Mechanistic Implications for the Ni(I)-Catalyzed Kumada Cross-Coupling Reaction


1
Anorganische Chemie I, Ruhr-Universität Bochum, Universitätsstraße 150, 44801 Bochum, Germany
2
Max-Planck-Institut für Chemische Energiekonversion, Stiftstraße 34-36, 45470 Mülheim, Germany
*
Author to whom correspondence should be addressed.
Inorganics 2017, 5(4), 78; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics5040078
Submission received: 9 October 2017
/
Revised: 7 November 2017
/
Accepted: 10 November 2017
/
Published: 14 November 2017
(This article belongs to the Special Issue Revealing Reaction Mechanisms in Homogeneous Transition Metal Catalysis)
Abstract
:Herein we report on the cross-coupling reaction of phenylmagnesium bromide with aryl halides using the well-defined tetrahedral Ni(I) complex, [(Triphos)NiICl] (Triphos = 1,1,1-tris(diphenylphosphinomethyl)ethane). In the presence of 0.5 mol % [(Triphos)NiICl], good to excellent yields (75–97%) of the respective coupling products within a reaction time of only 2.5 h at room temperature were achieved. Likewise, the tripodal Ni(II)complexes [(κ2-Triphos)NiIICl2] and [(κ3-Triphos)NiIICl](X) (X = ClO4, BF4) were tested as potential pre-catalysts for the Kumada cross-coupling reaction. While the Ni(II) complexes also afford the coupling products in comparable yields, mechanistic investigations by UV/Vis and electron paramagnetic resonance (EPR) spectroscopy indicate a Ni(I) intermediate as the catalytically active species in the Kumada cross-coupling reaction. Based on experimental findings and density functional theory (DFT) calculations, a plausible Ni(I)-catalyzed reaction mechanism for the Kumada cross-coupling reaction is presented.
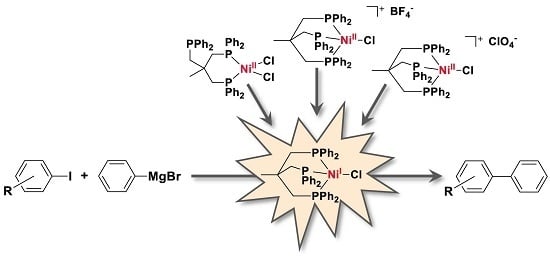
1. Introduction
Metal-catalyzed cross-coupling reactions are an essential tool for the formation of carbon–carbon bonds [1]. Coupling reactions of organometallic reagents with organic electrophiles are industrially utilized for the synthesis of pharmaceuticals, agrochemicals and polymers. Although the first cross-coupling reactions of organomagnesium bromides described by Kumada or Corriu utilized nickel catalysts, for example, [Ni(dppe)Cl2] and Ni(acac)2 [2,3], the most efficient and commonly employed catalysts for such C–C coupling reactions nowadays are based on noble metals [4]. In particular, palladium and its complexes are exhaustively reported for C–C bond formation reactions and mechanistic details of Pd-based catalysts are well understood [5]. Due to the high cost and low abundance of noble metals, the development of alternative catalytic systems based on earth-abundant and less expensive metals, such as nickel and iron, recently attracted much interest. Likewise, Ni(0), Ni(I), Ni(II) and Ni(III) complexes have been proposed as potential intermediates for various C–C coupling reactions including Heck, Hiyama, Kumada, Negishi, Suzuki–Miyaura, Sonogashira and Stille coupling techniques [6,7,8]. Along this line, Ni(I) as well as Ni(III) species were proposed as reactive intermediates in the nickel-catalyzed coupling of aryl halides by trans-ArNiBr(PEt3)2 [9]. Notably, nickel is substantially more nucleophilic as compared to Pd and Pt due to its smaller atomic size. Because of their higher nucleophilicity, nickel catalysts allow reactions under milder conditions and more challenging electrophiles than can be used in palladium catalysis.
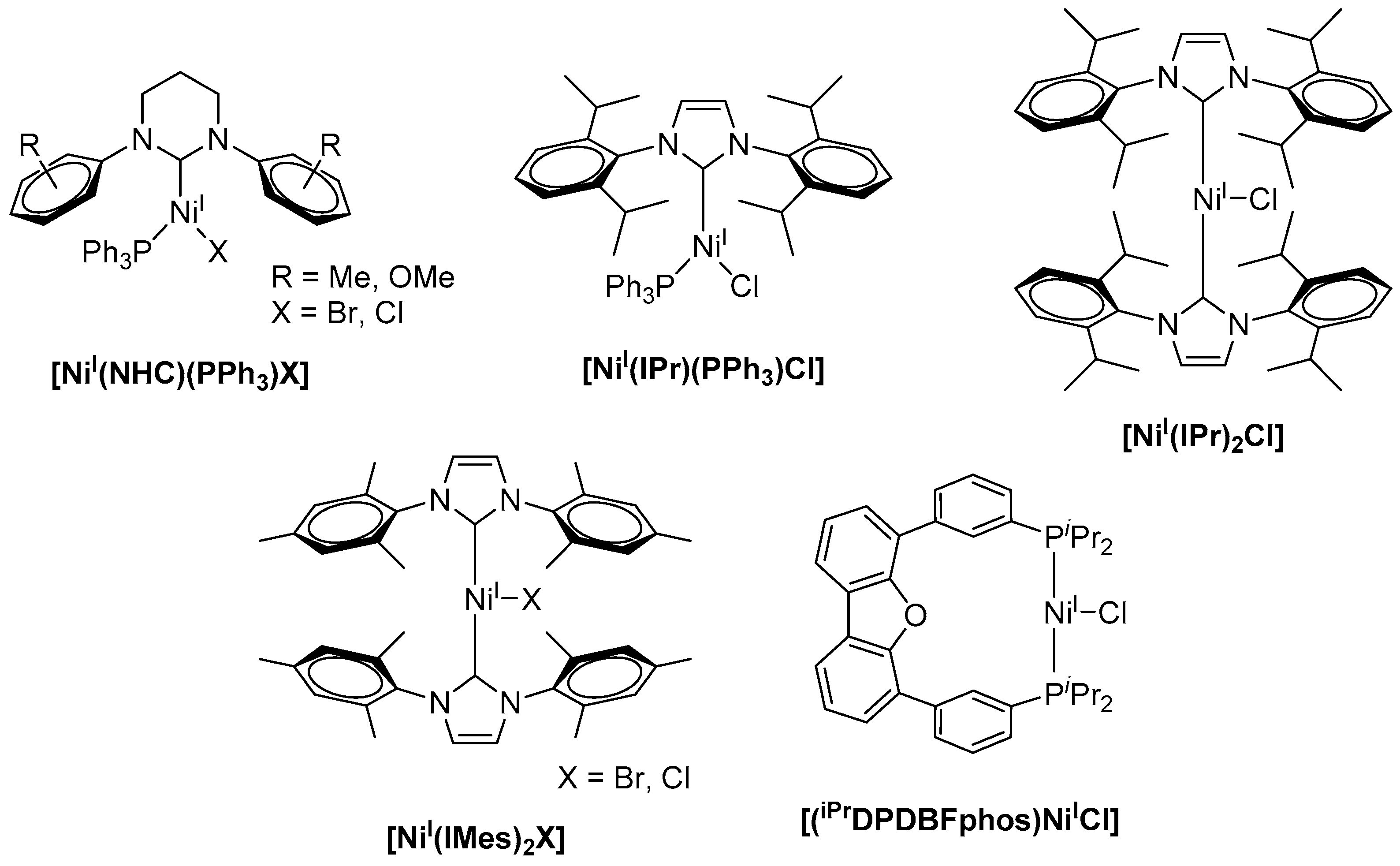
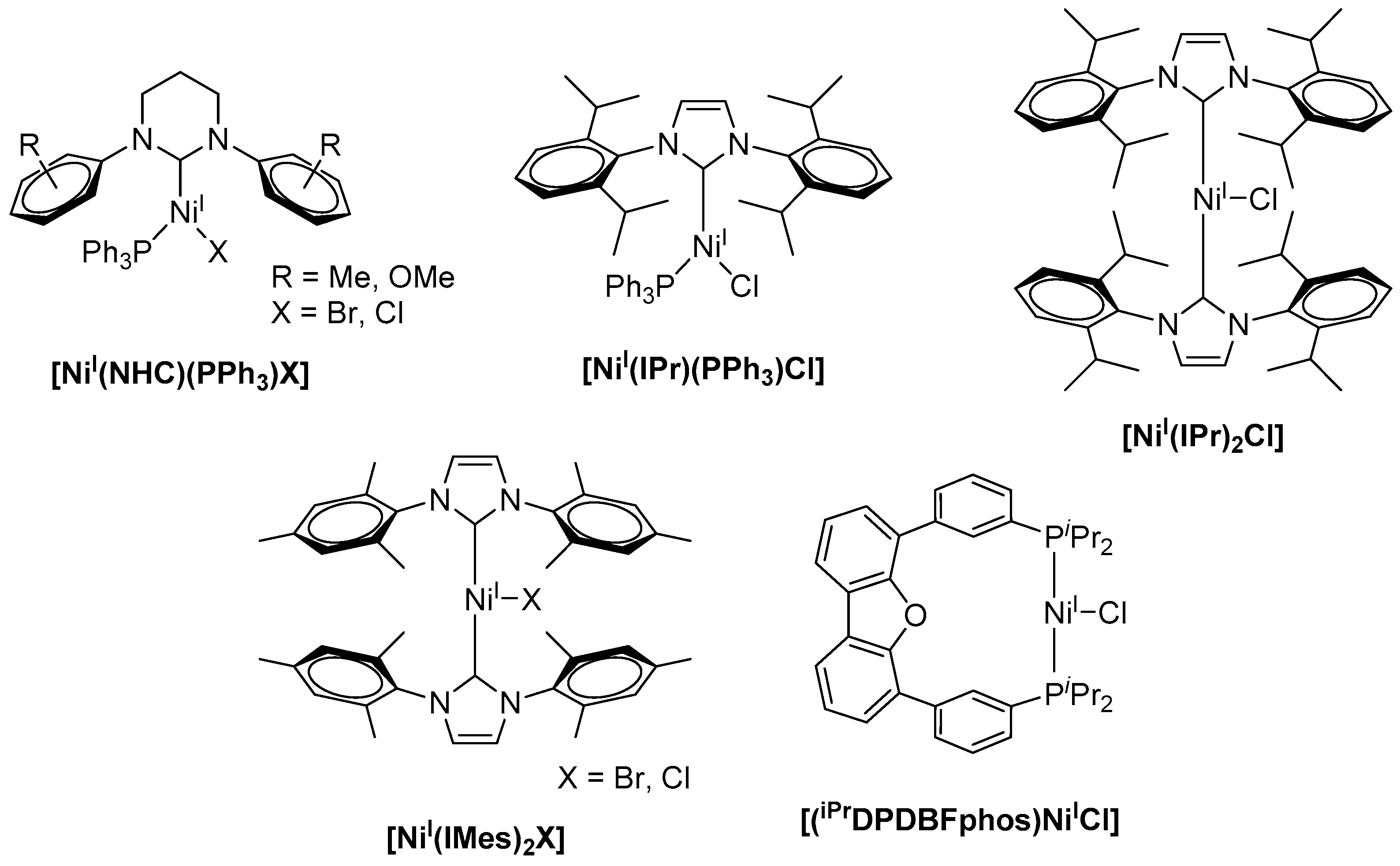
While numerous examples of low-valent nickel(I) complexes were reported, well-defined stable Ni(I) precursors as catalysts in cross-coupling reactions are rare [10]. Such Ni(I) complexes are frequently used as potent pre-catalysts in the Negishi coupling of alkyliodides and alkylzinc bromides [11,12,13], in the Suzuki–Miyaura reaction for the formation of C–C bonds from arylhalides and phenylboronic acid [14,15,16], as well as in Ni(I)-catalyzed Kumada coupling reactions [17,18]. Here, most commonly, bulky N-heterocyclic carbenes (NHCs) are utilized as a ligand platform for the stabilization of the monovalent nickel centers. Along this line, Whittlesey and coworkers reported a series of extremely air-sensitive [NiI(PPh3)(NHC)X] complexes (X = Br, Cl) (Scheme 1) comprising bulky NHC ligands [17]. Herein, the sterically demanding substitution at the central heteroaromatic ring is required to stabilize the low-coordinate and low-valent Ni(I)-complex and to avoid intermolecular reactions. The Ni(I) complexes were subsequently studied for the Kumada coupling of aryl chlorides, for example, chlorobenzene and 4-chlorotoluene, with phenylmagnesium chloride leading to 1,1′-biphenyl or 4-methyl-1,1′-biphenyl.
Notably, an increase in both the carbene ring size and steric demand of the N-substituents led to a decrease in catalytic activity. Likewise, bulkier cross-coupling partners like mesitylmagnesium bromide or more challenging substrates (e.g., aryl fluorides) led to a decrease in the yield of the bi-aryl products to less than 30%. Among all Ni(I) species used for these C–C bond formation studies, [NiI(6-Mes)(PPh3)Br] (6-Mes = 1,3-bis(2,4,6-trimethylphenyl)hexahydropyrimidine-2-ylidene) showed highest activity for the coupling of chlorobenzene and phenylmagnesium chloride with yields up to 83% of 1,1′-biphenyl [5]. The efficiency of the coupling reaction could further be increased when complexes [NiI(IPr)(PPh3)Cl] and [NiI(IPr)2Cl] (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene) were applied with catalyst loadings of 1 mol %, affording yields of up to 98% for the desired coupling product within reaction times of 3 and 18 h [18,19]. In analogy, [NiI(IMes)2X] (IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazole-2-ylidene, X = Br, Cl) acts as a potent catalyst for the Kumada cross-coupling reaction (Scheme 1) [20]. When compared to [Ni0(IMes)2] and [NiII(IMes)2X2], all three complexes revealed similar catalytic activity for the reaction of chlorobenzene and mesitylmagnesium bromide under the same experimental conditions and afforded 2,4,6-trimethyl-1,1′-biphenyl in 73–79% yield when 3 mol % catalyst was applied. It is worth mentioning that biscarbene Ni(I) complexes were shown to dimerize under the applied reaction conditions, forming dinuclear Ni(I)(µ-Cl)2Ni(I) intermediates that were suggested as the active catalyst, and the involvement of both [Ni(I)–Ni(I)] and [Ni(II)–Ni(II)] intermediates was subsequently suggested [21]. [(iPrDPDBFphos)NiICl] (iPrDPDBFphos = 4,6-bis(diisopropylphosphinophenyl)dibenzofuran) even allowed for the coupling of vinyl chloride and a phenyl Grignard reagent, allowing for the incorporation of a functional group into the target structure and affording styrene in about 85% yield within 22 h stirring at room temperature [22]. However, similar yields were obtained when the respective Ni(II) complex was utilized for the very same reaction.
We recently showed that Triphos (2-((diphenylphosphaneyl)methyl)-2-methylpropane-1,3-diyl)bis(diphenylphosphane)- and TriphosSi (((methylsilanetriyl)tris(methylene))-tris(diphenylphosphane))- derived Ni and Fe complexes are potential noble metal-free platforms to conduct the C–C cross-coupling of aryl iodides and alkynes [23,24]. Furthermore, the Ni(Triphos) complexes were shown to stabilize various oxidation states of Ni (Scheme 2) [25]. Notably, due to the high steric bulk of the Triphos ligand, such Ni complexes neither show any disproportionation of the respective Ni(I) complexes to Ni(0) and Ni(II), nor do they allow for dimerization to give a dinickel complex. The reported reactivity of such Ni(Triphos) complexes pointed our attention towards their application for Kumada cross-coupling reactions. Since Ni(0), Ni(I) and Ni(II) complexes were shown to be reactive complexes and can act as pre-catalysts in the C–C bond coupling reaction under Kumada conditions, we herein explore the role of the oxidation state based on well-defined [(Triphos)NiICl] for the C–C bond formation by UV/Vis and electron paramagnetic resonance (EPR) spectroscopy. The data is furthermore supported by density functional theory (DFT) calculations.
2. Results and Discussions
Following a recently reported synthetic route, the Ni(I) complex [(Triphos)NiICl] (1) was obtained by reduction of the square-planar complex [(Triphos)NiIICl2] (2) with cobaltocene in gram scale and good yield (87%). The molecular structure of 1 was previously unequivocally shown by single-crystal X-ray crystallography and the monovalent electronic configuration was confirmed by EPR spectroscopy, as well as its magnetic moment via the Evans method (µ = 1.9 µB) [25].
We subsequently investigated the catalytic potential of complex 1 for Kumada-type cross-coupling reactions. As a model reaction, we chose the coupling of para-substituted aryl iodides and phenylmagnesium bromide in THF at room temperature in the presence of 0.5 mol % complex 1.
Under those conditions, iodobenzene, 4-iodotoluene as well as 4-iodoanisole were successfully coupled to phenylmagnesium bromide and afforded the respective coupling products in 89%, 94% and 86% yield, respectively (Table 1). Likewise, we were able to utilize bromobenzene and 4-bromotoluene with phenylmagnesium bromide, showing that bromo-substituted arenes are suitable for the coupling with complex 1 and can be used instead of the usually expensive iodo-derivatives (Table 1). Here, 1,1′-biphenyl and 4-methyl-1,1′-biphenyl were obtained in 91% and 74% isolated yield, respectively. Following this line, we also tested the application of aryl chlorides and fluorides as substitutes in the cross-coupling reaction procedure. However, the respective coupling products were only obtained in 3% and 5% yield at room temperature, respectively (Table 1). When the reaction temperature was increased to 60 °C, for both aryl chlorides and fluorides, the expected coupling product 1,1′-biphenyl was detected via GC–MS analysis in 100% yield starting from chlorobenzene and in 44% yield for fluorobenzene (Table 1). Thus, aryl chlorides and fluorides are suitable starting materials at higher temperature for Kumada cross-coupling reactions when complex 1 is applied as potential catalyst, but not at room temperature. Notably, in the absence of complex 1, no formation of any respective bi-aryl product was observed, highlighting the role of the Ni(I) complex 1 as an important pre-catalyst herein. For the latter case, solely starting material was recovered quantitatively.
While the coupling of aryl halides and phenyl Grignard reagents proceeds rapidly and affords the coupling products in high yields, we set out to explore the possibility to perform coupling of alkyl halides and/or alkyl Grignard reagents. In a first step, we used phenylethynyl- and methylmagnesium bromide. All such reactions, however, did not yield any of the desired coupling products, and attempts to alter the reaction conditions by changing temperature, reaction time as well as the catalyst concentration failed as well in our hands. Likewise, complex 1 cannot facilitate the conversion of 1,2-diiodoethane and phenylmagnesium bromide. Neither (2-iodoethyl)benzene nor 1,2-diphenylethane were formed under our standard conditions, excluding alkyliodides for application in Kumada cross-coupling reactions with the investigated catalyst. These observations show that the monovalent Ni complex 1 is unsuitable for the coupling of C–C bonds from sp3-centers.
A common limitation of the Kumada cross-coupling reaction is the catalysts’ low tolerance towards functional groups within the substrate. To investigate the functional group tolerance of complex 1 as catalyst, we chose different substituted aryl iodides and also heterocyclic iodides as challenging substrates for the Kumada coupling (Table 2). Phenyl iodides with additional alkyl, alkoxy, aryl and fluoride substituents afforded the expected coupling products with yields between 86% and 94% (Table 2, Entries 1–6, 8). Notably, polysubstituted aryl iodides comprising substituents in the ortho-position, for example, 2-iodomesitylene and 1-iodonaphthalene, did not show formation of any coupling product (Table 2, Entry 7 and 9). These observations suggest that aryl iodides with substituents directly adjacent to iodine are unsuitable substrates for the cross-coupling reaction utilizing complex 1, due to steric hindrance of the oxidative addition of the aryl iodide to the Ni(I) center. Likewise, aryl iodides having amino, nitro, nitrile, hydroxyl, carbonyl or carboxylic acid groups did not allow for successful C–C bond formation and resulted in decomposition of complex 1 to a hitherto unknown product mixture (Table 2, Entry 11–17). Similarly, application of heteroaromatic substrates like iodopyridines (Table 2, Entry 18–20) did not result in the formation of the desired products, neither at a higher reaction temperature. Most likely, this lack of reactivity stems from an undesired coordination of the additional functional groups with complex 1. Thus, Ni(I) complex 1 is only suitable to perform the Kumada coupling of aryl Grignard compounds and alkyl-, alkoxy- and aryl-substituted sterically less-demanding aryl iodides, and has only little tolerance for other functional groups.
Since complexes featuring a Ni(II) ion were also reported for catalyzing Kumada cross-coupling reactions, we next tested the Ni(II) complexes 2–4 (1 and 0.5 mol % catalyst loading) as potential catalysts in our model reaction, namely the coupling of 4-iodotoluene and phenylmagnesium bromide under otherwise identical conditions as applied above. Ni(II) complex 2 was obtained in 94% yield according to literature-known procedures by complexation of Triphos with NiCl2, and Ni(II) complexes 3 and 4 were formed via reaction of [Ni(CH3CN)6](BF)2 or Ni(ClO4)·6H2O, respectively, to the Ni(II) complex 2 [23,25].
An overview of the observed product yields is provided in Table 3. Yields given for 4-methyl-1,1′-biphenyl were determined by GC–MS analysis using 1,1′-biphenyl as an internal calibration standard. In addition, isolated yields from experiments applying complex 1 are added for comparison. While application of complex 2 generally leads to a complete conversion of 4-iodotoluene to the desired coupling product for both 1 and 0.5 mol % catalyst loading, formation of 4-methyl-1,1′-biphenyl was observed in solely 72% (1 mol %) and 46% (0.5 mol %), as well as 59% (1 mol %) and 41% (0.5 mol %), yields for complexes 3 and 4, respectively. However, while the amount of 4-methyl-1,1′-biphenyl formed is lowered for 3 and 4, the results clearly show that Ni(II) complexes can likewise be utilized as potential pre-catalysts for the Kumada reaction, with complexes 1 and 2 showing the highest amounts of isolated product. Here, the choice of the counterions rather than the oxidation state of the nickel center seems to be of more importance. While complexes revealing only halogenide ligands like the tetrahedral Ni(I) complex 1 or the square planar Ni(II) complex 2 showed high activity for the coupling reaction, the formation of 4-methyl-1,1′-biphenyl by complexes 3 and 4 comprising non-coordinating BF4− or ClO4− counterions is hampered.
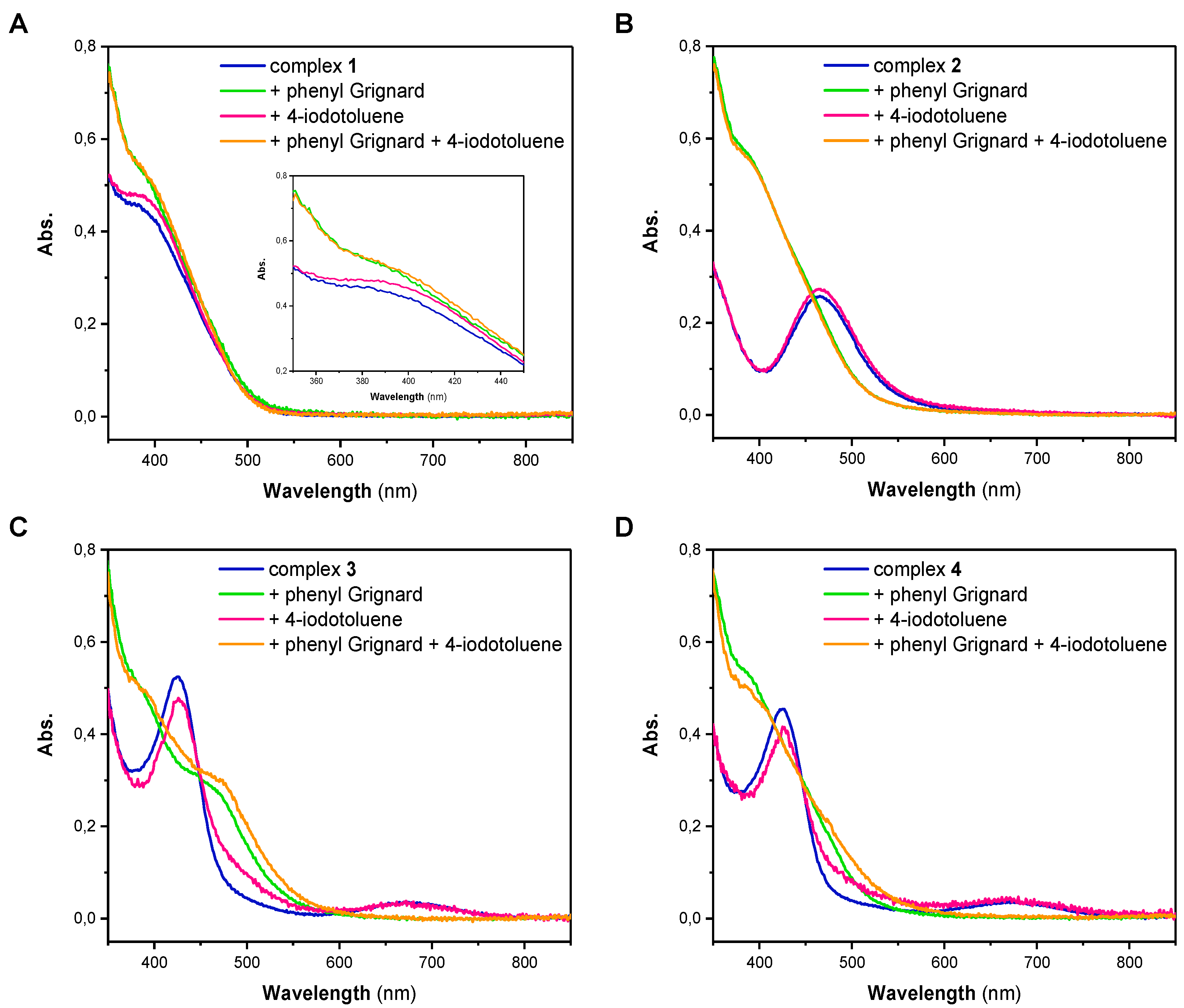
The high reactivity of both of the Ni(I) and Ni(II) complexes pointed our attention towards possible intermediates that are formed upon reaction with either phenylmagnesium bromide or 4-iodotoluene. Thus, we next performed stoichiometric reactions of the different Ni complexes with one equivalent of either reagent in THF. Subsequently, each solution was analyzed by UV/Vis spectroscopy (Figure 1). The UV/Vis spectrum of Ni(I) complex 1 has a broad absorption band at low wavelengths with a maximum at 385 nm (Figure 1A). Spectra of 2, 3 and 4 differ significantly from the spectrum of 1. While the UV/Vis spectrum of 2 has an absorption maximum at a wavelength of 465 nm, the spectra of 3 and 4, which are nearly identical, show an absorption maximum at 425 nm and a broader band with a lower intensity at 670 nm (Figure 1B–D). For complex 1, showing an absorption band at 385 nm, it is apparent that the addition of 4-iodotoluene did not lead to a change in the complexes’ electronic spectrum (Figure 1A). This observation illustrates that the oxidative addition of the aryl iodide is presumably not the first step in the coupling process and no formation of a Ni(III) intermediate takes place. Based on this observation, it can also be excluded that disproportionation of the Ni(I) complex 1 takes place. Addition of phenylmagnesium bromide to 1, similarly and regardless of whether the experiments were performed in the absence or presence of 4-iodotoluene, led only to minor alterations of the absorption band around 385 nm (Inset Figure 1A). Fundamentally different behavior, however, was observed when the Ni(II) complex 2 was investigated (Figure 1B). While addition of 4-iodotoluene also did not lead to any change of the UV/Vis spectrum, addition of phenylmagnesium bromide led to a significant change in the spectrum; the original band at 465 nm was lost and a new band centered at 385 nm appeared.
Identical trends were observed for the Ni(II) complexes 3 and 4 (Figure 1C,D). Complexes 3 and 4 reveal two characteristic absorption bands at around 430 and 670 nm in THF that disappear after treatment with phenylmagnesium bromide, suggesting an overall similar chemistry as observed for complexes 1 and 2. It is, however, notable that for complex 3 an additional band centered at 465 nm can be observed that was not observed for the other complexes. The origin of this additional band is hitherto unknown. Altogether it becomes obvious that Ni complexes 1–4 do not react with only the aryl iodide. A brief comparison of the electronic features of complex 1 and complexes 2, 3 and 4 after treatment with phenylmagnesium bromide clearly suggests similar electronic features. Thus, it becomes clear that phenylmagnesium additionally acts as a reducing agent.
In addition to our UV/Vis studies, we crystallized the products of the different solutions. Notably, crystals were obtained from the reaction of complex 1 with phenylmagnesium bromide as well as of complexes 2, 3 and 4 with phenylmagnesium bromide and 4-iodotoluene. For all experiments, we obtained a yellow crystalline material suitable for X-ray diffraction. The cell parameters of those crystals closely resembled those of complex 1, suggesting the formation of a complex with an overall similar structure (Table S1). In addition, the reaction of 4 with phenylmagnesium bromide and 4-iodotoluene afforded a second set of crystalline material with slightly enlarged cell parameters (Table S1). While the quality of this crystal was not sufficiently good, a structural motif was obtained showing a (Triphos)Ni(I) moiety with a coordinated iodine ligand (Figure S1, Table S2).
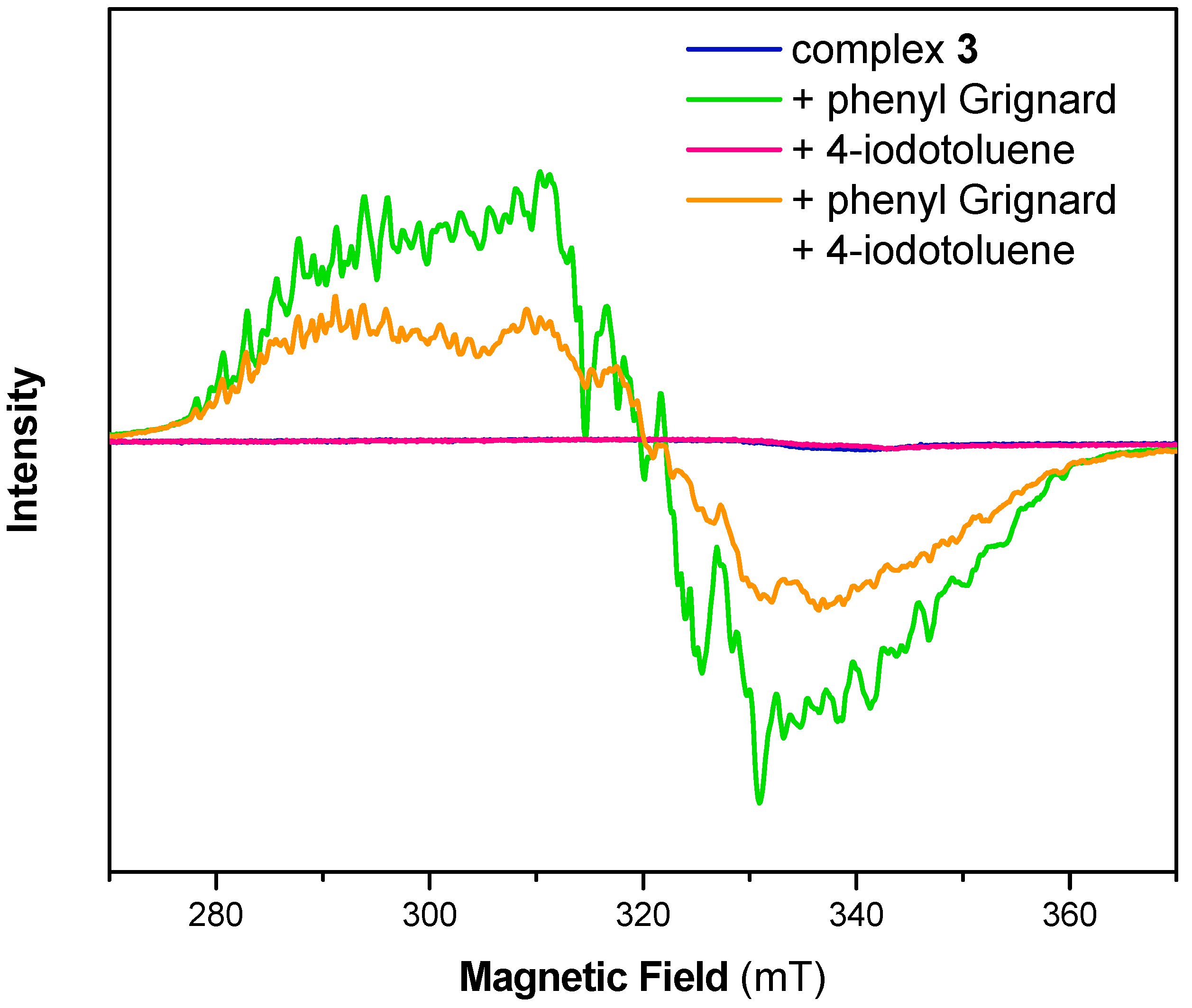
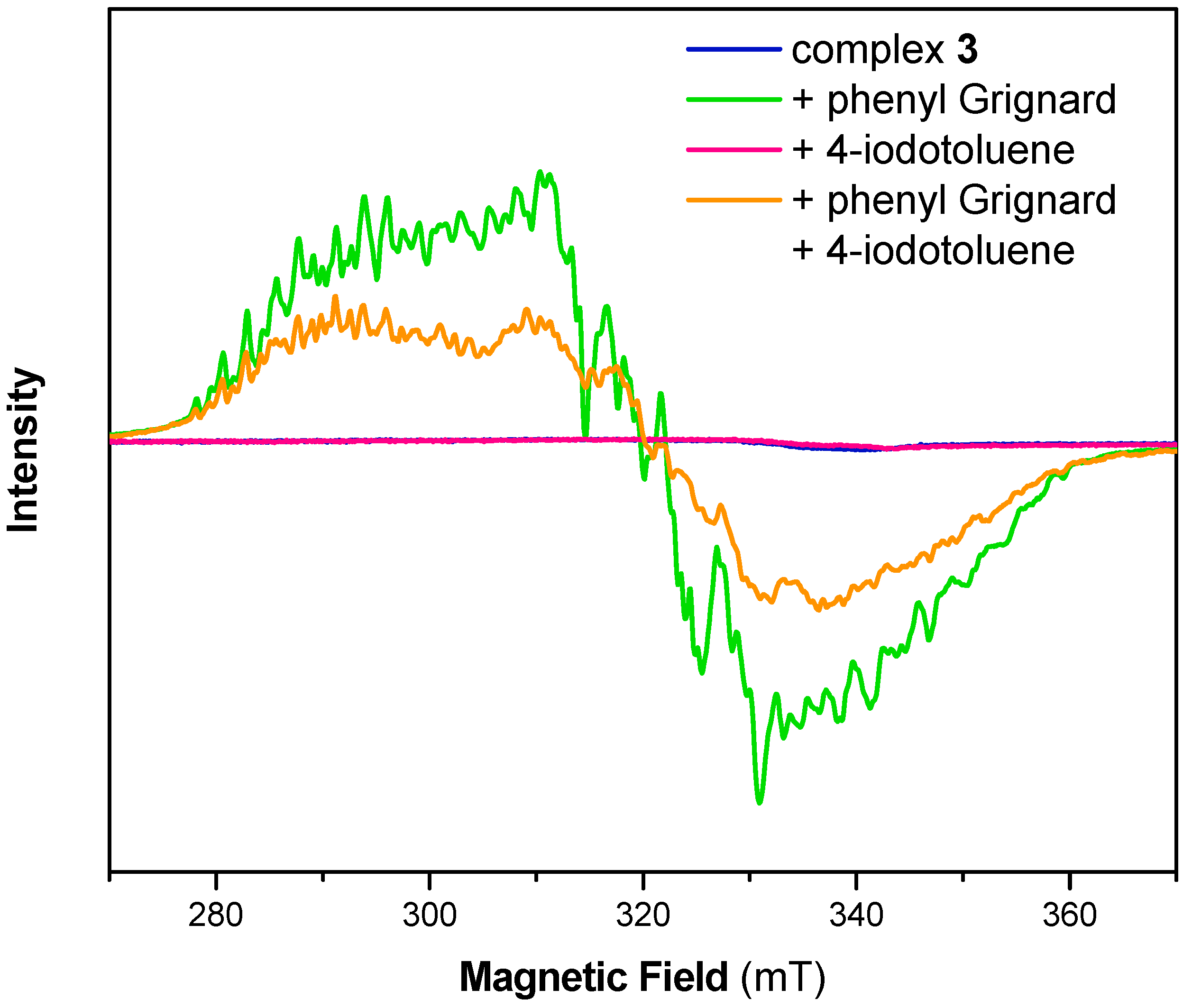
In order to clarify the formation of a Ni(I) species during C–C bond formation, we further conducted electron paramagnetic resonance (EPR) spectroscopy on the same reaction solutions. This technique is ideal to elucidate the presence of paramagnetic oxidation states of the transition metal. As previously reported, we found a very rich, albeit greatly overlapping, hyperfine structure for complex 1 in THF (Figure S2) [25]. The overall spectrum did not alter upon addition of 4-iodotoluene. Addition of phenylmagnesium bromide to the solution of 1, however, lead to broadening of the EPR signal and the hyperfine pattern slightly changed, while the g-value remained unaltered. The latter observation again confirms our hypothesis that complex 1 neither undergoes oxidative addition upon addition of aryl iodides nor shows any disproportionation to afford Ni(0) and Ni(II) species.
As was reported before, complexes 2, 3 and 4 are EPR silent (Figure 2, Figures S3 and S4) and no change was observed upon addition of 4-iodotoluene [23,25]. As expected from the experiments shown above, freeze-quenched reaction solutions of the Ni(II) complexes 2, 3 or 4 with added phenylmagnesium bromide revealed an EPR spectrum with identical g-values as for complex 1 with phenylmagnesium bromide but with minor changes to the complex hyperfine pattern. Notably, EPR spectra of reaction mixtures of the complexes and the Grignard reagent as well as 4-iodotoluene showed similar features as those without 4-iodotoluene. However, the signal intensity was significantly decreased, suggesting the formation of an EPR-silent species upon addition of 4-iodotoluene. While crystallographic data showed structurally related cell parameters, EPR analysis suggests that as well as [(Triphos)NiII] found during crystallization, at least one additional Ni(II) species might have been formed during such experiments.
As demonstrated by our experiments, the nickel complexes initially react with phenylmagnesium bromide and not with 4-iodotoluene. This finding suggests that in the Ni(I)-driven Kumada cross-coupling of aryl iodides and a Grignard reagent, the Grignard reagent initially acts as a reducing agent for Ni(II) to afford a Ni(I) species. Such a scenario would imply that phenyl radicals originating from the Grignard might form. While we were not able to detect such species in situ, we recognized that after 24 h, solutions of the Ni(II) complexes and phenylmagnesium bromide always contained small amounts of 1,1′-biphenyl (~1%) as analyzed by GC–MS. This assumption is supported by the fact that in the case of an additional treatment of complex 1 with phenylmagnesium bromide, we did not find any 1,1′-biphenyl.
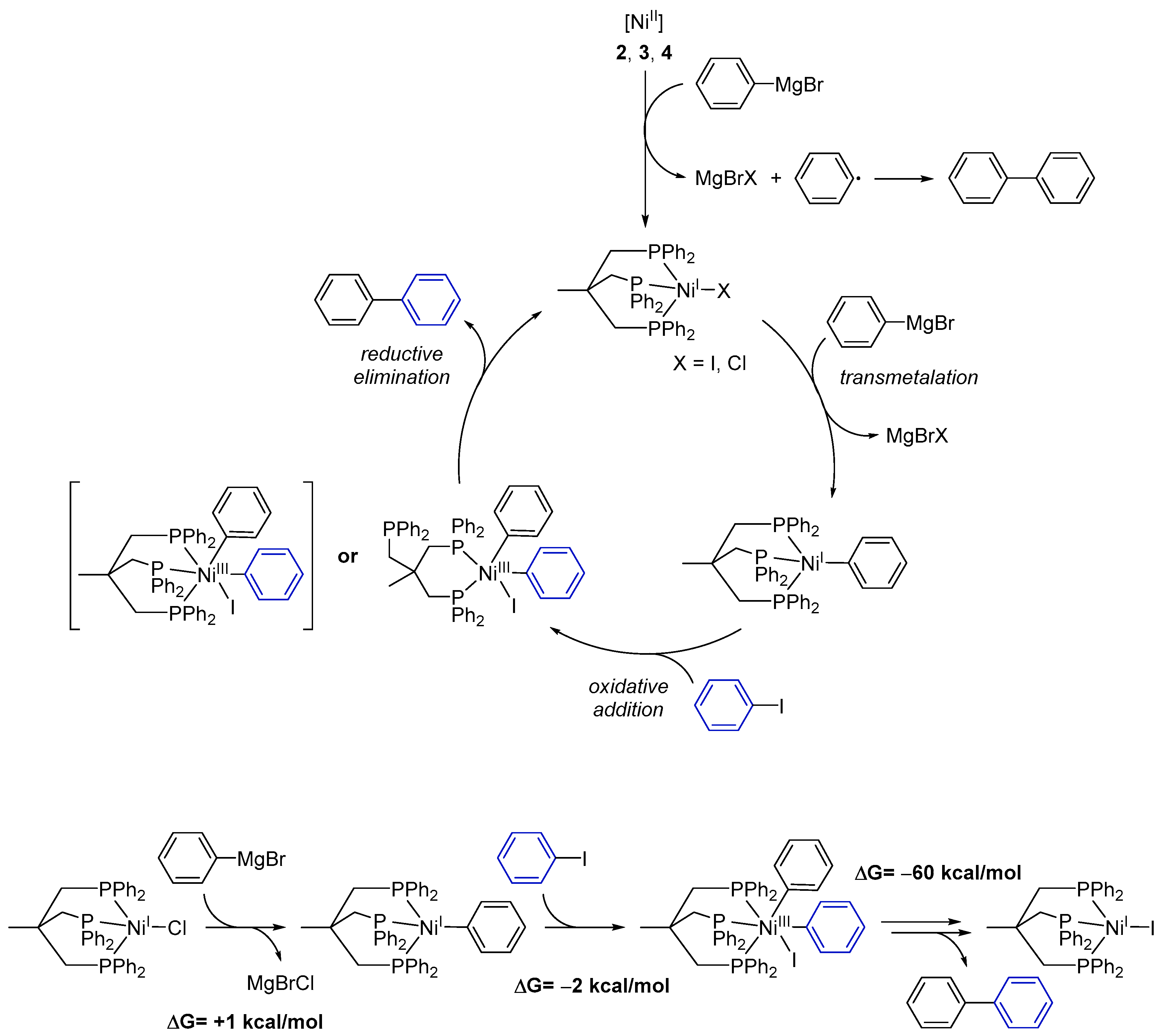
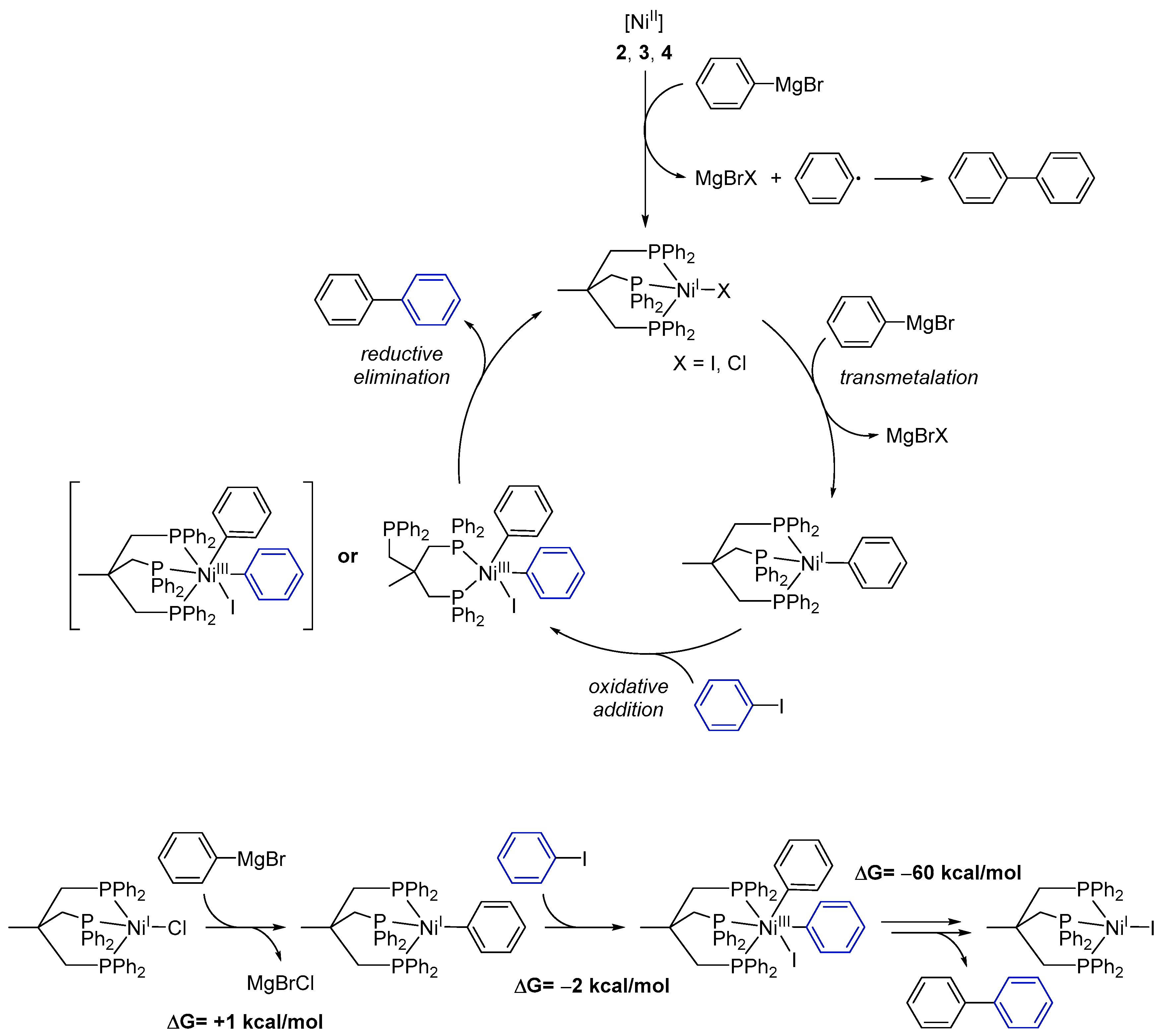
Based on our experimental results we propose the following mechanism for the catalytic cross-coupling reaction catalyzed by the Ni complexes 1–4 (Scheme 3):
- (1)
- Starting from Ni(II) complexes 2–4, a reduction to a Ni(I) species by a Grignard reagent as reducing agent occurs as initial reaction step. This reduction likewise leads to formation of 1,1′-biphenyl.
- (2)
- The Ni(I) species will then undergo transmetalation with a second phenylmagnesium bromide molecule. This proposition stems from the observation that the isolated Ni(I) complex 1 does not react with aryl iodides.
- (3)
- The reactive Ni(I)–phenyl intermediate formed upon transmetalation then reacts under an oxidative addition with the aryl halogenide. It is worth mentioning that the generated Ni(III) species may bear the Triphos ligand either in a κ2- or κ3-coordination mode. Occupation of a κ2-coordination mode seems an attractive option to reduce the steric hindrance at the Ni center and to allow for further reaction steps to proceed.
- (4)
- Subsequently, the coupling product is released by reductive elimination and the Ni(I) species, now bearing an iodide ligand, is regenerated.
Quantum-chemical calculations of the reaction enthalpies were performed in order to gain support for the proposed cycle. Indeed, the calculations indicated that transmetalation with the Grignard reagent to form [Triphos)NiI(phenylate)] is isothermic (+1 kcal/mol) within the accuracy of the calculations, and subsequent reaction with iodobenzene is thermodynamically favored for the κ2-coordination mode (Scheme 3, lower left) but endothermic for the κ3-intermediate. As such, κ2-conformation presents an ideal scaffold for the two phenylates to become co-coordinated to the same metal ion and in spatial vicinity. Moreover, the respective phenyl planes form an angle of 90 degrees in such a way that the two π-systems significantly overlap at the carbenes. The latter likely is of crucial importance as an onset for C–C bond formation. Release of biphenyl and subsequent re-coordination of the phosphine arm is highly exothermic (−60 kcal/mol) and gives rise to the biphenyl final product and [(Triphos)NiII], completely in line with experimental observations.
As such, besides giving theoretical support for the proposed mechanism, the calculations additionally underline the importance of the flexibility of the Triphos ligand as a κ2- or κ3-coordinated ligand: it can easily harbor a low-spin 3d8 Ni(II) halide species as a κ3-ligand, which is compatible with a preferred 4-coordinate square-planar coordination of low-spin Ni(II). Upon oxidation to Ni(III), 3d7, which prefers a five-coordinate square mono-pyramidal coordination, one phosphine easily dissociates in order to stabilize this oxidation state. Upon reductive elimination of biphenyl, re-coordination occurs.
3. Materials and Methods
General procedures. All reactions were performed under a dry N2 or Ar atmosphere using standard Schlenk techniques or in a glovebox. [(Triphos)NiICl] (1), [(Triphos)NiIICl2] (2), [(Triphos)NiIICl](BF4) and [(Triphos)NiIICl](ClO4) were synthesized according to previously reported procedures [23,25]. All other compounds were obtained from commercial vendors and used without further purification. All solvents were dried prior to use according to standard methods. 1H-, 13C{1H}-, 19F- and 31P{1H}-NMR spectra were recorded with a Bruker DPX-200 NMR spectrometer or a Bruker DPX-250 NMR spectrometer at room temperature. Peaks were referenced to residual 1H or 13C signals from the deuterated solvent and are reported in parts per million (ppm). Mass spectra were measured with a Shimadzu QP-2010 instrument. UV/Vis spectra were recorded with a Varian Cary 100 spectrometer at room temperature. EPR spectra were recorded at T = 10 K on a Bruker Elexsys E500 X-band spectrometer equipped with an Oxford CF935 flow cryostat and a ST9102 resonator. The microwave frequency amounted to 9.636 GHz in all experiments. A microwave power of 2 mW was used. The modulation amplitude of 0.3 mT was chosen to optimize the S/N ratio. A few additional experiments were performed at lower modulation amplitude, but did not reveal additional or more resolved hyperfine structure.
Quantum Chemistry. All calculations were performed with the ORCA program package [26], geometry optimization and frequency analysis was performed by using the BP86 functional [27] and def2-SVP basis set [28] and zeroth order regular approach (zora) [29,30] in order to take into account scalar relativistic effects at nickel and iodine.
General procedure for Kumada coupling reactions. The respective potential catalyst (0.5 mol %) was dissolved in anhydrous THF (6 mL) and the corresponding aryl halide (2.8 mmol) was subsequently added. The solution was stirred for 10 min before the addition of the corresponding Grignard reagent (5.6 mmol). The reaction mixture was stirred for 2.5 h at room temperature. Subsequently, methanol was added to quench the reaction. Silica was added to the solution and the solvent was removed. Purification via column chromatography allowed for isolation of the cross-coupling products P1–P7. 1H, 13C and 19F NMR spectra and GC-MS graphics for characterization of the coupling products are placed in Supplementary Materials.
1,1′-Biphenyl (P1): 1H NMR (200 MHz, CDCl3): δ = 7.64–7.59 (m, 4H, HAr), 7.50–7.32 (m, 6H, HAr). 13C NMR (50 MHz, CDCl3): δ = 141.4, 128.9, 129.4, 127.3. GC–MS (m/z): Calculated for [C12H10]+: 154, found: 154.
4-Methyl-1,1′-biphenyl (P2): 1H NMR (200 MHz, CDCl3): δ = 7.62–7.29 (m, 8H, HAr), 7.23 (m, 1H, HAr), 2.39 (s, 3H, CH3). 13C NMR (50 MHz, CDCl3): δ = 141.3, 138.5, 137.2, 129.6, 238.9, 128.8, 127.1, 127.1. GC–MS (m/z): Calculated for [C13H12]+: 168, found: 168.
4-Methoxy-1,1′-biphenyl (P3): 1H NMR (200 MHz, CDCl3): δ = 7.59–7.50 (m, 4H, HAr), 7.47–7.39 (m, 2H, HAr), 7.35–7.27 (m, 1H, HAr), 7.03–6.95 (m, 2H, HAr), 3.86 (s, 3H, CH3). 13C NMR (50 MHz, CDCl3): δ = 159.3, 141.0, 133.9, 128.9, 128.3, 126.9, 126.8, 114.4, 55.5. GC–MS (m/z): Calculated for [C13H12O]+: 184, found: 184.
1,4-Diphenylbenzene (P4): 1H NMR (200 MHz, CDCl3): δ = 7.69–7.62 (m, 8H, HAr), 7.51–7.32 (m, 6H, HAr). 13C NMR (50 MHz, CDCl3): δ = 140.9, 140.3, 129.0, 127.6, 127.5, 127.2. GC–MS (m/z): Calculated for [C18H14]+: 230, found: 230.
4-Fluoro-1,1′-biphenyl (P5): 1H NMR (200 MHz, CDCl3): δ = 7.59–7.52 (m, 4H, HAr), 7.49–7.33 (m, 3H, HAr), 7.19–7.08 (m, 2H, HAr). 13C NMR (50 MHz, CDCl3): δ = 162.6 (d, J = 246.2 Hz), 140.4, 137.5 (d, J = 3.3 Hz), 129.0, 128.8 (d, J = 8.0 Hz), 127.4, 127.2, 115.8 (d, J = 21.4 Hz). 19F NMR (235 MHz, CDCl3): –115.9. GC–MS (m/z): Calculated for [C12H9F]+: 172, found: 172.
3,5-Dimethyl-1,1′-biphenyl (P6): 1H NMR (200 MHz, CDCl3): δ = 7.62 (d, 2H, HAr), 7.49–7.35 (m, 5H, HAr), 7.03 (s, 1H, HAr), 2.42 (s, 6H, CH3). 13C NMR (50 MHz, CDCl3): δ = 141.6, 141.4, 138.3, 129.0, 128.7, 127.3, 127.2, 125.2, 21.5. GC–MS (m/z): Calculated for [C14H14]+: 182, found: 182.
2-Phenylnaphthalene (P7): 1H NMR (200 MHz, CDCl3): δ = 8.05 (s, 1H, HAr), 7.95–7.85 (m, 3H, HAr), 7.78–7.71 (m, 3H, HAr), 7.55–7.35 (m, 5H, HAr). 13C NMR (50 MHz, CDCl3): δ = 141.3 138.7, 133.8, 132.8, 129.0, 128.6, 128.3, 127.8, 127.6, 127.5, 126.4, 126.1, 125.9, 125.7. GC–MS (m/z): Calculated for [C16H12]+: 204, found: 204.
4. Conclusions
In conclusion, we herein showed that the Ni(I) complex 1 can indeed serve as a catalyst in C–C bond formation in Kumada cross-coupling reactions. Using attractive reaction conditions with short reaction times of 2.5 h at room temperature and very low catalytic loadings of 0.5 mol %, the coupling products were obtained in good to excellent yields. Owing to the relatively low tolerance of the Ni(I) complex 1 towards functional groups, the substrate scope for Kumada coupling reactions is limited to alkyl-, alkoxy- and aryl-substituted substrates. We also observed that Ni(II) complexes 2–4 are suitable to catalyze the coupling of aryl iodides with phenylmagnesium bromide. Investigating the reaction solutions of stoichiometric reactions of the Ni complexes with the coupling reagents by UV/vis measurements showed that reaction with the Grignard reagents leads to the same catalytic Ni(I) species for all complexes. Furthermore, the Ni complexes did not react under oxidative addition with aryl iodides. EPR measurements confirmed the formation of a Ni(I) species by adding Grignard reagent to the Ni(II) complexes. This is in line with the results from crystallization experiments, where for several reactions employing compounds 2, 3 or 4, Ni(I) complex 1 was isolated. Based on our findings, we propose that for Kumada cross-coupling reactions with Ni complexes bearing tripodal phosphine ligand Triphos, a monovalent Ni(I) complex acts as the catalytically active species.
Supplementary Materials
The following are available online at www.mdpi.com/2304-6740/5/4/78/s1. Cif and cif checked files. Details of the calculations. Figure S1: Structure of the asymmetric unit of compound 1, Figure S2: EPR Spectrum of complex 1 (blue) and its reaction solutions with 1 equiv. phenyl Grignard (green), 1 equiv. 4-iodotoluene (red) and with both 1 equiv. phenyl Grignard and 1 equiv. 4-iodotoluene (orange) in THF, Figure S3: EPR Spectrum of complex 2 (blue) and its reaction solutions with 1 equiv. phenyl Grignard (green), 1 equiv. 4-iodotoluene (red) and with both 1 equiv. phenyl Grignard and 1 equiv. 4-iodotoluene (orange) in THF, Figure S4: EPR Spectrum of complex 4 (blue) and its reaction solutions with 1 equiv. phenyl Grignard (green), 1 equiv. 4-iodotoluene (red) and with both 1 equiv. phenyl Grignard and 1 equiv. 4-iodotoluene (orange) in THF, Figure S5: 1H NMR (200 MHz) and 13C NMR (50 MHz) spectra and GC-MS of 1,1′-biphenyl (P1), Figure S6: 1H NMR (200 MHz) and 13C NMR (50 MHz) spectra and GC-MS of 4-methyl-1,1′-biphenyl (P2), Figure S7: 1H NMR (200 MHz) and 13C NMR (50 MHz) spectra and GC-MS of 4-methoxy-1,1′-biphenyl (P3), Figure S8: 1H NMR (200 MHz), 13C NMR (50 MHz) and 19F NMR (235 MHz) spectra and GC-MS of 4-fluoro-1,1′-biphenyl (P4), Figure S9: 1H NMR (200 MHz) and 13C NMR (50 MHz) and GC-MS of 3,5-dimethyl-1,1′-biphenyl (P5), Figure S10: 1H NMR (200 MHz) and 13C NMR (50 MHz) and GC-MS of 1,4-biphenylbenzene (P6), Figure S11: 1H NMR (200 MHz) and 13C NMR (50 MHz) and GC-MS of 2-phenylnaphthalene (P7), Table S1: Cell Parameters of complex 1 and of crystals obtained from the stoichiometric reactions solutions of 1 with 1 equiv. phenylmagnesium bromide, and of 2, 3 or 4 with both 1 equiv. phenylmagnesium bromide and 1, Table S2: Crystal data and structure refinement of [(Triphos)NiII], Data S1: Cartesian coordinates [Angstrom] of geometry-optimized models used in the calculations.
Acknowledgments
This work was supported by the Fonds der Chemischen Industrie (Liebig grant to Ulf-Peter Apfel) and through the Deutsche Forschungsgemeinschaft (Emmy Noether grant to Ulf-Peter Apfel, AP242/2-1).
Author Contributions
Linda Iffland, Anette Petuker and Ulf-Peter Apfel conceived and designed the experiments; Linda Iffland and Anette Petuker performed the experiments. All authors were involved in the analysis of the data; Maurice van Gastel performed DFT calculations and EPR spectroscopy. All authors were involved in writing the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- De Meijere, A.; Diederich, F. Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Tamao, K.; Sumitani, K.; Kumada, M. Selective carbon–carbon bond formation by cross-coupling of Grignard reagents with organic halides. Catalysis by nickel-phosphine complexes. J. Am. Chem. Soc. 1972, 94, 4374–4376. [Google Scholar] [CrossRef]
- Corriu, R.J.P.; Masse, J.P. Activation of Grignard reagents by transition-metal complexes. A new and simple synthesis of trans-stilbenes and polyphenyls. J. Chem. Soc. Chem. Commun. 1972, 144a. [Google Scholar] [CrossRef]
- Knappke, C.E.I.; von Wangelin, A.J. 35 years of palladium-catalyzed cross-coupling with Grignard reagents: How far have we come? Chem. Soc. Rev. 2011, 40, 4948. [Google Scholar] [CrossRef] [PubMed]
- Braga, A.A.C.; Ujaque, G.; Maseras, F. Mechanism of Palladium-Catalyzed Cross-Coupling Reactions. In Computational Modeling for Homogeneous and Enzymatic Catalysis: A Knowledge-Base for Designing Efficient Catalysts; Morokuma, K., Musaev, D.G., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 109–130. [Google Scholar]
- Rosen, B.M.; Quasdorf, K.W.; Wilson, D.A.; Zhang, N.; Resmerita, A.-M.; Garg, N.K.; Percec, V. Nickel-Catalyzed Cross-Couplings Involving Carbon–Oxygen Bonds. Chem. Rev. 2011, 111, 1346–1416. [Google Scholar] [CrossRef] [PubMed]
- Amatore, C.; Jutand, A. Rates and mechanism of biphenyl synthesis catalyzed by electrogenerated coordinatively unsaturated nickel complexes. Organometallics 1988, 7, 2203–2214. [Google Scholar] [CrossRef]
- Shi, S.; Meng, G.; Szostak, M. Synthesis of Biaryls through Nickel-Catalyzed Suzuki-Miyaura Coupling of Amides by Carbon–Nitrogen Bond Cleavage. Angew. Chem. Int. Ed. 2016, 55, 6959–6963. [Google Scholar] [CrossRef] [PubMed]
- Tsou, T.T.; Kochi, J.K. Mechanism of biaryl synthesis with nickel complexes. J. Am. Chem. Soc. 1979, 101, 7547–7560. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Power, P.P. Complexes of Ni(I): A “rare” oxidation state of growing importance. Chem. Soc. Rev. 2017, 46, 5347–5399. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.G.; McFarland, C.; Anderson, T.J.; Vicic, D.A. Analysis of key steps in the catalytic cross-coupling of alkyl electrophiles under Negishi-like conditions. Chem. Commun. 2005, 33, 4211–4213. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.G.; Martin, J.L.; McFarland, C.; Allen, O.R.; Hall, R.E.; Haley, A.D.; Brandon, R.J.; Konovalova, T.; Desrochers, P.J.; Pulay, P.; et al. Ligand Redox Effects in the Synthesis, Electronic Structure, and Reactivity od an Alkyl-Alkyl Cross-Coupling Catalyst. J. Am. Chem. Soc. 2006, 128, 13175–13183. [Google Scholar] [CrossRef] [PubMed]
- Phapale, V.B.; Buñuel, E.; García-Iglesias, M.; Cárdenas, D.J. Ni-Catalyzed Cascade Formation of C(sp3)–C(sp3) Bonds by Cyclization and Cross-Coupling Reactions of Iodoalkanes with Alkyl Zinc Halides. Angew. Chem. Int. Ed. 2007, 46, 8790–8795. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Nova, A.; Balcells, D.; Brudvig, G.W.; Dai, W.; Guard, L.M.; Hazari, N.; Lin, P.-H.; Pokhrel, R.; Takase, M.K. Nickel(I) Monomers and Dimers with Cyclopentadienyl and Indenyl Ligands. Chem. Eur. J. 2014, 20, 5327–5337. [Google Scholar] [CrossRef] [PubMed]
- Guard, L.M.; Mohadjer Beromi, M.; Brudvig, G.W.; Hazari, N.; Vinyard, D.J. Comparison of dppf-Supported Nickel Precatalysts for the Suzuki-Miyaura Reaction: The Observation and Activity of Nickel(I). Angew. Chem. Int. Ed. 2015, 54, 13352–13356. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, K.; Fukahori, Y.; Inatomi, T.; Tazaki, S.; Yamada, Y.; Koga, Y.; Kanegawa, S.; Nakamura, T. Monomeric Three-Coordinated N-Heterocyclic Carbene Nickel(I) Complexes: Synthesis, Structures, and Catalytic Applications in Cross-Coupling Reactions. Organometallics 2016, 35, 3281–3287. [Google Scholar] [CrossRef]
- Page, M.J.; Lu, W.Y.; Poulten, R.C.; Carter, E.; Algarra, A.G.; Kariuki, B.M.; Macgregor, S.A.; Mahon, M.F.; Cavell, K.J.; Murphy, D.M.; et al. Three-Coordinated Nickel(I) Complexes Stabilised by Six-, Seven-, and Eight-Membered Ring N-Heterocyclic Carbenes: Synthesis, EPR/DFT Studies and Catalytic Activity. Chem. Eur. J. 2013, 19, 2158–2167. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, S.; Koga, Y.; Matsumoto, T.; Matsubara, K. A new aspect of nickel-catalyzed Grignard cross-coupling reaction: Selective synthesis, structure, and catalytic behavior of T-shape three-coordinated nickel(I) chloride bearing a bulky NHC ligand. Chem. Commun. 2010, 46, 1932. [Google Scholar] [CrossRef] [PubMed]
- Nagao, S.; Matsumoto, T.; Koga, Y.; Matsubara, K. Monovalent Nickel Complex Bearing a Bulky N-Heterocyclic Carbene Catalyzes Buchwald-Hartwig Amination of Aryl Halides under Mild Conditions. Chem. Lett. 2011, 40, 1036–1038. [Google Scholar] [CrossRef]
- Zhang, K.; Conda-Sheridan, M.; Cooke, S.R.; Louie, J. N-Heterocyclic Carbene Bound Nickel(I) Complexes and Their Roles in Catalysis. Organometallics 2011, 30, 2546–2552. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, K.; Yamamoto, H.; Miyazaki, S.; Inatomi, T.; Nonaka, K.; Koga, Y.; Yamada, Y.; Veiros, L.F.; Kirchner, K. Dinuclear Systems in Efficient Nickel-Catalyzed Kumada-Tamao-Corriu Cross-Coupling of Aryl Halides. Organometallics 2017, 36, 255–265. [Google Scholar] [CrossRef]
- Marlier, E.E.; Tereniak, S.J.; Ding, K.; Mulliken, J.E.; Lu, C.C. First-Row Transition-Metal Chloride Complexes of the Wide Bite-Angle Diphosphine iPrDPDBFphos and Reactivity Studies of Monovalent Nickel. Inorg. Chem. 2011, 50, 9290–9299. [Google Scholar] [CrossRef] [PubMed]
- Petuker, A.; Merten, C.; Apfel, U.-P. Modulating Sonogashira Cross-Coupling Reactivity in Four-Coordinated Nickel Complexes by Using Geometric Control: Modulating Cross-Coupling Reactivity in Ni Complexes. Eur. J. Inorg. Chem. 2015, 2015, 2139–2144. [Google Scholar] [CrossRef]
- Petuker, A.; El-Tokhey, M.; Reback, M.L.; Mallick, B.; Apfel, U.-P. Towards Iron-Catalyzed Sonogashira Cross-Coupling Reactions. ChemistrySelect 2016, 1, 2717–2721. [Google Scholar] [CrossRef]
- Petuker, A.; Mebs, S.; Schuth, N.; Gerschel, P.; Reback, M.L.; Mallick, B.; van Gastel, M.; Haumann, M.; Apfel, U.-P. Spontaneous Si–C bond cleavage in (TriphosSi)-nickel complexes. Dalton Trans. 2017, 46, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software update: The ORCA program system, version 4.0: Software update. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, e1327. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Chen, X.-Y.; Landis, C.R.; Neese, F. All Electron Scalar relativistic Basic Sets for Third-Row Transition Metal Atoms. J. Chem. Theory Comput. 2008, 4, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Lenthe, E.; Snijders, J.G.; Baerends, E.J. The zero-order regular approximation for relativistic effects: The effect of spin-orbit coupling in closed shell molecules. J. Chem. Phys. 1996, 105, 6505–6516. [Google Scholar] [CrossRef] [Green Version]
- Van Lenthe, E.; van der Avoird, A.; Wormer, P.E.S. Density functional calculations of molecular hyperfine interactions in zero order regular approximation for relativistic effects. J. Chem. Phys. 1998, 108, 4783–4796. [Google Scholar] [CrossRef]
Scheme 1.
Literature-known monovalent nickel catalysts for the Kumada coupling [10].
Scheme 1.
Literature-known monovalent nickel catalysts for the Kumada coupling [10].
Scheme 2.
Nickel complexes [(Triphos)NiICl] (1), [(Triphos)NiIICl2] (2), [(Triphos)NiIICl](BF4) (3) and [(Triphos)NiIICl](ClO4) (4).
Scheme 2.
Nickel complexes [(Triphos)NiICl] (1), [(Triphos)NiIICl2] (2), [(Triphos)NiIICl](BF4) (3) and [(Triphos)NiIICl](ClO4) (4).

Figure 1.
UV/Vis spectra of complexes 1 (A), 2 (B), 3 (C) and 4 (D) (blue) and their reaction solutions with 1 equiv. phenylmagnesium bromide (green), 1 equiv. 4-iodotoluene (red) and with both substrates (orange) in THF.
Figure 1.
UV/Vis spectra of complexes 1 (A), 2 (B), 3 (C) and 4 (D) (blue) and their reaction solutions with 1 equiv. phenylmagnesium bromide (green), 1 equiv. 4-iodotoluene (red) and with both substrates (orange) in THF.
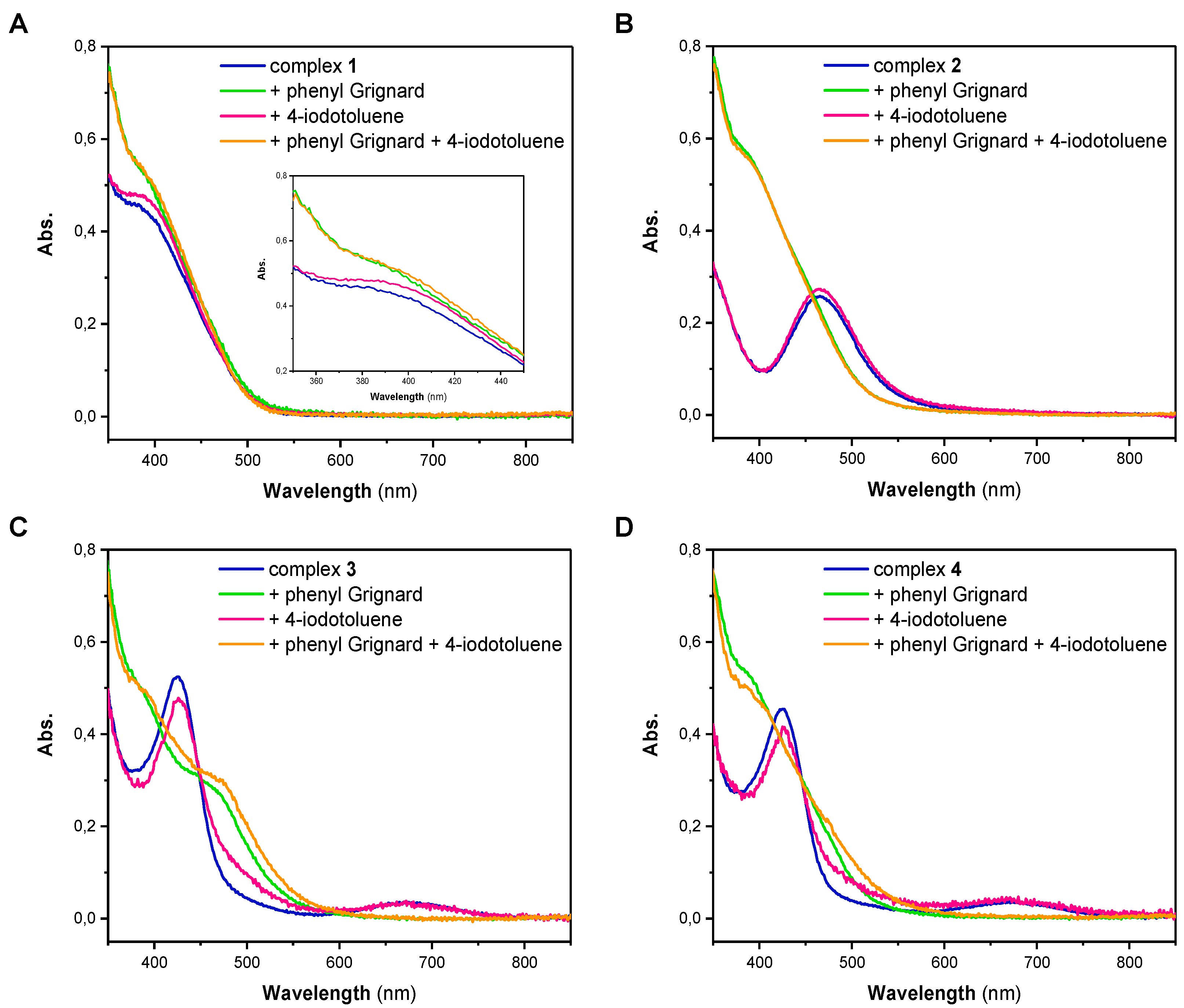
Figure 2.
Electron paramagnetic resonance (EPR) spectrum of complex 3 before (blue) and after reaction with 1 equiv. phenyl Grignard (green), 1 equiv. 4-iodotoluene (red) and with both 1 equiv. phenyl Grignard and 1 equiv. 4-iodotoluene (orange) in THF. Experimental conditions: T = 10 K, frequency 9.63 GHz, microwave power 2 mW, modulation amplitude 0.3 mT.
Figure 2.
Electron paramagnetic resonance (EPR) spectrum of complex 3 before (blue) and after reaction with 1 equiv. phenyl Grignard (green), 1 equiv. 4-iodotoluene (red) and with both 1 equiv. phenyl Grignard and 1 equiv. 4-iodotoluene (orange) in THF. Experimental conditions: T = 10 K, frequency 9.63 GHz, microwave power 2 mW, modulation amplitude 0.3 mT.
Scheme 3.
Proposed mechanism for the Ni-catalyzed Kumada cross-coupling reaction with complexes 1–4. While possible, DFT calculations suggest the formation of the NiIII intermediate shown in parentheses not to be a significant part of the reaction cycle. Additional reaction scheme for the formation of one 1,1′-biphenyl molecule starting with 1 and ending up with [(Triphos)NiII] with calculated ΔG values for each reaction step.
Scheme 3.
Proposed mechanism for the Ni-catalyzed Kumada cross-coupling reaction with complexes 1–4. While possible, DFT calculations suggest the formation of the NiIII intermediate shown in parentheses not to be a significant part of the reaction cycle. Additional reaction scheme for the formation of one 1,1′-biphenyl molecule starting with 1 and ending up with [(Triphos)NiII] with calculated ΔG values for each reaction step.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Kumada cross-coupling reaction of aryl halides with phenylmagnesium bromide catalyzed by 0.5 mol % 1 [a].
Table 1.
Kumada cross-coupling reaction of aryl halides with phenylmagnesium bromide catalyzed by 0.5 mol % 1 [a].

| Aryl Halide | Yield in (%) for X = I | Yield in (%) for X = Br | Yield in (%) for X = Cl | Yield in (%) for X = F |
|---|---|---|---|---|
 | 89 [a] | 91 [a] | 3 [a], 100 [c] | 3 [a], 44 [c] |
 | 94 [a] | 74 [a] | 0 [b] | – |
 | 86 [a] | – | 5 | – |
[a] Reaction conditions: RArI (2.8 mmol), ArMgBr (5.6 mmol), [Ni(I)] (0.5 mol %), THF (6 mL), RT, 2.5 h. Isolated yields; [b] Coupling product detected by TLC, but not isolated; [c] Reaction conditions: RArI (2.8 mmol), ArMgBr (5.6 mmol), [Ni(I)] (0.5 mol %), THF (6 mL), 60 °C, 2.5 h. Yield determined by GC–MS analysis using 4-methyl-1,1′-biphenyl as an internal standard.
Table 2.
Kumada cross-coupling reaction of aryl iodides with phenylmagnesium bromide catalyzed by 0.5 mol % 1 [a].
Table 2.
Kumada cross-coupling reaction of aryl iodides with phenylmagnesium bromide catalyzed by 0.5 mol % 1 [a].

| Entry | Product | Yield (%) [b] | Entry | Product | Yield (%) [b] | |
|---|---|---|---|---|---|---|
| 1 |  | P1 | 89 | 11 |  | 0 |
| 2 |  | P2 | 94 | 12 |  | 0 |
| 3 |  | P3 | 86 | 13 |  | 0 |
| 4 |  | P4 | 92 | 14 |  | 0 |
| 5 |  | P5 | 75 | 15 |  | 0 |
| 6 |  | P6 | 96 | 16 |  | 0 |
| 7 |  | 17 |  | 0 | ||
| 8 |  | P7 | 97 | 18 |  | 0 |
| 9 |  | 0 | 19 |  | 0 | |
| 10 |  | 0 | 20 |  | 0 | |
[a] Reaction conditions: RArI (2.8 mmol), ArMgBr (5.6 mmol), [Ni(I)] (0.5 mol %), THF (6 mL), RT, 2.5 h; [b] Isolated yields.
Table 3.
Ni(I)- and Ni(II)-catalyzed Kumada cross-coupling reaction of 4-iodotoluene with phenylmagnesium bromide.
Table 3.
Ni(I)- and Ni(II)-catalyzed Kumada cross-coupling reaction of 4-iodotoluene with phenylmagnesium bromide.

| [Ni] Complex | Catalytic Amount of [Ni] | Yield (%) |
|---|---|---|
| 1 | 1 mol % | 81 [a], 100 [b] |
| 1 | 0.5 mol % | 94 [a], 100 [b] |
| 2 | 1 mol % | 100 [b] |
| 2 | 0.5 mol % | 100 [b] |
| 3 | 1 mol % | 72 ± 2 [b] |
| 3 | 0.5 mol % | 46 ± 1 [b] |
| 4 | 1 mol % | 59 ± 1 [b] |
| 4 | 0.5 mol % | 41 ± 1 [b] |
[a] Isolated yields. [b] Yields determined by GC–MS analysis using 1,1′-biphenyl as an internal standard.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Iffland, L.; Petuker, A.; Van Gastel, M.; Apfel, U.-P. Mechanistic Implications for the Ni(I)-Catalyzed Kumada Cross-Coupling Reaction. Inorganics 2017, 5, 78. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics5040078
AMA Style
Iffland L, Petuker A, Van Gastel M, Apfel U-P. Mechanistic Implications for the Ni(I)-Catalyzed Kumada Cross-Coupling Reaction. Inorganics. 2017; 5(4):78. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics5040078
Chicago/Turabian StyleIffland, Linda, Anette Petuker, Maurice Van Gastel, and Ulf-Peter Apfel. 2017. "Mechanistic Implications for the Ni(I)-Catalyzed Kumada Cross-Coupling Reaction" Inorganics 5, no. 4: 78. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics5040078
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.