Properties and Applications of Metal (M) dodecahydro-closo-dodecaborates (Mn=1,2B12H12) and Their Implications for Reversible Hydrogen Storage in the Borohydrides


Abstract
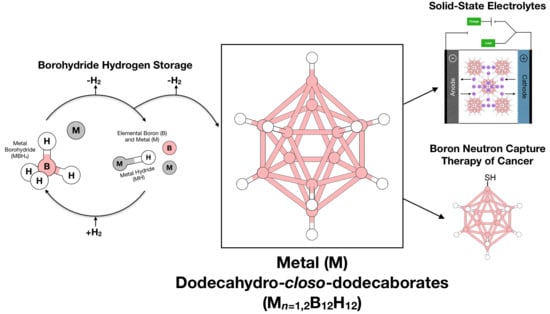
:
1. Introduction
2. Metal Borohydrides for Hydrogen Storage
2.1. Possible H2 Desorption Pathways
2.1.1. Alkali Borohydrides
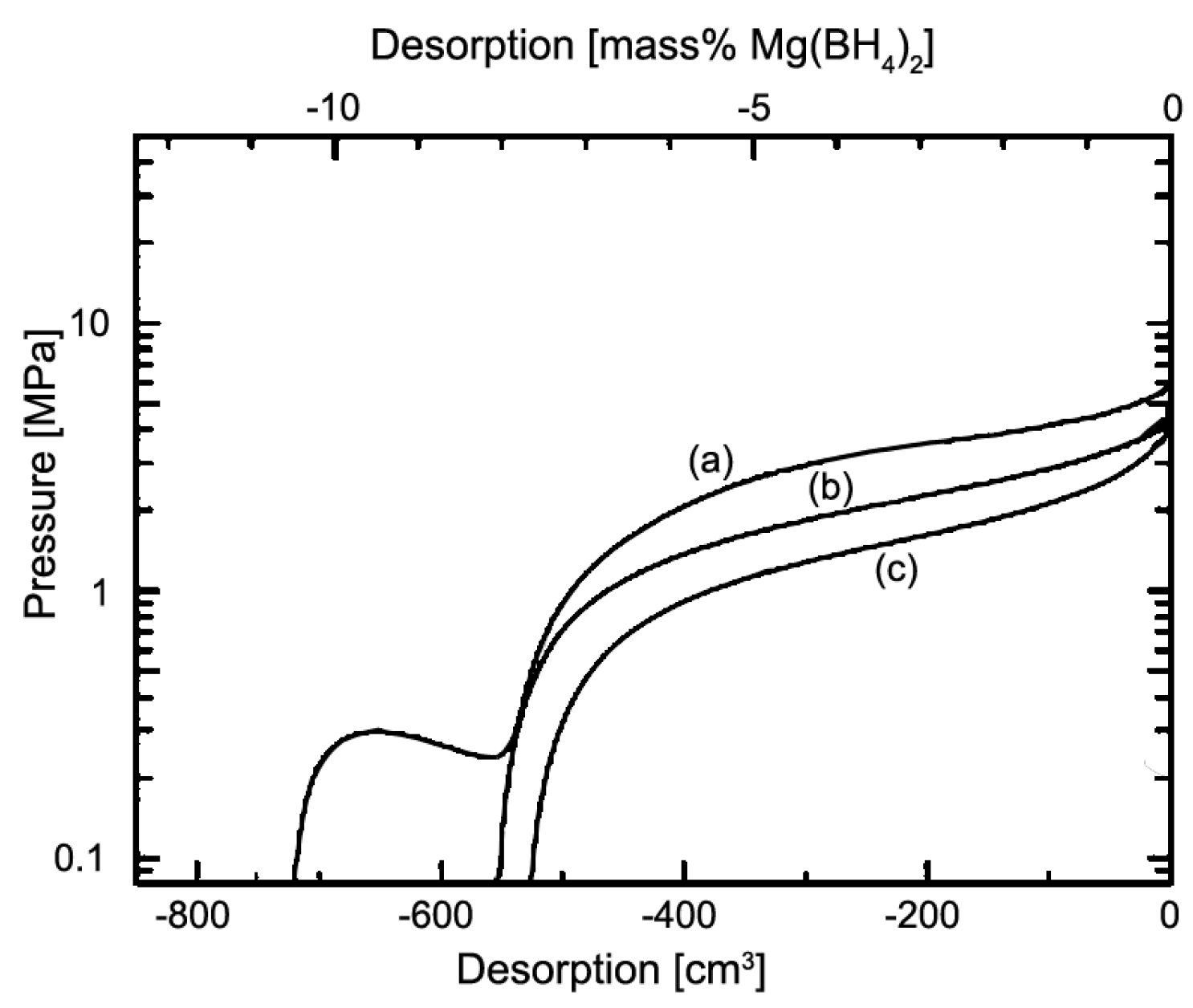
2.2. Alkaline Earth Borohydrides
3. Dodecaborates in the Borohydride System
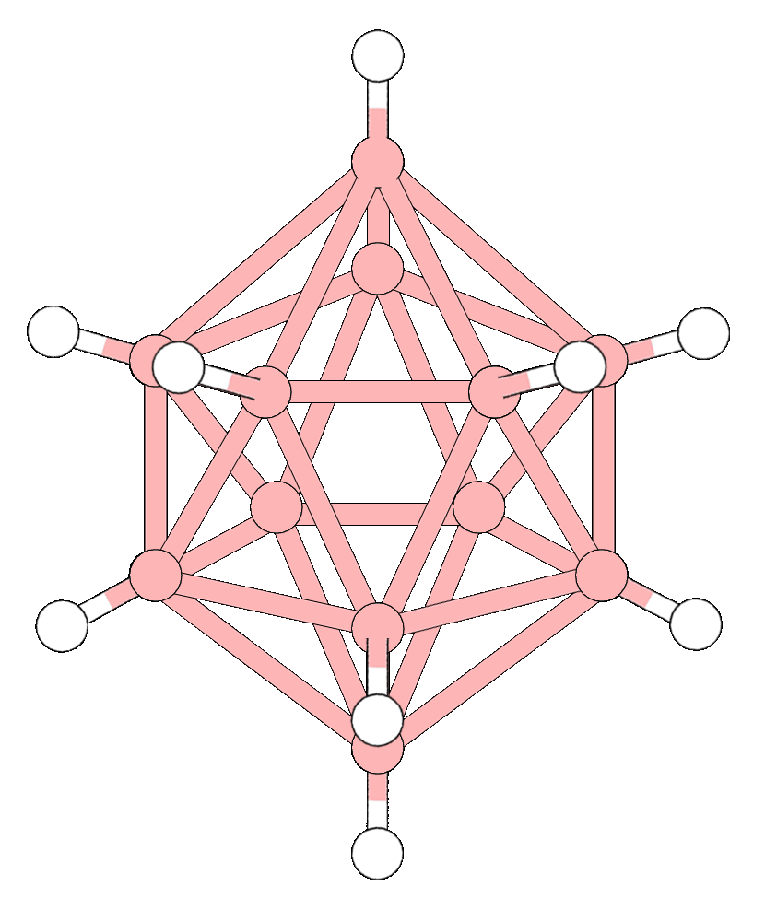
3.1. Chemical Structure and Properties
3.2. Synthesis Methods
3.3. Other BxHy Compounds
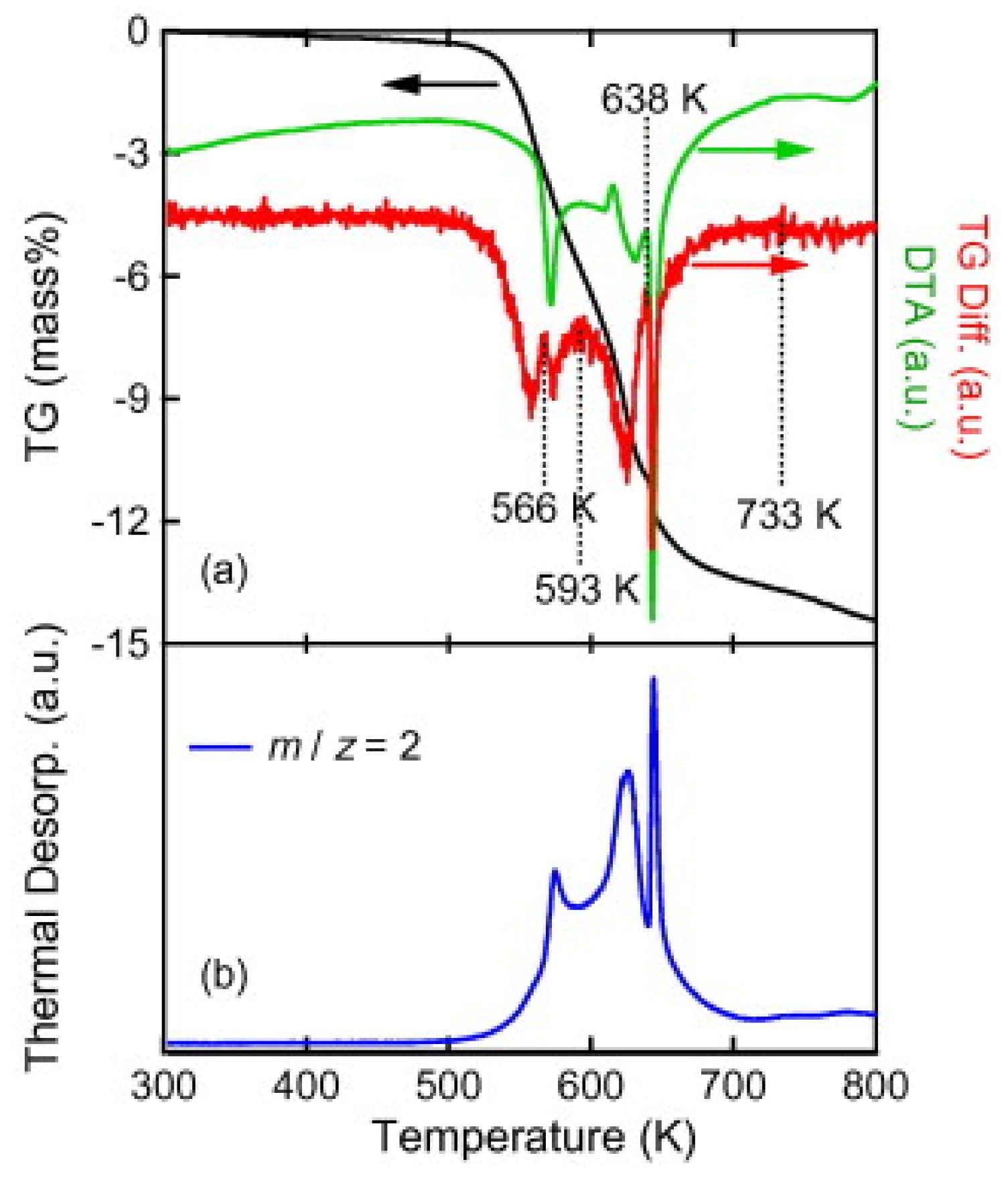
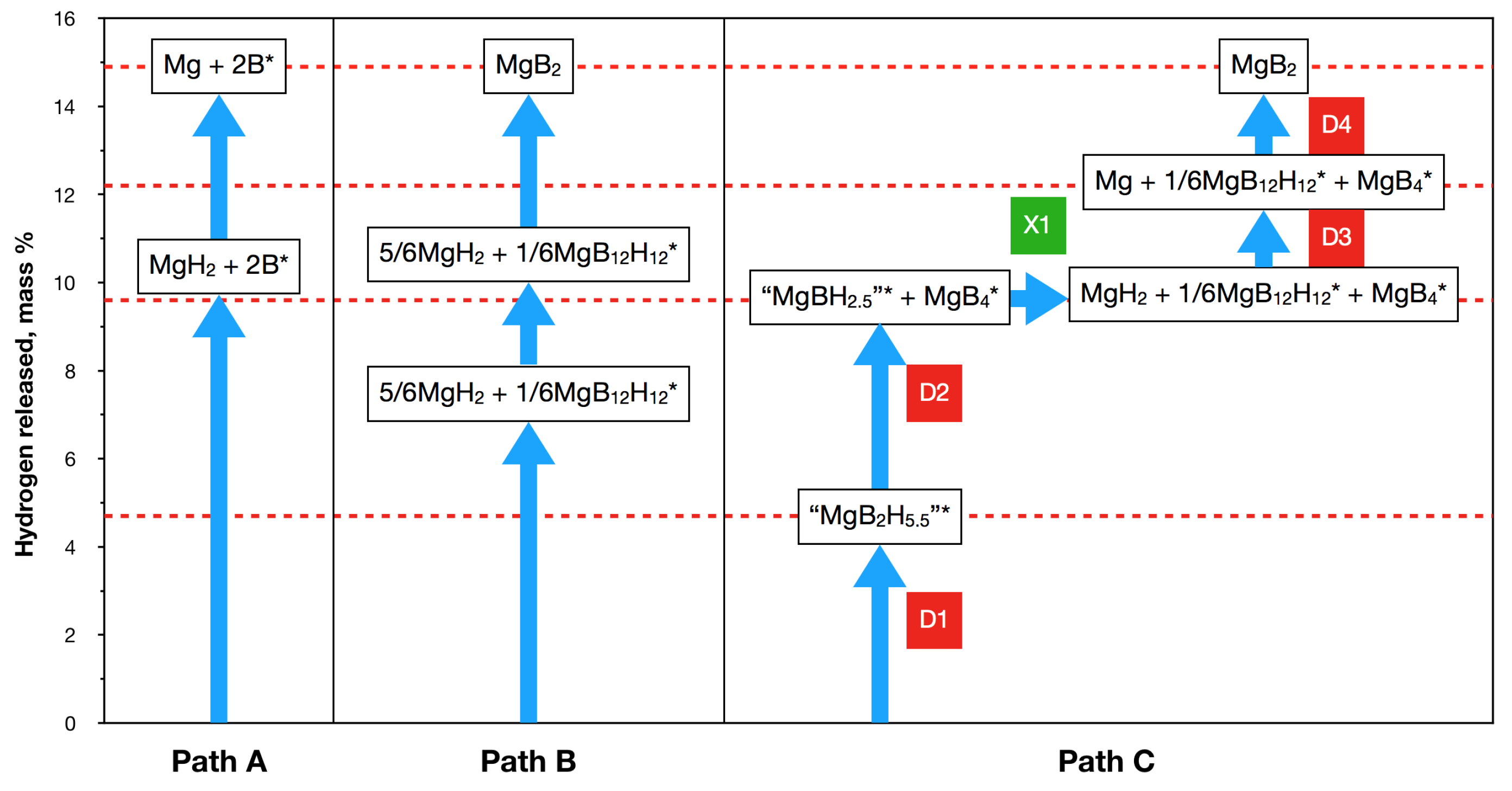
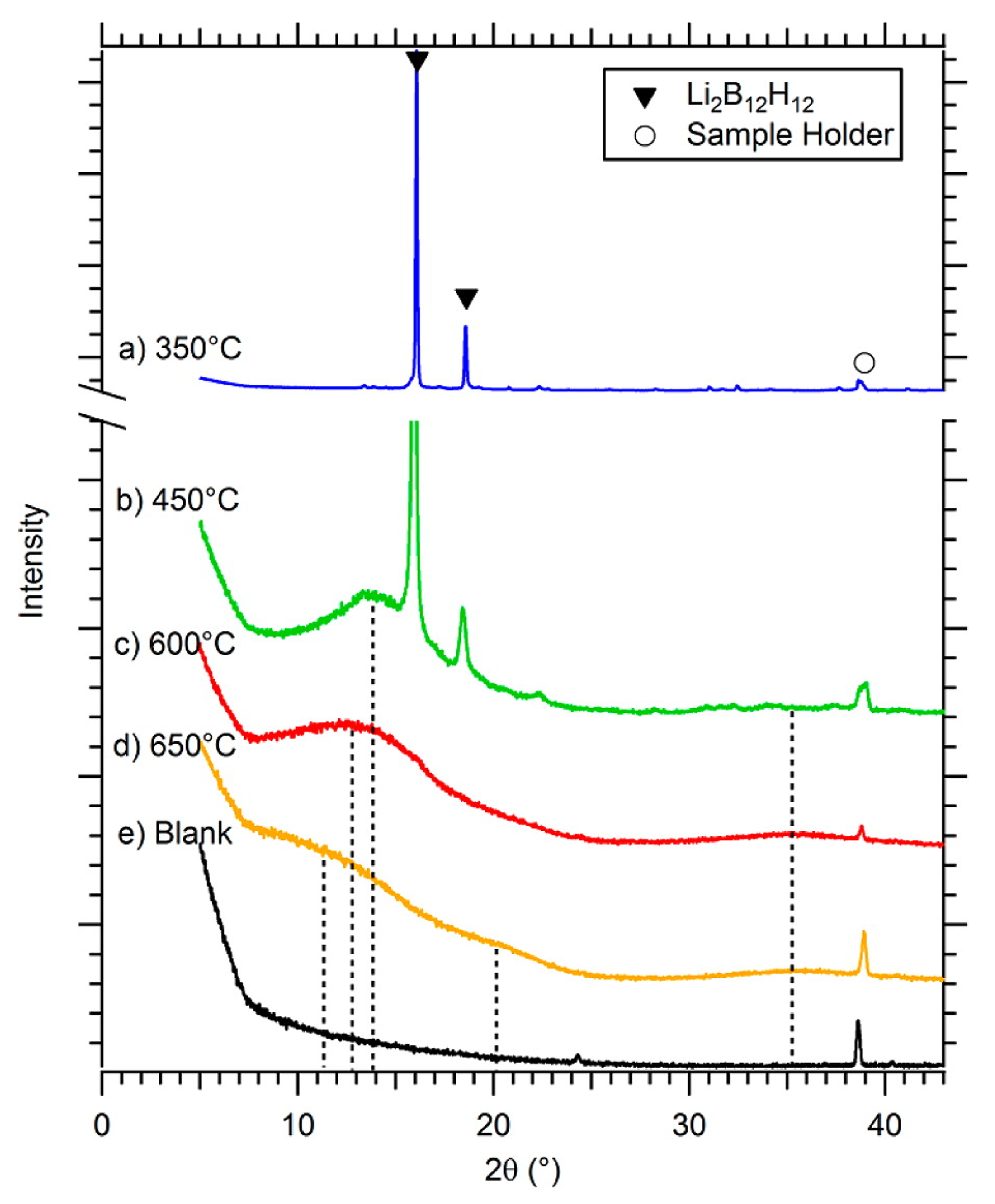
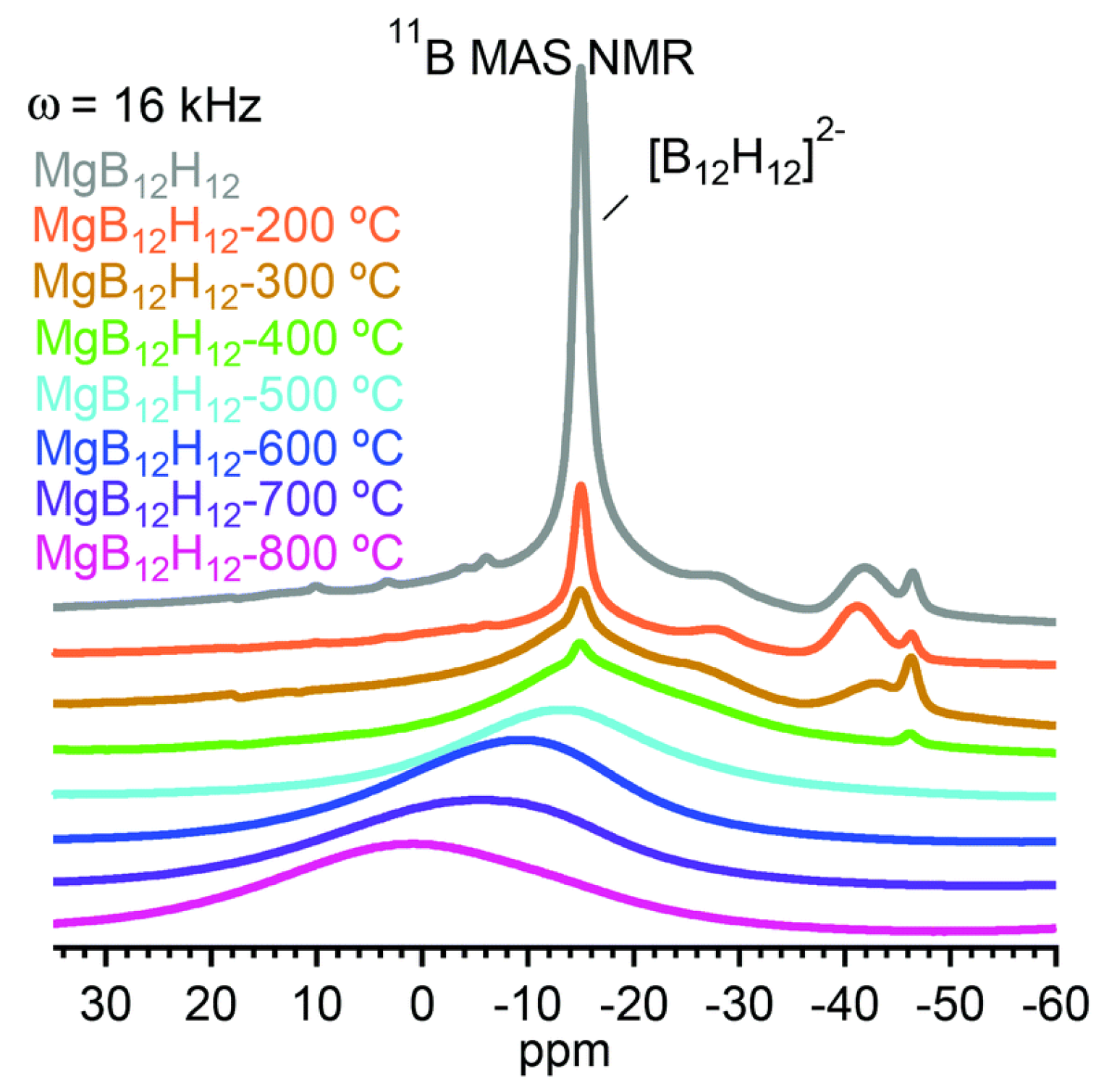
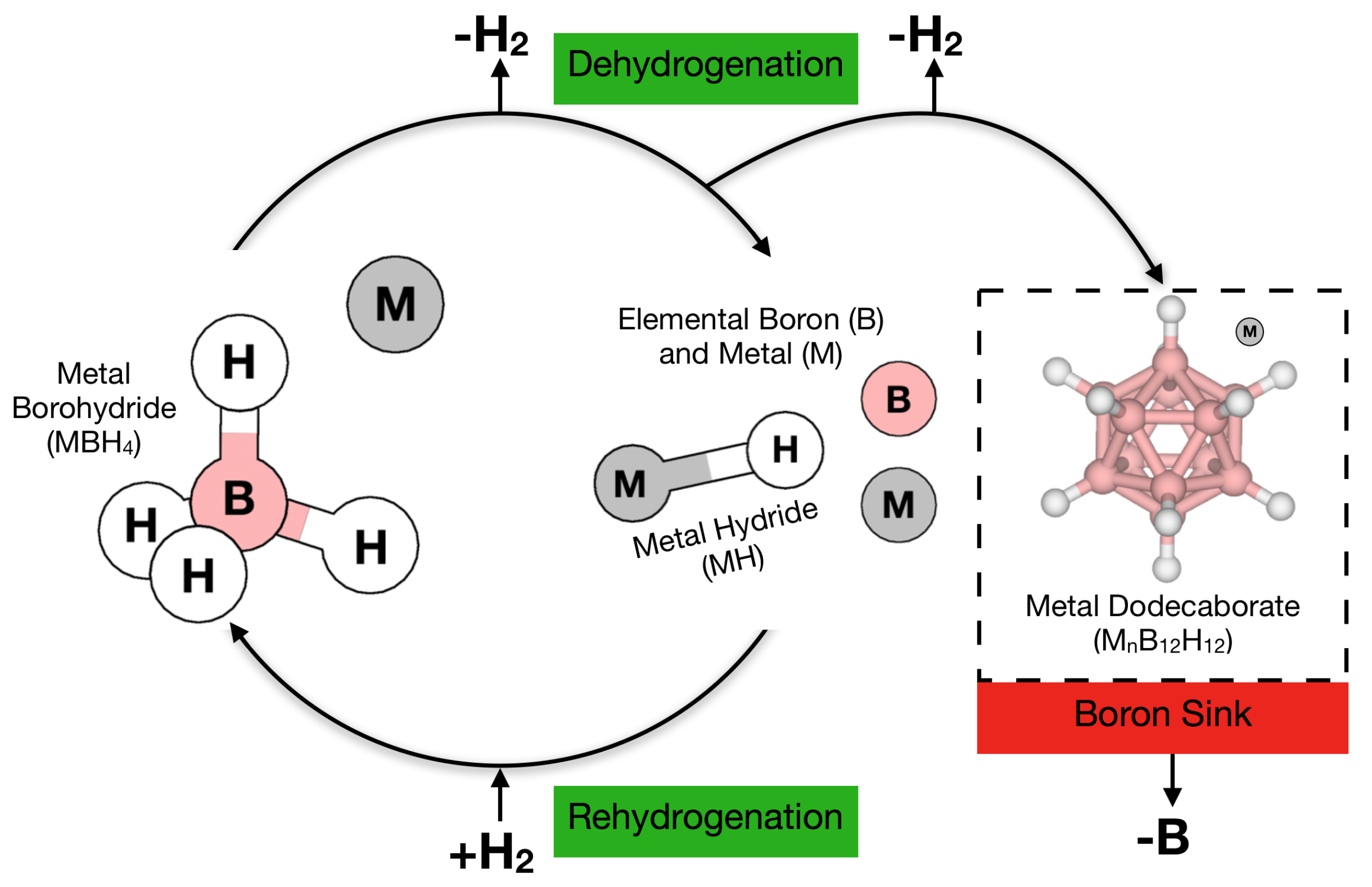
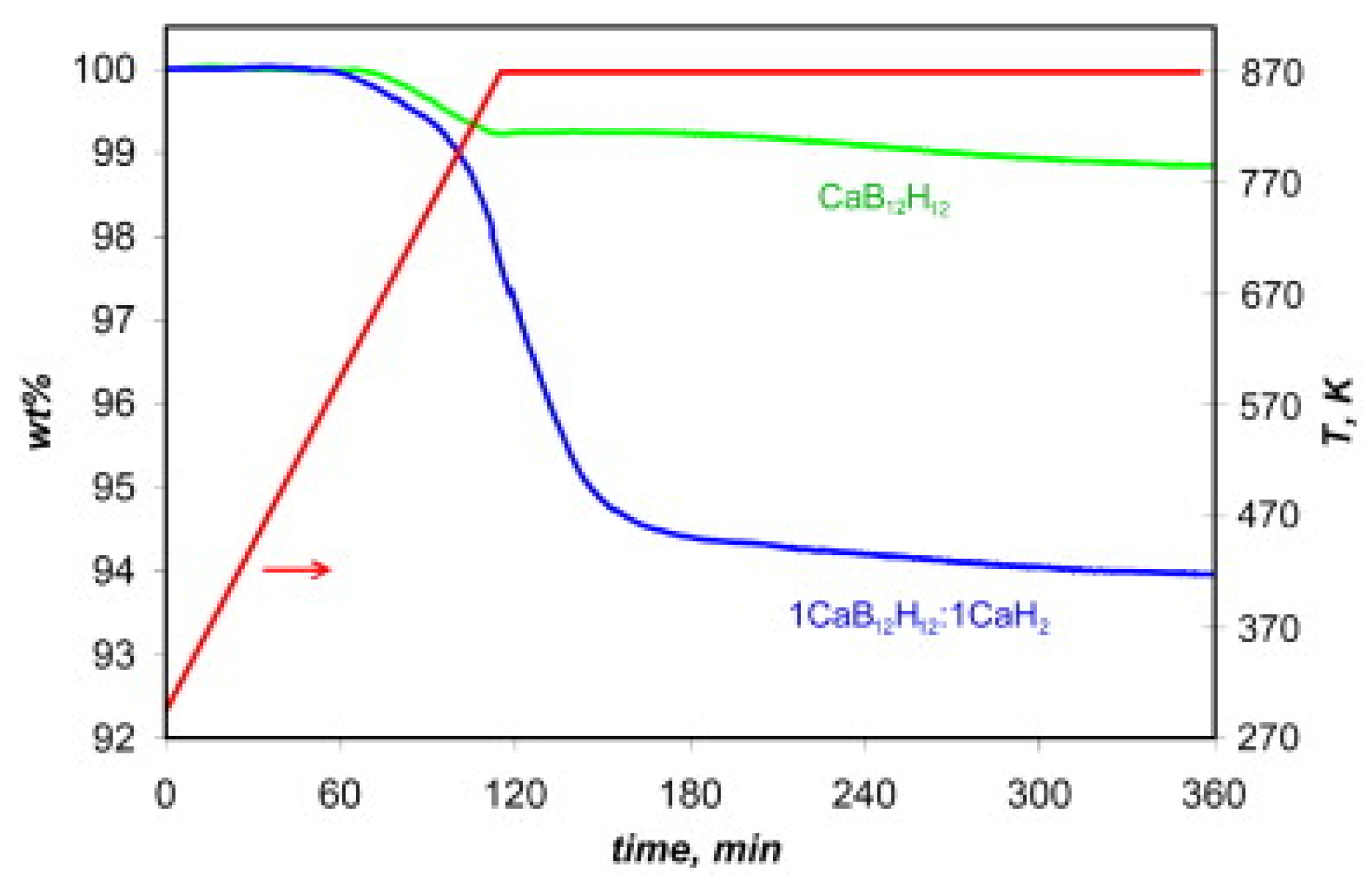
3.4. Role of Mn=1,2B12H12 in Borohydride Dehydrogenation
4. Approaches to the Mitigation of Mn=1,2B12H12 Formation
4.1. Catalysis of Mn=1,2B12H12 Dehydrogenation and Rehydrogenation
4.2. Reactive Hydride Composites
4.3. Nanoconfinement
4.4. Perspective
5. Applications of Dodecaborates and Their Derivatives
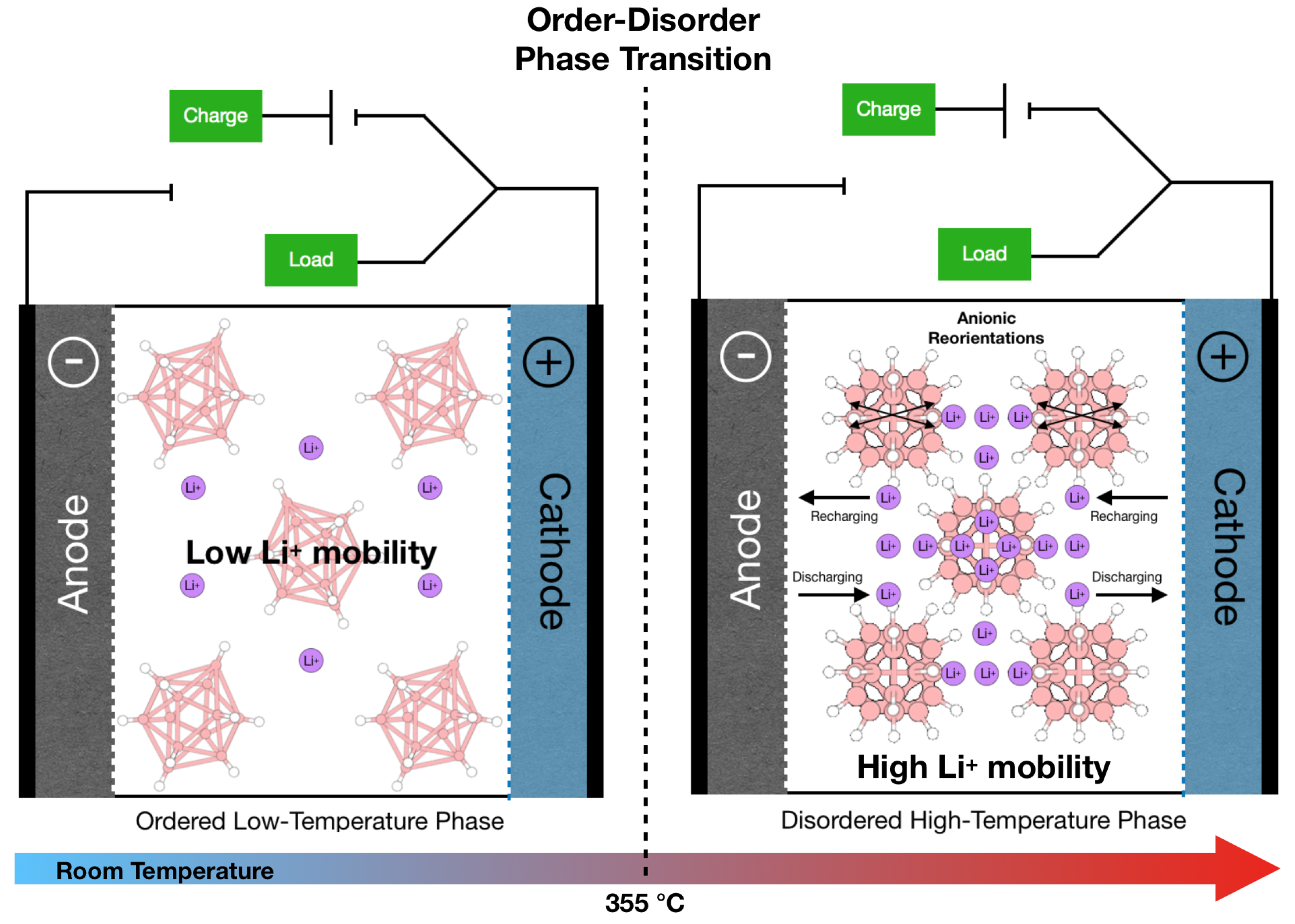
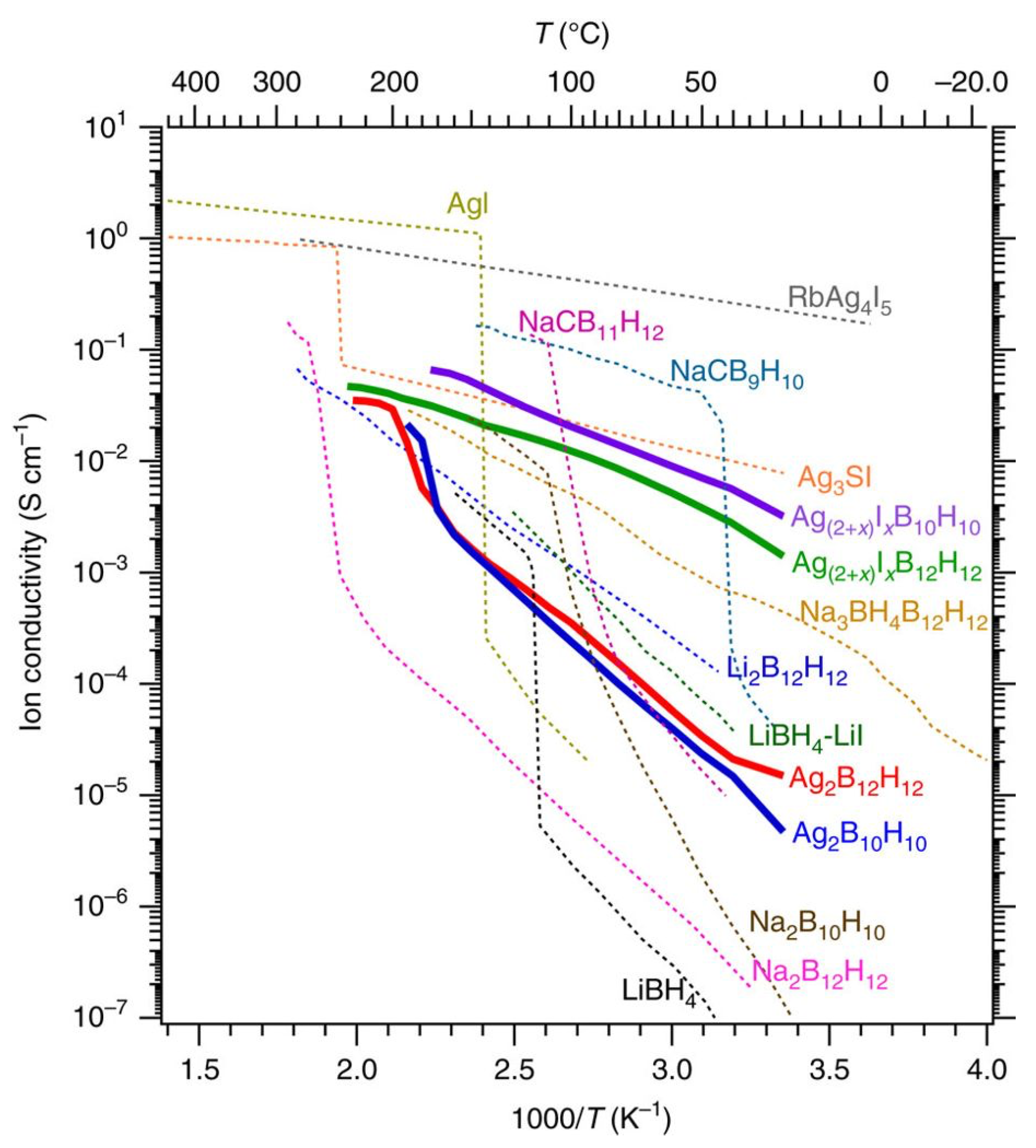
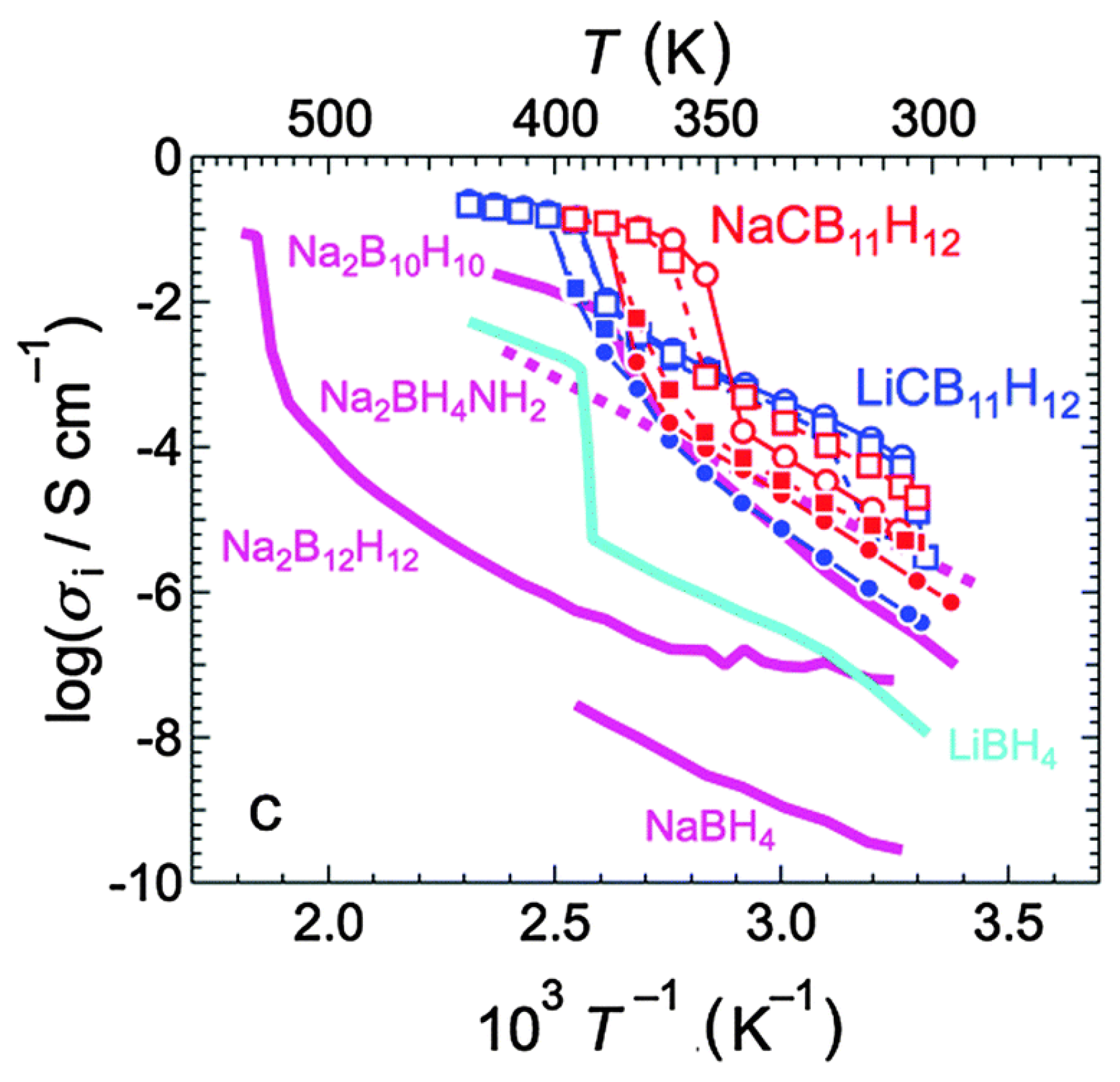
5.1. Lithium-Ion Battery Technology
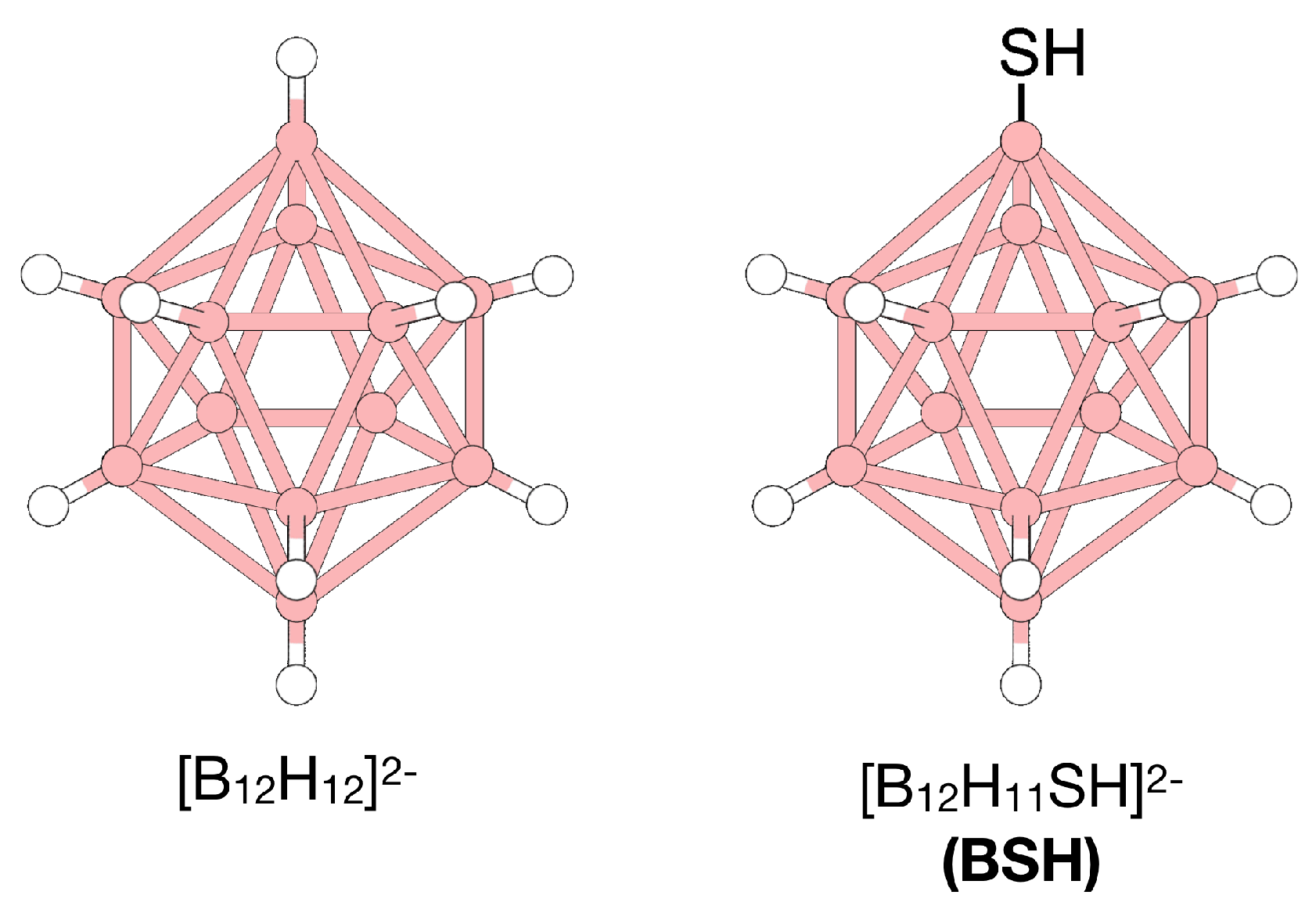
5.2. Other Applications
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lai, Q.; Paskevicius, M.; Sheppard Drew, A.; Buckley Craig, E.; Thornton Aaron, W.; Hill Matthew, R.; Gu, Q.; Mao, J.; Huang, Z.; Liu Hua, K.; et al. Hydrogen Storage Materials for Mobile and Stationary Applications: Current State of the Art. ChemSusChem 2015, 8, 2789–2825. [Google Scholar] [CrossRef] [PubMed]
- Bockris, J.O.M. The hydrogen economy: Its history. Int. J. Hydrog. Energy 2013, 38, 2579–2588. [Google Scholar] [CrossRef]
- Andújar, J.M.; Segura, F. Fuel cells: History and updating. A walk along two centuries. Renew. Sustain. Energy Rev. 2009, 13, 2309–2322. [Google Scholar] [CrossRef]
- Ley, M.B.; Jepsen, L.H.; Lee, Y.S.; Cho, Y.W.; Bellosta von Colbe, J.M.; Dornheim, M.; Rokni, M.; Jensen, J.O.; Sloth, M.; Filinchuk, Y.; et al. Complex hydrides for hydrogen storage: new perspectives. Mater. Today 2014, 17, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Bockris, J.O.; Appleby, A.J. The hydrogen economy: an ultimate economy? Environ. This Mon. 1972, 1, 29–35. [Google Scholar] [CrossRef]
- Makridis, S. Hydrogen storage and compression. In Methane and Hydrogen for Energy Storage; Carriveau, R., Ting, D.S.K., Eds.; IET Digital Library: Stevenage, UK, 2016; pp. 1–28. [Google Scholar] [Green Version]
- Lèon, A. Hydrogen Storage. In Hydrogen Technology: Mobile and Portable Applications.; Lèon, A., Ed.; Springer: Dordrecht, The Netherlands, 2008. [Google Scholar]
- Gray, E.M. Hydrogen storage status and prospects. Adv. Appl. Ceram. 2007, 106, 25–28. [Google Scholar] [CrossRef]
- Klell, M. Storage of Hydrogen in the Pure Form. In Handbook of Hydrogen Storage: New Materials for Future Energy Storage; Hirscher, M., Ed.; Wiley Online Books, Wiley-VCH: Weinheim, Germany, 2010; pp. 1–36. [Google Scholar]
- Krainz, G.; Bartlok, G.; Bodner, P.; Casapicola, P.; Doeller, C.; Hofmeister, F.; Neubacher, E.; Zieger, A. Development of Automotive Liquid Hydrogen Storage Systems. AIP Conf. Proc. 2004, 710, 35–40. [Google Scholar] [CrossRef] [Green Version]
- Orimo, S.I.; Nakamori, Y.; Eliseo, J.R.; Züttel, A.; Jensen, C.M. Complex Hydrides for Hydrogen Storage. Chem. Rev. 2007, 107, 4111–4132. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shen, C.; Lai, Q.; Liu, W.; Wang, D.W.; Aguey-Zinsou, K.F. Tailoring magnesium based materials for hydrogen storage through synthesis: Current state of the art. Energy Storage Mater. 2018, 10, 168–198. [Google Scholar] [CrossRef]
- Lai, Q.; Wang, T.; Sun, Y.; Aguey-Zinsou, K.F. Rational Design of Nanosized Light Elements for Hydrogen Storage: Classes, Synthesis, Characterization, and Properties. Adv. Mater. Technol. 2018, 3, 1700298. [Google Scholar] [CrossRef]
- Qiu, S.; Chu, H.; Zou, Y.; Xiang, C.; Xu, F.; Sun, L. Light metal borohydrides/amides combined hydrogen storage systems: composition, structure and properties. J. Mater. Chem. A 2017, 5, 25112–25130. [Google Scholar] [CrossRef]
- Møller, T.K.; Sheppard, D.; Ravnsbæk, B.D.; Buckley, E.C.; Akiba, E.; Li, H.W.; Jensen, R.T. Complex Metal Hydrides for Hydrogen, Thermal and Electrochemical Energy Storage. Energies 2017, 10. [Google Scholar] [CrossRef]
- Yu, X.; Tang, Z.; Sun, D.; Ouyang, L.; Zhu, M. Recent advances and remaining challenges of nanostructured materials for hydrogen storage applications. Prog. Mater. Sci. 2017, 88, 1–48. [Google Scholar] [CrossRef]
- Rusman, N.A.A.; Dahari, M. A review on the current progress of metal hydrides material for solid-state hydrogen storage applications. Int. J. Hydrog. Energy 2016, 41, 12108–12126. [Google Scholar] [CrossRef]
- Liu, B.H.; Li, Z.P. A review: Hydrogen generation from borohydride hydrolysis reaction. J. Power Sources 2009, 187, 527–534. [Google Scholar] [CrossRef]
- Çakanyildirim, C.; Gürü, M. Hydrogen cycle with sodium borohydride. Int. J. Hydrog. Energy 2008, 33, 4634–4639. [Google Scholar] [CrossRef]
- Demirci, U.B.; Akdim, O.; Andrieux, J.; Hannauer, J.; Chamoun, R.; Miele, P. Sodium Borohydride Hydrolysis as Hydrogen Generator: Issues, State of the Art and Applicability Upstream from a Fuel Cell. Fuel Cells 2010, 10, 335–350. [Google Scholar] [CrossRef]
- Ouyang, L.; Zhong, H.; Li, H.W.; Zhu, M. A Recycling Hydrogen Supply System of NaBH4 Based on a Facile Regeneration Process: A Review. Inorganics 2018, 6. [Google Scholar] [CrossRef]
- Kojima, Y.; Haga, T. Recycling process of sodium metaborate to sodium borohydride. Int. J. Hydrog. Energy 2003, 28, 989–993. [Google Scholar] [CrossRef]
- Hsueh, C.L.; Liu, C.H.; Chen, B.H.; Chen, C.Y.; Kuo, Y.C.; Hwang, K.J.; Ku, J.R. Regeneration of spent-NaBH4 back to NaBH4 by using high-energy ball milling. Int. J. Hydrog. Energy 2009, 34, 1717–1725. [Google Scholar] [CrossRef]
- Li, Z.P.; Liu, B.H.; Zhu, J.K.; Morigasaki, N.; Suda, S. NaBH4 formation mechanism by reaction of sodium borate with Mg and H2. J. Alloys Compd. 2007, 437, 311–316. [Google Scholar] [CrossRef]
- Suda, S.; Morigasaki, N.; Iwase, Y.; Li, Z.P. Production of sodium borohydride by using dynamic behaviors of protide at the extreme surface of magnesium particles. J. Alloys Compd. 2005, 404–406, 643–647. [Google Scholar] [CrossRef]
- Ouyang, L.; Chen, W.; Liu, J.; Felderhoff, M.; Wang, H.; Zhu, M. Enhancing the Regeneration Process of Consumed NaBH4 for Hydrogen Storage. Adv. Energy Mater. 2017, 7, 1700299. [Google Scholar] [CrossRef]
- Miwa, K.; Ohba, N.; Towata, S.I.; Nakamori, Y.; Orimo, S.I. First-principles study on lithium borohydride LiBH4. Phys. Rev. B 2004, 69, 245120. [Google Scholar] [CrossRef]
- Nakamori, Y.; Li, H.W.; Kikuchi, K.; Aoki, M.; Miwa, K.; Towata, S.; Orimo, S. Thermodynamical stabilities of metal-borohydrides. J. Alloys Compd. 2007, 446–447, 296–300. [Google Scholar] [CrossRef]
- Nakamori, Y.; Miwa, K.; Ninomiya, A.; Li, H.; Ohba, N.; Towata, S.I.; Züttel, A.; Orimo, S.I. Correlation between thermodynamical stabilities of metal borohydrides and cation electronegativites: First-principles calculations and experiments. Phys. Rev. B 2006, 74, 045126. [Google Scholar] [CrossRef]
- Miwa, K.; Ohba, N.; Towata, S.; Nakamori, Y.; Orimo, S. First-principles study on copper-substituted lithium borohydride, (Li1-xCux)BH4. J. Alloys Compd. 2005, 404–406, 140–143. [Google Scholar] [CrossRef]
- Vajeeston, P.; Ravindran, P.; Kjekshus, A.; Fjellvåg, H. Structural stability of alkali boron tetrahydrides ABH4 (A = Li, Na, K, Rb, Cs) from first principle calculation. J. Alloys Compd. 2005, 387, 97–104. [Google Scholar] [CrossRef]
- Paskevicius, M.; Jepsen, L.H.; Schouwink, P.; Cerny, R.; Ravnsbæk, D.B.; Filinchuk, Y.; Dornheim, M.; Besenbacherf, F.; Jensen, T.R. Metal borohydrides and derivatives: Synthesis, structure and properties. Chem. Soc. Rev. 2017, 46, 1565–1634. [Google Scholar] [CrossRef] [PubMed]
- Mauron, P.; Buchter, F.; Friedrichs, O.; Remhof, A.; Bielmann, M.; Zwicky, C.N.; Züttel, A. Stability and Reversibility of LiBH4. J. Phys. Chem. B 2008, 112, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Ohba, N.; Miwa, K.; Aoki, M.; Noritake, T.; Towata, S.I.; Nakamori, Y.; Orimo, S.I.; Züttel, A. First-principles study on the stability of intermediate compounds of LiBH4. Phys. Rev. B 2006, 74, 075110. [Google Scholar] [CrossRef]
- Martelli, P.; Caputo, R.; Remhof, A.; Mauron, P.; Borgschulte, A.; Züttel, A. Stability and Decomposition of NaBH4. J. Phys. Chem. C 2010, 114, 7173–7177. [Google Scholar] [CrossRef]
- Smith, M.B.; Bass, G.E. Heats and Free Energies of Formation of the Alkali Aluminum Hydrides and of Cesium Hydride. J. Chem. Eng. Data 1963, 8, 342–346. [Google Scholar] [CrossRef]
- Matsunaga, T.; Buchter, F.; Mauron, P.; Bielman, M.; Nakamori, Y.; Orimo, S.; Ohba, N.; Miwa, K.; Towata, S.; Züttel, A. Hydrogen storage properties of Mg[BH4]2. J. Alloys Compd. 2008, 459, 583–588. [Google Scholar] [CrossRef]
- Li, H.W.; Kikuchi, K.; Nakamori, Y.; Ohba, N.; Miwa, K.; Towata, S.; Orimo, S. Dehydriding and rehydriding processes of well-crystallized Mg(BH4)2 accompanying with formation of intermediate compounds. Acta Mater. 2008, 56, 1342–1347. [Google Scholar] [CrossRef]
- Mao, J.; Guo, Z.; Poh, C.K.; Ranjbar, A.; Guo, Y.; Yu, X.; Liu, H. Study on the dehydrogenation kinetics and thermodynamics of Ca(BH4)2. J. Alloys Compd. 2010, 500, 200–205. [Google Scholar] [CrossRef]
- Kim, Y.; Reed, D.; Lee, Y.S.; Lee, J.Y.; Shim, J.H.; Book, D.; Cho, Y.W. Identification of the Dehydrogenated Product of Ca(BH4)2. J. Phys. Chem. C 2009, 113, 5865–5871. [Google Scholar] [CrossRef]
- Severa, G.; Rönnebro, E.; Jensen, C.M. Direct hydrogenation of magnesium boride to magnesium borohydride: demonstration of >11 weight percent reversible hydrogen storage. Chem. Commun. 2010, 46, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Li, H.W.; Miwa, K.; Ohba, N.; Fujita, T.; Sato, T.; Yan, Y.; Towata, S.; Chen, M.W.; Orimo, S. Formation of an intermediate compound with a B12H12 cluster: experimental and theoretical studies on magnesium borohydride Mg(BH4)2. Nanotechnology 2009, 20, 204013. [Google Scholar] [CrossRef] [PubMed]
- Rönnebro, E.; Majzoub, E.H. Calcium Borohydride for Hydrogen Storage: Catalysis and Reversibility. J. Phys. Chem. B 2007, 111, 12045–12047. [Google Scholar] [CrossRef] [PubMed]
- Ngene, P.; van Zwienen, M.; de Jongh, P.E. Reversibility of the hydrogen desorption from LiBH4: a synergetic effect of nanoconfinement and Ni addition. Chem. Commun. 2010, 46, 8201–8203. [Google Scholar] [CrossRef] [PubMed]
- Orimo, S.; Nakamori, Y.; Kitahara, G.; Miwa, K.; Ohba, N.; Towata, S.; Züttel, A. Dehydriding and rehydriding reactions of LiBH4. J. Alloys Compd. 2005, 404–406, 427–430. [Google Scholar] [CrossRef]
- Wang, H.; Lin, H.J.; Cai, W.T.; Ouyang, L.Z.; Zhu, M. Tuning kinetics and thermodynamics of hydrogen storage in light metal element based systems: A review of recent progress. J. Alloys Compd. 2016, 658, 280–300. [Google Scholar] [CrossRef]
- Rude Line, H.; Nielsen Thomas, K.; Ravnsbæk Dorthe, B.; Bösenberg, U.; Ley Morten, B.; Richter, B.; Arnbjerg Lene, M.; Dornheim, M.; Filinchuk, Y.; Besenbacher, F.; et al. Tailoring properties of borohydrides for hydrogen storage: A review. Phys. Status Solidi A 2011, 208, 1754–1773. [Google Scholar] [CrossRef]
- Soulié, J.P.; Renaudin, G.; Černỳ, R.; Yvon, K. Lithium boro-hydride LiBH4: I. Crystal structure. J. Alloys Compd. 2002, 346, 200–205. [Google Scholar] [CrossRef]
- Pistorius Carl, W.F.T. Melting and Polymorphism of LiBH4 to 45 kbar. Z. Phys. Chem. 1974, 88, 253–263. [Google Scholar] [CrossRef]
- Filinchuk, Y.; Chernyshov, D.; Nevidomskyy, A.; Dmitriev, V. High-Pressure Polymorphism as a Step towards Destabilization of LiBH4. Angew. Chem. Int. Ed. 2007, 47, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, S.C.; Kalnajs, J. The Lattice Constants of the Alkali Borohydrides and the Low-Temperature Phase of Sodium Borohydride. J. Chem. Phys. 1954, 22, 434–436. [Google Scholar] [CrossRef]
- Allis, D.G.; Hudson, B.S. Inelastic neutron scattering spectra of NaBH4 and KBH4: reproduction of anion mode shifts via periodic DFT. Chem. Phys. Lett. 2004, 385, 166–172. [Google Scholar] [CrossRef]
- Filinchuk, Y.; Talyzin, A.V.; Chernyshov, D.; Dmitriev, V. High-pressure phase of NaBH4: Crystal structure from synchrotron powder diffraction data. Phys. Rev. B 2007, 76, 092104. [Google Scholar] [CrossRef]
- Renaudin, G.; Gomes, S.; Hagemann, H.; Keller, L.; Yvon, K. Structural and spectroscopic studies on the alkali borohydrides MBH4 (M = Na, K, Rb, Cs). J. Alloys Compd. 2004, 375, 98–106. [Google Scholar] [CrossRef]
- Kumar, R.S.; Kim, E.; Cornelius, A.L. Structural Phase Transitions in the Potential Hydrogen Storage Compound KBH4 under Compression. J. Phys. Chem. C 2008, 112, 8452–8457. [Google Scholar] [CrossRef]
- Züttel, A.; Wenger, P.; Rentsch, S.; Sudan, P.; Mauron, P.; Emmenegger, C. LiBH4 a new hydrogen storage material. J. Power Sources 2003, 118, 1–7. [Google Scholar] [CrossRef]
- Mosegaard, L.; Møller, B.; Jørgensen, J.E.; Filinchuk, Y.; Cerenius, Y.; Hanson, J.C.; Dimasi, E.; Besenbacher, F.; Jensen, T.R. Reactivity of LiBH4: In-Situ Synchrotron Radiation Powder X-ray Diffraction Study. J. Phys. Chem. C 2008, 112, 1299–1303. [Google Scholar] [CrossRef]
- Ozoliņš, V.; Majzoub, E.H.; Wolverton, C. First-Principles Prediction of Thermodynamically Reversible Hydrogen Storage Reactions in the Li-Mg-Ca-B-H System. J. Am. Chem. Soc. 2009, 131, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, O.; Borgschulte, A.; Kato, S.; Buchter, F.; Gremaud, R.; Remhof, A.; Züttel, A. Low-Temperature Synthesis of LiBH4 by Gas-Solid Reaction. Chem. A Eur. J. 2009, 15, 5531–5534. [Google Scholar] [CrossRef] [PubMed]
- Caputo, R.; Züttel, A. First-principles study of the paths of the decomposition reaction of LiBH4. Mol. Phys. 2010, 108, 1263–1276. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Sequeira, C.A.C. Sodium borohydride as a fuel for the future. Renew. Sustain. Energy Rev. 2011, 15, 3980–4001. [Google Scholar] [CrossRef]
- Muir, S.S.; Yao, X. Progress in sodium borohydride as a hydrogen storage material: Development of hydrolysis catalysts and reaction systems. Int. J. Hydrog. Energy 2011, 36, 5983–5997. [Google Scholar] [CrossRef]
- Kim, E.; Kumar, R.; Weck, P.F.; Cornelius, A.L.; Nicol, M.; Vogel, S.C.; Zhang, J.; Hartl, M.; Stowe, A.C.; Daemen, L.; et al. Pressure-Driven Phase Transitions in NaBH4: Theory and Experiments. J. Phys. Chem. B 2007, 111, 13873–13876. [Google Scholar] [CrossRef] [PubMed]
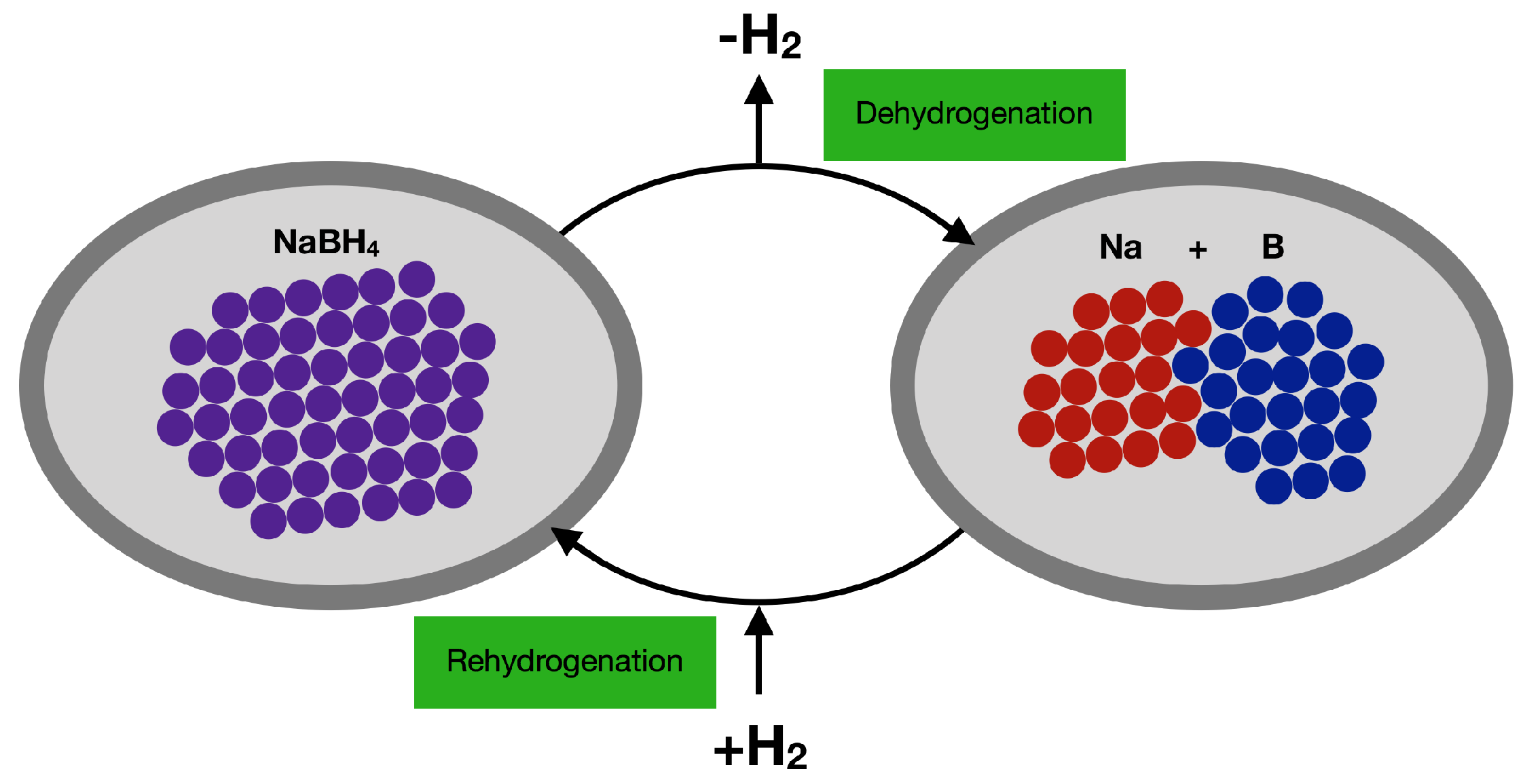
- Çakir, D.; de Wijs, G.A.; Brocks, G. Native Defects and the Dehydrogenation of NaBH4. J. Phys. Chem. C 2011, 115, 24429–24434. [Google Scholar] [CrossRef]
- Friedrichs, O.; Remhof, A.; Hwang, S.J.; Züttel, A. Role of Li2B12H12 for the Formation and Decomposition of LiBH4. Chem. Mater. 2010, 22, 3265–3268. [Google Scholar] [CrossRef]
- Urgnani, J.; Torres, F.J.; Palumbo, M.; Baricco, M. Hydrogen release from solid state NaBH4. Int. J. Hydrog. Energy 2008, 33, 3111–3115. [Google Scholar] [CrossRef]
- Mao, J.; Guo, Z.; Yu, X.; Liu, H. Improved Hydrogen Storage Properties of NaBH4 Destabilized by CaH2 and Ca(BH4)2. J. Phys. Chem. C 2011, 115, 9283–9290. [Google Scholar] [CrossRef]
- Garroni, S.; Milanese, C.; Pottmaier, D.; Mulas, G.; Nolis, P.; Girella, A.; Caputo, R.; Olid, D.; Teixdor, F.; Baricco, M.; et al. Experimental Evidence of Na2B12H12 and Na Formation in the Desorption Pathway of the 2NaBH4 + MgH2 System. J. Phys. Chem. C 2011, 115, 16664–16671. [Google Scholar] [CrossRef]
- Ngene, P.; van den Berg, R.; Verkuijlen, M.H.W.; de Jong, K.P.; de Jongh, P.E. Reversibility of the hydrogen desorption from NaBH4 by confinement in nanoporous carbon. Energy Environ. Sci. 2011, 4, 4108–4115. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.C.; Sholl, D.S. Crystal Structures and Thermodynamic Investigations of LiK(BH4)2, KBH4, and NaBH4 from First-Principles Calculations. J. Phys. Chem. C 2010, 114, 678–686. [Google Scholar] [CrossRef]
- Černỳ, R.; Filinchuk, Y.; Hagemann, H.; Yvon, K. Magnesium Borohydride: Synthesis and Crystal Structure. Angew. Chem. Int. Ed. 2007, 46, 5765–5767. [Google Scholar] [CrossRef] [PubMed]
- Her, J.H.; Stephens, P.W.; Gao, Y.; Soloveichik, G.L.; Rijssenbeek, J.; Andrus, M.; Zhao, J.C. Structure of unsolvated magnesium borohydride Mg(BH4)2. Acta Crystallogr. Sect. B 2007, 63, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Filinchuk, Y.; Richter, B.; Jensen Torben, R.; Dmitriev, V.; Chernyshov, D.; Hagemann, H. Porous and Dense Magnesium Borohydride Frameworks: Synthesis, Stability, and Reversible Absorption of Guest Species. Angew. Chem. Int. Ed. 2011, 50, 11162–11166. [Google Scholar] [CrossRef] [PubMed]
- Filinchuk, Y.; Rönnebro, E.; Chandra, D. Crystal structures and phase transformations in Ca(BH4)2. Acta Mater. 2009, 57, 732–738. [Google Scholar] [CrossRef]
- Buchter, F.; Lodziana, Z.; Remhof, A.; Friedrichs, O.; Borgschulte, A.; Mauron, P.; Züttel, A.; Sheptyakov, D.; Barkhordarian, G.; Bormann, R.; et al. Structure of Ca(BD4)2 beta-phase from combined neutron and synchrotron X-ray powder diffraction data and density functional calculations. J. Phys. Chem. B 2008, 112, 8042–8048. [Google Scholar] [CrossRef] [PubMed]
- Soloveichik, G.L.; Gao, Y.; Rijssenbeeka, J.; Andrusa, M.; Kniajanskia, S.; Bowman, R.C., Jr.; Hwang, S.J.; Zhao, J.C. Magnesium borohydride as a hydrogen storage material: Properties and dehydrogenation pathway of unsolvated Mg(BH4)2. Int. J. Hydrog. Energy 2009, 34, 916–928. [Google Scholar] [CrossRef]
- Chłopek, K.; Frommen, C.; Leon, A.; Zabara, O.; Fichtner, M. Synthesis and properties of magnesium tetrahydroborate, Mg(BH4)2. J. Mater. Chem. 2007, 17, 3496–3503. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, X.; Zheng, J.; Song, P.; Li, X. Decomposition pathway of Mg(BH4)2 under pressure: Metastable phases and thermodynamic parameters. Scr. Mater. 2011, 64, 225–228. [Google Scholar] [CrossRef]
- Paskevicius, M.; Pitt, M.P.; Webb, C.J.; Sheppard, D.A.; Filsø, U.; Gray, E.M.; Buckley, C.E. In-Situ X-ray Diffraction Study of Mg(BH4)2 Decomposition. J. Phys. Chem. C 2012, 116, 15231–15240. [Google Scholar] [CrossRef]
- Miwa, K.; Aoki, M.; Noritake, T.; Ohba, N.; Nakamori, Y.; Towata, S.I.; Züttel, A.; Orimo, S.I. Thermodynamical stability of calcium borohydride. Phys. Rev. B 2006, 74, 155122. [Google Scholar] [CrossRef]
- Kim, J.H.; Jin, S.A.; Shim, J.H.; Cho, Y.W. Thermal decomposition behavior of calcium borohydride Ca(BH4)2. J. Alloys Compd. 2008, 461, L20–L22. [Google Scholar] [CrossRef]
- Sahle, C.J.; Sternemann, C.; Giacobbe, C.; Yan, Y.; Weis, C.; Harder, M.; Forov, Y.; Spiekermann, G.; Tolan, M.; Krisch, M.; et al. Formation of CaB6 in the thermal decomposition of the hydrogen storage material Ca(BH4)2. Phys. Chem. Chem. Phys. 2016, 18, 19866–19872. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Remhof, A.; Rentsch, D.; Züttel, A.; Giri, S.; Jena, P. A novel strategy for reversible hydrogen storage in Ca(BH4)2. Chem. Commun. 2015, 51, 11008–11011. [Google Scholar] [CrossRef] [PubMed]
- Riktor, M.D.; Sørby, M.H.; Chłopek, K.; Fichtner, M.; Hauback, B.C. The identification of a hitherto unknown intermediate phase CaB2Hx from decomposition of Ca(BH4)2. J. Mater. Chem. 2009, 19, 2754–2759. [Google Scholar] [CrossRef]
- Aoki, M.; Miwa, K.; Noritake, T.; Ohba, N.; Matsumoto, M.; Li, H.W.; Nakamori, Y.; Towata, S.; Orimo, S. Structural and dehydriding properties of Ca(BH4)2. Appl. Phys. A 2008, 92, 601–605. [Google Scholar] [CrossRef]
- Wang, L.L.; Graham, D.D.; Robertson, I.M.; Johnson, D.D. On the Reversibility of Hydrogen-Storage Reactions in Ca(BH4)2: Characterization via Experiment and Theory. J. Phys. Chem. C 2009, 113, 20088–20096. [Google Scholar] [CrossRef]
- Kim, Y.; Hwang, S.J.; Shim, J.H.; Lee, Y.S.; Han, H.N.; Cho, Y.W. Investigation of the Dehydrogenation Reaction Pathway of Ca(BH4)2 and Reversibility of Intermediate Phases. J. Phys. Chem. C 2012, 116, 4330–4334. [Google Scholar] [CrossRef]
- Kim, Y.; Hwang, S.J.; Lee, Y.S.; Suh, J.Y.; Han, H.N.; Cho, Y.W. Hydrogen Back-Pressure Effects on the Dehydrogenation Reactions of Ca(BH4)2. J. Phys. Chem. C 2012, 116, 25715–25720. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Bregadze, V.I.; Sjöberg, S. Chemistry of closo-Dodecaborate Anion [B12H12]2−: A Review. Collect. Czech. Chem. Commun. 2002, 67. [Google Scholar] [CrossRef]
- Muetterties, E.L. Boron Hydride Chemistry; Academic Press Inc.: London, UK, 1975. [Google Scholar]
- Caputo, R.; Garroni, S.; Olid, D.; Teixidor, F.; Surinach, S.; Baro, M.D. Can Na2B12H12 be a decomposition product of NaBH4? Phys. Chem. Chem. Phys. 2010, 12, 15093–15100. [Google Scholar] [CrossRef] [PubMed]
- Pitt, M.P.; Paskevicius, M.; Brown, D.H.; Sheppard, D.A.; Buckley, C.E. Thermal Stability of Li2B12H12 and its Role in the Decomposition of LiBH4. J. Am. Chem. Soc. 2013, 135, 6930–6941. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Remhof, A.; Hwang, S.J.; Li, H.W.; Mauron, P.; Orimo, S.i.; Züttel, A. Pressure and temperature dependence of the decomposition pathway of LiBH4. Phys. Chem. Chem. Phys. 2012, 14, 6514–6519. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Li, H.W.; Akiba, E. Thermal Decomposition of Anhydrous Alkali Metal Dodecaborates M2B12H12 (M = Li, Na, K). Energies 2015, 8. [Google Scholar] [CrossRef]
- He, L.; Li, H.W.; Tumanov, N.; Filinchuk, Y.; Akiba, E. Facile synthesis of anhydrous alkaline earth metal dodecaborates MB12H12 (M = Mg, Ca) from M(BH4)2. Dalton Trans. 2015, 44, 15882–15887. [Google Scholar] [CrossRef] [PubMed]
- Remhof, A.; Yan, Y.; Rentsch, D.; Borgschulte, A.; Jensen, C.M.; Züttel, A. Solvent-free synthesis and stability of MgB12H12. J. Mater. Chem. A 2014, 2, 7244–7249. [Google Scholar] [CrossRef]
- Verdal, N.; Wu, H.; Udovic, T.J.; Stavila, V.; Zhou, W.; Rush, J.J. Evidence of a transition to reorientational disorder in the cubic alkali-metal dodecahydro-closo-dodecaborates. J. Solid State Chem. 2011, 184, 3110–3116. [Google Scholar] [CrossRef]
- Paskevicius, M.; Pitt, M.P.; Brown, D.H.; Sheppard, D.A.; Chumphongphan, S.; Buckley, C.E. First-order phase transition in the Li2B12H12 system. Phys. Chem. Chem. Phys. 2013, 15, 15825–15828. [Google Scholar] [CrossRef] [PubMed]
- Her, J.H.; Yousufuddin, M.; Zhou, W.; Jalisatgi, S.S.; Kulleck, J.G.; Zan, J.A.; Hwang, S.J.; Bowman, R.C.; Udovic, T.J. Crystal Structure of Li2B12H12: a Possible Intermediate Species in the Decomposition of LiBH4. Inorg. Chem. 2008, 47, 9757–9759. [Google Scholar] [CrossRef] [PubMed]
- Her, J.H.; Zhou, W.; Stavila, V.; Brown, C.M.; Udovic, T.J. Role of Cation Size on the Structural Behavior of the Alkali-Metal Dodecahydro-closo-Dodecaborates. J. Phys. Chem. C 2009, 113, 11187–11189. [Google Scholar] [CrossRef]
- Tiritiris, I.; Schleid, T. Die Dodekahydro-closo-Dodekaborate M2[B12H12] der schweren Alkalimetalle (M+ = K+, Rb+, NH4+, Cs+) und ihre formalen Iodid-Addukte M3I[B12H12] (MI-M2[B12H12]). Z. Anorg. Allg. Chem. 2003, 629, 1390–1402. [Google Scholar] [CrossRef]
- Stavila, V.; Her, J.H.; Zhou, W.; Hwang, S.J.; Kim, C.; Ottley, L.A.M.; Udovic, T.J. Probing the structure, stability and hydrogen storage properties of calcium dodecahydro-closo-dodecaborate. J. Solid State Chem. 2010, 183, 1133–1140. [Google Scholar] [CrossRef]
- Pitochelli, A.R.; Hawthorne, F.M. THE ISOLATION OF THE ICOSAHEDRAL B12H122− ION. J. Am. Chem. Soc. 1960, 82, 3228–3229. [Google Scholar] [CrossRef]
- Hansen, B.R.S.; Paskevicius, M.; Li, H.W.; Akiba, E.; Jensen, T.R. Metal boranes: Progress and applications. Coord. Chem. Rev. 2016, 323, 60–70. [Google Scholar] [CrossRef]
- Miller, H.C.; Miller, N.E.; Muetterties, E.L. Chemistry of Boranes. XX. Syntheses of Polyhedral Boranes. Inorg. Chem. 1964, 3, 1456–1463. [Google Scholar] [CrossRef]
- Brown, H.C.; Tierney, P.A. The Reaction of Lewis Acids of Boron with Sodium Hydride and Borohydride. J. Am. Chem. Soc. 1958, 80, 1552–1558. [Google Scholar] [CrossRef]
- Adams, R.M.; Siedle, A.R.; Grant, J. Convenient Preparation of the Dodecahydrododecaborate Ion. Inorg. Chem. 1964, 3, 461. [Google Scholar] [CrossRef]
- Safronov, A.V.; Jalisatgi, S.S.; Lee, H.B.; Hawthorne, M.F. Chemical hydrogen storage using polynuclear borane anion salts. Int. J. Hydrog. Energy 2011, 36, 234–239. [Google Scholar] [CrossRef]
- Yan, Y.; Remhof, A.; Rentsch, D.; Lee, Y.S.; Whan Cho, Y.; Züttel, A. Is Y2(B12H12)3 the main intermediate in the decomposition process of Y(BH4)3? Chem. Commun. 2013, 49, 5234–5236. [Google Scholar] [CrossRef] [PubMed]
- Chong, M.; Karkamkar, A.; Autrey, T.; Orimo, S.i.; Jalisatgi, S.; Jensen, C.M. Reversible dehydrogenation of magnesium borohydride to magnesium triborane in the solid state under moderate conditions. Chem. Commun. 2011, 47, 1330–1332. [Google Scholar] [CrossRef] [PubMed]
- Chong, M.; Autrey, T.; Jensen, M.C. Lewis Base Complexes of Magnesium Borohydride: Enhanced Kinetics and Product Selectivity upon Hydrogen Release. Inorganics 2017, 5. [Google Scholar] [CrossRef]
- Zhang, Y.; Majzoub, E.; Ozoliņš, V.; Wolverton, C. Theoretical Prediction of Metastable Intermediates in the Decomposition of Mg(BH4)2. J. Phys. Chem. C 2012, 116, 10522–10528. [Google Scholar] [CrossRef]
- Huang, Z.; Eagles, M.; Porter, S.; Sorte, E.G.; Billet, B.; Corey, R.L.; Conradi, M.S.; Zhao, J.C. Thermolysis and solid state NMR studies of NaB3H8, NH3B3H7, and NH4B3H8. Dalton Trans. 2013, 42, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Matus, M.H.; Dixon, D.A. Heats of Formation of Boron Hydride Anions and Dianions and Their Ammonium Salts [BnHmy−][NH4+]y with y = 1-2. Inorg. Chem. 2007, 46, 7561–7570. [Google Scholar] [CrossRef] [PubMed]
- Verdal, N.; Her, J.H.; Stavila, V.; Soloninin, A.V.; Babanova, O.A.; Skripov, A.V.; Udovic, T.J.; Rush, J.J. Complex high-temperature phase transitions in Li2B12H12 and Na2B12H12. J. Solid State Chem. 2014, 212, 81–91. [Google Scholar] [CrossRef]
- Udovic Terrence, J.; Matsuo, M.; Tang Wan, S.; Wu, H.; Stavila, V.; Soloninin Alexei, V.; Skoryunov Roman, V.; Babanova Olga, A.; Skripov Alexander, V.; Rush John, J.; et al. Exceptional Superionic Conductivity in Disordered Sodium Decahydro-closo-decaborate. Adv. Mater. 2014, 26, 7622–7626. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.S.; Dimitrievska, M.; Stavila, V.; Zhou, W.; Wu, H.; Talin, A.A.; Udovic, T.J. Order-Disorder Transitions and Superionic Conductivity in the Sodium nido-Undeca(carba)borates. Chem. Mater. 2017, 29, 10496–10509. [Google Scholar] [CrossRef]
- Bykov, A.Y.; Zhizhin, K.Y.; Kuznetsov, N.T. The chemistry of the octahydrotriborate anion [B3H8]−. Russ. J. Inorg. Chem. 2014, 59, 1539–1555. [Google Scholar] [CrossRef]
- Driess, M.; Nöth, H. Molecular Clusters of the Main Group Elements; Wiley-VCH: Berlin, Germany, 2008. [Google Scholar]
- Wade, K. Structural and Bonding Patterns in Cluster Chemistry. In Advances in Inorganic Chemistry and Radiochemistry; Emelèus, H.J., Sharpe, A.G., Eds.; Academic Press: Cambridge, MA, USA, 1976; Volume 18, pp. 1–66. [Google Scholar]
- Mingos, D.M.P. A General Theory for Cluster and Ring Compounds of the Main Group and Transition Elements. Nat. Phys. Sci. 1972, 236, 99–102. [Google Scholar] [CrossRef]
- Schubert, D.M. Boron Chemistry for Hydrogen Storage. In Boron Science: New Technologies and Applications; Hosmane, N.S., Ed.; CRC Press: Boca Raton, FL, USA, 2012; pp. 393–397. [Google Scholar]
- Hansen, B.R.S.; Ravnsbæk, D.B.; Skibsted, J.; Jensen, T.R. Hydrogen reversibility of LiBH4-MgH2-Al composites. Phys. Chem. Chem. Phys. 2014, 16, 8970–8980. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.R.S.; Ravnsbæk, D.B.; Reed, D.; Book, D.; Gundlach, C.; Skibsted, J.; Jensen, T.R. Hydrogen Storage Capacity Loss in a LiBH4-Al Composite. J. Phys. Chem. C 2013, 117, 7423–7432. [Google Scholar] [CrossRef]
- Kim, J.H.; Shim, J.H.; Cho, Y.W. On the reversibility of hydrogen storage in Ti- and Nb-catalyzed Ca(BH4)2. J. Power Sources 2008, 181, 140–143. [Google Scholar] [CrossRef]
- Kim, J.H.; Jin, S.A.; Shim, J.H.; Cho, Y.W. Reversible hydrogen storage in calcium borohydride Ca(BH4)2. Scr. Mater. 2008, 58, 481–483. [Google Scholar] [CrossRef]
- Rongeat, C.; D’Anna, V.; Hagemann, H.; Borgschulte, A.; Züttel, A.; Schultz, L.; Gutfleisch, O. Effect of additives on the synthesis and reversibility of Ca(BH4)2. J. Alloys Compd. 2010, 493, 281–287. [Google Scholar] [CrossRef]
- Gosalawit-Utke, R.; Suarez, K.; Bellosta von Colbe, J.M.; Bösenberg, U.; Jensen, T.R.; Cerenius, Y.; Bonatto Minella, C.; Pistidda, C.; Barkhordarian, G.; Schulze, M.; et al. Ca(BH4)2-MgF2 Reversible Hydrogen Storage: Reaction Mechanisms and Kinetic Properties. J. Phys. Chem. C 2011, 115, 3762–3768. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, Y.S.; Suh, J.Y.; Shim, J.H.; Cho, Y.W. Metal halide doped metal borohydrides for hydrogen storage: The case of Ca(BH4)2-CaX2 (X = F, Cl) mixture. J. Alloys Compd. 2010, 506, 721–727. [Google Scholar] [CrossRef]
- White, J.L.; Newhouse, R.J.; Zhang, J.Z.; Udovic, T.J.; Stavila, V. Understanding and Mitigating the Effects of Stable Dodecahydrocloso-dodecaborate Intermediates on Hydrogen-Storage Reactions. J. Phys. Chem. C 2016, 120, 25725–25731. [Google Scholar] [CrossRef]
- Jensen, S.R.H.; Paskevicius, M.; Hansen, B.R.S.; Jakobsen, A.S.; Møller, K.T.; White, J.L.; Allendorf, M.D.; Stavila, V.; Skibsted, J.; Jensen, T.R. Hydrogenation properties of lithium and sodium hydride—closo-borate, [B10H10]2− and [B12H12]2−, composites. Phys. Chem. Chem. Phys. 2018, 20, 16266–16275. [Google Scholar] [CrossRef] [PubMed]
- Bonatto Minella, C.; Garroni, S.; Olid, D.; Teixidor, F.; Pistidda, C.; Lindemann, I. Experimental Evidence of CaB12H12 Formation During Decomposition of a Ca(BH4)2 + MgH2 Based Reactive Hydride Composite. J. Phys. Chem. C 2011, 115, 18010–18014. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, H.; Zhu, M.; Cai, W.; Rentsch, D.; Remhof, A. Direct Rehydrogenation of LiBH4 from H-Deficient Li2B12H12-x. Crystals 2018, 8. [Google Scholar] [CrossRef]
- Tang, W.S.; Wu, G.; Liu, T.; Wee, A.T.S.; Yong, C.K.; Xiong, Z.; Hor, A.T.S.; Chen, P. Cobalt-catalyzed hydrogen desorption from the LiNH2-LiBH4 system. Dalton Trans. 2008, 2395–2399. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wu, F.; Bai, Y.; Yi, B.; Zhang, H. Cobalt boride catalysts for hydrogen generation from alkaline NaBH4 solution. Mater. Lett. 2005, 59, 1748–1751. [Google Scholar] [CrossRef]
- Paskevicius, M.; Ley, M.B.; Sheppard, D.A.; Jensen, T.R.; Buckley, C.E. Eutectic melting in metal borohydrides. Phys. Chem. Chem. Phys. 2013, 15, 19774–19789. [Google Scholar] [CrossRef] [PubMed]
- Dornheim, M.; Doppiu, S.; Barkhordarian, G.; Boesenberg, U.; Klassen, T.; Gutfleisch, O.; Bormann, R. Hydrogen storage in magnesium-based hydrides and hydride composites. Scr. Mater. 2007, 56, 841–846. [Google Scholar] [CrossRef]
- Vajo, J.J.; Skeith, S.L.; Mertens, F. Reversible Storage of Hydrogen in Destabilized LiBH4. J. Phys. Chem. B 2005, 109, 3719–3722. [Google Scholar] [CrossRef] [PubMed]
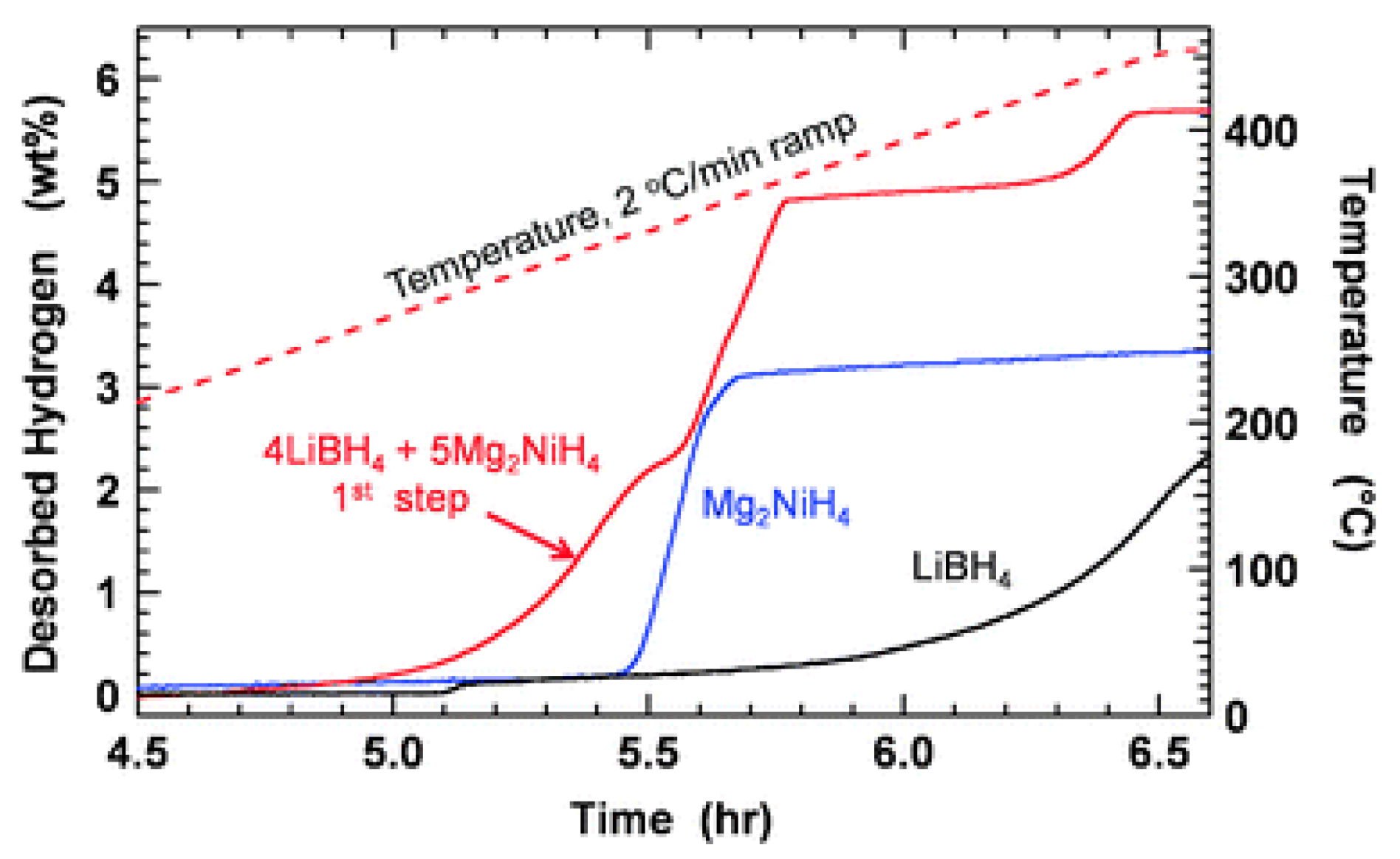
- Bergemann, N.; Pistidda, C.; Milanese, C.; Emmler, T.; Karimi, F.; Chaudhary, A.L.; Chierotti, M.R.; Klassen, T.; Dornheim, M. Ca(BH4)2-Mg2NiH4: On the pathway to a Ca(BH4)2 system with a reversible hydrogen cycle. Chem. Commun. 2016, 52, 4836–4839. [Google Scholar] [CrossRef] [PubMed]
- Vajo, J.J.; Li, W.; Liu, P. Thermodynamic and kinetic destabilization in LiBH4/Mg2NiH4: Promise for borohydride-based hydrogen storage. Chem. Commun. 2010, 46, 6687–6689. [Google Scholar] [CrossRef] [PubMed]
- Afonso, G.; Bonakdarpour, A.; Wilkinson, D.P. Hydrogen Storage Properties of the Destabilized 4NaBH4/5Mg2NiH4 Composite System. J. Phys. Chem. C 2013, 117, 21105–21111. [Google Scholar] [CrossRef]
- Bösenberg, U.; Doppiu, S.; Mosegaard, L.; Barkhordarian, G.; Eigen, N.; Borgschulte, A.; Jensen, T.R.; Cerenius, Y.; Gutfleisch, O.; Klassen, T.; et al. Hydrogen sorption properties of MgH2-LiBH4 composites. Acta Mater. 2007, 55, 3951–3958. [Google Scholar] [CrossRef]
- Maximilian, F. Properties of nanoscale metal hydrides. Nanotechnology 2009, 20, 204009. [Google Scholar]
- Zhao-Karger, Z.; Witter, R.; Bardaji, E.G.; Wang, D.; Cossement, D.; Fichtner, M. Altered reaction pathways of eutectic LiBH4-Mg(BH4)2 by nanoconfinement. J. Mater. Chem. A 2013, 1, 3379–3386. [Google Scholar] [CrossRef]
- Nale, A.; Catti, M.; Bardají, E.G.; Fichtner, M. On the decomposition of the 0.6LiBH4-0.4Mg(BH4)2 eutectic mixture for hydrogen storage. Int. J. Hydrog. Energy 2011, 36, 13676–13682. [Google Scholar] [CrossRef]
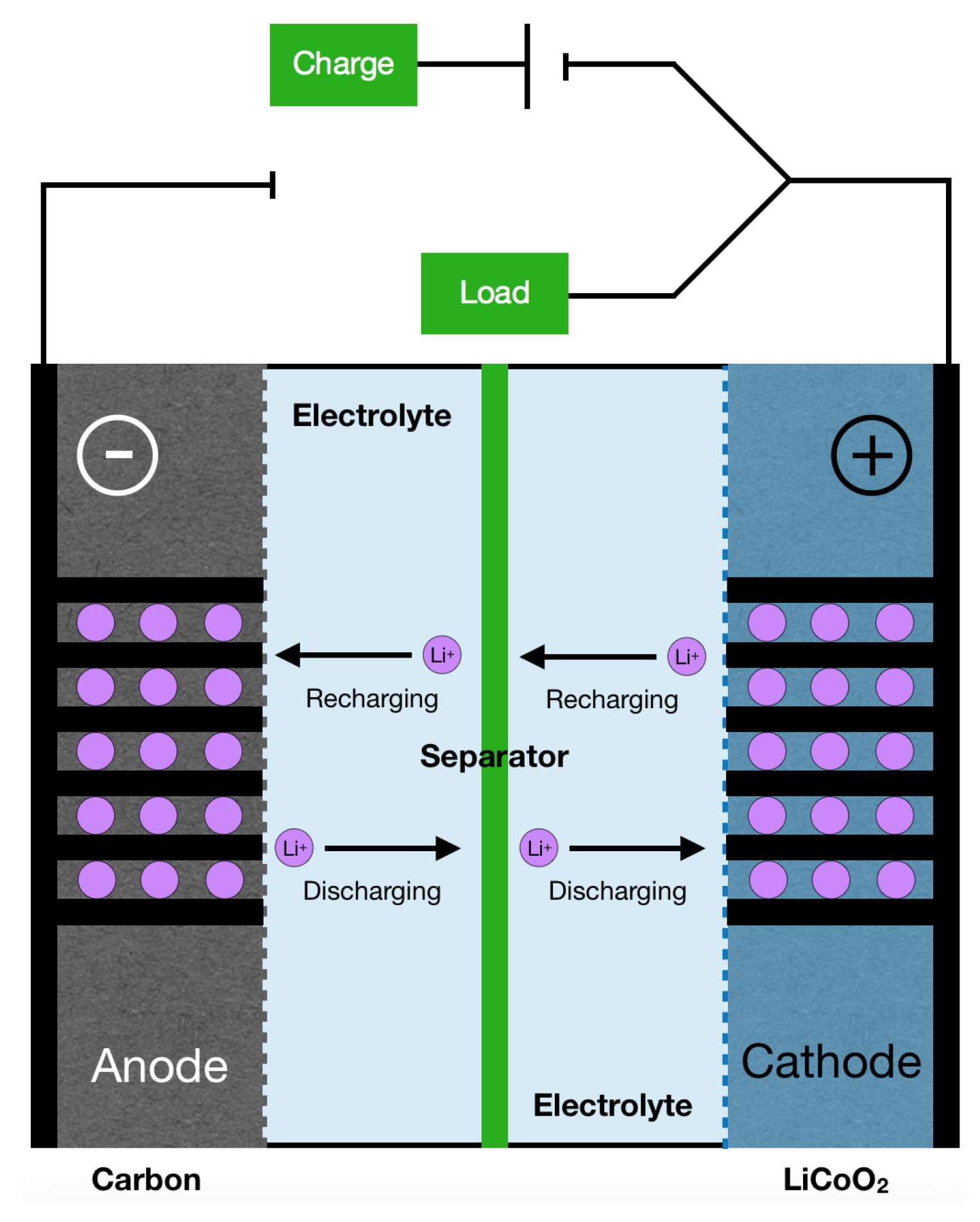
- Etacheri, V.; Marom, R.; Elazari, R.; Salitra, G.; Aurbach, D. Challenges in the development of advanced Li-ion batteries: a review. Energy Environ. Sci. 2011, 4, 3243–3262. [Google Scholar] [CrossRef]
- Goodenough, J.B.; Kim, Y. Challenges for Rechargeable Li Batteries. Chem. Mater. 2010, 22, 587–603. [Google Scholar] [CrossRef]
- Wang, Q.; Ping, P.; Zhao, X.; Chu, G.; Sun, J.; Chen, C. Thermal runaway caused fire and explosion of lithium ion battery. J. Power Sources 2012, 208, 210–224. [Google Scholar] [CrossRef]
- Xiayin, Y.; Bingxin, H.; Jingyun, Y.; Gang, P.; Zhen, H.; Chao, G.; Deng, L.; Xiaoxiong, X. All-solid-state lithium batteries with inorganic solid electrolytes: Review of fundamental science. Chin. Phys. B 2016, 25, 018802. [Google Scholar]
- Skripov, A.V.; Babanova, O.A.; Soloninin, A.V.; Stavila, V.; Verdal, N.; Udovic, T.J.; Rush, J.J. Nuclear Magnetic Resonance Study of Atomic Motion in A2B12H12 (A = Na, K, Rb, Cs): Anion Reorientations and Na+ Mobility. J. Phys. Chem. C 2013, 117, 25961–25968. [Google Scholar] [CrossRef]
- Kumar, P.P.; Yashonath, S. Ionic conduction in the solid state. J. Chem. Sci. 2006, 118, 135–154. [Google Scholar] [CrossRef]
- Chandra, A. Ion conduction in crystalline superionic solids and its applications. Eur. Phys. J.-Appl. Phys. 2014, 66, 30905. [Google Scholar] [CrossRef]
- Tiritiris, I.; Schleid, T.; Müller, K. Solid-State NMR Studies on Ionic closo-Dodecaborates. Appl. Magn. Reson. 2007, 32, 459–481. [Google Scholar] [CrossRef]
- Udovic, T.J.; Matsuo, M.; Unemoto, A.; Verdal, N.; Stavila, V.; Skripov, A.V.; Rush, J.J.; Takamura, H.; Orimo, S.I. Sodium superionic conduction in Na2B12H12. Chem. Commun. 2014, 50, 3750–3752. [Google Scholar] [CrossRef] [PubMed]
- Uvarov, N.F.; Ulikhin, A.S.; Iskakova, A.A.; Medvedev, N.N.; Anikeenko, A.V. Ionic conductivity in orientationally disordered phases. Russ. J. Electrochem. 2011, 47, 404. [Google Scholar] [CrossRef]
- Tang, W.S.; Unemoto, A.; Zhou, W.; Stavila, V.; Matsuo, M.; Wu, H.; Orimo, S.i.; Udovic, T.J. Unparalleled lithium and sodium superionic conduction in solid electrolytes with large monovalent cage-like anions. Energy Environ. Sci. 2015, 8, 3637–3645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paskevicius, M.; Hansen, B.R.S.; Jørgensen, M.; Richter, B.; Jensen, T.R. Multifunctionality of silver closo-boranes. Nat. Commun. 2017, 8, 15136. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Li, H.W.; Nakajima, H.; Tumanov, N.; Filinchuk, Y.; Hwang, S.J.; Sharma, M.; Hagemann, H.; Akiba, E. Synthesis of a Bimetallic Dodecaborate LiNaB12H12 with Outstanding Superionic Conductivity. Chem. Mater. 2015, 27, 5483–5486. [Google Scholar] [CrossRef]
- Duchene, L.; Kuhnel, R.S.; Rentsch, D.; Remhof, A.; Hagemann, H.; Battaglia, C. A highly stable sodium solid-state electrolyte based on a dodeca/deca-borate equimolar mixture. Chem. Commun. 2017, 53, 4195–4198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.W.; Thompson, A.H. Lithium Closoboranes II. Stable Nonaqueous Electrolytes for Elevated Temperature Lithium Cells. J. Electrochem. Soc. 1981, 128, 932–933. [Google Scholar] [CrossRef]
- Dey, A.N.; Miller, J. Primary Li/SOCl2 Cells: VII . Effect of Li2B10Cl10 and Li2B12Cl12 Electrolyte Salts on the Performance. J. Electrochem. Soc. 1979, 126, 1445–1451. [Google Scholar] [CrossRef]
- Hansen, B.R.S.; Paskevicius, M.; Jørgensen, M.; Jensen, T.R. Halogenated Sodium-closo-Dodecaboranes as Solid-State Ion Conductors. Chem. Mater. 2017, 29, 3423–3430. [Google Scholar] [CrossRef]
- Kweon, K.E.; Varley, J.B.; Shea, P.; Adelstein, N.; Mehta, P.; Heo, T.W.; Udovic, T.J.; Stavila, V.; Wood, B.C. Structural, Chemical, and Dynamical Frustration: Origins of Superionic Conductivity in closo-Borate Solid Electrolytes. Chem. Mater. 2017, 29, 9142–9153. [Google Scholar] [CrossRef]
- Oumellal, Y.; Rougier, A.; Nazri, G.A.; Tarascon, J.M.; Aymard, L. Metal hydrides for lithium-ion batteries. Nat. Mater. 2008, 7, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Meggiolaro, D.; Farina, L.; Silvestri, L.; Panero, S.; Brutti, S.; Reale, P. Lightweight Borohydrides Electro-Activity in Lithium Cells. Energies 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Mason, T.H.; Liu, X.; Hong, J.; Graetz, J.; Majzoub, E.H. First-Principles Study of Novel Conversion Reactions for High-Capacity Li-Ion Battery Anodes in the Li-Mg-B-N-H System. J. Phys. Chem. C 2011, 115, 16681–16687. [Google Scholar] [CrossRef]
- Valliant, J.F.; Guenther, K.J.; King, A.S.; Morel, P.; Schaffer, P.; Sogbein, O.O.; Stephenson, K.A. The medicinal chemistry of carboranes. Coord. Chem. Rev. 2002, 232, 173–230. [Google Scholar] [CrossRef]
- Barth, R.F.; Zhang, Z.; Liu, T. A realistic appraisal of boron neutron capture therapy as a cancer treatment modality. Cancer Commun. 2018, 38, 36. [Google Scholar] [CrossRef] [PubMed]
- Barth, R.F.; Mi, P.; Yang, W. Boron delivery agents for neutron capture therapy of cancer. Cancer Commun. 2018, 38, 35. [Google Scholar] [CrossRef] [PubMed]
- Mi, P.; Yanagie, H.; Dewi, N.; Yen, H.C.; Liu, X.; Suzuki, M.; Sakurai, Y.; Ono, K.; Takahashi, H.; Cabral, H.; et al. Block copolymer-boron cluster conjugate for effective boron neutron capture therapy of solid tumors. J. Control. Release 2017, 254, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Kanoh, D.; Sato, S.; Sakurai, Y.; Suzuki, M.; Nakamura, H. Maleimide-functionalized closo-dodecaborate albumin conjugates (MID-AC): Unique ligation at cysteine and lysine residues enables efficient boron delivery to tumor for neutron capture therapy. J. Control. Release 2016, 237, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Coffer, J.L.; Gillen, J.G.; Brewer, T.M. Incorporation of Cesium Borocaptate onto Silicon Nanowires as a Delivery Vehicle for Boron Neutron Capture Therapy. Chem. Mater. 2010, 22, 279–281. [Google Scholar] [CrossRef]
- Gao, Z.; Horiguchi, Y.; Nakai, K.; Matsumura, A.; Suzuki, M.; Ono, K.; Nagasaki, Y. Use of boron cluster-containing redox nanoparticles with ROS scavenging ability in boron neutron capture therapy to achieve high therapeutic efficiency and low adverse effects. Biomaterials 2016, 104, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, P.P.; Biswas, S.; Torchilin, V.P. Current trends in the use of liposomes for tumor targeting. Nanomedicine 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Ishida, O.; Kasaoka, S.; Takizawa, T.; Utoguchi, N.; Shinohara, A.; Chiba, M.; Kobayashi, H.; Eriguchi, M.; Yanagie, H. Intracellular targeting of sodium mercaptoundecahydrododecaborate (BSH) to solid tumors by transferrin-PEG liposomes, for boron neutron-capture therapy (BNCT). J. Control. Release 2004, 98, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H. Chapter 10—Liposomal Boron Delivery for Neutron Capture Therapy. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2009; Volume 465, pp. 179–208. [Google Scholar]
- Lee, J.D.; Ueno, M.; Miyajima, Y.; Nakamura, H. Synthesis of Boron Cluster Lipids: closo-Dodecaborate as an Alternative Hydrophilic Function of Boronated Liposomes for Neutron Capture Therapy. Org. Lett. 2007, 9, 323–326. [Google Scholar] [CrossRef] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
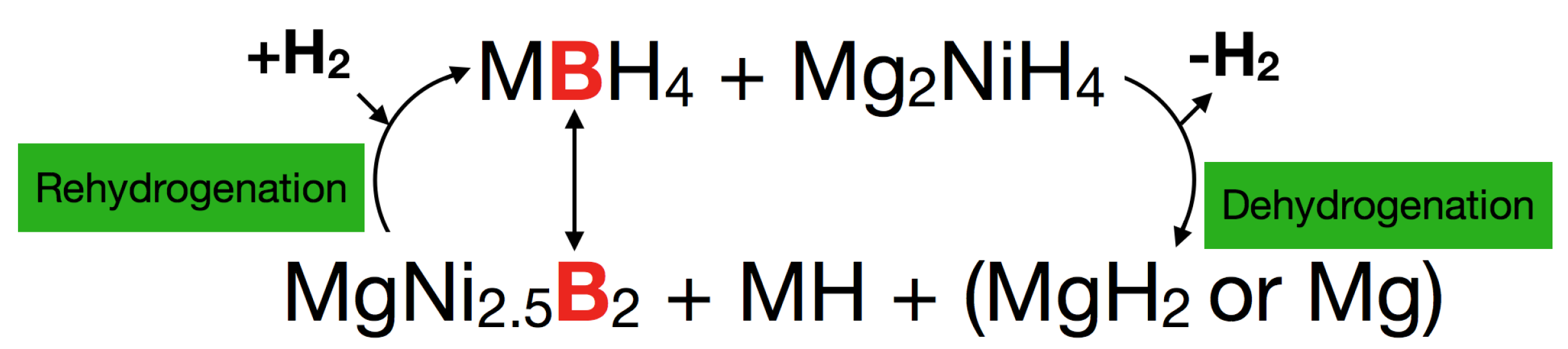{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Borohydride | Hdec (kJ mol−1 H2) | Tdec at 0.1 MPa H2 (C) | Reference |
|---|---|---|---|
| LiBH4 | 56–75 | 370 | [33,34] |
| NaBH4 | 89.6–108 | 539 | [35,36] |
| KBH4 | 113.9 | 826 | [36] |
| Mg(BH4)2 | 39.3–57 | 157 | [37,38] |
| Ca(BH4)2 | 40.6–87 | 278 | [39,40] |
| Metal Species | Polymorph | Crystal System | Space Group | Reference |
|---|---|---|---|---|
| Li | o-LiBH4 | Orthorhombic | Pnma | [48] |
| h–LiBH4 | Hexagonal | P63mc | [48] | |
| hp1–LiBH4 | Orthorhombic | Ama2 | [50] | |
| hp2–LiBH4 | Cubic | Fm3m | [50] | |
| Na | –NaBH4 | Cubic | Fm3m | [51] |
| –NaBH4 | Tetragonal | P421c or P42/nmc | [31,52] | |
| –NaBH4 | Orthorhombic | Pnma | [53] | |
| K | –KBH4 | Cubic | Fm3m | [51] |
| ’–KBH4 | Tetragonal | P42/nmc or P421c | [54] | |
| –KBH4 | Orthorhombic | Pnma | [55] |
| Metal Species | Polymorph | Crystal System | Space Group | Reference |
|---|---|---|---|---|
| Mg | –Mg(BH4)2 | Hexagonal | P6122 | [71] |
| –Mg(BH4)2 | Orthorhombic | Fddd | [72] | |
| –Mg(BH4)2 | Cubic | Ia3d | [73] | |
| –Mg(BH4)2 | Tetragonal | P42nm | [73] | |
| –Mg(BH4)2 | Hexagonal | P3112 | [73] | |
| Ca | –Ca(BH4)2 | Orthorhombic | F2dd | [74] |
| ’–Ca(BH4)2 | Tetragonal | I42d | [74] | |
| –Ca(BH4)2 | Tetragonal | P4 or P42/m | [74] | |
| –Ca(BH4)2 | Orthorhombic | Pbca | [75] |
| Species | H (kJ mol−1) | Crystal System | Space Group |
|---|---|---|---|
| Li2B12H12 | −945.95 [60] | Cubic [99] | Pa [99] |
| - | Monoclinic [34] | P21/n [34] | |
| Na2B12H12 | −1086.196 [91] | Cubic [91] | Pa [91] |
| −1086.381 [91] | Monoclinic [100] | P21/n [100] | |
| K2B12H12 | - | Cubic [101] | Fm [101] |
| CaB12H12 | - | Monoclinic [102] | C2/c [102] |
| MgB12H12 | - | Monoclinic [58] | C2/m [58] |
| Anion | H (kJ mol−1) | Tdec of Nan(BH)− (C) | Anion Geometry [112] |
|---|---|---|---|
| [B12H12]2− | −328.4 [114] | 612 (He flow) [115] |  |
| [B10H10]2− | −26.8 [114] | 577 (He flow) [116] |  |
| [B11H14]− | −200.4 [114] | 127 (He flow) [117] |  |
| [B3H8]− | −72.8 [114] | 100 (Ar flow) [113,118] |  |
| Metal Species | Reaction | H (kJ mol−1) | Reference |
|---|---|---|---|
| Li | LiBH4 → Li2B12H12 + LiH + H2 | 56 | [34] |
| 2LiBH4 + 5B2H6 → Li2B12H12 + 13H2 | Unspecified | [59] | |
| 12LiBH4 → Li2B12H12 + 10LiH + 13H2 | 56.139 | [60] | |
| 12LiBH4 → Li2B12H12 + 10Li + 18H2 | 122.50 | [60] | |
| 12LiBH4 → Li2B12H12 + 10LiH + 13H2 | 40.9 | [58] | |
| Na | 2NaBH4 + 5B2H6 → Na2B12H12 + 13H2 | Unspecified | [64] |
| 12 NaBH4 → 10Na + Na2B12H12 + 18H2 | 167.313 | [91] | |
| 12 NaBH4 → 10NaH + Na2B12H12 + 13H2 | 129.386 | [91] | |
| K | KBH4 →K2B12H12 + KH + H2 | 117.5 | [70] |
| Mg | Mg(BH4)2 → MgH2 + MgB12H12 + MgB4 | Unspecified | [76] |
| 6Mg(BH4)2 → MgB12H12 + 5MgH2 + 13H2 | 25 | [58] | |
| Ca | 6Ca(BH4)2 → CaB12H12 + 5CaH2 + 13H2 | 34.2 | [58] |
| Ca(BH4)2 → CaH2 + CaB6 + H2 | 35.2 | [58] |
| Species | Transition Temperature (C) | Ionic Conductivity (S cm−1) |
|---|---|---|
| NaCB11H12 | 107 [156] | 0.15 (130 C) [156] |
| LiCB11H12 | 127 [156] | 0.12 (110 C) [156] |
| Ag2B12H12 | 200 [157] | 0.035 (227 C) [157] |
| LiNaB12H12 | 215 [158] | 0.79 (277 C) [158] |
| Na2B12H12 | 247 [97] | 0.1 (267 C) [154] |
| Li2B12H12 | 355 [98] | 0.07 (277 C) [158] |
| Na2(B12H12)0.5(B10H10)0.5, | N/A | 0.0009 (20 C) [159] |
| Ag(2+x)IxB12H12 | N/A | 0.002 (25 C) [157] |
| Cs2B12H12 | 256 [97] | - |
| Rb2B12H12 | 469 [97] | - |
| K2B12H12 | 538 [97] | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grahame, A.; Aguey-Zinsou, K.-F. Properties and Applications of Metal (M) dodecahydro-closo-dodecaborates (Mn=1,2B12H12) and Their Implications for Reversible Hydrogen Storage in the Borohydrides. Inorganics 2018, 6, 106. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6040106
Grahame A, Aguey-Zinsou K-F. Properties and Applications of Metal (M) dodecahydro-closo-dodecaborates (Mn=1,2B12H12) and Their Implications for Reversible Hydrogen Storage in the Borohydrides. Inorganics. 2018; 6(4):106. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6040106
Chicago/Turabian StyleGrahame, Aiden, and Kondo-François Aguey-Zinsou. 2018. "Properties and Applications of Metal (M) dodecahydro-closo-dodecaborates (Mn=1,2B12H12) and Their Implications for Reversible Hydrogen Storage in the Borohydrides" Inorganics 6, no. 4: 106. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6040106