Tetraruthenium Metallamacrocycles with Potentially Coordinating Appended Functionalities


Fachbereich Chemie der Universität Konstanz, Universitätsstraße 10, D-78457 Konstanz, Germany
*
Author to whom correspondence should be addressed.
Inorganics 2018, 6(3), 73; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6030073
Submission received: 26 June 2018
/
Revised: 13 July 2018
/
Accepted: 20 July 2018
/
Published: 24 July 2018
(This article belongs to the Special Issue Organometallic Macrocycles and Their Applications)
Abstract
:We present four new tetraruthenium macrocycles built from two 1,4-divinylphenylene diruthenium and two isophthalic acid building blocks with peripheral, potentially mono- or tridentate donor functions attached to the isophthalic linkers. These macrocycles are characterized by multinuclear NMR spectroscopy, mass spectrometry and, in the case of the thioacetyl-appended complex 4, by X-ray crystallography. Cyclic and square wave voltammetry establish that the macrocycles can be oxidized in four consecutive redox steps that come as two pairs of two closely spaced one-electron waves. Spectroscopic changes observed during IR and UV/Vis/NIR spectroelectrochemical experiments (NIR = near infrared) show that the isophthalate linkers insulate the electroactive divinylphenylene diruthenium moieties against each other. The macrocycles exhibit nevertheless pronounced polyelectrochromism with highly intense absorptions in the Vis (2+/4+ states) and the NIR (2+ states) with extinction coefficients of up to >100,000 M−1·cm−1. The strong absorptivity enhancement with respect to the individual divinylphenylene diruthenium building blocks is attributed to conformational restrictions imposed by the macrocycle backbone. Moreover, the di- and tetracations of these macrocycles are paramagnetic as revealed by EPR spectroscopy.
1. Introduction
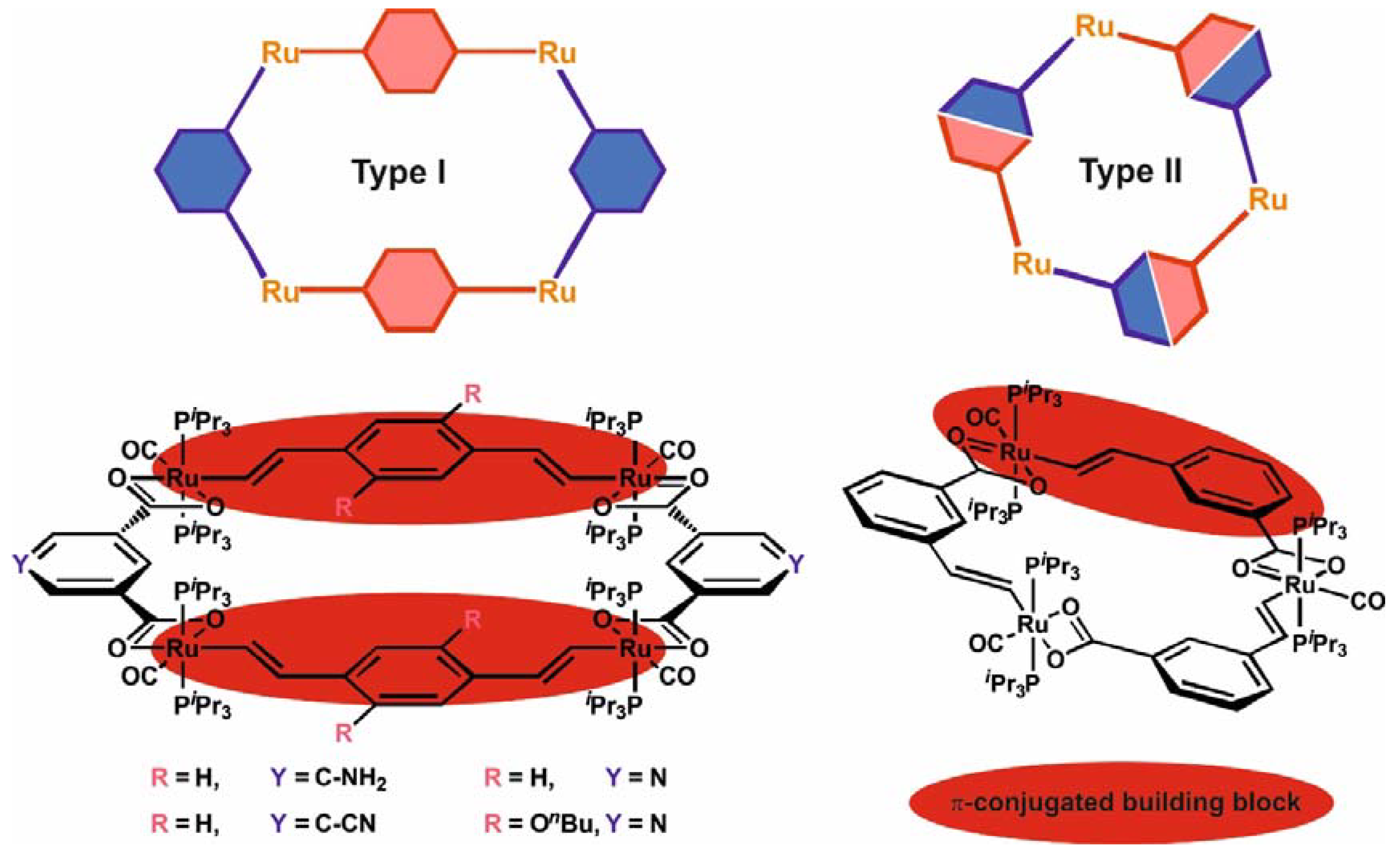
We have recently reported on some representatives of new types of metallamacrocyclic ruthenium complexes based on alkenylarylene and carboxylate linkers [1,2,3,4], which add to the rapidly growing class of redox-active metallamacrocycles [5,6,7]. Metallacycles of type I in Figure 1 are constructed from bis(alkenyl)arylene diruthenium complexes and isophthalic acid derivatives or pyridine-3,5-dicarboxylate as two different kinds of building blocks [1,2,4]. Metallacycles of type II, in contrast, are made from self-complimentary building blocks that incorporate the alkenylarylene and the carboxylate functionalities within the same bridging ligand [3,4]. A feature common to both types of macrocycles is their redox activity with one reversible one-electron oxidation per alkenyl ruthenium moiety. By peripherally appending triarylamine tags, their redox activity can be even further enhanced to the reversible loss of up to eight electrons per macrocycle [2].
Despite this unifying principle, ruthenium macrocycles of types I or II strongly differ with respect to the pattern of the individual redox waves and their properties. Thus, members of the type I family are characterized by two pairs of closely spaced one-electron voltammetric waves that are mutually split by ca. 300 mV, representing the stepwise oxidation of first the one and then the other divinylarylene diruthenium moiety to their associated radical cationic (first pair of waves) and then to the dicationic states (second pair of waves). The spatial extension of the π-conjugated metal-organic pathway renders tetraruthenium macrocycles of this architecture highly efficient polyelectrochromic dyes [8,9,10,11,12]. Another interesting property is the paramagnetism of the di- and tetraoxidized forms, which feature two or four uncoupled spins, even in the presence of 1,4-divinylphenylene linkages [1]. Tri- or tetraruthenium macrocycles of family II, however, offer three or four individually resolved, rather evenly spaced, consecutive one-electron redox waves in electrochemical experiments. Moreover, they show clear signs of electronic coupling between the individual monoruthenium constituents in their mixed-valent +/2+ or +/2+/3+ states as revealed by the presence of weak intervalence charge transfer (IVCT) transitions. They thus resemble electrically conductive molecular loops. This, however, comes at the expense of a significantly attenuated performance as polyelectrochromics, [13,14,15,16,17,18,19] due to fact that the π-conjugated chromophores are restricted to just a monoruthenium alkenylarylene building block.
One formidable challenge en route to responsive materials made from such macrocycles is to interconnect individual molecules to larger, ordered arrays [20]. Assembling individual macrocycles via coordinative or covalent bonds or other specific intermolecular interactions might ultimately provide more complex structures with anisotropic electrical or electronic properties, if the type of bonding interactions between individual macrocycles differs for different directions in space. Such a level of structural complexity would go beyond that of traditional MOFs. As a first step to such materials, we here report on four new tetraruthenium macrocycles of type I with either terpyridine- (terpy), 1,3-(dipyrid-2-yl)phenyl-(phpy2), (4-ethynylpyridine)- or 4-thioacetyl-functionalized isophthalate linkers along with their electrochemical and spectroscopic properties.
2. Results and Discussion
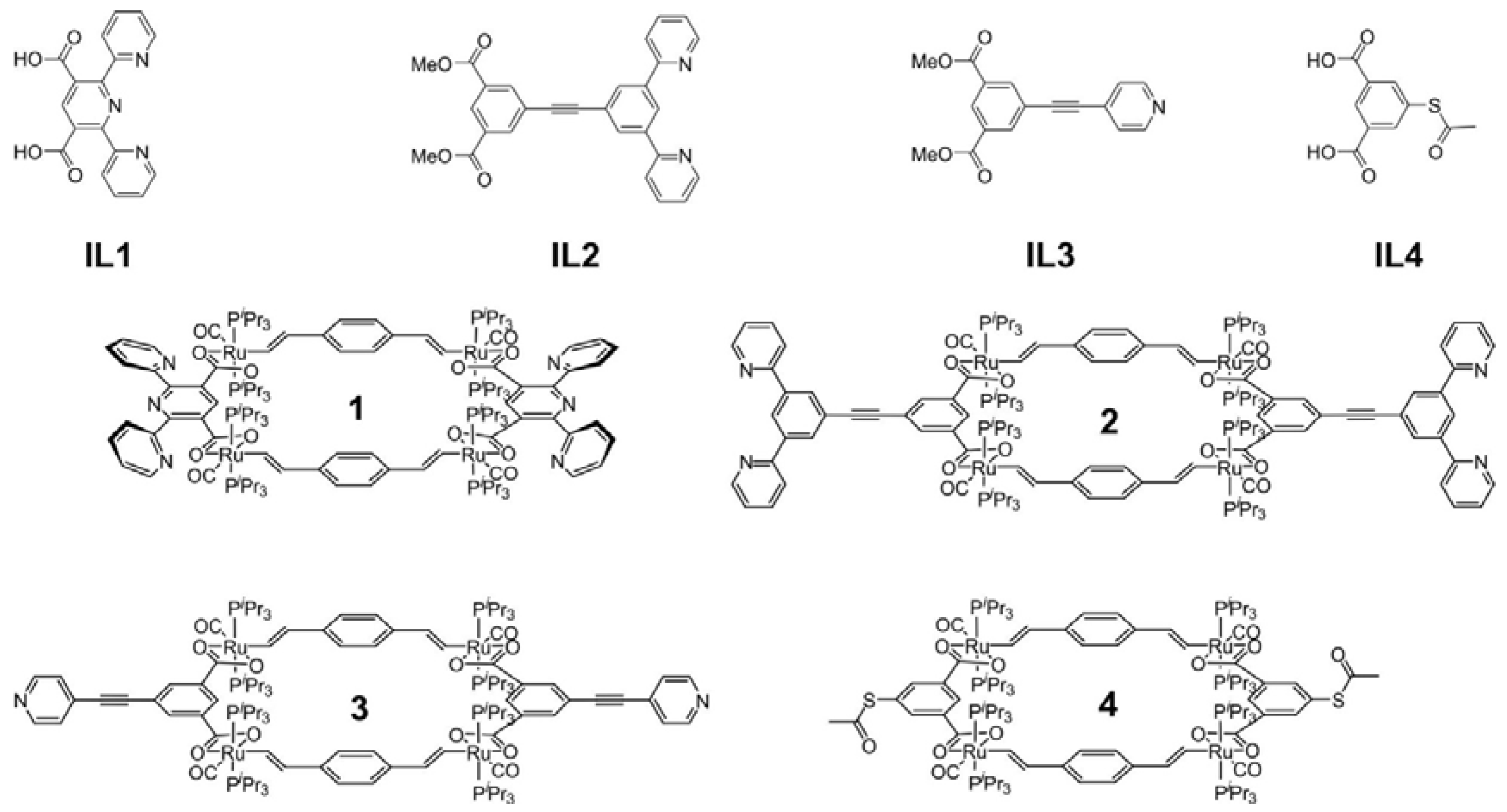
As the functionalized isophthalate linkers of this study we employed 2,6-di(pyrid-2-yl)-pyridine-3,5-dicarboxylate (IL1), the 3,5-bis(pyrid-2-yl)phenylethynyl-appended isophthalate IL2, the 5-ethynylpyridine-appended isophthalate IL3, and 5-thioacetyl-modified isophthalate IL4 (see Figure 2). IL1 was already reported in 1961 and made by condensation of picolinoylacetate with ammonia followed by oxidation of the cyclic 1,4-dihydropyridine with nitrosulphuric acid and subsequent basic hydrolysis of the formed diethyl ester [21]. In our present work we employed permanganate oxidation of 3′,5′-dimethyl-2,2′,6′,2′′-terpyridine [22] to prepare the dimethyl ester of IL1. The latter was saponified in situ before macrocycle synthesis. Linkers IL2 to IL4 are new. IL2 was made by Sonogashira coupling of 1-bromo-3,5-di(pyrid-2yl)benzene [23] with 5-ethynylisophthalic acid dimethyl ester. The latter was prepared by a Sonogashira reaction of the corresponding 5-bromo derivative with trimethylsilylacetylene (TMSA) followed by deprotection of the product with K2CO3/MeOH. Sonogashira coupling of 5-ethynylisophthalic acid dimethyl ester with the hydrochloride of 4-bromopyridine afforded linker IL3. IL4 was prepared from acetylation of the known 5-mercaptoisophthalic acid [24] with acetic acid anhydride. Details of the synthesis and the characterization of IL1 to IL4 and of new synthetic intermediates are given in the Experimental Section. NMR spectra of all new isophthalate ligands are collected as Figures S1 to S8 in the Supplementary Materials.
The four new tetraruthenium macrocycles 1–4 shown in Figure 2 were generated in yields of 46% to 87% by stirring equimolar amounts of the divinylphenylene-bridged diruthenium complex {Ru(CO)Cl(PiPr3)2}2(µ-CH=CH–C6H4–CH=CH-1,4) [8] and the respective isophthalate linkers IL1 to IL4 in a MeOH/CH2Cl2 mixture (ca. 1:1, vol:vol), subsequent filtration of the soluble materials over Celite®, and washing of the solid residue that remained after removal of the solvents with methanol, ether and pentane. Reaction times varied between 3 h and 3 days. In the latter cases, monitoring of the reaction solutions by 31P NMR spectroscopy revealed the presence of several species during intermediate stages of the reaction. These multicomponent mixtures gradually turned uniform, leaving just one principal, dissolved species besides variable quantities of insoluble, solid material at the end of the reactions. The success of these self-assembly reactions obviously rests on the reversible binding of the carboxylate linkers and a thermodynamic competitiveness of the cyclic architectures with respect to linear, polymeric structures due to enthalpy-entropy compensation [25,26,27] and the stabilities of the as-formed macrocycles [28,29,30]. Similar observations of equilibria involving reversible ligand binding en route to closed metallamacrocycles were reported on earlier occasions, including diruthenium complexes [31]. However, even closed macrocycles may undergo ligand scrambling in the presence of an excess of a ditopic ligand [30,32,33]. Gratifyingly, the angular dispositions of the coordinating functionalities at the divinylphenylene and the isophthalate linkers of ca. 160° and of ca. 120° [2,4] grant selective formation of rectangular, unstrained 2 + 2 tetraruthenium macrocyclic structures as opposed to larger cyclic arrays (vide infra).
The new macrocycles are characterized by the appropriate NMR resonances of the two different kinds of ditopic building blocks—the divinylarylene-bridged diruthenium complex and the isophthalate linker—in a 1:1 ratio, with distinct peak shifts when compared to the isolated precursors (for graphical representations of the NMR spectra see Figures S9 to S21 in the Supplementary Materials). As usual, substitution of the chlorido ligand at the ruthenium atoms by a bidentate carboxylate goes along with low-field shifts of the vinylic protons Ru–CH and Ru–CH=CH and of those at the 1,4-phenylene linker by ca. 0.6 to 0.7 ppm, 0.8 to 1.0 ppm, and 0.6 to 0.7 ppm, respectively [1,2,11,34]. Significant shifts are also observed for the protons at the isophthalate ligands, in particular for those which are poised to the central cavity. This is exemplified by macrocycle 1 where the corresponding 1H NMR resonance changes from 8.29 ppm in IL1 to 9.80 ppm, or by the 2/IL2 pair of compounds, where a shift from 8.65 ppm to 9.28 ppm is observed. Chloride substitution by bidentate carboxylate donors is indicated by low-field shifts of the COO 13C NMR resonances by ca. 10 ppm when compared to the parent isophthalic esters and, concomitant with the increase of the valence electron count at the ruthenium atom from 16 to 18, by a 9 cm−1 shift of the CO stretch of the Ru-bonded carbonyl ligands to lower energy.
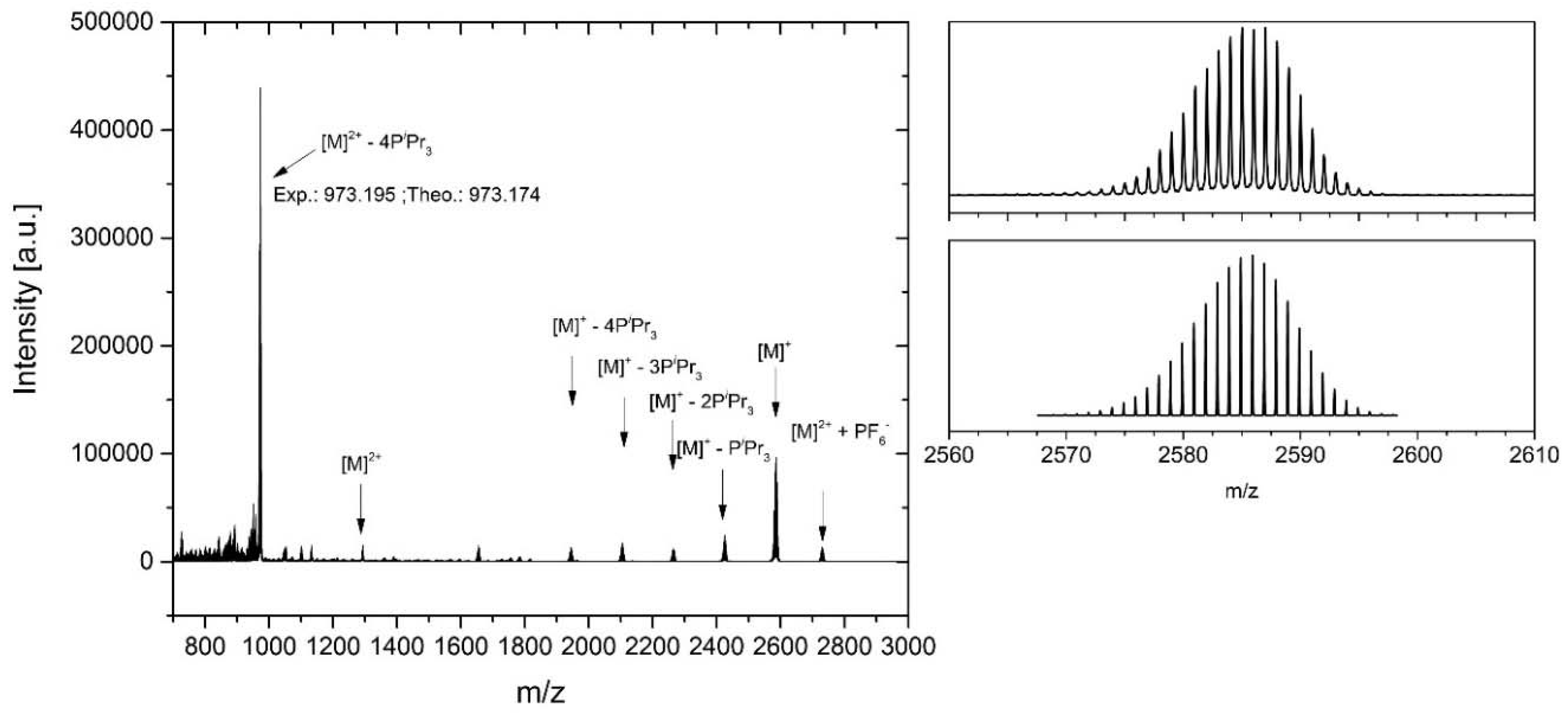
While these observations generally confirm the presence of well-defined macrocyclic structures, they are indeterminate with respect to their nuclearity. Such information can, however, be gleaned from the corresponding mass spectra. The latter show clear peaks corresponding to the mono- and dications of the intact macrocycles with series of additional peaks corresponding to the stepwise loss of up to four PiPr3 ligands. In most cases, the mass peak for the [M2+ − 4PiPr3] ion constitutes the base peak of the mass spectrum, confirming stability of even the higher oxidized, dicationic states. For preoxidized samples, mass peaks corresponding to even the trications were observed. Figure 3 depicts a representative mass spectrum of the ethynylpyridyl-appended macrocycle 3 and demonstrates the excellent match between the calculated and experimentally observed peak positions and isotope patterns. Mass spectra of the other metallamacrocycles can be retrieved from Figures S22 to S27 of the Supplementary Materials.
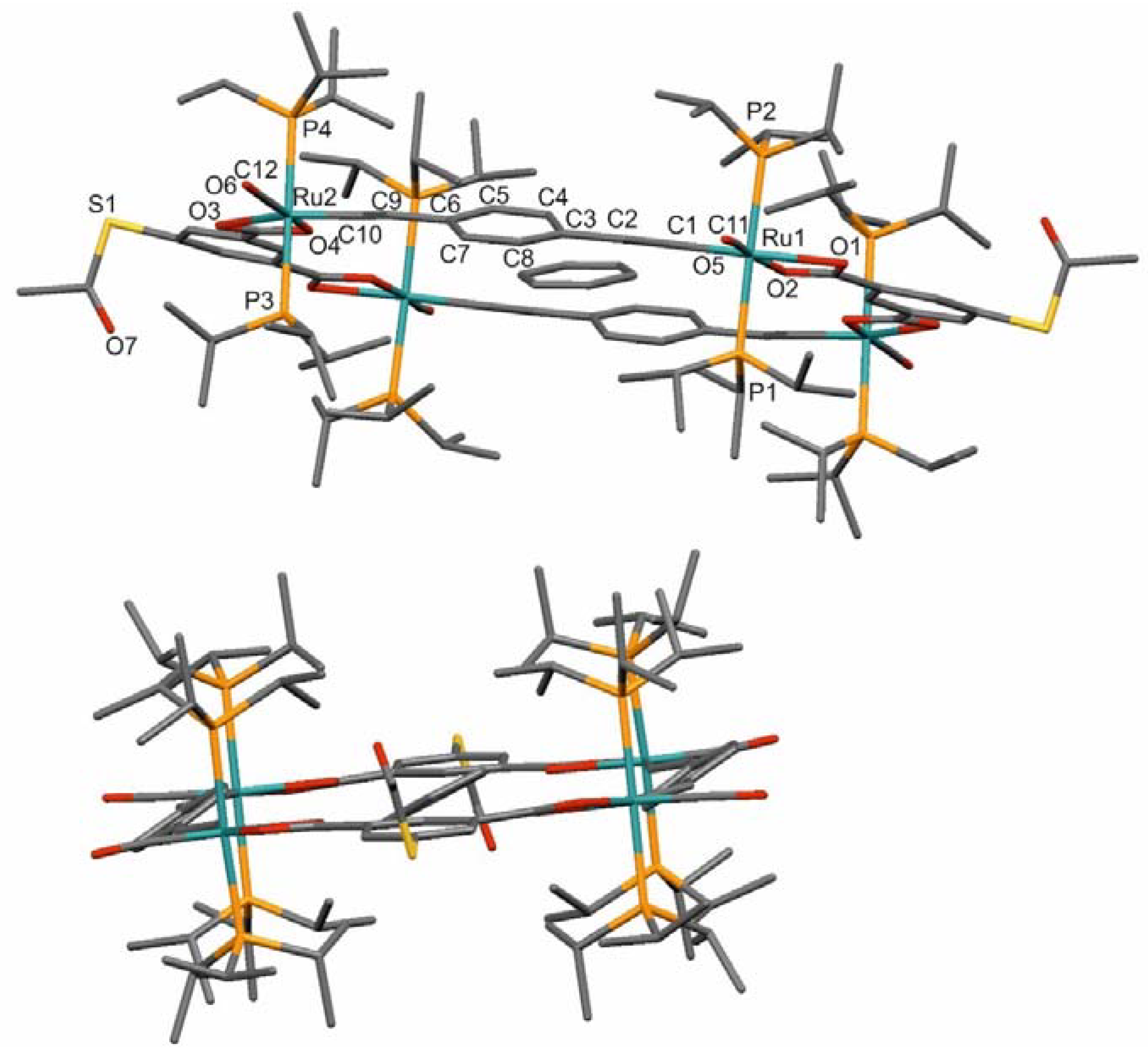
Final confirmation of the formation of tetraruthenium macrocycles was obtained by an X-ray diffraction experiment on a single crystal of macrocycle 4 grown from a solution of the complex in a benzene/MeOH mixture. Details to the data collection, the structure solution and refinement, as well as listings of the bond lengths and angles are provided as Tables S30 to S32 in the Supplementary Materials. Complex 4 crystallizes in the triclinic space group P−1 with two half macrocycles and eleven benzene solvate molecules in the unit cell. One benzene solvate molecule rests within the inner cavity of every macrocycle and, at an interplanar angle of 8.0°, is in a close to parallel orientation with the arene rings of the divinylphenylene linkers.
Figure 4 shows front and rear views of an individual molecule of 4 with the numbering of the most important atoms and with the incorporated benzene solvate molecule. The other ten solvate molecules occupy hydrophobic voids between parallel displaced macrocycle molecules (see Figure S28 of the Supplementary Materials). Individual solvate molecules do not display any notable interactions with each other or the metallamacrocycles. Thus, the shortest C–H⋅⋅⋅C distances between individual benzene molecules are 3.0 Å, which is by 0.1 Å longer than what is usually considered as a secondary bonding interaction. The only exception is a weak contact of 2.638 Å between one benzene solvate hydrogen atom and atom O1 of a carboxylate ligand. The absence of any intermolecular interactions also explains why single crystals of 4 rapidly lose their solvate molecules on removal from the mother liquor. This posed considerable difficulties in collecting a specimen for data collection. Thus, it was necessary to immediately freeze the crystal in a stream of liquid nitrogen in order to avoid a rapid loss of transparency and crystallinity. Some solvent loss was nevertheless encountered during the X-ray diffraction experiment, even at liquid nitrogen temperature. As is shown in Figure S29 of the Supplementary Materials, individual macrocycles weakly associate along the c axis of the unit cell by two pairs of hydrogen bonding interactions of 2.578 Å and 2.636 Å between atom O7 at the thioacetyl moiety and methyl protons at the PiPr3 ligands.
While the rather poor quality of the data set in terms of intensity-to-noise ratio does not allow for a detailed discussion of the bond parameters, a qualitative discussion and some statements on the macrocycle conformation are nevertheless possible. Of note are the large differences in the Ru–O bond lengths at the bidentate carboxylate linkers. Thus, the Ru–O bonds to the carboxylate O donors trans to the divinylphenylene ligands (Ru1–O1 = 2.316(6) Å, Ru2–O3 = 2.287(6) Å) are by 0.13 or 0.08 Å longer than those to the oxygen atoms trans to the carbonyl ligand (Ru1–O2 = 2.183(6) Å, Ru2–O4 = 2.205(6) Å). Likewise, the trans-angles C1–Ru1–O1 and C10–Ru2–O3 of 156.3(3)° or 160.5(3)° show considerably larger deviations from ideality than the angles C11–Ru1–O2 and C12–Ru2–O4 of 172.2(4)° and 168.9(4)° involving the C atoms of the carbonyl ligands. These differences, however, do not seem to be imposed by conformational constraints of macrocycle formation, since very similar observations were also made for similar mono- and diruthenium complexes with terminal benzoate ligands, e.g., Ru–O = 2.305(2) Å and 2.200(2) Å, O–Ru–Cst = 157.83(10)°, O–Ru–CCO = 170.39(11)° for the closely related complex Ru(CO)(PiPr3)2(CH=CH–C6H5)(κO,O′-OOCC6H4-4SC(=O)CH3) [11]. The macrocycles therefore seem to be largely devoid of ring strain. This is also indicated by an only minute twisting of the P–Ru vectors, as indicated by torsional angles P–Ru⋅⋅⋅Ru–P of 0.2° or 3.0° along the divinylphenylene, and of 4.8° along the isophthalate linkers. The phenylene rings at the divinylphenylene linkers are rotated by 27.9° and 26.4° out of the coordination planes defined by the Ru atoms and the equatorial ligands. As was found for other tetraruthenium macrocycles of the same general architecture, the vinyl linkers at the Ru atoms are rotated outward from the interior cavity [2,4]. This conformation generates the most regular cavity shape, and was also found to be the most stable one by quantum chemical calculations [1]. Cavity dimensions in 4 are 12.7 Å × 8.3 Å as measured between opposite hydrogen atoms, which is very similar to the values obtained for other structurally characterized tetraruthenium macrocycles with isophthalate linkers [2]. Together with the rotation of the phenyl rings, the outward rotation at the phenylene linkers generates the necessary space within the central cavity to incorporate one benzene solvate molecule.
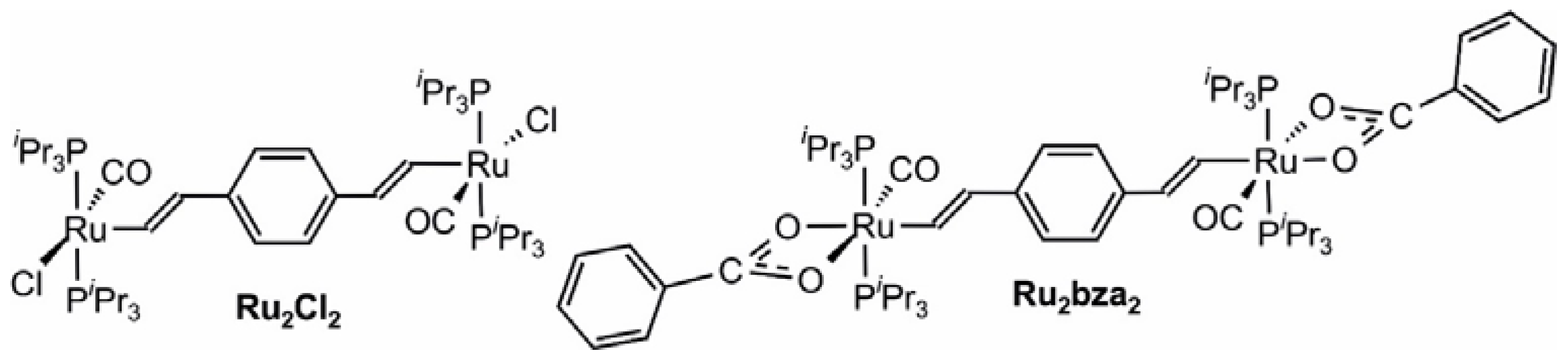
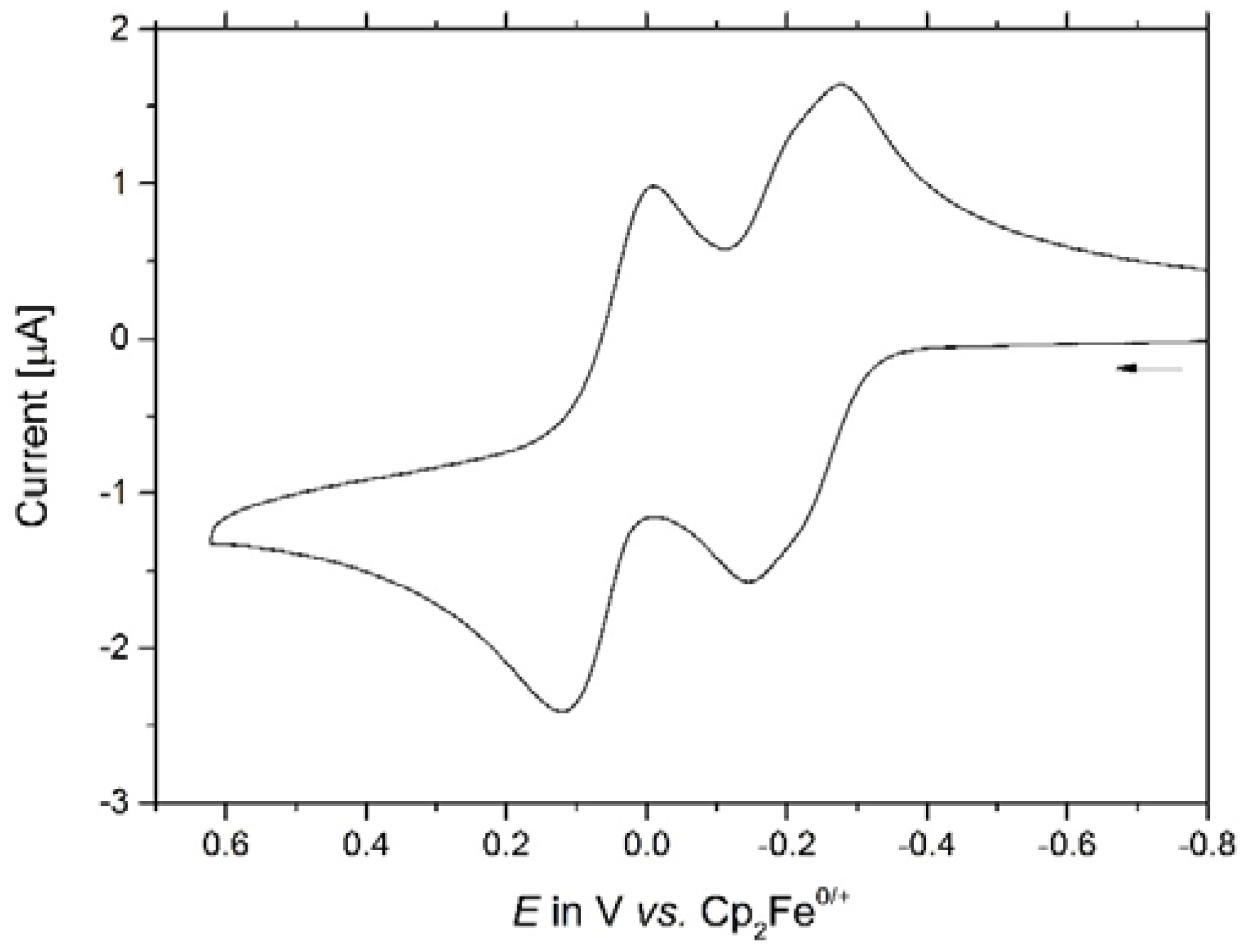
The redox properties of macrocycles 1–4 were explored by cyclic and square wave voltammetric measurements in 0.1 M NBu4PF6/CH2Cl2 solutions and, in the case of 1 and 4, also in the NBu4+B{C6H3(CF3)2-3,5}4− supporting electrolyte. Table 1 summarizes the relevant data from electrochemical experiments on the present complexes 1–4 and compares them to those for the dinuclear chloro-substituted precursor (PiPr3)2Cl(CO)Ru–CH=CH–C6H4–CH=CH–Ru(CO)Cl(PiPr3)2 (Ru2Cl2) [1] and its bis(benzoato) derivative (PhCOO-κO,O′)(PiPr3)2(CO)Ru–CH=CH–C6H4–CH=CH–Ru(CO)(PiPr3)2–(κO,O′-OOCPh) (Ru2bza2 [11]; for their structures see Figure 5). All macrocycles are oxidized in two consecutive redox processes with potential splittings of 195 mV to 248 mV. Figure 6 shows a representative voltammogram of macrocycle 3; those of the other complexes can be found as Figures S30 to S34 of the Supplementary Materials. Each of these processes involves two electrons in total and corresponds to the stepwise loss of one electron from every conjugated divinylphenylene diruthenium side. Some small splitting ∆E1/2 between the individual one-electron waves is evident for the first composite redox process constituted by the 0/+ and +/2+ redox couples, in particular in square wave experiments. The latter also made it possible to derive individual half-wave potentials E1/2 for the individual processes by digital simulation of the overall peak shape (see Figures S35 to S40 of the Supplementary Materials). No such splitting is, however, seen for the second composite wave, corresponding to the overall 2+/4+ redox event. All oxidations involve the π-conjugated Ru–CH=CH–C6H4–CH=CH–Ru sides, which are largely identical for every macrocycle and only vary by the substituents at the isophthalate linker. Since the isophthalate linkers essentially act as insulators between the conjugated sides, they only exert an inductive influence. This also explains that variations of E1/2 values for equivalent redox processes between the individual macrocycles are rather small. Changing the anion of the supporting electrolyte from PF6− to the even more weakly ion pairing B{C6H3(CF3)2-3,5}4− anion leaves the splitting between the first and second one-electron steps unaltered (Table 1), while increasing the half-wave potential differences between the second and third oxidations (i.e., for the second charging of a conjugated side) by ca. 100 mV. Some further minor splitting between the 2+/3+ and the 3+/4+ waves is indicated by a broadening of the corresponding CV wave (see Figures S31 and S34 of the Supplementary Materials) and the square wave peaks. Digital simulation of the square wave voltammograms now required four separate peaks (Figures S36 and S40 of the Supplementary Materials), and the corresponding potentials are also provided in Table 1. It becomes evident that increasing the electron count at the ruthenium atoms from 16 to 18 does not only influence the energies of the υ(CO) stretching vibrations of the carbonyl ligands but also shifts the redox potentials cathodically by ca. 150 to 180 mV for the first, and by 90 to 130 mV for the second oxidation of an individual {Ru}–CH=CH–C6H4–CH=CH–{Ru} entity.
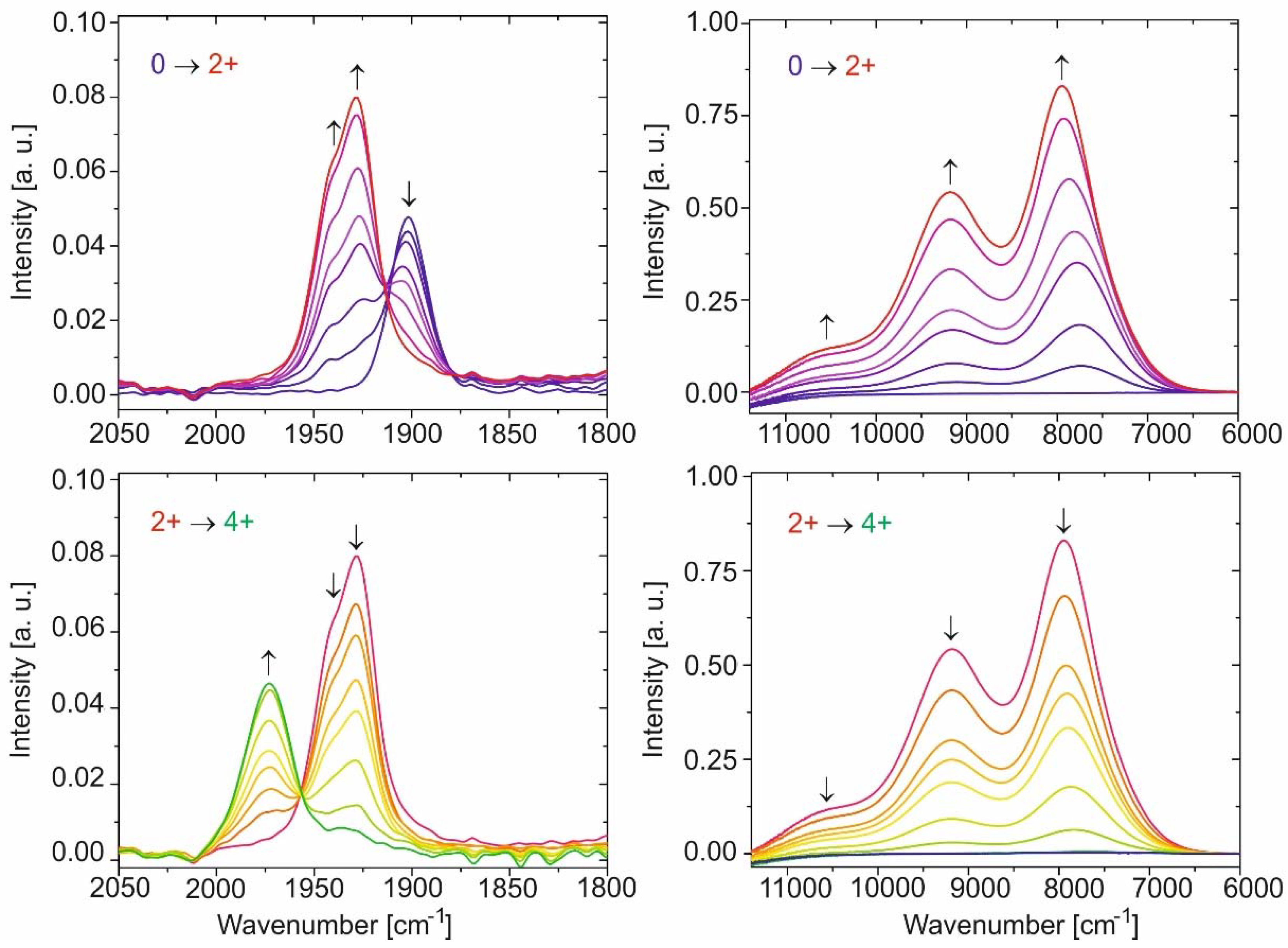
Previous studies on similar 2 + 2 tetraruthenium macrocycles assembled from divinylphenylene and isophthalate building blocks had indicated that the small splitting of the 0/+ and +/2+ redox couples, which correspond to the stepwise charging of first the one and then the other divinylphenylene diruthenium sides of a macrocycle, are entirely due to electrostatic and inductive effects, with no indication for through-space or through-bond electronic interactions between them. This also holds here, as is indicated by the results of our IR and UV/Vis/NIR spectroelectrochemical experiments. Figure 7 provides a graphical account of the spectroscopic changes for the Ru(CO) stretching vibrations and in the near infrared (NIR) for macrocycle 1; the results for the other macrocycles are included as Figures S41 to S52 in the Supplementary Materials. The corresponding data are summarized in Table 2.
On gradual oxidation of the macrocycles to their dications, we observe the expected blue shift of the Ru(CO) bands and the formation of a pattern of a weaker band at a higher energy of 1944 cm−1 and a more intense, main feature at a lower energy of 1927 or 1928 cm−1. This behavior is identical to that of linear divinylarylene-bridged diruthenium complexes [1,8,11,12]. The initially puzzling observation of two Ru(CO) bands (suggesting that the two Ru(CO) sites are electronically inequivalent) despite complete charge delocalization as indicated by detailed quantum chemical calculations and EPR studies was finally traced to the lifting of the degeneracy between the symmetric and antisymmetric combinations of the Ru(CO) stretches [1,35]. Oxidation to the dications is also accompanied by the growth of highly intense, structured and sharp NIR bands at ca. 10,500 cm−1, 9200 cm−1, and 8000 cm−1 (ca. 950 nm, 1085 nm and 1250 nm), with half-widths of the order of 800 cm−1, 1100 cm−1, and 1000–1430 cm−1. These bands correspond to the electronic β-HOSO → β-LUSO excitation within the open-shell divinylphenylene–diruthenium chromophore(s). No clear distinction between the 0/+ and +/2+ processes was evident in any of these experiments. This is, however, due to the absence of any specific absorption band of the monocations of these macrocycles with one monooxidized and one neutral divinylphenylene diruthenium side, and not to an inherent failure of our optically transparent thin layer electrolysis (OTTLE) cell to resolve two close-lying one-electron processes. In this respect we note that similar experiments on the cis- and trans-isomers of bis(styryl)ethene-bridged diruthenium complexes {Ru}–CH=CH–C6H4–CH=CH–C6H4–CH=CH–{Ru} had clearly identified the intermediate radical cations despite similar or even smaller half-wave potential splittings (74 mV to 49 mV) than in the present cases [10].
Simple thermodynamic considerations indicate that half-wave potential splittings of 45 mV to 82 mV correspond to equilibrium constants Kc of 6 to 24 for the comproportionation reaction according to Equation (1), where MC represents an individual macrocycle molecule.
MC2+ + MC ⇄ 2 MC+
This in turn means that, after stoichiometric release of one electron per macrocycle (which point must be passed in every experiment), the composition of the solution varies between 55% of the monocation and 22.5% of each the dicationic and neutral forms (Kc = 6), and 71% of the monocation and 14.5% of each the dicationic and neutral forms (Kc = 24). Thus, in every instance, the radical cation must be formed and is even the dominant species at intermediate stages of the spectroelectrochemical experiment. The monocations thus contain no specific Ru(CO) and electronic NIR bands apart from those of the isolated divinylphenylene diruthenium chromophores in their respective oxidation states. Hence, the (hypothetical) spectra of the pure monocations would essentially be indistinguishable from those of a 1:1 mixture of the dicationic and the neutral macrocycles. This in turn indicates the absence of any electronic interaction through the isophthalate linkers (i.e., through bond) or through space.
On close inspection, however, we note a gradual blue shift of the most intense and most red-shifted NIR band on continuous oxidation. This is likely due to a slight change of the electron-donating capabilities of the isophthalate linkers, which are bound to one oxidized and one reduced divinylphenylene diruthenium entity at initial stages of the electrolysis, but to two oxidized ones near the point of full conversion to the dications. The electronic properties of the coligands at ruthenium will affect the energies of the frontier molecular orbitals β-HOSO and β-LUSO that are involved in the corresponding electronic transition and their energy differences, which explains the slight positional changes from the initial to the later stages of the IR/NIR spectroelectrochemical experiment. These changes in inductive effects relate to the contribution of the ∆Gind term to the free enthalpy change associated with two consecutive redox processes and will therefore also contribute to the observed half-wave potential splittings [36,37,38,39,40].
Oxidation of the dications to the tetracations is accompanied by a further blue shift of the Ru(CO) bands along with an intensity decrease. This is particularly true for the symmetric combination of the individual Ru(CO) stretches, such that only one Ru(CO) band remains. Likewise, the NIR absorptions of the open-shell divinylphenylene diruthenium chromophores bleach completely (see Figure 7 and Figures S44, S48 and S52 of the Supplementary Materials). Again, the intermittently formed trications have no specific spectroscopic signatures that would distinguish them from the dications (the remaining singly oxidized divinylphenylene diruthenium side) or the tetracations (the doubly oxidized divinylphenylene diruthenium side).
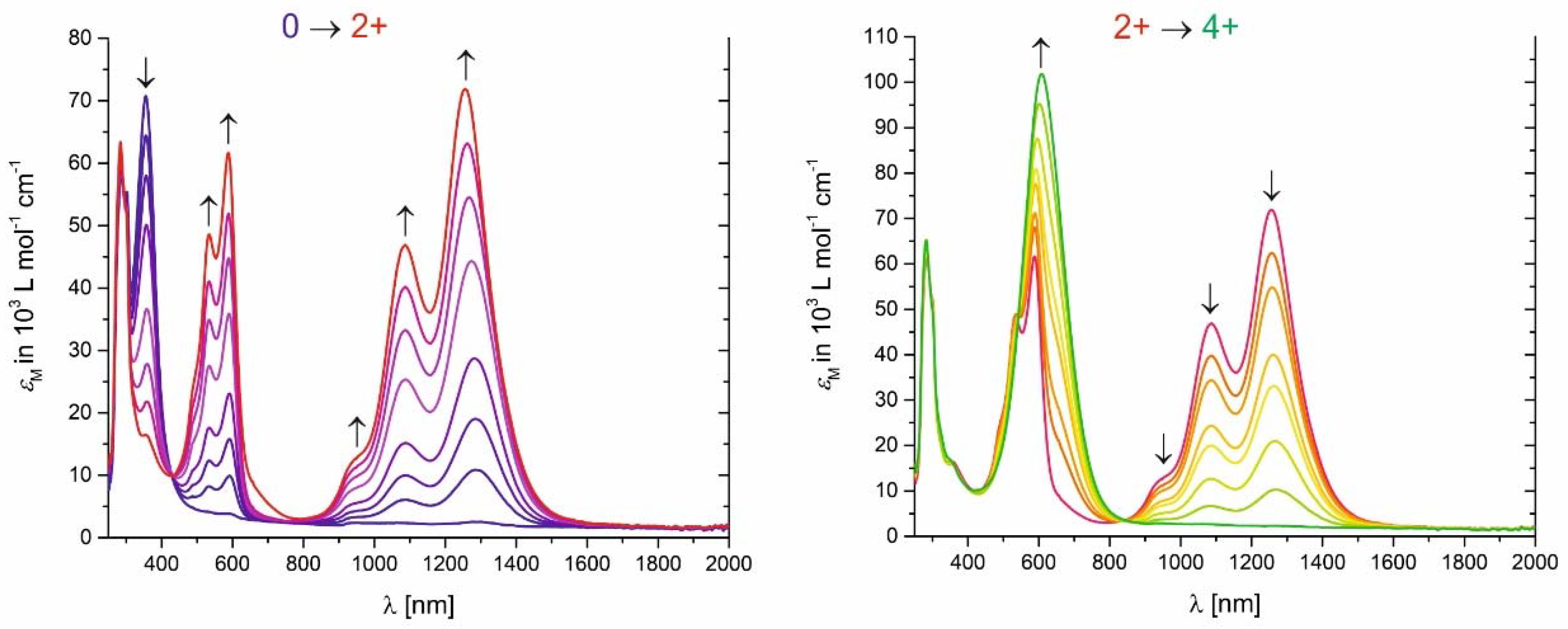
Changes in the electronic spectra of the macrocycles are exemplified for complex 3 in Figure 8. Representative spectra for all other macrocycles are displayed in Figures S53 to S58 of the Supplementary Materials while pertinent data are collected in Table 2. Visual impressions of 200 µM solutions of macrocycle 1 in its neutral, dicationic and tetracationic states are displayed in Figure 9. On stepwise oxidation, the faint yellow hue of the neutral macrocycles gives way to a deep purple coloration of the dications. The latter is due to structured prominent absorptions that cover the range of 500–600 nm with extinction coefficients larger than 6 × 104 M−1·cm−1. Concomitantly, the equally intense NIR features that were already observed in the IR/NIR experiments appear. Both kinds of absorptions can be described as π → π* type transitions within the conjugated metal–organic chromophores [1,4]. On further oxidation to the associated tetracations, the colour changes to deep royal blue or blue-green. Electronic spectra indicate a red shift and broadening of the prominent Vis absorptions together with a loss of the fine structure of the absorption bands and a further gain in absorptivity to ultimately reach extinction coefficients of >1 × 105 M−1·cm−1. In line with our observations from the IR/NIR experiments, oxidation of the di- to the tetracations is accompanied by a complete bleach of the NIR bands.
While the electronic bands of the di- and tetracations of the present macrocycles are, in terms of their position and general appearance, very similar to those of the radical cations and dications of analogous divinylphenylene-bridged diruthenium complexes like Ru2Cl2 or Ru2bza2 (see Figure 5), their extinction coefficients clearly surpass those of their open, linear analogues by a factor of >2 (see Table 2; a value of 2 would be expected for the effect of simply doubling the number of the chromophores). This is probably due to restricted conformational freedom of the individual chromophores once they are embedded into rigidified macrocyclic structures. Their linear diruthenium analogues can exist as mixtures of cisoid and transoid conformers with respect to the orientation of the Ru–CO vectors and may also exhibit larger torsions around the bonds connecting the vinyl groups to the ruthenium atoms or the phenyl rings. These observations qualify the present tetraruthenium macrocycles as particularly powerful polyelectrochromic dyes for the Vis and the NIR.
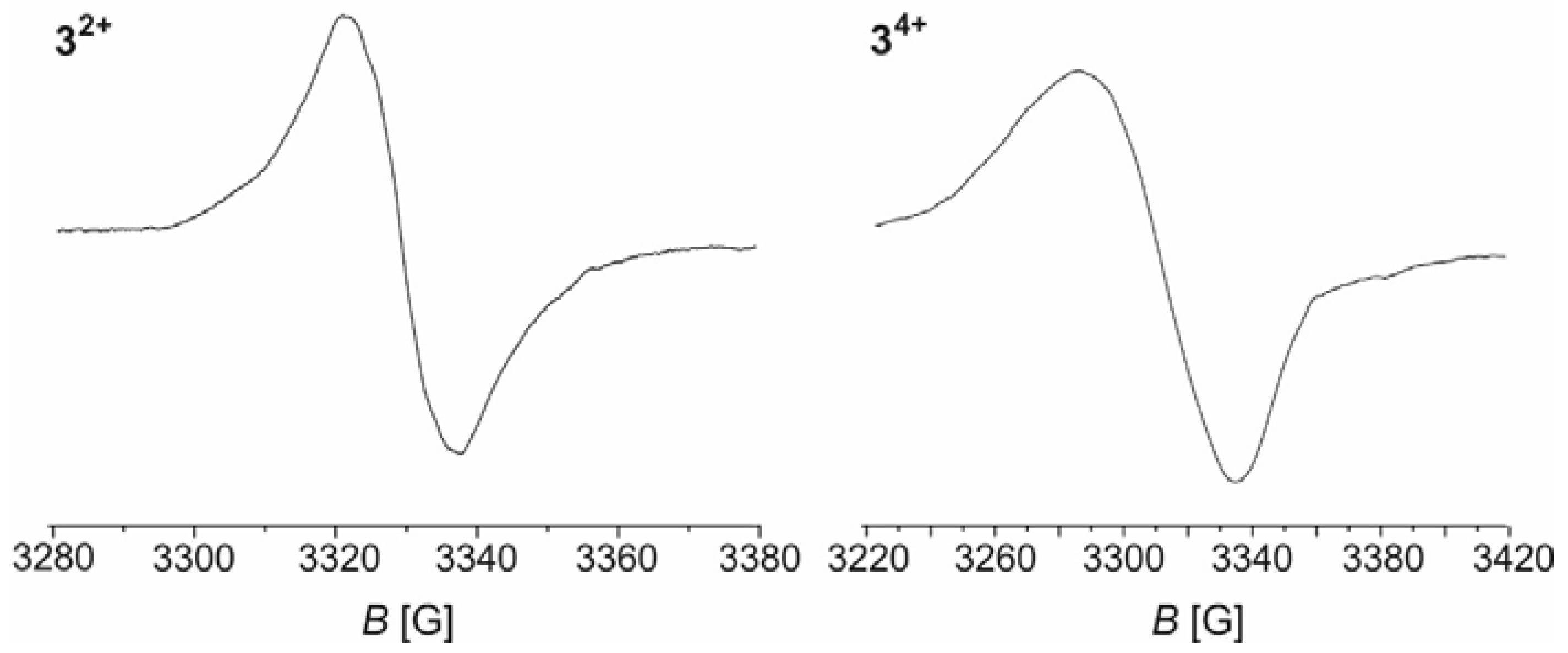
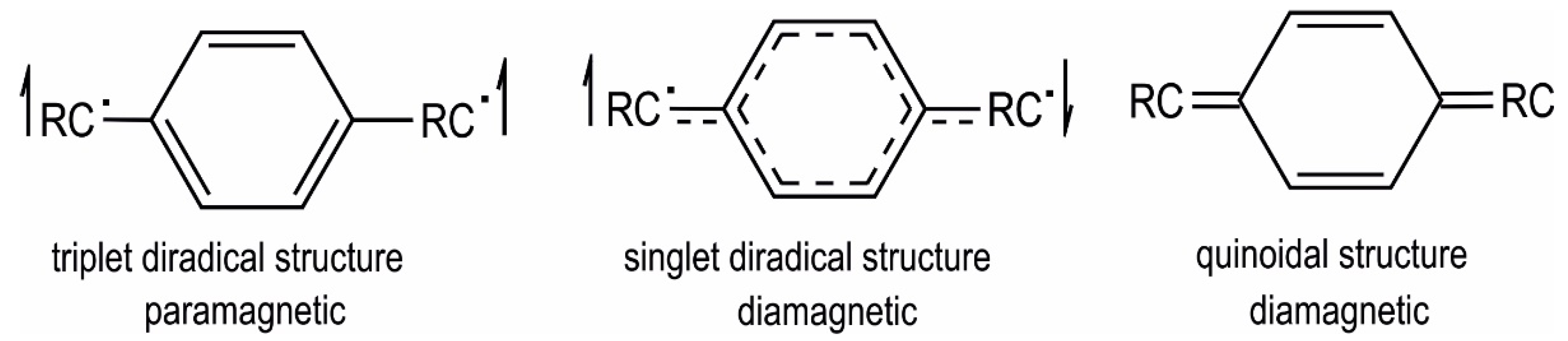
Samples of the di- and tetracations of macrocycles 1–4 for EPR investigations were prepared by selective oxidation of the neutral precursors with ferrocenium (E1/2 = 0 mV) or acetylferrocenium (E1/2 = 270 mV) hexafluorophosphate in CH2Cl2 solution [41]. Both kinds of species are EPR active and provide broadened isotropic signals with no resolved hyperfine splittings in fluid solution at r.t. and in frozen solutions. g values are close to 2.02 for the dications and slightly higher, ca. 2.03, for the tetracations (see Table 3). We also note that, for similar concentration levels, signal intensities of the tetracations are appreciably smaller than those for the dications. Figure 10 displays representative EPR spectra of 32+ and 34+ at r.t.; additional spectra at T = 123 K and those for the other complexes can be found as Figures S59 to S70 of the Supplementary Materials. In no case could a half-field signal corresponding to a ∆ms ± 2 transition be observed. This indicates that (i) every individual spin of the dications is delocalized over one divinylphenylene diruthenium side with major contributions of the π-conjugated organic linker and does not interact with the other over the isophthalate linkers to a detectable extent; and (ii) the two spins at an individual divinylphenylene diruthenium side in the tetracationic state also act independently from each other and do not couple antiferromagnetically, although the para-phenylene linkage normally supports such coupling [42,43,44,45,46]. This kind of behaviour was also observed for the dication of the similar linear diruthenium complex (PiPr3)2Cl(CO)Ru–C6H2(2,5-OBu)2–Ru(CO)Cl(PiPr3)2 and studied in detail by variable-temperature EPR spectroscopy down to 4 K [1]. Particularly well represented among the few examples of paramagnetic compounds with two spin centers interconnected by para-phenylene linkers are those with two Ar2N•+-aminiumyl-type paramagnetic centers interlinked by long π-conjugated spacers with para-phenylene motifs [47,48,49]. Quite interestingly, dicationic complexes [(PiPr3)2Cl(CO)Ru–C6H4–N(C6H4R-4)2]2+ (R = CHO, COCH3, COOMe, Me, OMe), which combine both motifs in one compound, show the same kind of behaviour [50]. Common to all these systems is that the ability of the terminal entities to accommodate the unpaired spin density allows the arylene linkers to retain their aromatic Clar’s sextets rather than sacrificing them in favour of quinoidal structures. The latter are necessary prerequisites for achieving antiferromagnetic coupling (see Figure 11). In the absence of appreciable magnetic interactions between the terminal spin-bearing sites the paramagnetic triplet and the diamagnetic singlet diradical structures will coexist in solution.
Initial attempts to utilize functionalized macrocycles 1–4 in a complexes-as-ligands approach [51,52] for further metal coordination were frustrated by their rather poor solubilities and their sensitivity towards even mildly Lewis-acidic and oxophilic cations like Zn2+ or Cu2+. Thus, addition of zinc(II) or copper(II) salts to complexes 1 and 2 led to rapid decomposition as indicated by a colour change to brown, the deposition of brownish solids, and the loss of the Ru(CO) stretches in the solution phase and these solid deposits. We assume that these cations compete with the Ru centers for the carboxylate linkers, which may ultimately lead to the opening of the macrocycles. Further attempts to engage macrocycle 2 in typical cyclometalation reactions, e.g., with PtCl2(dmso)2, were thwarted by their insufficient thermal stability to withstand the typical reaction conditions that are required to induce ligand substitution of such Pt complexes. Structural models of macrocycle 1 also indicate that the outer pyridyl rings may be spatially too close to the isophthalate carboxylate functionalities to allow for the proper orientation for providing a tridentate binding pocket. We note, however, that macrocycle 4, due to its easily deprotected Au-binding thiolate functions, has the potential to bridge Au nanogap electrodes for measuring molecular conductivities in an STM-based setup. Work along these lines is presently being pursued, and the results will be reported on a separate occasion.
3. Materials, Methods, Syntheses and Characterization Data
3.1. Materials and General Methods
All manipulations were carried out under a nitrogen atmosphere. Solvents were dried and distilled prior to use. All reagents from commercial sources were used without further purification. 1H NMR (400 MHz), 13C{1H} NMR (101 MHz) and 31P{1H}-NMR (162 MHz) spectra were measured on a Bruker AvanceIII 400 spectrometer (Bruker, Billerica, MA, USA), and 1H-NMR (600 MHz) and 13C{1H} NMR (151 MHz) spectra on a Bruker AvanceIII 600 spectrometer (Bruker, Billerica, MA, USA). {Ru(CO)Cl(PiPr3)2}2(1,4-(CH=CH)2–C6H4) [8], 3′,5′-dimethyl-2,2′:6′,2′′-terpyridine [22] and 5-mercaptoisophthalic acid [24] were prepared according to literature methods. ESI-MS data was acquired on a Bruker micrOTOF II mass spectrometer (Bruker, Billerica, MA, USA) from partially oxidized analyte/CHCl3 solutions employing [Cp2Fe]+PF6− as the oxidant. Combustion analysis was conducted with an Elementar vario MICRO cube CHN-analyzer from Heraeus (Heraeus, Hanau, Germany).
X-ray diffraction analysis of a single crystal of macrocycle 4 was performed at 100 K on a STOE IPDS-II diffractometer (STOE, Darmstadt, Germany) equipped with a graphite-monochromated radiation source (λ = 0.71073 Å) and an image plate detection system. A crystal mounted on a fine glass fiber with silicon grease was employed. If not indicated otherwise, the selection, integration, and averaging procedure of the measured reflection intensities, the determination of the unit cell dimensions and a least-squares fit of the 2θ values as well as data reduction, LP-correction and space group determination were performed using the X-Area software package delivered with the diffractometer. A semiempirical absorption correction was performed [53]. All structures were solved by the heavy-atom methods (SHELXS-97, [54] SHELXS-2014, [55] and OLEX2 [56]). Structure solutions were completed with difference Fourier syntheses and full-matrix last-squares refinements using SHELXL-97, [54] SHELXS-2014, [55] and OLEX2 [56] minimizing ω(Fo2 − Fc2)2. The weighted R factor (wR2) and the goodness of fit GOF are based on F2. All non-hydrogen atoms were refined with anisotropic displacement parameters, while hydrogen atoms were treated in a riding model. Molecular structures in this work are plotted with the Mercury program [57].
The crystallographic data file for macrocycle 4 has been deposited at the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, [Fax: + 44 1223/336-033; E-mail [email protected].] under the deposition number CCDC 1846386 and can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html.
3.2. Electrochemical and Spectroelectrochemical Measurements
All electrochemical experiments were performed in a home-built cylindrical vacuum-tight one-compartment cell equipped with a spiral-shaped Pt-wire and an Ag-wire as the counter and reference electrodes. The electrodes are sealed into glass capillaries that connect via Quickfit screws at opposite sides of the cell. A platinum electrode is used as the working electrode and is attached from the top port via a Teflon screw cap with a suitable fitting. Before measurements, the working electrode was polished with 1 μm and then 0.25 μm diamond pastes (Buehler-Wirtz, Lake Bluff, IL, USA). Measurements were carried out in argon-saturated solutions containing CH2Cl2 and nBu4N+PF6− or nBu4N+B{C6H3(CF3)2-3,5}4− as the supporting electrolyte. Referencing was done with addition of cobaltocenium hexafluorophosphate as an internal standard. Representative sets of scans were repeated with the added standard. The final referencing was done against the ferrocene/ferrocenium (Cp2Fe0/+) redox couple with E1/2(Cp2Co+/0) = −1330 mV vs. Cp2Fe0/+. Electrochemical data were acquired BASi potentiostat (Bioanalytical Systems, West Lafayette, IN, USA).
The OTTLE cell was home-built and comprises of a Pt-mesh working and counter electrode and a thin Ag/AgCl wire as a pseudoreference electrode sandwiched between the CaF2 windows of a conventional liquid IR cell. Its design is based on that of Hartl et al. [58]. The working electrode is positioned in the beam the spectrometer. For the spectroelectrochemical experiments a Wenking POS2 potentiostat (Bank Elektronik-Intelligent Controls GmbH, Pohlheim, Germany) was used. FT-IR spectra were recorded using a Bruker Tensor II FT-IR spectrometer (Bruker, Billerica, MA, USA). UV/Vis/NIR spectra were obtained on a TIDAS fiberoptic diode array spectrometer (combined MCS UV/NIR and PGS NIR instrumentation) from j&m Analytik AG, Essingen, Germany. EPR spectra were obtained using an X-Band tabletop spectrometer (MiniScope MS 400) build by Magnettech GmbH, Berlin, Germany. All measurements were performed at room temperature and at −150 °C. Macrocycles were oxidized chemically employing ferrocenium hexafluorophosphate or acetylferrocenium hexafluoroantimonate.
3.3. Syntheses and Characterization
3.3.1. Syntheses and Characterization of 2,6-Di(pyrid-2-yl)-pyridine-3,5-dicarboxylic acid dimethyl ester (IL1)

3′,5′-Dimethyl-[2,2′,6′,2′′]terpyridine (738 mg, 2.83 mmol) and KMnO4 (2.23 g, 14.13 mmol) were suspended in 200 mL of water and heated to reflux for 24 h. Upon heating, the purple colour of the suspension slowly faded. Subsequent filtration and washing with 40 mL of water and 80 of MeOH followed by evaporation of the solvents gave the crude oxidized product. The solid was dissolved in methanolic sulfuric acid (200 mL, 4% vol.) and heated to reflux for 24 h. After cooling to room temperature, aqueous NaHCO3 was added until the pH of the solution reached 7. Subsequent evaporation of the solvents and extraction of the residue with 140 mL of CHCl3/water, separation and drying of the organic phase over Na2SO4 and solvent removal gave the crude product. Purification on silica gel (ethyl acetate:petroleum benzene 3:2) yielded a colorless powder. Yield 310 mg, 31%.
1H NMR(400 MHz, CDCl3): δ (ppm) = 8.63 (ddd, 3JHH = 4.8 Hz, 4JHH = 1.8 Hz, 5JHH = 0.9 Hz, 2H, H9), 8.29 (ddd, 3JHH = 7.9 Hz, 4JHH = 1.0, 5JHH = 0.9 Hz, H6), 8.23 (s, 1H, H3), 7.85 (td, 3JHH = 7.8 Hz, 4JHH = 1.8 Hz, 2H, H7), 7.34 (ddd, 3JHH = 7.8 Hz, 3JHH = 4.8 Hz, 4JHH = 1.0. Hz, 2H, H8), 3.82 (s, 6H, H10).
13C{1H} NMR (100 MHz, CDCl3): δ (ppm) = 168.8 (s, C1), 155.6 (s, C4), 155.3 (s, C5), 148.7 (s, C9), 138.4 (s, C3), 136.9 (s, C7), 127.4 (s, C2), 124.2 (s, C8), 123.2 (s, C6), 52.6 (s, C10).
Elemental analysis (CHN):(C19H15N3O4) Calc. (found): C, 65.32 (65.19); H, 4.33 (4.53); N, 12.03 (12.03).
3.3.2. Syntheses and Characterization of 3,5-Bis(pyrid-2-yl)-phenylethynyl isophthalic acid dimethyl ester (IL2)

1,3-Dimethyl-5-ethynyl-isophthalic acid (2.07 mmol), 1-bromo-3,5-bis(2-pyridinyl)-benzene (653, 2.10 mmol), Pd(PPh3)4 (97 mg, 4 mol.%) and CuI (16 mg, 4 mol.%) were dissolved in 90 mL of THF/NEt3 1:1. The mixture was stirred and heated to reflux for 3 days. The solvents were removed in vacuum. The residue was suspended in 20 mL DCM and filtered through silica gel. The solvent was removed and the crude product was further purified via column chromatography on silica gel (petroleum benzene:ethyl acetate 3:1). A beige powder was obtained. Yield 263 mg, 28%.
1H NMR(400 MHz, CDCl3): δ(ppm) = 8.75 (ddd, 3JHH = 4.8 Hz, 4JHH = 1.9 Hz, 5JHH = 0.9 Hz, 2H, H16), 8.68 (t, 4JHH = 1.7 Hz, 1H, H11), 8.65 (t, 4JHH = 1.6 Hz, 1H, H4), 8.40 (d, 4JHH = 1.6 Hz, 2H, H, H3), 8.28 (d, 4JHH = 1.7 Hz, 2H, H9), 7.89 (dt, 3JHH = 7.9 Hz, 4JHH = 1.1 Hz, 2H, H13), 7.81 (ddd, 3JHH = 7.9 Hz, 3JHH = 7.7 Hz, 4JHH = 1.9 Hz, 2H, H14), 7.30 (ddd, 3JHH = 7.7 Hz,3JHH = 4.8 Hz, 4JHH = 0.9 Hz, 2H, H15), 3.98 (s, 6H, COOCH3).
13C{1H}NMR (100 MHz, CDCl3): δ(ppm) = 166.1 (s,C1), 156.8 (s, C12), 150.3 (s, C16), 140.8 (s, C10), 137.4 (s, C14), 137.1 (s, C3), 131.6 (s, C2), 131.1 (s, C9),130.7 (s, C4), 126.4 (s, C11), 124.9 (s, C8), 124.1 (s, C5), 123.2 (s, C15), 121.2 (s, C13), 91.6 (s, C7), 88.3 (s, C6), 53.4 (s, CH3).
3.3.3. Syntheses and Characterization of 1,3-Dimethyl-5-(ethynyl-4-pyridyl)isophthalate(IL3)

4-Bromopyridine hydrochloride (450 mg, 2.31 mmol), 1,3-dimethyl-5-ethynylisophthalic acid (480 mg, 2.2 mmol), Pd(PPh3)4 (100 mg, 4 mol.%) and CuI (17 mg, 4 mol.%) were dissolved in 60 mL of NEt3:THF (1:1) and heated to 80 °C for 3 days. Upon heating, the yellow solution turned light brown. Subsequently, the solvents were removed in vacuum and the residue was suspended in DCM and filtered over Celite®. Chromatography on silica (CH2Cl2:ethyl acetate, 2:1) followed by solvent removal yielded IL3 as a colorless solid. Yield: 545 mg, 80%.
1H NMR (400 MHz, CD2Cl2): δ(ppm) = 8.63 (t, 4JHH = 1.6 Hz, 1H, H4), 8.61 (d, 2H, 3JHH = 4.6 Hz, H10), 8.38 (s, 2H, H3), 7.42 (d, 3JHH = 4.6 Hz, 2H, H9), 3.95 (s, 6H, H11).
13C{1H} NMR (100 MHz, CD2Cl2): δ(ppm) = 165.9 (s, C1), 150.5 (s, C10), 137.1 (s, C3), 131.2 (s, C2), 131.9 (s, C4), 130.0 (s, C8), 126.0 (s, C9), 123.8 (s, C5), 91.9 (s, C6), 88.7 (s, C7), 53.1(s, C11).
Elemental analysis (CHN): (C17H13NO4) Calc. (found): C, 69.15 (69.06); H, 4.44 (4.95); N, 4.74 (4.55).
3.3.4. Syntheses and Characterization of 5-Acetylthioisophthalic acid (IL4)

5-Mercaptoisophthalic acid 150 mg (1 eq., 0.76 mmol) was dissolved in CH2Cl2 and pyridine (2 eq., 0.12 mL, 1.51 mmol) were cooled to 0 °C. To the mixture 0.13 mL of Ac2O (1.7 eq., 1.3 mmol) were added and the mixture was stirred for 12h at room temperature. Then 50 mL of ethyl acetate was added and the organic layer was washed 3 times with 1M HCl and water each. Finally, the organic layer was dried over MgSO4 and the solvents were removed in vacuum. A beige powder was obtained. Yield 155 mg, 84%.
1H NMR (400 MHz, dmso-d6): δ(ppm) = 13.5 (s(br), 2H, H8), 8.49 (t, 4JHH = 1.6 Hz, 1H, H4), 8.12 (d, 4JHH = 1.6, 2H, H3), 2.49 (s, 3H, H7).
13C{1H} NMR (100 MHz, dmso-d6): δ(ppm) = 192.7 (s, C6), 165.8 (s, C1), 138.6 (s, C3), 132.38 (s, C4), 130.6 (s, C2), 129.3 (s, C5), 30.4 (s, C7).
Elemental analysis (CHS): (C10H8O5S) Calc. (found): C, 50.00 (50.02); H, 3.36 (3.62); S, 13.35 (13.48).
3.3.5. Syntheses and Characterization of Macrocycle 1

3′,5′-Dimethylcarboxy-2,2′,6′,2′′-terpyridine (30 mg, 0.086 mmol) was dissolved in 40 mL of THF/MeOH and 3.45 mL of 0.05 M NaOH and heated to reflux until TLC (ethyl acetate/petroleum benzene) indicated complete saponification. The solvents were removed in vacuo and the residue was dissolved in 30 mL of MeOH. A purple solution of {Ru(CO)Cl(PiPr3)2}2{µ-1,4-(CH=CH)2-C6H4} (90 mg, 0.082 mmol) in 40 mL of CH2Cl2 was added slowly. A yellow-orange suspension was obtained and stirred at room temperature for 30 min. The solvents were removed and the residue was dissolved in CH2Cl2 and filtered through Celite®. The solvent was stripped of the resulting orange solution and the obtained yellow solid was washed with MeOH, Et2O and pentane and then dried in vacuum. Yield 96 mg, 87%.
1H NMR (600 MHz, C6D6): δ(ppm) = 9.80 (s, 2H, H6), 8.96 (dt, 3JHH = 15.3 Hz, 4JHP = 1.7 Hz, 4H, H1), 8.36 (ddd, 3JHH = 8.4 Hz, 4JHH = 1.8 Hz, 5JHH = 1.0 Hz, 4H, H13), 7.44 (s, 8H, H4), 7.34 (dvt, 3JHH = 7.8 Hz, 4JHH = 1.0 Hz, H10), 7.17 (overlapped by solvent signal, H11), 6.73 (ddd, 3JHH = 7.7 Hz, 3JHH = 4.9 Hz, 3JHH = 1.2 Hz, H12), 6.67 (dt, 3JHH = 15.3 Hz, 4JHP = 2.1 Hz, H2), 2.38–2.28 (m, 24H, H15), 1.32 (vq, 3JHH/HP = 6.7 Hz, 72H, H16), 1.20 (vq, 3JHH/HP = 6.7 Hz, 72H, H16′).
13C{1H} NMR (150 MHz, C6D6): δ(ppm) = 209.8 (s, C14), 174.5 (s, C5), 160.7 (s, C7), 160.3 (s, C8), 156.5 (t, C1), 148.1 (s, C13), 142 (s, C6), 137.2 (s, C3), 136.0 (s, C2), 135.3 (s, C9), 126.8 (s, C11), 124.6 (s, C4), 123.7 (s, C10), 122.3 (s, C11), 25.1 (vt, 3JCP = 1JCP = 9.4 Hz, C15), 20.2 (s, C16‘), 19.8 (s, C16).
31P{1H} NMR (162 MHz, C6D6): δ(ppm) = 38.53.
Elemental analysis (CHN): (C130H202N6O12P8Ru4) Calc. (found): C, 57.98 (56.57); H, 7.56 (7.37); N, 3.12 (3.08).
3.3.6. Syntheses and Characterization of Macrocycle 2

1,3-Dimethyl-(5-ethynylphenyl-3,5:2,2′-bispyridyl)-isophthalic acid (35 mg, 0.063 mmol), 80 mL of MeOH and 2.52 mL of 0.05M NaOH were heated to reflux for 24 h. After cooling to room temperature, the solvent was removed and the residue was dissolved in 25 mL of MeOH. A solution of {Ru(CO)Cl(PiPr3)2}2{µ-1,4-(CH=CH)2–C6H4} (90 mg, 0.082 mmol) in 30 mL of CH2Cl2 was slowly added to give a yellow solution. The reaction mixture was stirred for 3 days at r.t. Then the solvent was removed in vacuo and the yellow residue was dissolved in CH2Cl2 and filtered through Celite®. Evaporation of the solvent and washing the residue with MeOH, Et2O and n-pentane and drying in vacuum afforded a yellow powder. Yield: 42 mg, 46%.
1H NMR (600 MHz, C6D6): δ(ppm) = 9.28 (s, 2H, H6), 9.24 (t, 4JHH = 1.7 Hz, 2H, H15), 9.07 (d, 3JHH = 15.2 Hz, 4H, H1), 8.90 (s, 4H, H8), 8.58 (d, 3JHH = 4.7 Hz, 4H, H17), 8.26 (d, 4JHH = 1.7 Hz, H13), 7.53 (s, 8H, H4), 7.22 (d, 3JHH = 7.7 Hz, H4, H20), 7.08 (td, 3JHH = 7.7 Hz, 3JHH = 7.7 Hz, 4JHH = 1.9 Hz, 4H, H19), 6.74 (td, 3JHH = 15.3 Hz, 4JHP = 2.1 Hz, 4H, H2), 6.67 (dd, 3JHH = 7.4 Hz, 3JHH = 4.7 Hz, 4H, H18), 2.39–2.26 (m, H24, H22), 1.35–1.21 (m, H144, H23/H23′).
13C{1H} NMR (150 MHz, C6D6): δ(ppm) = 209.8 (t, 3JCP = 15 Hz, C21), 175.4 (s, C5), 156.3 (m, C1/C16), 149.9 (s, C17), 140.4 (s, C14), 137.3 (s, C3), 136.4 (s, C19), 135.4 (s, C2), 135.2 (s, C9), 135.0 (s, C7), 131.0 (s, C13), 129.8 (s, C6), 126.4 (s, C15), 137.3 (s, C3), 123.9 (s, C12), 122.4 (s, C18), 120.4 (s, C20), 91.6 (s, C11), 88.9 (s, C10), 25.1 (vt, 3JCP = 1JCP = 10.0 Hz, C22), 19.9 (s, C23′), 20.1 (s, C23).
31P{1H} NMR (162 MHz, C6D6): δ(ppm) = 38.12.
Elemental analysis (CHN): (C148H212N4O12P8Ru4) Calc. (found): C, 61.48 (58.71); H, 7.39 (7.35); N, 1.94 (1.80).
3.3.7. Syntheses and Characterization of Macrocycle 3

1,3-Phenyl-dicarboxylmethylester-5-ethynyl-4-pyridine (60 mg, 1.1 eq., 0.20 mmol), 8.2 mL of 0.05 M NaOH (2.2 eq., 0.41 mmol) and 15 mL of THF were heated to reflux for 4 h. The solvents were removed in vacuum and the remaining solid was dissolved in 120 mL of MeOH. To the colorless solution, a purple solution of {Ru(CO)Cl(PiPr3)2}2{µ-1,4-(CH=CH)2–C6H4} (203 mg, 0.18 mmol) in 120 mL of CH2Cl2 was added over 10 min. A yellow solution was obtained and stirred at room temperature for 12 h. Subsequently, the solvents were removed in vacuum and the yellow crude product was dissolved in CH2Cl2 and filtered through Celite®. The yellow filtrate was dried and washed with MeOH and n-hexane (3 × 10 mL). Drying in vacuum afforded macrocycle 3 as an orange solid. Yield: 169 mg, 71%
1H NMR (600 MHz, C6D6): δ(ppm) = 9.28 (t, 4JHH = 1.5 Hz, 2H, H6), 9.03 (dt, 3JHH = 15.3 Hz, 4JPH = 1.7 Hz, 4H, H1), 8.78 (d, 4JHH = 1.5 Hz, 2H, H8), 8.36 (m, 4H, H14) 7.51 (s, 8H, 4H), 6.75 (m, 4H, H13), 6.72 (dt, 3JHH = 15.3 Hz, 4JPH = 2.1 Hz, 4H, H2), 2.37–2.23 (m, 24H, H16), 1.32–1.21 (m, 144H, H17/H17′).
13C{1H} NMR (150 MHz, C6D6): δ(ppm) = 209.7 (t, 2JCP = 13 Hz, C15), 175.0 (s, C5), 156.1 (t, 2JCP = 11 Hz, C1), 150.2 (s, C14), 137.3 (s, C3), 135.6 (s, C2), 135.3 (s, C9), 135.0 (s, C8), 130.5 (s, C7) 130.3 (s,C6), 125.6 (s, C13), 124.7 (s, C4), 123.5 (s, C12), 92.4 (s, C10), 88.7 (s, C11), 25.1 (vt, 3JCP = 1JCP = 9.0 Hz, C16), 20.0 (s, C17) 19.8 (s, C17′).
31P{1H} NMR (C6D6): δ(ppm) = 37.97.
Elemental analysis (CHN): (C126H198N2O12P8Ru4) Calc. (found): C, 58.54 (58.58); H, 7.72 (8.17); N, 1.08 (1.01).
3.3.8. Syntheses and Characterization of Macrocycle 4

5-Acetylthioisophthalic acid 78 mg (1.1 eq., 33 mmol), K2CO3 45 mg (1.1 eq., 33 mmol) and 50 mL of MeOH were stirred for 2 h at room temperature. To the colorless solution, a solution of {Ru(CO)Cl(PiPr3)2}2{µ-1,4-(CH=CH)2-C6H4} (325 mg, 30 mmol) in 140 mL of CH2Cl2 was added over 5 min. After the yellow suspension was stirred for 30 min, the solvents were evaporated. The yellow solid was then dissolved in CH2Cl2 and filtered through Celite®. Subsequently, the solvent was removed, the yellow solid was washed with methanol and hexane. Drying in vacuum gave 4 as a yellow solid. Yield 300 mg, 78%.
1H NMR (600 Mhz, C6D6): δ(ppm) = 9.27 (t, 4JHH = 1.2 Hz, 2H, H6), 9.03 (d, 3JHH = 15.3 Hz, 4H, H1), 8.61 (d, 4JHH = 1.2 Hz, 4H, H8), 7.50 (s, 8H, H4), 6.71 (dt, 3JHH = 15.3 Hz, 4JHP = 2.2 Hz, 4H, H2), 2.37–2.25 (m, 24H, H13), 1.62 (s, 6H, H11), 1.36–1.19 (m, 144H, H14/H14′).
13C{1H} NMR (150 MHz, C6D6): δ(ppm) = 209.7 (t, 2JCP = 13 Hz, C12), 190.8 (s, C10), 175.3 (s, C5), 156.2 (t, 2JCP = 11 Hz, C1), 137.6 (s, C8), 137.3 (s, C3), 135.52 (s, C2), 135.50 (s, C9), 130.4 (s, C7), 129.8 (s, C6), 124.7 (s, C4), 29.5 (s, C11) 25.1 (vt, 3JCP = 1JCP = 9.0 Hz, C13), 20.0/19.9 (s, C14/C14′).
31P{1H} NMR (162 MHz, C6D6): δ(ppm) = 38.09 (s).
Elemental analysis (CHN): (C116H196O14P8Ru4S2) Calc. (found): C, 55.05 (54.81); H, 7.81 (7.64); S, 2.53 (2.32).
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2304-6740/6/3/73/s1, Figures S1–S21: Graphical representations of the 1H and 13C{1H} and 31P{1H} NMR spectra of all compounds, Figures S22–S27: ESI-MS spectra of complexes 1, 2, and 4, Figure S28: Additional packing diagrams of macrocycle 4, Figure S29: Association of macrocycle 4 in the crystal by hydrogen bonding, Tables S1–S3: Crystal and refinement data and full lists of bond parameters of 4, Figures S30–S40: Graphical representations of the cyclic and square wave voltammograms, Figures S41–S58: the results of IR/NIR as well as UV/Vis/NIR spectroelectrochemical experiments; Figures S59–S70: EPR spectra of the di- and tetracations of the macrocycles at r.t. and at 78 K.
Author Contributions
Experimental work P.A. and M.R.R.; electrochemistry and spectroelectrochemistry: P.A.; X-ray structure analysis P.A. and M.L. Original Draft Preparation, R.F.W. and P.A.
Funding
We thank the University of Konstanz and the state of Baden-Württemberg for financial support of this work.
Acknowledgments
We thank Gernot Haug for the synthesis of IL4.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Scheerer, S.; Linseis, M.; Wuttke, E.; Weickert, S.; Drescher, M.; Tröppner, O.; Ivanović-Burmazović, I.; Irmler, A.; Pauly, F.; Winter, R.F. Redox-Active Tetraruthenium Macrocycles Built from 1,4-Divinylphenylene-Bridged Diruthenium Complexes. Chem. Eur. J. 2016, 22, 9574–9590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, D.; Weibert, B.; Winter, R.F. Redox-active tetraruthenium metallacycles: Reversible release of up to eight electrons resulting in strong electrochromism. Chem. Commun. 2016, 52, 6103–6106. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.; Bodensteiner, M.; Linseis, M.; Winter, R.F. Macrocyclic Triruthenium Complexes Having Electronically Coupled Mixed-Valent States. Chem. Eur. J. 2018, 24, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.; Linseis, M.; Winter, R.F. Constitutional Isomers of Macrocyclic Tetraruthenium Complexes with Vastly Different Spectroscopic and Electrochemical Properties. Organometallics 2018, 37, 1817–1820. [Google Scholar] [CrossRef]
- Sadhukhan, N.; Patra, S.K.; Sana, K.; Bera, J.K. Novel Heterobimetallic Metallamacrocycles Based on the 1,1′-Bis(1,8-naphthyrid-2-yl)ferrocene (FcNP2) Ligand: Structural Characterization of the Complexes [{M(FcNP2)}2]2+ (M = CuI, AgI) and {MCl2(FcNP2)}4 (M = ZnII, CoII). Organometallics 2006, 25, 2914–2916. [Google Scholar] [CrossRef]
- Croue, V.; Goeb, S.; Salle, M. Metal-driven self-assembly: The case of redox-active discrete architectures. Chem. Commun. 2015, 51, 7275–7289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, R.F. The molecular electrochemistry of metal–organic metallamacrocycles. Curr. Opin. Electrochem. 2018, 8, 14–23. [Google Scholar] [CrossRef]
- Maurer, J.; Sarkar, B.; Schwederski, B.; Kaim, W.; Winter, R.F.; Záliš, S. Divinylphenylene bridged diruthenium complexes bearing Ru(CO)Cl(PiPr3)2 entities. Organometallics 2006, 25, 3701–3712. [Google Scholar] [CrossRef]
- Linseis, M.; Winter, R.F.; Sarkar, B.; Kaim, W.; Záliš, S. Multistep Electrochromic Behaviour from an Organometallic Tetranuclear Complex of a Tetradonor-Substituted Olefin. Organometallics 2008, 27, 3321–3324. [Google Scholar] [CrossRef]
- Linseis, M.; Záliš, S.; Zabel, M.; Winter, R.F. Ruthenium Stilbenyl and Diruthenium Distyrylethene Complexes: Aspects of Electron Delocalization and Electrocatalyzed Isomerization of the Z-Isomer. J. Am. Chem. Soc. 2012, 134, 16671–16692. [Google Scholar] [CrossRef] [PubMed]
- Wuttke, E.; Hervault, Y.-M.; Polit, W.; Linseis, M.; Erler, P.; Rigaut, S.; Winter, R.F. Divinylphenylene-and Ethynylvinylphenylene-Bridged Mono-, Di-, and Triruthenium Complexes for Covalent Binding to Gold Electrodes. Organometallics 2014, 33, 4672–4686. [Google Scholar] [CrossRef]
- Pfaff, U.; Hildebrandt, A.; Korb, M.; Oßwald, S.; Linseis, M.; Schreiter, K.; Spange, S.; Winter, R.F.; Lang, H. Electronically Strongly Coupled Divinylheterocyclic-Bridged Diruthenium Complexes. Chem. Eur. J. 2016, 22, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.D. Near-infrared electrochromic materials for optical attenuation based on transition-metal coordination complexes. J. Solid State Electrochem. 2005, 9, 778–787. [Google Scholar] [CrossRef]
- Mortimer, R.J.; Dyer, A.L.; Reynolds, J.R. Electrochromic organic and polymeric materials for display applications. Org. Disp. 2006, 27, 2–18. [Google Scholar] [CrossRef] [Green Version]
- Sreejith, S.; Carol, P.; Chithra, P.; Ajayaghosh, A. Squaraine dyes: A mine of molecular materials. J. Mater. Chem. 2008, 18, 264–274. [Google Scholar] [CrossRef]
- Astruc, D.; Ornelas, C.; Ruiz, J. Dendritic Molecular Electrochromic Batteries Based on Redox-Robust Metallocenes. Chem. Eur. J. 2009, 15, 8936–8944. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, R.J. Electrochromic Materials. Annu. Rev. Mater. Res. 2011, 41, 241–268. [Google Scholar] [CrossRef]
- D’Alessandro, D.M. Exploiting redox activity in metal-organic frameworks: Concepts, trends and perspectives. Chem. Commun. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Chen, S.; Lu, B.; Xu, J. Hybrid π-conjugated polymers from dibenzo pentacyclic centers: Precursor design, electrosynthesis and electrochromics. Sci. China Chem. 2017, 60, 38–53. [Google Scholar] [CrossRef]
- Dai, F.; Dou, J.; He, H.; Zhao, X.; Sun, D. Self-Assembly of Metal—Organic Supramolecules: From a Metallamacrocycle and a Metal—Organic Coordination Cage to 1D or 2D Coordination Polymers Based on Flexible Dicarboxylate Ligands. Inorg. Chem. 2010, 49, 4117–4124. [Google Scholar] [CrossRef] [PubMed]
- Case, F.H.; Butte, W.A. Further Preparation of Substituted 2,6-Bis(2′-pyridyl)pyridines. J. Org. Chem. 1961, 26, 4415–4418. [Google Scholar] [CrossRef]
- Wang, H.-B.; Mudraboyina, B.P.; Li, J.; Wisner, J.A. Minimal complementary hydrogen-bonded double helices. Chem. Commun. 2010, 46, 7343–7345. [Google Scholar] [CrossRef] [PubMed]
- Kazuki, O.; Daisuke, S.; Tomoya, Y.; Katsuhiro, I.; Ryota, Y.; Toshio, T.; Hirofumi, S.; Tetsuya, O.; Hiroki, K.; Nobuhiro, Y.; et al. Synthesis and Self-Assembly of NCN-Pincer Pd-Complex-Bound Norvalines. Chem. Eur. J. 2013, 19, 12356–12375. [Google Scholar]
- Evans, B.J.; Doi, J.T.; Musker, W.K. The aqueous periodate oxidation of aromatic and carboxylic acid disufides. Phosphorus Sulfur Silicon Relat. Elem. 1992, 73, 5–13. [Google Scholar] [CrossRef]
- Lawrence, D.S.; Jiang, T.; Levett, M. Self-Assembling Supramolecular Complexes. Chem. Rev. 1995, 95, 2229–2260. [Google Scholar] [CrossRef]
- Swiegers, G.F.; Malefetse, T.J. New Self-Assembled Structural Motifs in Coordination Chemistry. Chem. Rev. 2000, 100, 3483–3538. [Google Scholar] [CrossRef] [PubMed]
- Oliveri, C.G.; Ulmann, P.A.; Wiester, M.J.; Mirkin, C.A. Heteroligated Supramolecular Coordination Complexes Formed via the Halide-Induced Ligand Rearrangement Reaction. Acc. Chem. Res. 2008, 41, 1618–1629. [Google Scholar] [CrossRef] [PubMed]
- Psomas, G.; Stemmler, A.J.; Dendrinou-Samara, C.; Bodwin, J.J.; Schneider, M.; Alexiou, M.; Kampf, J.W.; Kessissoglou, D.P.; Pecoraro, V.L. Preparation of Site-Differentiated Mixed Ligand and Mixed Ligand/Mixed Metal Metallacrowns. Inorg. Chem. 2001, 40, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.V.; Raymond, K.N. The Big Squeeze: Guest Exchange in an M4L6 Supramolecular Host. J. Am. Chem. Soc. 2005, 127, 7912–7919. [Google Scholar] [CrossRef] [PubMed]
- Garci, A.; Marti, S.; Schurch, S.; Therrien, B. Insight into the dynamic ligand exchange process involved in bipyridyl linked arene ruthenium metalla-rectangles. RSC Adv. 2014, 4, 8597–8604. [Google Scholar] [CrossRef]
- Garci, A.; Gupta, G.; Dalvit, C.; Therrien, B. Investigating the Formation Mechanism of Arene Ruthenium Metallacycles by NMR Spectroscopy. Eur. J. Inorg. Chem. 2014, 2014, 5651–5661. [Google Scholar] [CrossRef]
- Hiraoka, S.; Sakata, Y.; Shionoya, M. Ti(IV)-Centered Dynamic Interconversion between Pd(II), Ti(IV)-Containing Ring and Cage Molecules. J. Am. Chem. Soc. 2008, 130, 10058–10059. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.-R.; Stang, P.J. Direct and Quantitative Characterization of Dynamic Ligand Exchange between Coordination-Driven Self-Assembled Supramolecular Polygons. J. Am. Chem. Soc. 2009, 131, 3487–3489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pevny, F.; Winter, R.F.; Sarkar, B.; Záliš, S. How to elucidate and control the redox sequence in vinylbenzoate and vinylpyridine bridged diruthenium complexes. Dalton Trans. 2010, 8000–8011. [Google Scholar] [CrossRef] [PubMed]
- Záliš, S.; Winter, R.F.; Kaim, W. Quantum chemical interpretation of redox properties of ruthenium complexes with vinyl and TCNX type non-innocent ligands. Coord. Chem. Rev. 2010, 254, 1383–1396. [Google Scholar] [CrossRef] [Green Version]
- Richardson, D.E.; Taube, H. Mixed-valence molecules: Electronic delocalization and stabilization. Coord. Chem. Rev. 1984, 60, 107–129. [Google Scholar] [CrossRef]
- De la Rosa, R.; Chang, P.J.; Salaymeh, F.; Curtis, J.C. Redox asymmetry and metal–metal coupling in pyrazine-bridged ruthenium dimers. Inorg. Chem. 1985, 24, 4229–4231. [Google Scholar] [CrossRef]
- Crutchley, R.J. Intervalence Charge Transfer and Electron Exchange Studies of Dinuclear Ruthenium Complexes. In Advances in Inorganic Chemistry; Sykes, A.G., Ed.; Academic Press: Cambridge, MA, USA, 1994; Volume 41, pp. 273–325. [Google Scholar]
- Winter, R.F. Half-Wave Potential Splittings ΔE1/2 as a Measure of Electronic Coupling in Mixed-Valent Systems: Triumphs and Defeats. Organometallics 2014, 33, 4517–4536. [Google Scholar] [CrossRef]
- Hildebrandt, A.; Lang, H. (Multi)ferrocenyl Five-Membered Heterocycles: Excellent Connecting Units for Electron Transfer Studies. Organometallics 2013, 32, 5640–5653. [Google Scholar] [CrossRef]
- Connelly, N.G.; Geiger, W.E. Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, A.A. Multiplicity of the ground state of large alternant organic molecules with conjugated bonds (do organic ferromagnets exist?). Theor. Chim. Acta 1978, 47, 297–304. [Google Scholar] [CrossRef]
- Yoshizawa, K.; Hoffmann, R. Potential Linear-Chain Organic Ferromagnets. Chem. Eur. J. 1995, 1, 403–413. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Wang, L.; Wang, R.; Wang, L. Effect of configuration and conformation on the spin multiplicity in xylylene type biradicals. Sci. China Ser. B Chem. 2000, 43, 524–530. [Google Scholar] [CrossRef]
- Baumgarten, M. High Spin Molecules Directed Towards Molecular Magnets. In EPR of Free Radicals in Solids II; Lund, A., Shiotani, M., Eds.; Springer: Dordrecht, The Netherlands, 2012; Volume 25, pp. 205–244. [Google Scholar]
- Abe, M. Diradicals. Chem. Rev. 2013, 113, 7011–7088. [Google Scholar] [CrossRef] [PubMed]
- Barlow, S.; Risko, C.; Chung, S.-J.; Tucker, N.M.; Coropceanu, V.; Jones, S.C.; Levi, Z.; Brédas, J.-L.; Marder, S.R. Intervalence Transitions in the Mixed-Valence Monocations of Bis(triarylamines) Linked with Vinylene and Phenylene—Vinylene Bridges. J. Am. Chem. Soc. 2005, 127, 16900–16911. [Google Scholar] [CrossRef] [PubMed]
- Barlow, S.; Risko, C.; Odom, S.A.; Zheng, S.; Coropceanu, V.; Beverina, L.; Brédas, J.-L.; Marder, S.R. Tuning Delocalization in the Radical Cations of 1,4-Bis[4-(diarylamino)styryl]benzenes, 2,5-Bis[4-(diarylamino)styryl]thiophenes, and 2,5-Bis[4-(diarylamino)styryl]pyrroles through Substituent Effects. J. Am. Chem. Soc. 2012, 134, 10146–10155. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.-J.; Yao, C.-J.; Shao, J.-Y.; Yao, J.; Zhong, Y.-W. Oligotriarylamines with a Pyrene Core: A Multicenter Strategy for Enhancing Radical Cation and Dication Stability and Tuning Spin Distribution. Chem. Eur. J. 2014, 20, 17454–17465. [Google Scholar] [CrossRef] [PubMed]
- Hassenrück, C.; Winter, R.F. Manipulation and Assessment of Charge and Spin Delocalization in Mixed-Valent Triarylamine–Vinylruthenium Conjugates. Inorg. Chem. 2017, 56, 13517–13529. [Google Scholar] [CrossRef] [PubMed]
- Garnovskii, A.D.; Kharisov, B.I.; Blanco, L.M.; Sadimenko, A.P.; Uraev, A.I.; Vasilchenko, I.S.; Garnovskii, D.A. Metal Complexes as Ligands. J. Coord. Chem. 2002, 55, 1119–1134. [Google Scholar] [CrossRef]
- Serroni, S.; Campagna, S.; Puntoriero, F.; Di Pietro, C.; McClenaghan, N.D.; Loiseau, F. Dendrimers based on ruthenium(II) and osmium(II) polypyridine complexes and the approach of using complexes as ligands and complexes as metals. Chem. Soc. Rev. 2001, 30, 367–375. [Google Scholar] [CrossRef]
- Herrendorf, W.; Bärnighausen, W. HABITUS. Program for the Optimization of the Crystal Shape for Numerical Absorption Correction in X-SHAPE, version 1.06; Fa. STOE: Darmstadt, Germany, 1999. [Google Scholar]
- Sheldrick, G.M. SHELXL-97, Program for Crystal Structure Solution and Refinement; Universität Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Van De Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Krejcik, M.; Danicek, M.; Hartl, F. Simple construction of an infrared optically transparent thin-layer cell: Applications to the redox reactions of ferrocene, Mn2(CO)10 and Mn(CO)3(3,5-di-t-butylcatcholate)−. J. Eletcroanal. Chem. 1991, 317, 179–187. [Google Scholar] [CrossRef]
Figure 1.
Vinylarylene-bridged ruthenium macrocycles of types I and II.

Figure 2.
Ligands IL1 to IL4 and the derived tetraruthenium macrocycles 1–4.

Figure 3.
(Left): HR ESI Mass spectrum of macrocycle 3; (right): experimental (top) and simulated (bottom) isotope peak pattern for the [M+] peak.
Figure 3.
(Left): HR ESI Mass spectrum of macrocycle 3; (right): experimental (top) and simulated (bottom) isotope peak pattern for the [M+] peak.

Figure 4.
Two different views of an individual molecule of 4 with the benzene solvate molecule incorporated into the central cavity. Selected bond lengths in Å: Ru1–P1: 2.398(4), Ru1–P2: 2.413(4), Ru2–P3: 2.402(4), Ru2–P4: 2.402(4), Ru1–O1: 2.316(6), Ru1–O2: 2.183(6), Ru2–O3: 2.287(6), Ru2–O4: 2.205(6), Ru1–C1: 1.976(9), Ru1–C11: 1.764(10), Ru2–C10: 1.982(10), Ru2–C12: 1.763(10). Selected bond angles in °: P1–Ru1–P2: 179.72(12), P3–Ru2–P4: 176.83(12), O1–Ru1–O2: 58.7(2), O3–Ru2–O4: 59.1(2), O1–Ru1–C1: 156.3(3), O2–Ru1–C11: 172.2(4), O3–Ru2–C10: 160.5(3), O4–Ru2–C12: 168.9(4).
Figure 4.
Two different views of an individual molecule of 4 with the benzene solvate molecule incorporated into the central cavity. Selected bond lengths in Å: Ru1–P1: 2.398(4), Ru1–P2: 2.413(4), Ru2–P3: 2.402(4), Ru2–P4: 2.402(4), Ru1–O1: 2.316(6), Ru1–O2: 2.183(6), Ru2–O3: 2.287(6), Ru2–O4: 2.205(6), Ru1–C1: 1.976(9), Ru1–C11: 1.764(10), Ru2–C10: 1.982(10), Ru2–C12: 1.763(10). Selected bond angles in °: P1–Ru1–P2: 179.72(12), P3–Ru2–P4: 176.83(12), O1–Ru1–O2: 58.7(2), O3–Ru2–O4: 59.1(2), O1–Ru1–C1: 156.3(3), O2–Ru1–C11: 172.2(4), O3–Ru2–C10: 160.5(3), O4–Ru2–C12: 168.9(4).

Figure 5.
The diruthenium reference complexes Ru2Cl2 and Ru2bza2.

Figure 6.
Cyclic voltammogram of macrocycle 3 (CH2Cl2, NBu4+PF6−, 0.1 M, at v = 100 mV/s, r.t. diruthenium reference complexes Ru2Cl2 and Ru2bza2.
Figure 6.
Cyclic voltammogram of macrocycle 3 (CH2Cl2, NBu4+PF6−, 0.1 M, at v = 100 mV/s, r.t. diruthenium reference complexes Ru2Cl2 and Ru2bza2.

Figure 7.
Spectroscopic changes of the Ru(CO) bands (left) and in the near infrared (right) during the oxidation of the neutral macrocycle 1 to its dication 12+ (top) and from dication 12+ to the tetracation 14+ (bottom) in 1,2-C2H4Cl2/0.1 M NBu4+PF6−, at r.t.
Figure 7.
Spectroscopic changes of the Ru(CO) bands (left) and in the near infrared (right) during the oxidation of the neutral macrocycle 1 to its dication 12+ (top) and from dication 12+ to the tetracation 14+ (bottom) in 1,2-C2H4Cl2/0.1 M NBu4+PF6−, at r.t.

Figure 8.
Changes in the UV/Vis/NIR spectra on oxidation of neutral macrocycle 3 to the dicationic state 32+ (left) and from the di- to the tetracationic state 34+ (right, 1,2-C2H4Cl2, NBu4+PF6−, r.t.).
Figure 8.
Changes in the UV/Vis/NIR spectra on oxidation of neutral macrocycle 3 to the dicationic state 32+ (left) and from the di- to the tetracationic state 34+ (right, 1,2-C2H4Cl2, NBu4+PF6−, r.t.).

Figure 9.
Visual impressions of the neutral (left), dicationic (middle) and tetracationic (right) forms of macrocycle 1 at concentration levels of 200 µM in CH2Cl2 solution.
Figure 9.
Visual impressions of the neutral (left), dicationic (middle) and tetracationic (right) forms of macrocycle 1 at concentration levels of 200 µM in CH2Cl2 solution.

Figure 10.
EPR spectra of the di- (left) and tetracation (right) of macrocycle 3 (CH2Cl2, r.t.).

Figure 11.
Possible structures and spin multiplicities for compounds with two paramagnetic redox centers RC• bridged by a para-phenylene spacer.
Figure 11.
Possible structures and spin multiplicities for compounds with two paramagnetic redox centers RC• bridged by a para-phenylene spacer.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Electrochemistry data for macrocycles 1–4 and the diruthenium analogs Ru2bza2 and Ru2Cl2 1.
Table 1.
Electrochemistry data for macrocycles 1–4 and the diruthenium analogs Ru2bza2 and Ru2Cl2 1.
| Complex | E1/20/+ (mV) | E1/21+/2+ (mV) | E1/2+2/4+ (mV) | ΔE1/2+1/2+ (mV) | ΔE1/22+/4+ (mV) |
|---|---|---|---|---|---|
| 12 | −244 | −147 | 82 | 97 | 229 |
| 13 | −330 | −241 | 74/125 4 | 89 | 315 |
| 22 | −256 | −173 | 59 | 83 | 232 |
| 32 | −256 | −175 | 73 | 81 | 248 |
| 42 | −220 | −150 | 45 | 70 | 195 |
| 43 | −330 | −255 | 57/114 4 | 75 | 312 |
| Ru2bza2 | −250 | - | 20 5 | - | 270 |
| Ru2Cl2 | −75 | - | 175 5 | - | 250 |
1 In CH2Cl2 at r.t. and v = 100 mV/s or at a step width of 2 mV and a square wave frequency of 20 Hz in square wave voltammetry. Potentials are provided against the ferrocene/ferrocenium redox scale. 2 With 0.1 M NBu4+PF6− as the supporting electrolyte. 3 With 0.02 M NBu4+B{C6H3(CF3)2-3,5}4− as the supporting electrolyte. 4 Separate potentials for the 2+/3+ and the 3+/4+ waves. 5 Half-wave potential for the +/2+ wave, which phenomenologically corresponds to the 2+/3+ wave of a macrocyclic analog.
Table 2.
Energies of the υ(CO) stretches and UV/Vis/NIR data for macrocycles 1–4 and the analogous diruthenium complex Ru2bza2 in their various oxidation states 1,2.
Table 2.
Energies of the υ(CO) stretches and UV/Vis/NIR data for macrocycles 1–4 and the analogous diruthenium complex Ru2bza2 in their various oxidation states 1,2.
| Complex | υ(CO) 1 | λmax(εmax) 1 |
|---|---|---|
| 1 | 1901 | 354(85,000) |
| 12+ | 1927, 1944 | 279(59,300), 492(35,700), 534(69,000), 587(83,000), 963(15,900), 1089(62,700), 1259(95,000) |
| 14+ | 1973 | 279(79,200), 602(126,000) |
| 2 | 1901 | 354(85,000) |
| 22+ | 1927, 1943 | 279(59,500), 495(37,000), 535(69,000), 587(83,000), 963(15,600), 1088(62,700), 1257(95,000) |
| 24+ | 1973 | 279(79,600), 602(127,000) |
| 3 | 1902 | 354(70,700) |
| 32+ | 1928, 1944 | 283(63,300), (495(25,900), 535(48,600), 587(61,600) 936(13,400), 1085(46,900), 1256(72,000) |
| 34+ | 1972 | 282(65,300), 608 (102,000) |
| 4 | 1901 | 356(60,600) |
| 42+ | 1928, 1944 | 279(46,400), 537(49,700), 589(61,000), 963(12,100), 1086(45,100), 1258(70,000) |
| 44+ | 1973 | 283(116,000), 364(42,700), 592(109,000) |
| Ru2bza2 | 1904 | 362(19,000) |
| Ru2bza2+ | 1924, 1944 | 357(19300), 487(6600), 536(6800), 587(8000), 1016(9200), 1084(1900), 1272 (5800) |
| Ru2bza22+ | 1971 | 375 (9200), 427(9000), 606(13000) |
1 υ(CO) in cm−1; λmax in nm, ε in M−1 cm−1; in 1,2-C2H4Cl2/0.1 M NBu4+PF6−. 2 Data for Ru2bza2n+ from ref. [11].
Table 3.
g values for oxidized complexes 1–4 in CH2Cl2.
| Complex | Dication | Tetracation | ||
|---|---|---|---|---|
| r.t. | 123 K | r.t. | 123 K | |
| 1 | 2.019 | 2.019 | 2.030 | 2.036 |
| 2 | 2.019 | 2.018 | 2.031 | 2.028 |
| 3 | 2.019 | 2.018 | 2.031 | 2.028 |
| 4 | 2.019 | 2.018 | 2.032 | 2.029 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Anders, P.; Rapp, M.R.; Linseis, M.; Winter, R.F. Tetraruthenium Metallamacrocycles with Potentially Coordinating Appended Functionalities. Inorganics 2018, 6, 73. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6030073
AMA Style
Anders P, Rapp MR, Linseis M, Winter RF. Tetraruthenium Metallamacrocycles with Potentially Coordinating Appended Functionalities. Inorganics. 2018; 6(3):73. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6030073
Chicago/Turabian StyleAnders, Patrick, Mario Robin Rapp, Michael Linseis, and Rainer F. Winter. 2018. "Tetraruthenium Metallamacrocycles with Potentially Coordinating Appended Functionalities" Inorganics 6, no. 3: 73. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6030073
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.