Effect on Oxygen Reduction Reaction Activity. Nano Lett. 2011, 11, 3714–3719. [Google Scholar] [CrossRef] [PubMed]
- Bocquet, M.-L.; Cerdà, J.; Sautet, P. Transformation of Molecular Oxygen on a Platinum Surface: A theoretical Calculation of STM images. Phys. Rev. B 1999, 59, 15437–15445. [Google Scholar] [CrossRef]
- Eichler, A.; Hafner, J. Molecular Precursors in the Dissociative Adsorption of O2 on Pt(111). Phys. Rev. Lett. 1997, 79, 4481–4484. [Google Scholar] [CrossRef]
- McEwen, J.S.; Bray, J.M.; Wucb, C.; Schneider, W.F. How Low Can You Go? Minimum Energy Pathways for O2 Dissociation on Pt(111). Phys. Chem. Chem. Phys. 2012, 14, 16677–16685. [Google Scholar] [CrossRef] [PubMed]
- Gland, J.L.; Sexton, B.A.; Fisher, G.B. Oxygen Interactions with the Pt(111) Surface. Surf. Sci. 1980, 95, 587–602. [Google Scholar] [CrossRef]
- Steininger, H.; Lehwald, S.; Ibach, H. Adsorption of Oxygen on Pt(111). Surf. Sci. 1982, 123, 1–17. [Google Scholar] [CrossRef]
- Bray, J.M.; Schneider, W.F. Potential Energy Surfaces for Oxygen Adsorption, Dissociation, and Diffusion at the Pt(321) Surface. Langmuir 2011, 27, 8177. [Google Scholar] [CrossRef] [PubMed]
- Jennings, P.C.; Aleksandrov, H.A.; Neyman, K.M.; Johnston, R.L. DFT Studies of Oxygen Dissociation on the 116-Atom Platinum Truncated Octahedron Particle. Phys. Chem. Chem. Phys. 2014, 16, 26539–26545. [Google Scholar] [CrossRef] [PubMed]
- Jennings, P.C.; Aleksandrow, H.A.; Neyman, K.M.; Johnston, R.L. O2 Dissociation on M@Pt Core–Shell Particles for 3d, 4d, and 5d Transition Metals. J. Phys. Chem. C 2015, 119, 11031. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, Z.; Cao, L.; Chen, Y.; Zhu, E.; Lin, Z.; Li, M.; Yan, A.; Zettl, A.; Morris Wang, Y.; et al. High-Performance Transition Metal-Doped Pt3Ni Octahedra for Oxygen Reduction Reaction. Science 2015, 348, 1230–1234. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Fernandez, P.; Masini, F.; McCarthy, D.N.; Strebel, C.E.; Friebel, D.; Deiana, D.; Malacrida, P.; Nierhoff, A.; Bodin, A.; Wise, A.M.; et al. Mass-selected Nanoparticles of PtxY as Model Catalysts for Oxygen Electroreduction. Nat. Chem. 2014, 6, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Norskov, J.K.; Bligaard, T.; Hvolbaek, B.; Abild-Pedersen, F.; Chorkendorff, I.; Christensen, C.H. The Nature of the Active Site in Heterogeneous Metal Catalysis. Chem. Soc. Rev. 2008, 37, 2163–2171. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structural models of PtNi, Pt and Ni clusters supported on MgO(100) used to study O adsorption. Geometries range from: truncated octahedra (TO), cuboctahedral (CO), icosahedra (Ih) and decahedra (Dh). Two different cluster/oxide interfaces are considered; namely (100) and (111) cluster epitaxies.
Figure 1.
Structural models of PtNi, Pt and Ni clusters supported on MgO(100) used to study O adsorption. Geometries range from: truncated octahedra (TO), cuboctahedral (CO), icosahedra (Ih) and decahedra (Dh). Two different cluster/oxide interfaces are considered; namely (100) and (111) cluster epitaxies.

Figure 2.
O adsorption sites considered for supported PtNi clusters involving TO and CO geometries, in the size range 25–55 atoms. A distinction is made if the cluster is in contact with the substrate via one of its (111) or (100) facets. E values are displayed in eV, and dispersion corrected (DFT-D) values are shown in brackets. An asterisk (*) indicate those configurations where spontaneous O dissociation occurred during PBE-based relaxations. The corresponding generalized coordination number (GCN) value at the adsorption site is provided in bold. Colour labelling: light grey (Pt), orange (Ni), green (Mg) and red (O).
Figure 2.
O adsorption sites considered for supported PtNi clusters involving TO and CO geometries, in the size range 25–55 atoms. A distinction is made if the cluster is in contact with the substrate via one of its (111) or (100) facets. E values are displayed in eV, and dispersion corrected (DFT-D) values are shown in brackets. An asterisk (*) indicate those configurations where spontaneous O dissociation occurred during PBE-based relaxations. The corresponding generalized coordination number (GCN) value at the adsorption site is provided in bold. Colour labelling: light grey (Pt), orange (Ni), green (Mg) and red (O).

Figure 3.
O adsorption sites considered for supported PtNi clusters involving Ih and Dh geometries, in the size range 33–58 atoms. The cluster/oxide interface involves the supported clusters (111) facets. E values are displayed in eV, while dispersion corrected (DFT-D) values are shown in brackets. The corresponding GCN value at the adsorption site is provided in bold. Colour labelling: light grey (Pt), orange (Ni), green (Mg) and red (O).
Figure 3.
O adsorption sites considered for supported PtNi clusters involving Ih and Dh geometries, in the size range 33–58 atoms. The cluster/oxide interface involves the supported clusters (111) facets. E values are displayed in eV, while dispersion corrected (DFT-D) values are shown in brackets. The corresponding GCN value at the adsorption site is provided in bold. Colour labelling: light grey (Pt), orange (Ni), green (Mg) and red (O).

Figure 4.
Adsorption map involving all the different O adsorption sites for supported PtNi, Pt and Ni clusters. DFT (PBE) reference values are included for O adsorption on the flat Pt(111) surface (0.46–0.86 eV) [43,45,46,47], including experimental values (0.4–0.5 eV) [48,49], as well as for those for the stepped Pt(321) surface (1.23 to 1.56 eV) [50]. PtNi gas-phase clusters’ O adsorption value range (0.5–2.5 eV) [24] is highlighted by dashed green lines. Colour circles labelling: black (Pt), orange (Ni), blue (PtNi).
Figure 4.
Adsorption map involving all the different O adsorption sites for supported PtNi, Pt and Ni clusters. DFT (PBE) reference values are included for O adsorption on the flat Pt(111) surface (0.46–0.86 eV) [43,45,46,47], including experimental values (0.4–0.5 eV) [48,49], as well as for those for the stepped Pt(321) surface (1.23 to 1.56 eV) [50]. PtNi gas-phase clusters’ O adsorption value range (0.5–2.5 eV) [24] is highlighted by dashed green lines. Colour circles labelling: black (Pt), orange (Ni), blue (PtNi).

Figure 5.
(a) Calculated GCN values plotted as a function of all inequivalent sites—after O adsorption—on PtNi (blue), Pt (black) and Ni (orange) supported clusters; (b) Calculated O adsorption energies (E) PBE values plotted as a function of the generalized coordination number (GCN).
Figure 5.
(a) Calculated GCN values plotted as a function of all inequivalent sites—after O adsorption—on PtNi (blue), Pt (black) and Ni (orange) supported clusters; (b) Calculated O adsorption energies (E) PBE values plotted as a function of the generalized coordination number (GCN).

Figure 6.
Structural analysis of all O adsorption sites on supported PtNi, Pt, Ni clusters, displaying calculated E values (eV) as function of: (a) the metal–metal (M–M) bond length at the adsorption site; (b) the O molecule bond length; (c,d) cluster core and shell strain; (e) calculated cluster Bader charge transfer (both strongest and weakest adsorption sites); and (f) the cluster surface roughness.
Figure 6.
Structural analysis of all O adsorption sites on supported PtNi, Pt, Ni clusters, displaying calculated E values (eV) as function of: (a) the metal–metal (M–M) bond length at the adsorption site; (b) the O molecule bond length; (c,d) cluster core and shell strain; (e) calculated cluster Bader charge transfer (both strongest and weakest adsorption sites); and (f) the cluster surface roughness.

Figure 7.
Calculated average d-band center values () for all supported PtNi clusters (25 to 58 atoms), for the strongest (a) and weakest (b), namely FCC O adsorption sites, plotted as a function of E (eV) values. The red line represents the best linear fitting, while blue empty dots refer to those values calculated for dissociated O configurations.
Figure 7.
Calculated average d-band center values () for all supported PtNi clusters (25 to 58 atoms), for the strongest (a) and weakest (b), namely FCC O adsorption sites, plotted as a function of E (eV) values. The red line represents the best linear fitting, while blue empty dots refer to those values calculated for dissociated O configurations.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Paz-Borbón, L.O.; Baletto, F. A DFT Study on the O2 Adsorption Properties of Supported PtNi Clusters. Inorganics 2017, 5, 43. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics5030043
AMA Style
Paz-Borbón LO, Baletto F. A DFT Study on the O2 Adsorption Properties of Supported PtNi Clusters. Inorganics. 2017; 5(3):43. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics5030043
Chicago/Turabian StylePaz-Borbón, Lauro Oliver, and Francesca Baletto. 2017. "A DFT Study on the O2 Adsorption Properties of Supported PtNi Clusters" Inorganics 5, no. 3: 43. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics5030043
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.