(2-Pyridyloxy)silanes as Ligands in Transition Metal Coordination Chemistry


1
Institut für Anorganische Chemie, TU Bergakademie Freiberg, D-09596 Freiberg, Germany
2
Institut für Analytische Chemie, TU Bergakademie Freiberg, D-09596 Freiberg, Germany
*
Author to whom correspondence should be addressed.
Inorganics 2018, 6(4), 119; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6040119
Submission received: 29 September 2018
/
Revised: 24 October 2018
/
Accepted: 26 October 2018
/
Published: 31 October 2018
(This article belongs to the Special Issue Coordination Chemistry of Silicon)
Abstract
:Proceeding our initial studies of compounds with formally dative TM→Si bonds (TM = Ni, Pd, Pt), which feature a paddlewheel arrangement of four (N,S) or (N,N) bridging ligands around the TM–Si axis, the current study shows that the (N,O)-bidentate ligand 2-pyridyloxy (pyO) is also capable of bridging systems with TM→Si bonds (shown for TM = Pd, Cu). Reactions of MeSi(pyO)3 with [PdCl2(NCMe)2] and CuCl afforded the compounds MeSi(µ-pyO)4PdCl (1) and MeSi(µ-pyO)3CuCl (2), respectively. In the latter case, some crystals of the Cu(II) compound MeSi(µ-pyO)4CuCl (3) were obtained as a byproduct. Analogous reactions of Si(pyO)4, in the presence of HpyO, with [PdCl2(NCMe)2] and CuCl2, afforded the compounds [(HpyO)Si(µ-pyO)4PdCl]Cl (4), (HpyO)2Si[(µ-pyO)2PdCl2]2 (5), and (HpyO)2Si[(µ-pyO)2CuCl2]2 (6), respectively. Compounds 1–6 and the starting silanes MeSi(pyO)3 and Si(pyO)4 were characterized by single-crystal X-ray diffraction analyses and, with exception of the paramagnetic compounds 3 and 6, with NMR spectroscopy. Compound 2 features a pentacoordinate Si atom, the Si atoms of the other complexes are hexacoordinate. Whereas compounds 1–4 feature a TM→Si bond each, the Si atoms of compounds 5 and 6 are situated in an O6 coordination sphere, while the TMCl2 groups are coordinated to pyridine moieties in the periphery of the molecule. The TM–Si interatomic distances in compounds 1–4 are close to the sum of the covalent radii (1 and 4) or at least significantly shorter than the sum of the van-der-Waals radii (2 and 3). The latter indicates a noticeably weaker interaction for TM = Cu. For the series 1, 2, and 3, all of which feature the Me–Si motif trans-disposed to the TM→Si bond, the dependence of the TM→Si interaction on the nature of TM (Pd(II), Cu(I), and Cu(II)) was analyzed using quantum chemical calculations, that is, the natural localized molecular orbitals (NLMO) analyses, the non-covalent interaction (NCI) descriptor, Wiberg bond order (WBO), and topological characteristics of the bond critical points using the atoms in molecules (AIM) approach.
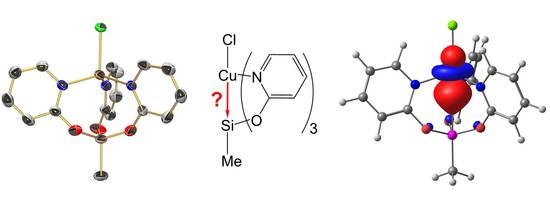
1. Introduction
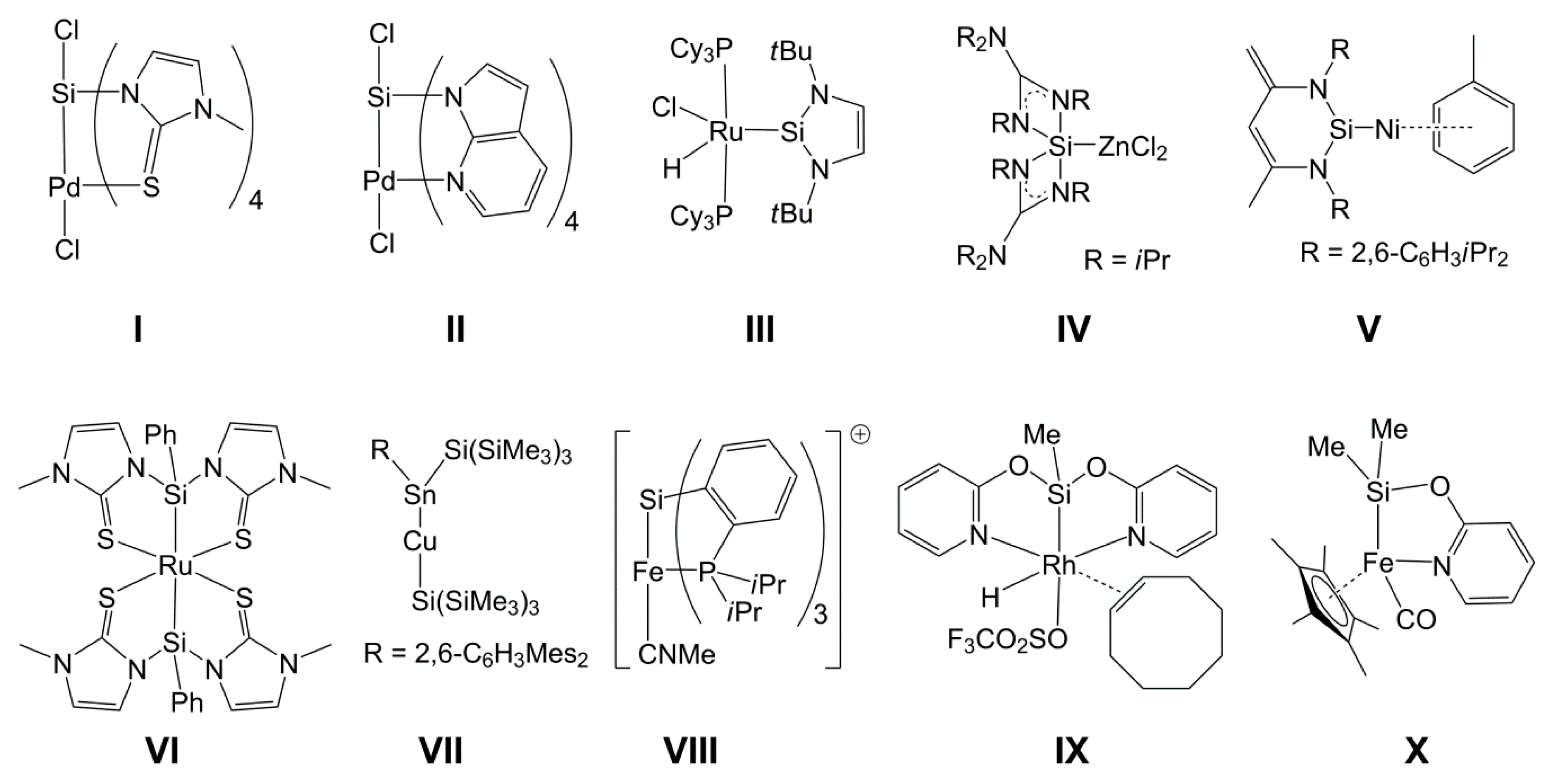
The coordination number of tetravalent silicon can easily be enhanced (up to five or six) with the aid of monodentate or chelating ligands [1,2,3,4]. In some of our studies, we have also shown that late transition metals (Ni(II), Pd(II), and Pt(II)) may serve the role of a lone pair donor at hexacoordinate silicon, for example, in I and II (Chart 1) [5,6,7,8]. That kind of complexes with silicon as a lone pair acceptor in the coordination sphere of a transition metal (TM) thus complements TM silicon complexes with, e.g., silylene ligands, in which the Si atom is the formal lone pair donor, such as III, IV, and V [9,10,11,12,13], and the silyl complexes, in which the TM–Si bond is one out of four bonds to a tetravalent Si atom (e.g., VI, VII, VIII, and IX) [14,15,16,17,18]. In complex IX and some other compounds with group 9 metals [19,20,21,22], the 2-pyridyloxy (pyO) ligand was successfully utilized for stabilizing the TM–Si bond by forming two bridges over the heterodinuclear core. Some other compounds have been reported, in which one pyO ligand bridges the TM–Si bond (e.g., X, and some others) [23,24,25]. In all of these Si(µ-pyO)TM compounds, the ambidentate pyO ligand is bound to silicon via the Si–O bond, while the softer Lewis base (pyridine N atom) is TM bound. This structural motif should enable access to a new class of paddlewheel-shaped complexes with formally dative TM→Si bonds, in which the Si atom may carry a sterically more demanding group or alkyl group, because of the rather poor steric demand of the surrounding donor atoms (i.e., O atoms), whereas in the previously reported TM→Si paddlewheel complexes (such as I and II), the donor atom situation at silicon merely allowed for the presence of a small electronegative H-acceptor moiety (i.e., a halide).
2. Results and Discussion
2.1. Syntheses and Characterization of Silanes MeSi(pyO)3 and Si(pyO)4
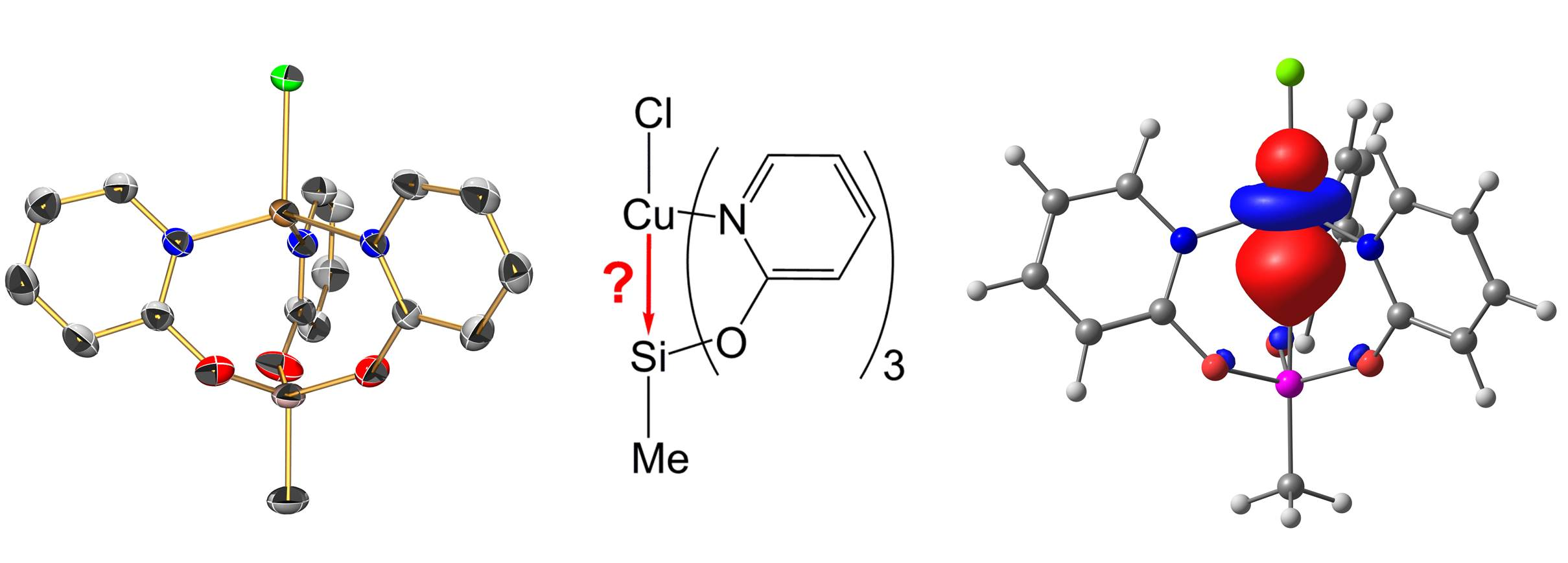
For the starting materials, we synthesized MeSi(pyO)3 via triethylamine supported reaction of MeSiCl3 and 2-hydroxypyridine, and Si(pyO)4, via transsilylation, with the preceding synthesis of Me3Si(pyO), which is known in the literature [26] (Scheme 1). Both of the silanes formed crystals suitable for single-crystal X-ray diffraction analysis (Figure 1 and Table 1). In both of the compounds, the Si atom is essentially tetracoordinate and the coordination sphere may be described as (4 + 3) in MeSi(pyO)3 and (4 + 4) in Si(pyO)4, because of the pyridine N atoms, which are capping the faces of the tetrahedral coordination spheres from distances close to the sum of the van-der-Waals radii (ranging between 2.91 and 3.03 Å). This tetracoordination of Si in Si(pyO)4 is in contrast to the Si hexacoordination in the thio analog Si(pyS)4 [27] (and other pyS-bearing Si compounds [28]), in which two of the 2-mercaptopyridyl ligands form four-membered chelates, and in complexes of the N-oxide of the pyO system, which forms five-membered chelates [29,30].
The 29Si NMR shifts (MeSi(pyO)3 in CDCl3: −46.5; Si(pyO)4 in solid state: −87.9, in CDCl3: −97.2) are in support of tetracoordination, as they are similar to (and even more downfield shifted than) the 29Si NMR shifts of the related phenoxysilanes MeSi(OPh)3 (−54.0) [31] and Si(OPh)4 (−101.1) [32], respectively. Some effect of the three- or four-fold capped coordination spheres is evident from the bond angles of the Si atoms, which exhibit notable deviations from the ideal tetrahedral angle (in MeSi(pyO)3, O1–Si1–O3 97.39(5)° and O2–Si1–O3 114.68(5)°; in Si(pyO)4, two sets of O–Si–O angles of 101.42(9)° and 113.64(5)°). In general, the Si tetracoordination in these silanes, in combination with the vacant pyridine N atoms, should be favorable for complex formation with additional metal atoms. The same feature, tetracoordinate Si atom and vacant additional donor atoms, has also been encountered with methimazolylsilanes [5,33] and 7-azaindolylsilanes [8], which turned out to be suitable starting materials for compounds such as I and II.
2.2. Choice of Metals: Pd(II) and Cu(I)
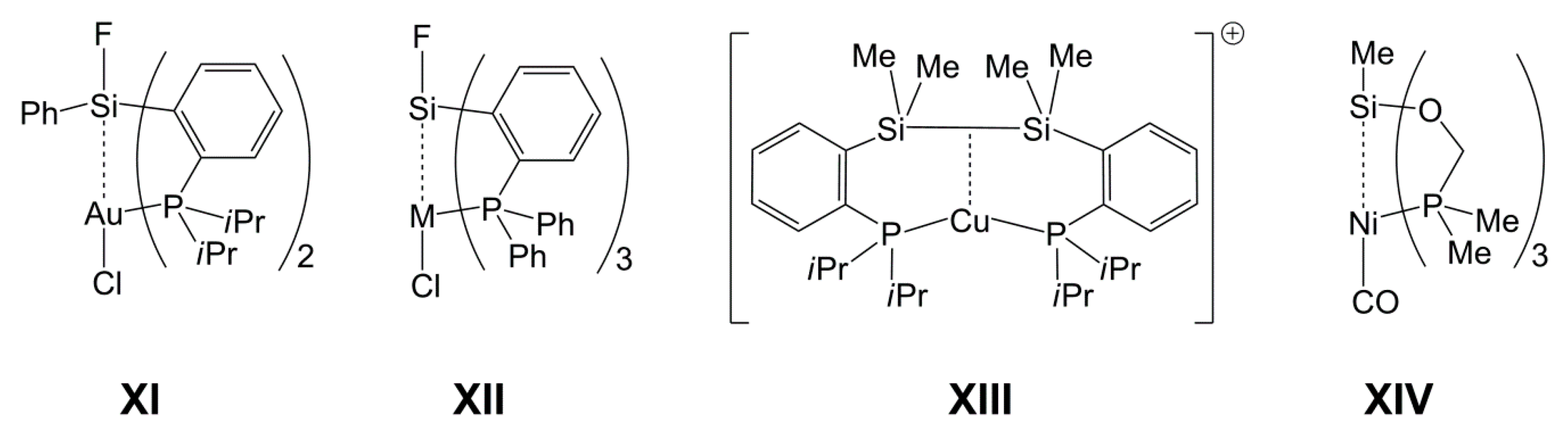
In reactions with 7-azaindolylsilanes, Pd(II) already proved to be a suitable candidate for forming paddlewheel-complexes with TM→Si bonds and TM-bound pyridine moieties (e.g., in II). In addition to d8 systems, other electron rich TMs may also be capable of forming complexes with TM→Si bonds, as shown by Bourissou et al. for the Au(I)→Si system (e.g., XI, Chart 2), in which the d10 metal is the electron pair donor [34,35,36]. Furthermore, for compounds XII (M = Cu, Ag, Au), Kameo et al. have shown that Cu(I) and Ag(I) are weaker donors [37]. In this context, we need to note that a previous study by Bourissou et al. [38] revealed an interaction between the Si–Si σ-bond electron pair of a disilane (as donor) and Cu(I) (as acceptor) (XIII). Similar systems (with rather weak d10 metal–silicon interaction) have been investigated for Ni(0), Pd(0), and Pt(0) by Grobe et al. (e.g., XIV) [39,40,41]. Whereas Au(I), Ni(0), Pd(0), and Pt(0) are more susceptible to phosphine ligands, Cu(I) represents a d10 system likely to bind to more than one or two N-donor ligands, and thus we included Cu(I) (as CuCl) in our investigations.
2.3. Reactions of MeSi(pyO)3 with [PdCl2(NCMe)2] and CuCl
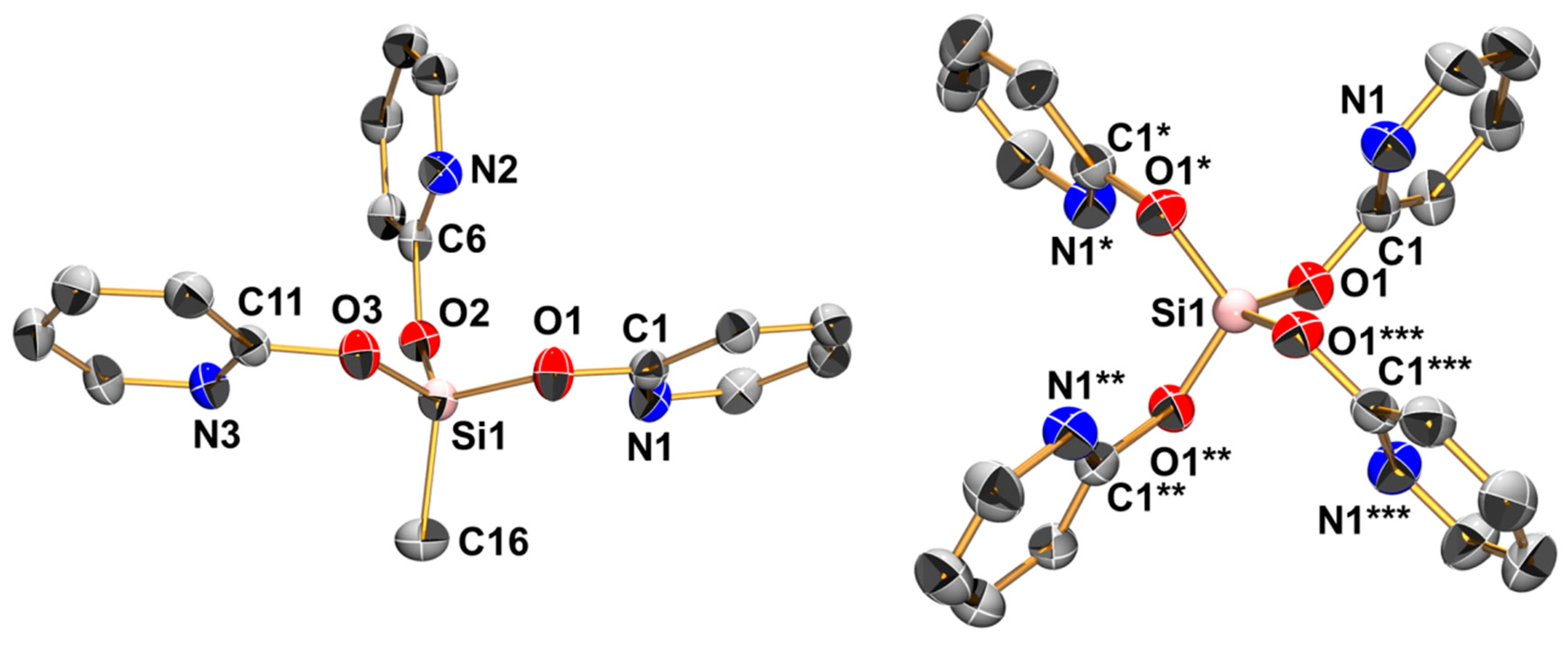
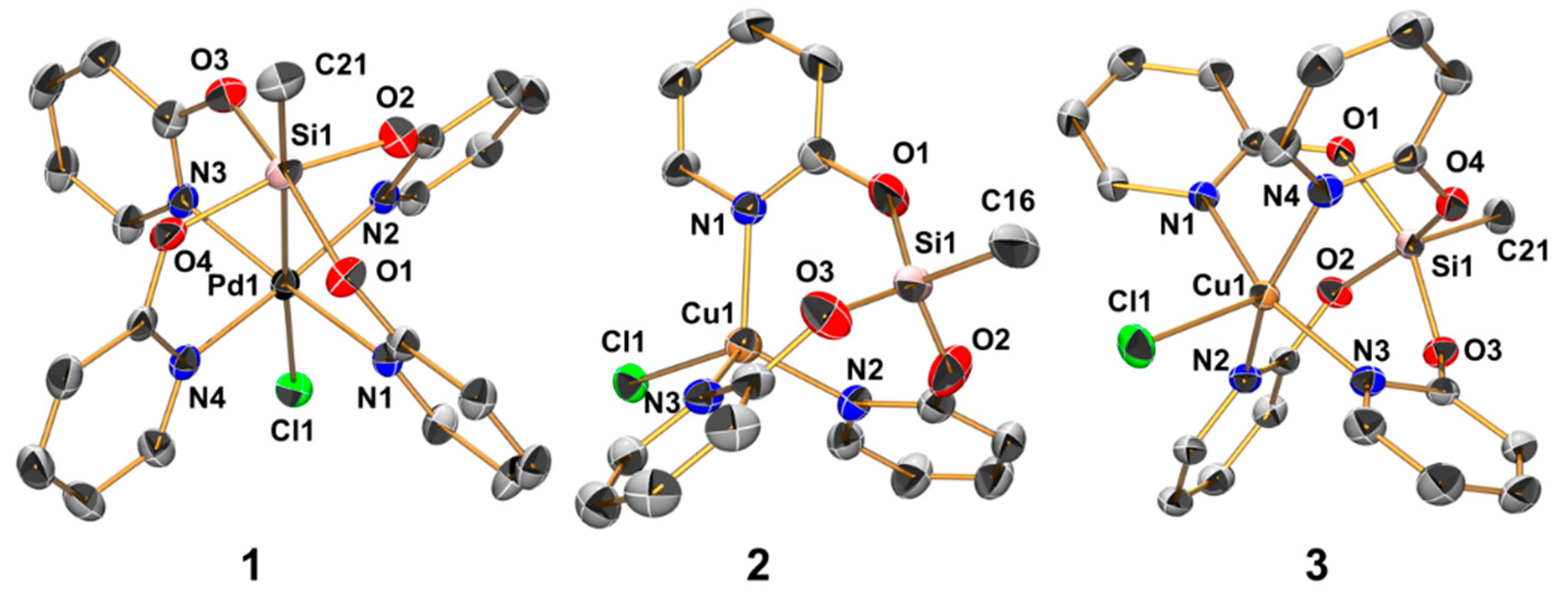
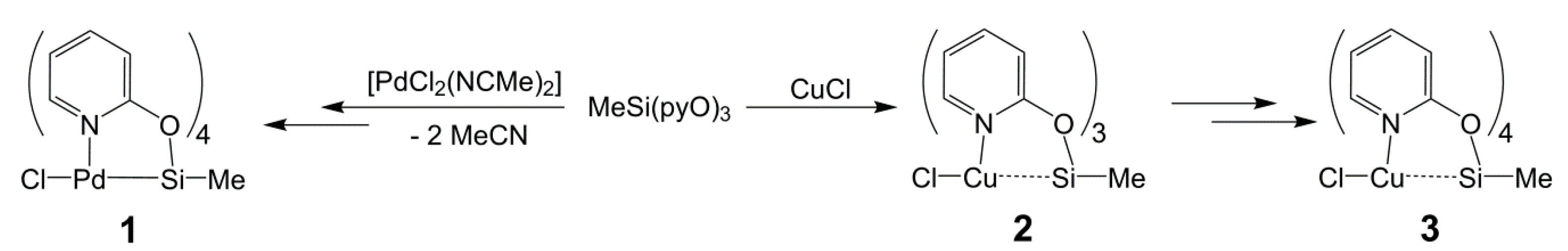
The reactions of MeSi(pyO)3 and [PdCl2(NCMe)2] (in 1:1 molar ratio) in chloroform proceeded with the partial dissolution of [PdCl2(NCMe)2] and the formation of a clear (slightly yellow, almost colorless) solution, from which the crystals of the chloroform solvate of compound 1 formed within one day (Scheme 2). The formal loss of Cl and the addition of a fourth pyO-bridge indicate ligand scrambling in the course of this reaction. The addition of excess MeSi(pyO)3 (as sacrificial pyO source) eventually led to the complete dissolution of [PdCl2(NCMe)2] and the formation of crystals of compound 1 · 2 CHCl3 in good yield. From such a crystal, the molecular structure of 1 was determined using X-ray diffraction analysis (Figure 2 and Table 1). In principle, the molecule has paddlewheel architecture with four pyO ligands attached to Si (via Si–O bonds) and Pd (via Pd–N bonds) of a Me–Si–Pd–Cl axis. The idealized planes of the pyO ligands are slightly tilted against the Si–Pd axis (Pd–Si–O–C torsion angles ranging between 34.6(3)° and 26.3(3)°), and the axial angles (C21–Si1–Pd1 and Si1–Pd1–Cl1) exhibit some deviation from linearity (177.88(12)° and 177.36(3)°, respectively). The Si and Pd atoms are displaced from the O4 and N4 least-squares planes, respectively (into opposite directions), by 0.252(1) and 0.165(1) Å, respectively. The Pd–Si bond (2.6268(2) Å) is slightly longer than in the methimazolyl bridged paddlewheel complexes (where the Pd–Si bond lengths in the range 2.53–2.60 Å were observed) [7], and slightly shorter than in the 7-azaindolyl bridged paddlewheels [8]. The Si–C bond (1.853(4) Å) is only marginally longer than in the starting silane MeSi(pyO)3 (1.837(2) Å), thus hinting at only a weak Pd→Si lone pair donor action. At hexacoordinate silicon with trans-disposed stronger donor (e.g., another hydrocarbyl group [42,43], 2-pyridinethiolato N atom [28] or 8-oxyquinolinyl N atom [44,45]), one would expect a Si–C bond lengthening beyond 1.90 Å. The Pd–Cl bond (2.891(1) Å), however, is unexpectedly long, thus hinting at ionic dissociation. This is supported by four H···Cl contacts with the pyO-H6 atoms. The 29Si NMR shift of compound 1 (−116.9 ppm in CDCl3) is notably more upfield with respect to MeSi(pyO)3 and Si(pyO)4, thus indicating the hypercoordination of the Si atom. This 29Si NMR shift, however, may be representative of either penta- or hexacoordinate silicon, and therefore the role of the sixth donor moiety (Pd→Si) requires further elucidation (vide infra).
The reaction of MeSi(pyO)3 with CuCl (Scheme 2) proceeds in the expected straightforward manner in a 1:1 molar ratio, i.e., CuCl dissolves in chloroform and in tetrahydrofuran (THF) in the presence of one mol equivalent of MeSi(pyO)3, to afford an almost colorless (slightly greenish, by traces of Cu(II)) solution. Also, whereas the starting material produces a 29Si NMR signal at −46.5 ppm (in CDCl3), the resonance is shifted upfield for the solutions of MeSi(pyO)3 with CuCl (−49.6 ppm for the THF solution, −64.1 ppm in CDCl3). From the THF solution, some colorless crystals of the expected product 2 formed, which were suitable for single-crystal X-ray diffraction analysis (Figure 2 and Table 1). The crystal structure is comprised of two independent molecules that exhibit similar conformation, that is, a propeller with a Cl–Cu–Si–CH3 axis and three pyO-bridges (bound to Si via Si–O bonds and to Cu via Cu–N bonds). The Cu···Si separation is rather long (ca. 3.2 Å), but it is shorter than in complex XII (M = Cu, Cu···Si 3.48 Å) [37], and the effect of Cu(I) on the Si coordination sphere is evident from the flattening of the SiO3 pyramidal base (sum of angles ca. 337°). Furthermore, the 29Si NMR shift of this solid is significantly upfield with respect to the starting silane (−70.0 and −71.4 ppm for the two crystallographically independent Si sites). In the CDCl3 solution, compound 2 produces one set of broad 1H NMR signals for the pyO moieties. Thus, this 1H NMR signal broadening and the less pronounced upfield shift of the 29Si NMR signal in the solution indicate conformational changes, such as coordination equilibria between isomers MeSi(µ-pyO)3CuCl and Me(κO-pyO)2Si(µ-pyO)CuCl, the latter with a weaker or absent Si···Cu interaction. Upon repeated opening/closing of the Schlenk flasks with solutions of 2 in THF or chloroform (e.g., for drawing NMR samples), some blue crystals of the related copper(II) compound 3 formed (Scheme 2) as solvent free crystals (from THF) and chloroform solvate (from the chloroform solution). The crystal structures of both compounds were determined using single-crystal X-ray diffraction (Table 2), and the molecular structure of 3 in the solvent free crystals (Figure 2) is included in the discussion as a representative example. This molecule combines features of both compounds 1 and 2, as the Si···Cu separation is rather long (2.92 Å), thus being similar to compound 2, but the axis of this paddlewheel complex is bridged by four pyO ligands. The Si–C bond length is intermediate between those of 1 and 2, but the Si–O bonds are almost as long as in 1, presumably as a result of the trans-arrangement of the Si–O bonds, but with a somewhat greater deviation of the O–Si–O axes from linearity in 3 (by ca. 25°). The Cu–N bond lengths in 3 are similar to those in 2, and we attribute this similarity to a combination of two antagonist effects, that is, bond shortening by a higher oxidation state of the Cu atom and bond lengthening by trans-arrangement along the N–Cu–N axes. Deviations of the O–Si–O and N–Cu–N axes from linearity as well as the rather long Si···Cu separations are combined with displacement of Si and Cu atoms from the O4 and N4 least-squares planes, respectively, into opposite directions by 0.379(1) and 0.371(1) Å, respectively. Thus, the coordination spheres of both the Si and Cu atoms are best described as square–pyramidal. Unfortunately, the deliberate synthesis of larger amounts of 3 (by deliberate exposure of solutions of 2 to air) failed, and therefore we have not been able to isolate pure 3 for further spectroscopic or other investigation. Nonetheless, this compound represents a welcome link between compounds 1 and 2, and therefore we performed computational analyses of the electronic situations in 1, 2, and 3.
2.4. Computational Analyses of the Pd→Si and Cu→Si Interactions in Compounds 1, 2, and 3
For the following investigations, we used the crystallographically determined molecular structures of 1, 2, and 3 (Figure 2) as a starting point, followed by the optimization of the H atom positions for the isolated molecules in the gas phase.
Natural localized molecular orbital (NLMO) analyses were performed using DFT-(RO)B3LYP functional with an SDD basis set for Pd and Cu, 6-311+G(d) for C, Cl, H, N, O, and Si (for details see experimental section). Figure 3 shows the NLMOs identified for the TM→Si donor–acceptor σ-interaction, and Table 3 lists the selected features of these NLMOs. In all three of the cases, the NLMO analysis treated this interaction as a donation of a mainly TM localized lone pair into a vacant orbital at Si. The latter has a σ*(Si–Me) character, which reasons its rather poor acceptor qualities. Thus, in contrast to Pd→Si–Cl systems such as I and II, which feature Pd/Si contributions of 84%/12% [6] and 83%/15% [8], respectively, the corresponding σ-donor electron pair in 1 is more TM localized (91% Pd contribution). In Cu–Si complexes 2 and 3, the corresponding lone pair is even more metal localized (ca. 98%), and has less than a 1% Si contribution. Thus, it resembles a d(z2) orbital more closely. Regardless of the different oxidation states of the Cu atoms and the different Cu···Si separations, the characteristics of the Cu→Si NLMOs of these two compounds are surprisingly similar. As it was shown for the related Pd and Ni systems that TM→Si interactions of related complexes from first and second row TMs may be very similar (83% Ni, 13% Si contribution to the Ni→Si NLMO in the Ni-analog of compound I) [6], we attribute the poor donicity of Cu in compounds 2 and 3 to the enhanced effective nuclear charge (group 11 instead of group 10 element) rather than to Cu being a first row transition metal.
As the TM–Si interactions in compounds 1 and especially in 2 and 3 appear to be of an electrostatic/polar nature rather than covalent, the non-covalent interactions (NCI) descriptor of these compounds was analyzed (Figure 4). In compound 1, a strong electrostatic non-covalent interaction between Pd and Si is detected, represented by a deep blue disc- or toroid-shaped area of the NCI along the Pd–Si bond. This feature is similar to the NCI along the Pd–Cl bond in this compound (and similar to the Cu–Cl bonds in compounds 2 and 3). In sharp contrast, the light blue color of the NCI encountered along the Cu–Si paths in 2 and 3 hints at significantly weaker interactions, and their nature seems to be more closely related to the polar interactions of the pyO6-hydrogen atoms with the metal bound chloride (i.e., weak hydrogen contacts).
In order to quantify these non-covalent interactions (TM···Si vs. C–H···Cl), topological analyses of the electron density distributions in compounds 1, 2, and 3 were performed using the atoms in molecules (AIM) approach. This AIM analysis of the wave function detected the bond critical points (BCPs) between Si and TM (TM = Pd, Cu) in all three of the complexes. Some of their features are listed in Table 4. For compounds 2 and 3, the electron density ρ(rb) and the positive Laplacian ∇2ρ(rb) are of low magnitude, and the ratio |V(rb)|/G(rb) slightly above 1 is indicative of an intermediate closed shell interaction with pronounced ionic contribution [46] (in accordance with the low Wiberg bond order (WBO) [47]). The |V(rb)|/G(rb) ratio in complex 1 is slightly greater than 2, and in combination with the negative Laplacian, it is indicative of a strong polarized shared shell interaction [48,49,50]. According to Espinosa et al. [51], the ratio H(rb)/ρ(rb) can be utilized as a covalence degree parameter (for systems where d < dcov, |V(rb)| > G(rb), H(rb) < 0), the greater magnitude of which indicates the stronger atom–atom interaction (leading to an order of increasing strength 2 < 3 < 1). For the series under investigation, the analysis of the estimated interaction energies (Eint) [52,53] yields an order of increasing Eint(TM–Si) in compounds 2 ≅ 3 < 1. The estimated Eint and the ratio H(rb)/ρ(rb) indicate a slightly stronger interaction in 3 relative to 2. In accordance with the NCI, the Cu···Si interactions in 2 and 3 are indeed similar to the C–H···Cl contacts in these molecules in terms of energetics. Whereas the Eint(Cu···Si) in 2 and 3 were estimated to −2.3 and −3.7 kcal·mol−1, respectively, the same approach of the analysis of V(rb) at the BCPs the C–H···Cl contacts in 1, 2, and 3 yielded an average Eint of −1.90, −1.65, and −2.02 kcal mol−1, respectively.
Furthermore, for the paramagnetic compound 3, this analysis afforded a Mulliken spin density distribution of 65.9% Cu-localization and 6.9–7.3%, located at each of the four nitrogen atoms. This is in accordance with the ligand field theory, which would assign the unpaired electron of a d9 system in the square pyramidal coordination sphere to the d(x2−y2) orbital, thus providing a d(z2)-located lone pair for potential donor–acceptor interactions perpendicular to the Cu(II)N4 plane in this particular case of compound 3.
2.5. Reactions of Si(pyO)4 with [PdCl2(NCMe)2] and CuCl2
In analogy to the formation of I and II from the respective silane Si(L)4 (L = bridging ligand) and [PdCl2(NCMe)2], Si(pyO)4 should be capable of forming paddlewheel complexes of the type ClSi(µ-pyO)4TMCl upon reaction with TMCl2, or suitable complexes thereof. Thus, we aimed at synthesizing ClSi(µ-pyO)4PdCl (4’) through the reaction of [PdCl2(NCMe)2] and Si(pyO)4 in chloroform (Scheme 3). As the main product, a beige solid of very poor solubility formed. In the dispersion of this fine solid in chloroform, some coarse crystals (beige, almost colorless) formed in the course of some days of storage at room temperature. Single-crystal X-ray diffraction analysis (Table 2, Figure 5) revealed the identity of this compound as the HpyO-adduct of the intended product (compound 4, Scheme 3). Presumably, the intended product 4’ had formed initially and then reacted further with traces of free HpyO. The deliberate synthesis of 4 by reacting [PdCl2(NCMe)2], Si(pyO)4, and HpyO in a 1:1:1 molar ratio in chloroform, eventually afforded this compound in good yield. The molecular structure of the cation [ClPd(µ-pyO)4Si(HpyO)]+ of 4 resembles the paddlewheel architecture of compound 1, and notable differences that arise from the different substituent at Si (trans to Pd) are the following: The Pd–Si bond is noticeably shorter (2.50 Å) because of the more electronegative Si-bound substituent. As a consequence, the Si atom is less displaced from the O4 least-squares plane (by 0.101(1) Å), whereas the displacement of the Pd atom from the N4 plane (into the opposite direction, by 0.167(1) Å) is not altered. Interestingly, the Si1–O5 bond (trans to Pd) is significantly shorter than the equatorial Si–O bonds, thus hinting at still rather poor Pd→Si donor action. In spite of the NH group of the trans-Pd–Si located HpyO moiety, the C–O bonds of all five of the pyO and HpyO ligands are very similar, ranging between 1.313(2) and 1.325(2) Å. The 29Si NMR shift of this compound (δ29Si −147.9 ppm in CD2Cl2) is in accordance with the hexacoordination of the Si atom, and the 1H and 13C NMR spectra feature two sets of pyO-signals in a 4:1 intensity ratio, reflecting the four bridging and the dangling pyO moieties, respectively, and the retention of this molecular architecture in solution.
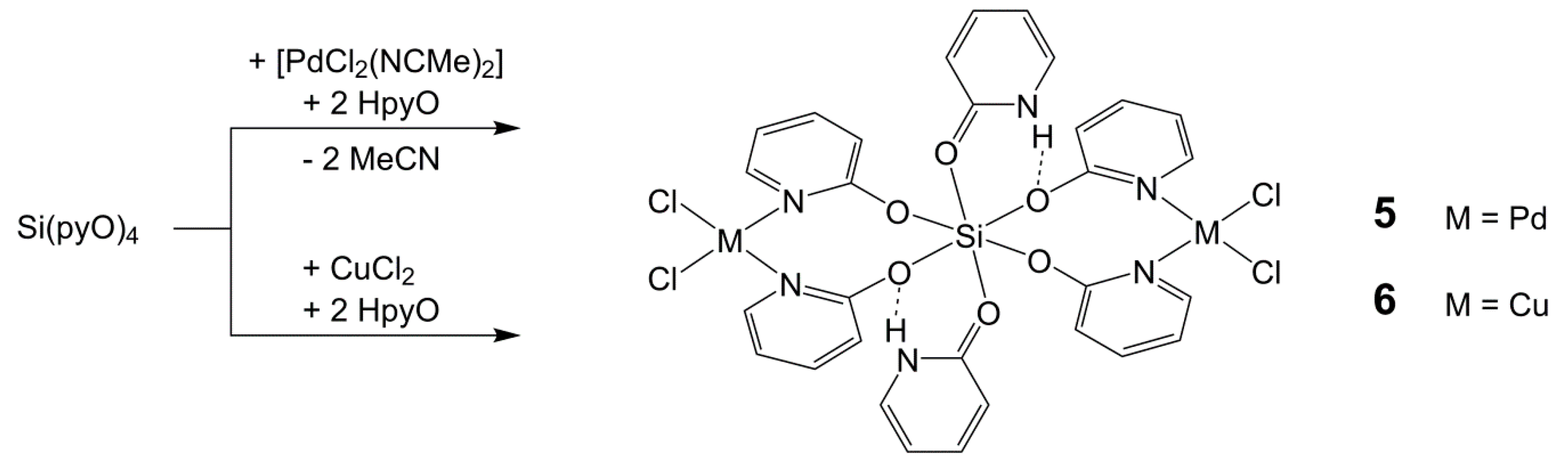
Upon harvesting the crystalline solid 4 · 4 CHCl3, some yellow crystals of another compound formed in the filtrate. Using single-crystal X-ray diffraction analysis, they were identified as the chloroform solvate 5 · 6 CHCl3 of the complex 5 (Scheme 4). The deliberate synthesis of this complex, by using [PdCl2(NCMe)2], Si(pyO)4, and HpyO in a 2:1:2 molar ratio in chloroform, afforded compound 5 in good yield. Interestingly, the crystals that were initially formed during the synthesis consisted of a different solvate (5 · 8 CHCl3), and upon filtration, some more crystals of solvate 5 · 2 CHCl3 formed in the filtrate. The elemental analysis of the final product upon drying was in agreement with the composition of the solvate 5 · 2 CHCl3. Even though all three solvates (5 · 8, 6, 2 CHCl3, respectively) were characterized crystallographically (Table 5), only the molecular structure of 5 in the solvate 5 · 6 CHCl3 is discussed as a representative example (Figure 6), as there are only marginal differences between the molecular conformations in the three different solvates.
The Si atom of compound 5 is, in the crystal structures, located on a center of inversion, thus the trans angles are 180°, and the very similar Si–O bond lengths and cis angles close to 90° (maximum deviations of ca. 1°) furnish an Si atom almost perfect octahedrally coordinated by six pyO oxygen atoms. Two sets of mutually cis situated anionic pyO ligands, the four O atoms of which are located in one plane, act as (N,N)-chelate donors toward PdCl2, and the axial positions of the Si coordination sphere are occupied by the O atoms of HpyO, each of which establishes an N–H···O contact to an adjacent pyO oxygen atom (as indicated in Scheme 4). In spite of the very similar Si–O bond lengths, the C–O bonds of the HpyO moieties (1.303(3) Å) are significantly shorter than the C–O bonds of the bridging pyO moieties (1.328(3) Å), and thus exhibit a pronounced double bond character, as expected for HpyO. In the solvate 5 · 8 CHCl3, these differences are even more pronounced with 1.296(3) vs. 1.331(3) and 1.336(3) Å. As expected, the Pd atoms are situated in a square planar coordination sphere (with a sum of cis angles of 360.3(1)°). Compound 5 exhibited too poor of a solubility for solution NMR characterization, but the 29Si cross-polarization magic-angle-spinning (CP/MAS) NMR spectroscopy of the solid unequivocally confirmed the hexacoordination of the central Si atom (δ29Si −190.2 ppm). This chemical shift is similar to other hexacoordinate Si complexes with SiO6 coordination sphere (e.g., Si(acetylacetonate)2(salicylate) δ29Si −191.7 ppm) [54].
In an attempt at synthesizing a paddlewheel complex of the composition ClCu(µ-pyO)4SiCl, in comparison to the attempted synthesis of 4’ (Scheme 3, left), anhydrous CuCl2 and Si(pyO)4 were dispersed in chloroform. Whereas most of the reactants remained unchanged, the solution phase became blue and some blue crystals formed upon storage within one week. The crystals were identified as compound 6 by single-crystal X-ray diffraction analysis. Apparently, traces of HpyO in the sample gave rise to the formation of this Cu-analog of compound 5. Deliberate synthesis, by using CuCl2, Si(pyO)4, and HpyO in a 2:1:2 molar ratio in chloroform, afforded compound 6 in good yield (Scheme 4). Because of the very poor solubility and the paramagnetic Cu(II) sites in this compound, NMR spectroscopic characterization was no option, and therefore we only discuss the molecular structure of this complex. The central parts of the molecule, that is, the bond lengths and angles of the hexacoordinate Si atom as well as the equatorial arrangement of two Si(µ-pyO)2M clamps and two axially situated HpyO ligands, which establish N–H···O hydrogen bridges to adjacent pyO oxygen atoms, are very similar to the arrangement in compound 5. The noteworthy difference is associated with the Cu(II) coordination sphere, which is distorted and intermediate between the tetrahedral and square planar. Then sum of the cis angles of Cu (367.9(2)°) deviates significantly from planarity. The N–Cu–N and Cl–Cu–Cl angles in particular are wider than 90°, and the angle between the CuN2 and CuCl2 planes is 30.7(2)°. Thus, in spite of the notable distortion, this coordination sphere is still closer to the square rather than tetrahedral.
3. Experimental Section
3.1. General Considerations
The commercially available chemicals (2-hydroxypyridine, anhydrous CuCl2, Me3SiCl, MeSiCl3, and SiCl4) were used as received without further purification. Chloroform (CDCl3 stabilized with silver, CHCl3 stabilized with amylenes) and CD2Cl2 were stored over activated molecular sieves (3 Å) for at least seven days. THF, diethyl ether, toluene, and triethylamine were distilled from sodium benzophenone. All of the reactions were carried out under an atmosphere of dry argon utilizing standard Schlenk techniques. 2-Trimethylsiloxypyridine [26] was synthesized according to a literature procedure. [PdCl2(NCMe)2] [8] and CuCl [55] were available in the laboratory from previous studies. The solution NMR spectra (1H, 13C, 29Si) were recorded on Bruker Avance III 500 MHz and Bruker Nanobay 400 MHz spectrometers (Bruker Biospin, Rheinstetten, Germany) and Me4Si was used as internal standard. The 29Si (CP/MAS) NMR spectra were recorded on a Bruker Avance HD 400 WB spectrometer with 7 mm zirconia (ZrO2) rotors and KelF inserts (compound 2) or 4 mm zirconia rotors (compounds Si(pyO)4 and 5) at an MAS frequency of νspin = 5 kHz. The elemental analyses were performed on an Elementar Vario MICRO cube (Elementar, Hanau, Germany). The single-crystal X-ray diffraction data were collected on a Stoe IPDS-2T diffractometer (Stoe, Darmstadt, Germany) using Mo Kα-radiation. The structures were solved by direct methods using SHELXS-97, and were refined with the full-matrix least-squares methods of F2 against all reflections with SHELXL-2014 [56,57,58]. All of the non-hydrogen atoms were anisotropically refined. The C-bound hydrogen atoms were isotropically refined in an idealized position (riding model), the N-bound H atoms of compounds 4 · 4 CHCl3, 5 · 2 CHCl3, 5 · 6 CHCl3, and 5 · 8 CHCl3 were refined isotropically without restraints, and in the case of compound 6, the isotropic displacement parameter of the N-bound H atom was set at the 1.2-fold mean displacement parameter of the pivot atom for stable refinement (because of the rather poor data set). The graphics of the molecular structures were generated with ORTEP-3 [59] and POV-Ray 3.6 [60]. CCDC 1869874 (MeSi(pyO)3), 1869875 (Si(pyO)4), 1869876 (3 · CHCl3), 1869877 (3), 1869878 (6), 1869879 (5 · 2 CHCl3), 1869880 (2), 1869881 (5 · 8 CHCl3), 1869882 (1· 2 CHCl3), 1869883 (5 · 6 CHCl3), and 1869884 (4 · 4 CHCl3) contain the supplementary crystal data for this article (see Supplementary Materials). These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. For the computational analyses of compounds 1, 2, and 3, the atomic coordinates from the crystallographically determined molecular structures were used for the non-hydrogen atoms. The geometry optimization of the H atom positions was carried out with Gaussian09 [60] using a DFT-PBEPBE functional and def2tzvpp basis set for all of the atoms. Subsequently, starting from these molecular structures, the NBO (natural bond orbital) and NLMO (natural localized bond orbital) calculations were performed using Gaussian09 [61] with the NBO6.0 package [62] using DFT-(RO)B3LYP functional (for Cu, Pd with SDD basis set; for C, H, N, O, Cl, and Si, with the 6-311+G(d) basis set including Douglas–Kroll–Hess second order scalar relativistic). The NLMO graphics were generated using ChemCraft [63]. The NCI [64], AIM [65], and Wiberg bond order [47] calculations were carried out using MultiWFN [66], using the same wave function that had been used for the NBO/NLMO calculations. The graphical representations of the NCI results were created using VMD [67].
3.2. Syntheses
MeSi(pyO)3. A Schlenk flask with magnetic stirring bar was charged with 2-hydroxypyridine (1.50 g, 15.6 mmol), evacuated, and set under Ar atmosphere prior to adding THF (50 mL) and triethylamine (1.89 g, 18.7 mmol), with stirring to afford a colorless solution. With continuous stirring at room temperature, methyltrichlorosilane (0.82 g, 5.5 mmol) was added dropwise via syringe, while the simultaneous formation of a white precipitate (Et3NHCl) was observed. Upon the complete addition of the silane stirring at room temperature was continued for 1 h, whereupon the hydrochloride precipitate was filtered off and washed with THF (2 × 5 mL). From the combined filtrate and washings, the solvent was removed under reduced pressure (condensed into a cold trap) to afford a crystalline residue, which was dissolved in hot THF (5 mL) and filtered prior to the addition of hexane (10 mL), and was stored at 6 °C. In the course of three days, colorless crystals of the product formed, which were separated by decantation (while cold) and were dried in vacuo (yield 0.90 g (2.7 mmol, 50%)). The crystals were suitable for X-ray diffraction analysis. The elemental analysis for C16H15N3O3Si (325.39 g·mol−1) was as follows: C, 59.06; H, 4.65; N, 12.91; found C, 56.37; H, 5.29; and N, 12.28. The composition found indicates hydrolysis upon sample preparation. The following was calculated for C16H18N4O5.5Si (i.e., MeSi(pyO)3 · 1.5 H2O) (352.42 g·mol−1): C, 54.53; H, 5.15; N, 11.92; multiplied with a mass correction factor of 1.035, which accounts for the uptake of similar amounts of water prior to and after weighing of the sample (C, 56.44; H, 5.33; N, 12.34). 1H NMR (CDCl3): δ (ppm) 0.92 (s, 3H, SiCH3), 6.84–6.88 (m, 6H, H3 and H5), 7.53–7.57 (m, 3H, H4), 8.07–8.08 (m, 3H, H6); 13C{1H} NMR (CDCl3): δ (ppm) −2.6 (SiCH3), 113.1 (C5), 118.0 (C3), 139.2 (C4), 147.4 (C6), 160.6 (C2); 29Si{1H} NMR (CDCl3): δ (ppm) −46.5.
Si(pyO)4. In a Schlenk flask with magnetic stirring bar 2-trimethylsiloxypyridine (3.00 g, 18.0 mmol) was dissolved in chloroform (4 mL), whereupon SiCl4 (0.82 g, 4.8 mmol) was added (via syringe) with stirring. The resultant solution was heated with stirring under reflux for 2 h, to afford a white precipitate of the product. The mixture was allowed to attain room temperature, the solid was filtered, washed with chloroform (2 × 5 mL), and dried in vacuo. The yield was 1.08 g (2.67 mmol, 57%). The single-crystals suitable for X-ray diffraction analysis were obtained by recrystallization in THF. The elemental analysis for C20H16N4O4Si (404.45 g·mol−1) was as follows: C, 59.39; H, 3.99; N, 13.85; found C, 57.19; H, 4.90; N, 13.36. The composition found indicates hydrolysis upon sample preparation. The following was calculated for C20H20N4O6Si (i.e., Si(pyO)4 · 2 H2O) (440.48 g·mol−1): C, 54.53; H, 4.58; N, 12.72; multiplied with a mass correction factor of 1.05, which accounts for uptake of similar amounts of water prior to and after weighing of the sample: C, 57.26; H, 4.81; N, 13.36. 1H NMR (CDCl3): δ (ppm) 6.86–6.98 (m, 8H, H3 and H5), 7.54–7.58 (m, 4H, H4), 8.01 (m, 4H, H6); 13C{1H} NMR (CDCl3): δ (ppm) 113.2 (C5), 118.4 (C3), 139.2 (C4), 147.3 (C6), 159.7 (C2); 29Si{1H} NMR (CDCl3): δ (ppm) −97.2, (CP/MAS): δ (ppm) −87.9.
ClPd(µ-pyO)4SiMe · 2 CHCl3 (complex 1 · 2 CHCl3). A Schlenk flask was charged with a magnetic stirring bar, MeSi(pyO)3 (0.40 g, 1.2 mmol) and [PdCl2(NCMe)2] (0.16 g, 0.62 mmol), evacuated, and set under Ar atmosphere prior to adding chloroform (3 mL). Upon brief stirring at room temperature (within two minutes), the starting materials dissolved completely to afford a light-yellow solution. Immediately after dissolution, the stirring was stopped and the solution was stored undisturbed at room temperature. In the course of one day, colorless crystals of the product formed, but the solution was stored for another week in order to complete crystallization, whereupon the crystals were separated from the supernatant by decantation and then briefly dried in vacuo. A single-crystal suitable for X-ray diffraction analysis was taken out of the mother liquor. The yield was 0.31 g (0.39 mmol, 65%). The elemental analysis for C23H21Cl7N4O4SiPd (800.11 g·mol−1) was as follows: C, 34.53; H, 2.65; N, 7.00; found C, 37.81; H, 2.85; N, 8.35. The composition found indicates a loss of solvent upon drying and storage, the C, H, N values found correspond to the composition ClPd(µ-pyO)4SiMe · 1.2 CHCl3 (for C22.2H20.2Cl4.6N4O4SiPd (704.61 g·mol−1): C, 37.84; H, 2.89; N, 7.95). 1H NMR (CDCl3): δ (ppm) 0.92 (s, 3H, CH3) 6.66–6.67 (m, 8H, H3 and H5), 7.47–7.51 (m, 4H, H4), 9.22–9.24 (m, 4H, H6); 13C{1H} NMR (CDCl3): δ (ppm) 3.5 (SiCH3), 114.2 (C5), 116.0 (C3), 141.4 (C4), 148.8 (C6), 162.7 (C2); 29Si{1H} NMR (CDCl3): δ (ppm) −116.9.
ClCu(µ-pyO)3SiMe (complex 2). Procedure A: A Schlenk flask was charged with a magnetic stirring bar, MeSi(pyO)3 (0.44 g, 1.4 mmol) and CuCl (0.14 g, 1.4 mmol), evacuated, and set under Ar atmosphere prior to adding THF (1.5 mL). Upon brief stirring at room temperature (within five minutes), the starting materials dissolved completely to afford a light turquoise (almost colorless) solution. 29Si NMR spectroscopic analysis of this solution (with D2O capillary used as lock) revealed one signal (at −49.6 ppm). In the course of one day, colorless crystals of the product formed, whereupon the crystals were separated from the supernatant by decantation, and then briefly dried in vacuo. A single-crystal suitable for X-ray diffraction analysis was taken out of the mother liquor. The yield was 0.12 g (0.28 mmol, 20%). The elemental analysis for C16H15ClCuN3O3Si (424.39 g·mol−1) was as follows: C, 45.28; H, 3.56; N, 9.90; found C, 43.44; H, 3.67; N, 9.44. The composition found indicates the presence of ca. 4% of “inert” materials (free of C, H, N), such as CuCl, as C, H, and N were found in the expected ratio. 29Si{1H} NMR (CP/MAS): δ (ppm) −70.0, −71.4 (intensity ratio 1:1).
Procedure B: A Schlenk flask was charged with a magnetic stirring bar, MeSi(pyO)3 (0.30 g, 0.92 mmol) and CuCl (0.09 g, 0.92 mmol), evacuated, and set under Ar atmosphere prior to adding CDCl3 (1.0 mL). Upon brief stirring at room temperature (within five minutes), the starting materials dissolved completely to afford a light turquoise (almost colorless) solution, which was used for the 1H and 29Si NMR spectroscopic analysis. Compound 2 did not crystallize from this solution. 1H NMR (CDCl3): δ (ppm) 0.71 (s, 3H, CH3) 6.87–6.95 (m, 6H, H3 and H5), 7.66 (m, 3H, H4), 8.78 (m, 3H, H6); 29Si{1H} NMR (CDCl3): δ (ppm) −64.1.
ClCu(µ-pyO)4SiMe (complex 3). In the Schlenk flasks with the crude product solutions of complex 2, upon opening, some deep blue crystals formed over the course of some days. In both cases, the crystals were suitable for single-crystal X-ray diffraction analysis. From Procedure A, the solvent free variety 3 crystallized. From Procedure B, some crystals of the mono-chloroform solvate of 3 formed. The amount of crystals of solvent free 3 obtained was sufficient for elemental analysis. The elemental analysis for C21H19ClCuN4O4Si (518.48 g·mol−1) was as follows: C, 48.65; H, 3.69; N, 10.81; found C, 48.29; H, 3.92; N, 10.63.
[ClPd(µ-pyO)4Si(HpyO)]Cl · 4 CHCl3 (complex 4 · 4 CHCl3). A Schlenk flask was charged with a magnetic stirring bar; Si(pyO)4 (0.30 g, 0.74 mmol), [PdCl2(NCMe)2] (0.19 g, 0.74 mmol), and 2-hydroxypyridine (0.07 g, 0.74 mmol); evacuated; and set under Ar atmosphere prior to adding chloroform (3 mL). Upon stirring at room temperature, a beige dispersion formed, and the supernatant was orange. Over the course of one week, the finely dispersed powder transformed into a beige coarse crystalline product. A single-crystal suitable for X-ray diffraction analysis was taken out of the mother liquor. This solid was filtered, washed with chloroform (2 × 2 mL), and briefly dried in vacuo. The yield was 0.40 g (0.30 mmol, 47%). From the filtrate, some yellow crystals of compound 5 · 6 CHCl3 formed, which were suitable for single-crystal X-ray diffraction analysis. The elemental analysis for C29H25Cl14N5O5SiPd (1154.38 g·mol−1) was as follows: C, 30.17; H, 2.18; N, 6.07; found C, 30.43; H, 2.10; N, 6.94. 1H NMR (CD2Cl2): δ (ppm) 6.82 (ddd, 4H, H5, 7.1 Hz, 6.1 Hz, 1.3 Hz), 6.85 (ddd, 4H, H3, 8.4 Hz, 1.3 Hz, 0.6 Hz), 7.40 (ddd, 1H, H5, 7.2 Hz, 6.1 Hz, 1.1 Hz) 7.62 (ddd, 4H, H4, 8.5 Hz, 7.0 Hz, 1.8 Hz), 7.76 (ddd, 1H, H3, 8.8 Hz, 1.0 Hz, 0.7 Hz), 8.25 (ddd, 1H, H4, 8.8 Hz, 7.2 Hz, 2.0 Hz), 8.53 (ddd, 1H, H6, 6.1 Hz, 2.0 Hz, 0.7 Hz), 9.29 (ddd, 4H, H6, 6.2 Hz, 1.8 Hz, 0.6 Hz), 15.82 (s, 1H, NH); 13C{1H} NMR (CD2Cl2): δ (ppm) bridging pyO: 114.2 (C5), 117.0 (C3), 142.3 (C4), 148.5 (C6), 162.1 (C2), Si-bound HpyO: 117.7 (C5), 118.6 (C3), 139.2 (C4), 145.8 (C6), 158.3 (C2); 29Si{1H} NMR (CD2Cl2): δ (ppm) −147.9, (CP/MAS): δ (ppm) −146.3.
Cl2Pd(µ-pyO)2(HpyO)Si(HpyO)(µ-pyO)2PdCl2 · 2 CHCl3 (complex 5 · 2 CHCl3). A Schlenk flask was charged with a magnetic stirring bar; Si(pyO)4 (0.40 g, 0.99 mmol), [PdCl2(NCMe)2] (0.51 g, 2.0 mmol), and 2-hydroxypyridine (0.19 g, 2.0 mmol); evacuated; and set under Ar atmosphere prior to adding chloroform (3 mL). Upon stirring at room temperature, an orange dispersion formed. Within three days of undisturbed storage at room temperature, some yellow crystals formed, which were identified as the solvate 5 · 8 CHCl3 by single-crystal X-ray diffraction analysis. To the mixture, further chloroform (2 mL) was added, and was briefly stirred prior to filtration and washing with chloroform (2 × 3 mL). The solid was dried in vacuo to afford a yellow powdery solid. In spite of the solvent rich solvate found in the crude mixture, the elemental analysis of the dried product corresponds to the approximate composition of 5 · 2 CHCl3. The yield was 0.83 g (0.70 mmol, 71%). In the filtrate some yellow crystals formed which were identified as the solvate 5 · 2 CHCl3 by single-crystal X-ray diffraction analysis. The elemental analysis for C32H28Cl10N6O6SiPd2 (1188.05 g·mol−1) was as follows: C, 32.35; H, 2.38; N, 7.07; found C, 31.68; H, 2.23; N, 7.10. The solubility of the product in various organic solvents was not sufficient for solution NMR spectroscopic characterization. 29Si{1H} NMR (CP/MAS): δ (ppm) −190.2.
Cl2Cu(µ-pyO)2(HpyO)Si(HpyO)(µ-pyO)2CuCl2 (complex 6). A Schlenk flask was charged with a magnetic stirring bar; Si(pyO)4 (0.38 g, 0.95 mmol), CuCl2 (0.25 g, 1.9 mmol), and 2-hydroxypyridine (0.18 g, 1.9 mmol); evacuated; and set under Ar atmosphere prior to adding chloroform (2.5 mL). Upon stirring at room temperature, a turquoise dispersion formed. After three days of storage at room temperature, further chloroform (3 mL) was added and was briefly stirred prior to filtration and washing with chloroform (2 × 3 mL). The solid was dried in vacuo to afford a blue powdery solid. The yield was 0.70 g (0.74 mmol, 78%). The elemental analysis indicates the presence of solvent (0.7 CHCl3) in the product. The following was calculated for C30H26Cl4N6O6SiCu2 (947.12 g·mol−1): C, 38.93; H, 2.84; N, 8.87; found C, 38.70; H, 3.08; N, 8.86. Some single-crystals of compound 6 were obtained from a mixture of Si(pyO)4 (0.10 g, 0.25 mmol) and CuCl2 (0.03 g, 0.025 mmol), which was layered with chloroform (1 mL) and stored undisturbed at room temperature for one week.
4. Conclusions
Two different 2-pyridyloxysilanes, MeSi(pyO)3 and Si(pyO)4, proved to be suitable starting materials for the syntheses of heteronuclear complexes, in which the ambidentate pyO ligand binds to Si via Si–O, and to transition metals via TM–N bonds. The chelation of the Si atom by this ligand has not been encountered, thus the pyO nitrogen atoms are readily available for complex formation.
As shown for a d8 and a d10 system (with formation of ClPd(µ-pyO)4SiMe 1 and ClCu(µ-pyO)3SiMe 2, respectively), pyridyloxysilanes may support the formation of different paddlewheel structures, that is, with four and three bridging ligands, respectively. The pyO buttresses do not force Si and TM into close proximity, as it is evident from the different Pd–Si and Cu–Si separations in 1 and 2, respectively. A higher coordination of the Si atom by the transition metal site still depends on the donicity of the TM site and the acceptor qualities of Si. As shown in the current study, the Si–Me moiety trans to an electron rich TM (Pd(II) in this particular case) causes weaker TM–Si interactions with respect to previously reported complexes with related TM→Si–Cl motif. Furthermore, group 11 metals (Cu(I) and Cu(II)) were shown to be significantly weaker lone pair donors toward Si(IV) than group 10 metals (e.g., Pd(II) and, according to previous studies, Ni(II) [6,7]). A systematic comparison of this series was enabled by the unintended formation of the decomposition product ClCu(µ-pyO)4SiMe 3. To our knowledge, compound 3 represents the first complex with a Cu(II)–Si(IV) bond (or at least with such a short (2.9 Å) Cu(II)···Si(IV) separation). As shown by the quantum chemical calculations, the Cu···Si interactions in compounds 2 and 3 are slightly stronger than the C–H···Cl hydrogen contacts encountered in the same molecules.
The reactions of Si(pyO)4 with [PdCl2(NCMe)2] and CuCl2, in the presence of 2-hydroxypyridine HpyO, gave rise to the formation of an entirely different class of hypercoordinate Si-complexes of the type (HpyO)2Si[(µ-pyO)2TMCl2]2, 5 and 6, respectively. As the coordination spheres about TM vary between square planar and distorted tetrahedral in compounds 5 and 6, respectively, this kind of complex architecture may turn out to be suitable for the complexation of various further TMX2 moieties (X = halide, pseudo-halide etc.).
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2304-6740/6/4/119/s1: the crystallographic data of the compounds reported in this paper in CIF format.
Author Contributions
L.E. and J.W. conceived and designed the experiments; L.E. performed the experiments (syntheses); R.G. the computational analyses; J.W. the single-crystal X-ray diffraction analyses; E.B. the solid state NMR spectroscopic analyses; J.W. wrote the paper.
Funding
This research received no external funding.
Acknowledgments
We are grateful to Ute Groß and Brunhilde Süßner for performing the elemental analyses and to Beate Kutzner and Erik Wächtler for the solution NMR spectroscopic measurements.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Tacke, R.; Ribbeck, T. Bis(amidinato)- and bis(guanidinato) silylenes and silylenes with one sterically demanding amidinato or guanidinato ligand: Synthesis and reactivity. Dalton Trans. 2017, 46, 13628–13659. [Google Scholar] [CrossRef] [PubMed]
- Peloquin, D.M.; Schmedake, T.A. Recent advances in hexacoordinate silicon with pyridine-containing ligands: Chemistry and emerging applications. Coord. Chem. Rev. 2016, 323, 107–119. [Google Scholar] [CrossRef]
- Wagler, J.; Böhme, U.; Kroke, E. Higher-Coordinated Molecular Silicon Compounds. In Structure and Bonding; Scheschkewitz, D., Ed.; Springer: Berlin, Germany, 2014; Volume 155, pp. 29–105. [Google Scholar]
- Levason, W.; Reid, G.; Zhang, W. Coordination complexes of silicon and germanium halides with neutral ligands. Coord. Chem. Rev. 2011, 255, 1319–1341. [Google Scholar] [CrossRef]
- Wagler, J.; Brendler, E. Metallasilatranes: Palladium(II) and Platinum(II) as Lone-Pair Donors to Silicon(IV). Angew. Chem. Int. Ed. 2010, 49, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Truflandier, L.A.; Brendler, E.; Wagler, J.; Autschbach, J. 29Si DFT/NMR Observation of Spin-Orbit Effect in Metallasilatrane Sheds Some Light on the Strength of the Metal → Si Interaction. Angew. Chem. Int. Ed. 2011, 50, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Autschbach, J.; Sutter, K.; Truflandier, L.A.; Brendler, E.; Wagler, J. Atomic Contributions from Spin-Orbit Coupling to 29Si NMR Chemical Shifts in Metallasilatrane Complexes. Chem. Eur. J. 2012, 18, 12803–12813. [Google Scholar] [CrossRef] [PubMed]
- Wahlicht, S.; Brendler, E.; Heine, T.; Zhechkov, L.; Wagler, J. 7-Azaindol-1-yl(organo)silanes and Their PdCl2 Complexes: Pd-Capped Tetrahedral Silicon Coordination Spheres and Paddlewheels with a Pd-Si Axis. Organometallics 2014, 33, 2479–2488. [Google Scholar] [CrossRef]
- Cade, I.A.; Hill, A.F.; Kämpfe, A.; Wagler, J. Five-Coordinate Hydrido-Ruthenium(II) Complexes Featuring N-Heterocyclic Silylene and Carbene Ligands. Organometallics 2010, 29, 4012–4017. [Google Scholar] [CrossRef]
- Baus, J.A.; Mück, F.M.; Schneider, H.; Tacke, R. Iron(II), Cobalt(II), Nickel(II), and Zinc(II) Silylene Complexes: Reaction of the Silylene [iPrNC(NiPr2)NiPr]2Si with FeBr2, CoBr2, NiBr2·MeOCH2CH2OMe, ZnCl2, and ZnBr2. Chem. Eur. J. 2017, 23, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Junold, K.; Baus, J.A.; Burschka, C.; Vent-Schmidt, T.; Riedel, S.; Tacke, R. Five-Coordinate Silicon(II) Compounds with Si−M Bonds (M = Cr, Mo, W, Fe): Bis[N,N′-diisopropylbenzamidinato(−)]silicon(II) as a Ligand in Transition-Metal Complexes. Inorg. Chem. 2013, 52, 11593–11599. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, S.; Köppe, R.; Roesky, P.W. Investigations of the Nature of ZnII–SiII Bonds. Chem. Eur. J. 2016, 22, 7127–7133. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, A.; Präsang, C.; Milsmann, C.; Driess, M. The Striking Stabilization of Ni0(η6-Arene) Complexes by an Ylide-Like Silylene Ligand. Angew. Chem. Int. Ed. 2009, 48, 3170–3173. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F.; Neumann, H.; Wagler, J. Bis(methimazolyl)silyl Complexes of Ruthenium. Organometallics 2010, 29, 1026–1031. [Google Scholar] [CrossRef]
- Klett, J.; Klinkhammer, K.W.; Niemeyer, M. Ligand Exchange between Arylcopper Compounds and Bis(hypersilyl)tin or Bis(hypersilyl)lead: Synthesis and Characterization of Hypersilylcopper and a Stannanediyl Complex with a Cu–Sn Bond. Chem. Eur. J. 2009, 5, 2531–2536. [Google Scholar] [CrossRef]
- Rittle, J.; Peters, J.C. N−H Bond Dissociation Enthalpies and Facile H Atom Transfers for Early Intermediates of Fe−N2 and Fe−CN Reductions. J. Am. Chem. Soc. 2017, 139, 3161–3170. [Google Scholar] [CrossRef] [PubMed]
- Suess, D.L.M.; Tsay, C.; Peters, J.C. Dihydrogen Binding to Isostructural S = 1/2 and S = 0 Cobalt Complexes. J. Am. Chem. Soc. 2012, 134, 14158–14164. [Google Scholar] [CrossRef] [PubMed]
- Garcés, K.; Lalrempuia, R.; Polo, V.; Fernández-Alvarez, F.J.; García-Orduña, P.; Lahoz, F.J.; Pérez-Torrente, J.J.; Oro, L.A. Rhodium-Catalyzed Dehydrogenative Silylation of Acetophenone Derivatives: Formation of Silyl Enol Ethers versus Silyl Ethers. Chem. Eur. J. 2016, 22, 14717–14729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalrempuia, R.; Iglesias, M.; Polo, V.; Sanz Miguel, P.J.; Fernández-Alvarez, F.J.; Pérez-Torrente, J.J.; Oro, L.A. Effective Fixation of CO2 by Iridium-Catalyzed Hydrosilylation. Angew. Chem. Int. Ed. 2012, 51, 12824–12827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julián, A.; Jaseer, E.A.; Garcés, K.; Fernández-Alvarez, F.J.; García-Orduña, P.; Lahoz, F.J.; Oro, L.A. Tuning the activity and selectivity of iridium-NSiN catalyzed CO2 hydrosilylation processes. Catal. Sci. Technol. 2016, 6, 4410–4417. [Google Scholar] [CrossRef]
- Julián, A.; Guzmán, J.; Jaseer, E.A.; Fernández-Alvarez, F.J.; Royo, R.; Polo, V.; García-Orduña, P.; Lahoz, F.J.; Oro, L.A. Mechanistic Insights on the Reduction of CO2 to Silylformates Catalyzed by Ir-NSiN Species. Chem. Eur. J. 2017, 23, 11898–11907. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ou, C.; Wang, C.; Uchiyama, M.; Deng, L. Silane-Functionalized N-Heterocyclic Carbene−Cobalt Complexes Containing a Five-Coordinate Silicon with a Covalent Co−Si Bond. Organometallics 2015, 34, 1546–1551. [Google Scholar] [CrossRef]
- Sato, T.; Okazaki, M.; Tobita, H.; Ogino, H. Synthesis, structure, and reactivity of novel iron(II) complexes with a five-membered chelate ligand κ2(Si,N)-SiMe2O(2-C5H4N). J. Organomet. Chem. 2003, 669, 189–199. [Google Scholar] [CrossRef]
- Kwok, W.-H.; Lu, G.-L.; Rickard, C.E.F.; Roper, W.R.; Wright, L.J. Tethered silyl complexes from nucleophilic substitution reactions at the Si–Cl bond of the chloro(diphenyl)silyl ligand in Ru(SiClPh2)(κ2-S2CNMe2)(CO)(PPh3)2. J. Organomet. Chem. 2004, 689, 2979–2987. [Google Scholar] [CrossRef]
- Kanno, Y.; Komuro, T.; Tobita, H. Direct Conversion of a Si−C(aryl) Bond to Si−Heteroatom Bonds in the Reactions of η3-α-Silabenzyl Molybdenum and Tungsten Complexes with 2-Substituted Pyridines. Organometallics 2015, 34, 3699–3705. [Google Scholar] [CrossRef]
- Motherwell, W.B.; Storey, L.J. Some studies on nucleophilic trifluoromethylation using the shelf-stable trifluoromethylacetophenone-N,N-dimethyltrimethylsilylamine adduct. J. Fluor. Chem. 2005, 126, 491–498. [Google Scholar] [CrossRef]
- Wächtler, E.; Gericke, R.; Kutter, S.; Brendler, E.; Wagler, J. Molecular structures of pyridinethiolato complexes of Sn(II), Sn(IV), Ge(IV), and Si(IV). Main Group Met. Chem. 2013, 36, 181–191. [Google Scholar] [CrossRef]
- Baus, J.A.; Burschka, C.; Bertermann, R.; Fonseca Guerra, C.; Bickelhaupt, F.M.; Tacke, R. Neutral Six-Coordinate and Cationic Five-Coordinate Silicon(IV) Complexes with Two Bidentate Monoanionic N,S-Pyridine-2-thiolato(-) Ligands. Inorg. Chem. 2013, 52, 10664–10676. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.G.; Brennessel, W.W.; Kraft, B.M. Neutral and Cationic Bis-Chelate Monoorganosilicon(IV) Complexes of 1-Hydroxy-2-pyridinone. Organometallics 2017, 36, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Kraft, B.M.; Brennessel, W.W. Chelation and Stereodynamic Equilibria in Neutral Hypercoordinate Organosilicon Complexes of 1-Hydroxy-2-pyridinone. Organometallics 2014, 33, 158–171. [Google Scholar] [CrossRef]
- Schraml, J.; Chvalovsky, V.; Magi, M.; Lippmaa, E. NMR study of organosilicon compounds. XI. The role of electronic and steric effects in silicon-29 NMR spectra of compounds with a silicon-oxygen-carbon group. Collect. Czechoslov. Chem. Commun. 1981, 46, 377–390. [Google Scholar] [CrossRef]
- Schraml, J.; Brezny, R.; Cermak, J.; Chvalovsky, V. Silicon-29 and carbon-13 NMR spectra of some substituted bis(trimethylsiloxy)benzenes. Collect. Czechoslov. Chem. Commun. 1990, 55, 2033–2037. [Google Scholar] [CrossRef]
- Wagler, J.; Heine, T.; Hill, A.F. Poly(methimazolyl)silanes: Syntheses and Molecular Structures. Organometallics 2010, 29, 5607–5613. [Google Scholar] [CrossRef]
- Gualco, P.; Mallet-Ladeira, S.; Kameo, H.; Nakazawa, H.; Mercy, M.; Maron, L.; Amgoune, A.; Bourissou, D. Coordination of a Triphosphine-Silane to Gold: Formation of a Trigonal Pyramidal Complex Featuring Au→Si Interaction. Organometallics 2015, 34, 1449–1453. [Google Scholar] [CrossRef]
- Gualco, P.; Mercy, M.; Ladeira, S.; Coppel, Y.; Maron, L.; Amgoune, A.; Bourissou, D. Hypervalent Silicon Compounds by Coordination of Diphosphine-Silanes to Gold. Chem. Eur. J. 2010, 16, 10808–10817. [Google Scholar] [CrossRef] [PubMed]
- Gualco, P.; Lin, T.-P.; Sircoglou, M.; Mercy, M.; Ladeira, S.; Bouhadir, G.; Pérez, L.M.; Amgoune, A.; Maron, L.; Gabbaï, F.P.; Bourissou, D. Gold–Silane and Gold–Stannane Complexes: Saturated Molecules as σ-Acceptor Ligands. Angew. Chem. Int. Ed. 2009, 48, 9892–9895. [Google Scholar] [CrossRef] [PubMed]
- Kameo, H.; Kawamoto, T.; Bourissou, D.; Sakaki, S.; Nakazawa, H. Evaluation of the σ-Donation from Group 11 Metals (Cu, Ag, Au) to Silane, Germane, and Stannane Based on the Experimental/Theoretical Systematic Approach. Organometallics 2015, 34, 1440–1448. [Google Scholar] [CrossRef]
- Gualco, P.; Amgoune, A.; Miqueu, K.; Ladeira, S.; Bourissou, D. A Crystalline σ Complex of Copper. J. Am. Chem. Soc. 2011, 133, 4257–4259. [Google Scholar] [CrossRef] [PubMed]
- Grobe, J.; Lütke-Brochtrup, K.; Krebs, B.; Läge, M.; Niemeyer, H.-H.; Würthwein, E.-U. Alternativ-Liganden XXXVIII. Neue Versuche zur Synthese von Pd(0)- und Pt(0)-Komplexen des Tripod-Phosphanliganden FSi(CH2CH2PMe2)3. Z. Naturforsch. 2007, 62, 55–65. [Google Scholar] [CrossRef]
- Grobe, J.; Wehmschulte, R.; Krebs, B.; Läge, M. Alternativ-Liganden. XXXII Neue Tetraphosphan-Nickelkomplexe mit Tripod-Liganden des Typs XM’(OCH2PMe2)n(CH2CH2PR2)3−n (M’ = Si, Ge; n = 0–3). Z. Anorg. Allg. Chem. 1995, 621, 583–596. [Google Scholar] [CrossRef]
- Grobe, J.; Krummen, N.; Wehmschulte, R.; Krebs, B.; Läge, M. Alternativ-Liganden. XXXI Nickelcarbonylkomplexe mit Tripod-Liganden des Typs XM’(OCH2PMe2)n(CH2CH2PR2)3–n (M’ = Si, Ge; n = 0–3). Z. Anorg. Allg. Chem. 1994, 620, 1645–1658. [Google Scholar] [CrossRef]
- Wagler, J.; Roewer, G.; Gerlach, D. Photo-Driven Si–C Bond Cleavage in Hexacoordinate Silicon Complexes. Z. Anorg. Allg. Chem. 2009, 635, 1279–1287. [Google Scholar] [CrossRef]
- Gerlach, D.; Brendler, E.; Wagler, J. Hexacoordinate Silicon Compounds with a Dianionic Tetradentate (N,N′,N,N′)-Chelating Ligand. Inorganics 2016, 4, 8. [Google Scholar] [CrossRef]
- Wächtler, E.; Kämpfe, A.; Krupinski, K.; Gerlach, D.; Kroke, E.; Brendler, E.; Wagler, J. New Insights into Hexacoordinated Silicon Complexes with 8-Oxyquinolinato Ligands: 1,3-Shift of Si-Bound Hydrocarbyl Substituents and the Influence of Si-Bound Halides on the 8-Oxyquinolinate Coordination Features. Z. Naturforsch. 2014, 69, 1402–1418. [Google Scholar] [CrossRef]
- Brendler, E.; Wächtler, E.; Wagler, J. Hypercoordinate Silacycloalkanes: Step-by-Step Tuning of N→Si Interactions. Organometallics 2009, 28, 5459–5465. [Google Scholar] [CrossRef]
- Dinda, S.; Samuelson, A.G. The Nature of Bond Critical Points in Dinuclear Copper(I) Complexes. Chem. Eur. J. 2012, 18, 3032–3042. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, K.B. Application of the Pople-Santry-Segal CNDO Method to the Cyclopropylcarbinyl and Cyclobutyl cation and to Bicyclobutane. Tetrahedron 1967, 24, 1083–1096. [Google Scholar] [CrossRef]
- Bianchi, R.; Gervasio, G.; Marabello, D. Experimental Electron Density Analysis of Mn2(CO)10: Metal-Metal and Metal-Ligand Bond Characterization. Inorg. Chem. 2000, 39, 2360–2366. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, R.; Gervasio, G.; Marabello, D. The experimental charge density in transition metal compounds. C. R. Chim. 2005, 8, 1392–1399. [Google Scholar] [CrossRef]
- Macchi, P.; Proserpio, D.M.; Sironi, A. Experimental Electron Density in a Transition Metal Dimer: Metal-Metal and Metal-Ligand Bonds. J. Am. Chem. Soc. 1998, 120, 13429–13435. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Lepetit, C.; Fau, P.; Fajerwerg, K.; Kahn, M.L.; Silvi, B. Topological analysis of the metal-metal bond: A tutorial review. Coord. Chem. Rev. 2017, 345, 150–181. [Google Scholar] [CrossRef] [Green Version]
- Seiler, O.; Burschka, C.; Fenske, T.; Troegel, D.; Tacke, R. Neutral Hexa- and Pentacoordinate Silicon(IV) Complexes with SiO6 and SiO4N Skeletons. Inorg. Chem. 2007, 46, 5419–5424. [Google Scholar] [CrossRef] [PubMed]
- Kämpfe, A.; Brendler, E.; Kroke, E.; Wagler, J. Tp*Cu(I)–CN–SiL2–NC–Cu(I)Tp*—A hexacoordinate Si-complex as connector for redox active metals via π-conjugated ligands. Dalton Trans. 2015, 44, 4744–4750. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Program for the Solution of Crystal Structures; shelxs-97; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. Program for the Refinement of Crystal Structures; shelxl-2014/7; University of Göttingen: Göttingen, Germany, 2014. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. ORTEP-3 for windows—A version of ORTEP-III with a graphical user interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- POV-RAY (Version 3.6), Trademark of Persistence of Vision Raytracer Pty. Ltd., Williamstown, Victoria (Australia). Copyright Hallam Oaks Pty. Ltd., 1994–2004. Available online: http://www.povray.org/download/ (accessed on 22 December 2011).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scurseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, A.; et al. Gaussian09; revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 6.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2013; Available online: http://nbo6.chem.wisc.edu/ (accessed on 16 August 2016).
- Chemcraft ver. 1.8 (Build 164). 2016. Available online: http://www.chemcraftprog.com (accessed on 8 April 2016).
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, R.F.W. Atoms in Molecules; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K.J. VMD: visual molecular dynamics. J. Mol. Gr. 1996, 14, 33–38. Available online: http://www.ks.uiuc.edu/Research/vmd/ (accessed on 5 April 2018). [CrossRef]
Chart 1.
Selected metal–silicon complexes.

Scheme 1.
Syntheses of starting materials MeSi(pyO)3 and Si(pyO)4.

Figure 1.
Molecular structures of MeSi(pyO)3 and Si(pyO)4 in the crystal; thermal displacement ellipsoids are drawn at the 50% probability level; H atoms are omitted for clarity and selected atoms are labeled. Because of the special crystallographic position of the Si atom of Si(pyO)4 in the solid (S4 symmetry), the asymmetric unit consists of ¼ of the molecule. The asterisked labels indicate the symmetry equivalents. Selected interatomic distances (Å) and angles (deg) are as follows, for MeSi(pyO)3: Si1–O1 1.645(1), Si1–O2 1.638(1), Si1–O3 1.652(1), Si1–C16 1.837(2), Si1···N1 2.998(1), Si1···N2 3.028(1), Si1···N3 2.920(1), O1–Si1–O2 114.68(5), O2–Si1–O3 112.74(5), O1–Si1–O3 97.77(5), O1–Si1–C16 111.96(7), O2–Si1–C16 106.46(6), O3–Si1–C16 113.72(6); for Si(pyO)4: Si1–O1 1.630(1), Si1···N1 2.913(2); O1–Si1–O1* = O1*–Si1–O1** = O1**–Si1–O1*** = O1–Si1–O1*** 113.64(5), O1–Si1–O1** = O1*–Si1–O1*** 101.42(9).
Figure 1.
Molecular structures of MeSi(pyO)3 and Si(pyO)4 in the crystal; thermal displacement ellipsoids are drawn at the 50% probability level; H atoms are omitted for clarity and selected atoms are labeled. Because of the special crystallographic position of the Si atom of Si(pyO)4 in the solid (S4 symmetry), the asymmetric unit consists of ¼ of the molecule. The asterisked labels indicate the symmetry equivalents. Selected interatomic distances (Å) and angles (deg) are as follows, for MeSi(pyO)3: Si1–O1 1.645(1), Si1–O2 1.638(1), Si1–O3 1.652(1), Si1–C16 1.837(2), Si1···N1 2.998(1), Si1···N2 3.028(1), Si1···N3 2.920(1), O1–Si1–O2 114.68(5), O2–Si1–O3 112.74(5), O1–Si1–O3 97.77(5), O1–Si1–C16 111.96(7), O2–Si1–C16 106.46(6), O3–Si1–C16 113.72(6); for Si(pyO)4: Si1–O1 1.630(1), Si1···N1 2.913(2); O1–Si1–O1* = O1*–Si1–O1** = O1**–Si1–O1*** = O1–Si1–O1*** 113.64(5), O1–Si1–O1** = O1*–Si1–O1*** 101.42(9).

Chart 2.
Selected metal–silicon complexes with donor–acceptor-interactions between the Si atom of a tetravalent silane and a d10 metal atom.
Chart 2.
Selected metal–silicon complexes with donor–acceptor-interactions between the Si atom of a tetravalent silane and a d10 metal atom.

Scheme 2.
Reactions of MeSi(pyO)3 with [PdCl2(NCMe)2] and CuCl.

Figure 2.
Molecular structures of compounds 1 (in 1 · 2 CHCl3), 2, and 3 in the crystal; thermal displacement ellipsoids are drawn at the 50% probability level; H atoms are omitted for clarity and selected atoms are labeled. For compound 2, only one of the two crystallographically independent (but conformationally similar) molecules is depicted. The selected interatomic distances (Å) and angles (deg) for 1 are as follows: Pd1–Si1 2.627(1), Pd1–Cl1 2.891(1), Pd1–N1 2.023(3), Pd1–N2 2.039(3), Pd1–N3 2.028(3), Pd1–N4 2.013(3), Si1–O1 1.785(2), Si1–O2 1.775(2), Si1–O3 1.789(2), Si1–O4 1.780(2), Si1–C21 1.853(4), Pd1–Si1–C21 177.88(12), Cl1–Pd1–Si1 177.36(3), N1–Pd1–N3 170.10(11), N2–Pd1–N4 170.91(11), O1–Si1–O3 163.66(12), O2–Si1–O4 163.72(12); for 2: Cu1–Cl1 2.361(1), Cu1–N1 2.038(2), Cu1–N2 2.039(2), Cu1–N3 2.023(2), Cu1···Si1 3.204(1), Si1–O1 1.626(2), Si1–O2 1.629(2), Si1–O3 1.618(2), Si1–C16 1.834(3); O1–Si1–C16 106.11(12), O2–Si1–C16 106.90(14), O3–Si1–C16 106.05(14), O1–Si1–O2 111.41(13), O1–Si1–O3 113.91(12), O2–Si1–O3 111.89(12); and for 3: Cu1···Si1 2.919(1), Cu1–Cl1 2.403(1), Cu1–N1 2.023(2), Cu1–N2 2.055(2), Cu1–N3 2.025(2), Cu1–N4 2.049(2), Si1–O1 1.753(2), Si1–O2 1.757(2), Si1–O3 1.753(2), Si1–O4 1.755(2), Si1–C21 1.847(2); Cu1–Si1–C21 177.51(6), Cl1–Cu1–Si1 178.24(2), N1–Cu1–N3 159.32(5), N2–Cu1–N4 158.66(5), O1–Si1–O3 154.22(6), and O2–Si1–O4 155.83(6).
Figure 2.
Molecular structures of compounds 1 (in 1 · 2 CHCl3), 2, and 3 in the crystal; thermal displacement ellipsoids are drawn at the 50% probability level; H atoms are omitted for clarity and selected atoms are labeled. For compound 2, only one of the two crystallographically independent (but conformationally similar) molecules is depicted. The selected interatomic distances (Å) and angles (deg) for 1 are as follows: Pd1–Si1 2.627(1), Pd1–Cl1 2.891(1), Pd1–N1 2.023(3), Pd1–N2 2.039(3), Pd1–N3 2.028(3), Pd1–N4 2.013(3), Si1–O1 1.785(2), Si1–O2 1.775(2), Si1–O3 1.789(2), Si1–O4 1.780(2), Si1–C21 1.853(4), Pd1–Si1–C21 177.88(12), Cl1–Pd1–Si1 177.36(3), N1–Pd1–N3 170.10(11), N2–Pd1–N4 170.91(11), O1–Si1–O3 163.66(12), O2–Si1–O4 163.72(12); for 2: Cu1–Cl1 2.361(1), Cu1–N1 2.038(2), Cu1–N2 2.039(2), Cu1–N3 2.023(2), Cu1···Si1 3.204(1), Si1–O1 1.626(2), Si1–O2 1.629(2), Si1–O3 1.618(2), Si1–C16 1.834(3); O1–Si1–C16 106.11(12), O2–Si1–C16 106.90(14), O3–Si1–C16 106.05(14), O1–Si1–O2 111.41(13), O1–Si1–O3 113.91(12), O2–Si1–O3 111.89(12); and for 3: Cu1···Si1 2.919(1), Cu1–Cl1 2.403(1), Cu1–N1 2.023(2), Cu1–N2 2.055(2), Cu1–N3 2.025(2), Cu1–N4 2.049(2), Si1–O1 1.753(2), Si1–O2 1.757(2), Si1–O3 1.753(2), Si1–O4 1.755(2), Si1–C21 1.847(2); Cu1–Si1–C21 177.51(6), Cl1–Cu1–Si1 178.24(2), N1–Cu1–N3 159.32(5), N2–Cu1–N4 158.66(5), O1–Si1–O3 154.22(6), and O2–Si1–O4 155.83(6).

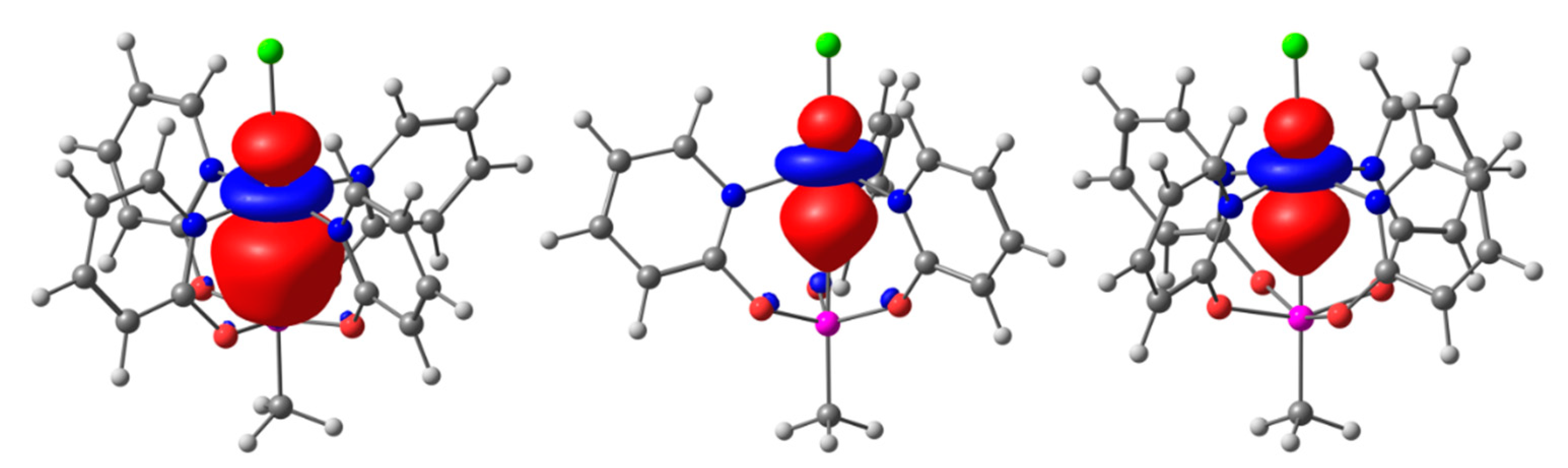
Figure 3.
Natural localized molecular orbital (NLMO) representations of the TM→Si interaction in (from left) compounds 1, 2, and 3. For compound 3, the α-spin contribution is shown. NLMOs are depicted with an isosurface value of 0.02, the atom color code is consistent with Figure 2.
Figure 3.
Natural localized molecular orbital (NLMO) representations of the TM→Si interaction in (from left) compounds 1, 2, and 3. For compound 3, the α-spin contribution is shown. NLMOs are depicted with an isosurface value of 0.02, the atom color code is consistent with Figure 2.

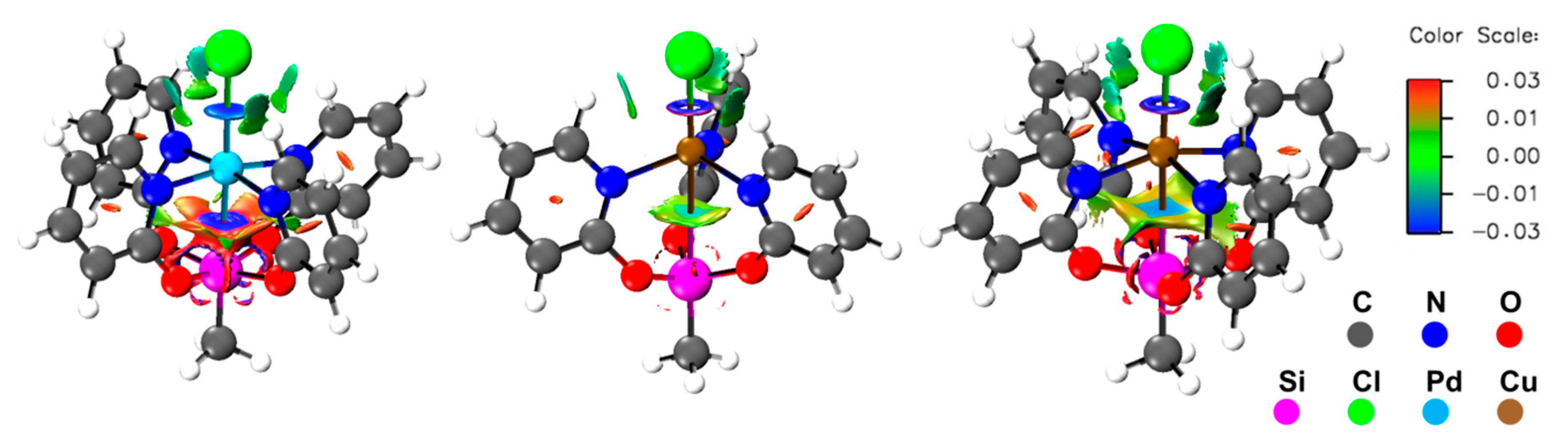
Figure 4.
Non-covalent interaction (NCI) representations of (from left) compounds 1, 2, and 3 depicted with an isosurface value of 0.4 and a color range from −0.03 (blue, attractive) to 0.03 (red, repulsive).
Figure 4.
Non-covalent interaction (NCI) representations of (from left) compounds 1, 2, and 3 depicted with an isosurface value of 0.4 and a color range from −0.03 (blue, attractive) to 0.03 (red, repulsive).

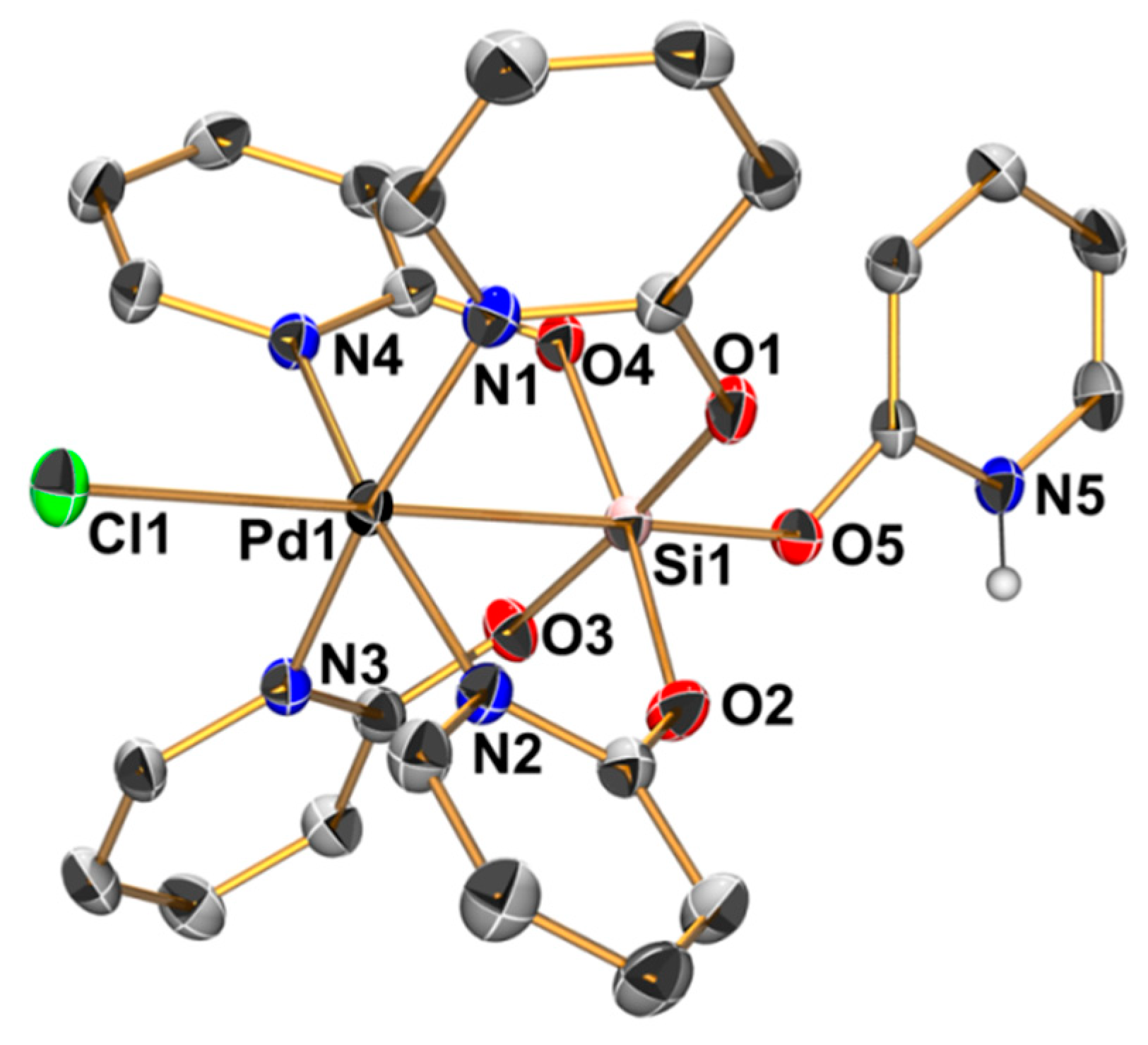
Figure 5.
Molecular structure of the cation [ClPd(µ-pyO)4Si(HpyO)]+ in the crystal structure of compound 4 · 4 CHCl3; thermal displacement ellipsoids are drawn at the 50% probability level; C-bound H atoms are omitted for clarity and the selected atoms are labeled. Selected bond lengths (Å) and angles (deg) are as follows: Pd1–Si1 2.496(1), Pd1–Cl1 2.785(1), Pd1–N1 2.033(1), Pd1–N2 2.036(1), Pd1–N3 2.019(1), Pd1–N4 2.031(1), Si1–O1 1.765(1), Si1–O2 1.751(1), Si1–O3 1.758(1), Si1–O4 1.759(1), Si1–O5 1.709(1), Pd1–Si1–O5 178.57(5), Cl1–Pd1–Si1 178.87(2), N1–Pd1–N3 170.88(6), N2–Pd1–N4 170.04(5), O1–Si1–O3 173.01(6), and O2–Si1–O4 173.77(6).
Figure 5.
Molecular structure of the cation [ClPd(µ-pyO)4Si(HpyO)]+ in the crystal structure of compound 4 · 4 CHCl3; thermal displacement ellipsoids are drawn at the 50% probability level; C-bound H atoms are omitted for clarity and the selected atoms are labeled. Selected bond lengths (Å) and angles (deg) are as follows: Pd1–Si1 2.496(1), Pd1–Cl1 2.785(1), Pd1–N1 2.033(1), Pd1–N2 2.036(1), Pd1–N3 2.019(1), Pd1–N4 2.031(1), Si1–O1 1.765(1), Si1–O2 1.751(1), Si1–O3 1.758(1), Si1–O4 1.759(1), Si1–O5 1.709(1), Pd1–Si1–O5 178.57(5), Cl1–Pd1–Si1 178.87(2), N1–Pd1–N3 170.88(6), N2–Pd1–N4 170.04(5), O1–Si1–O3 173.01(6), and O2–Si1–O4 173.77(6).

Scheme 3.
Reactions of Si(pyO)4 with [PdCl2(NCMe)2].

Scheme 4.
Reactions of Si(pyO)4 and HpyO with [PdCl2(NCMe)2] and CuCl2.

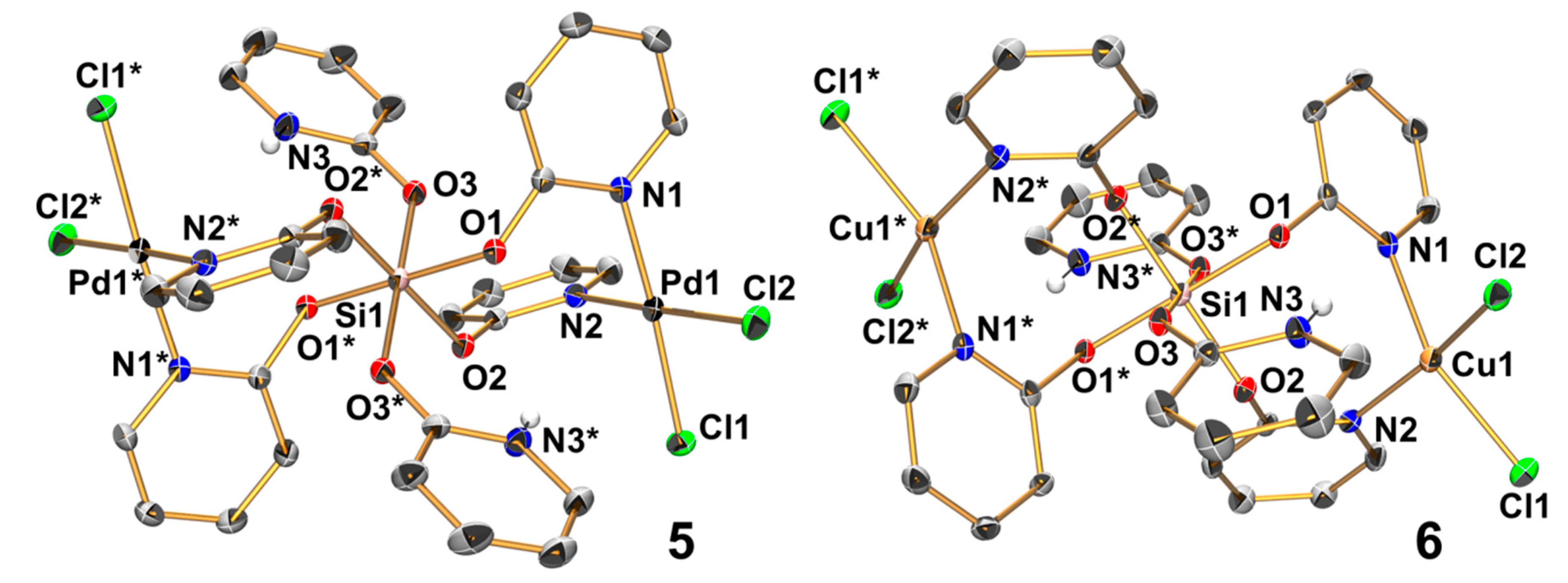
Figure 6.
Molecular structures of 5 and 6 in the crystal (5 in the structure of solvate 5 · 6 CHCl3); thermal displacement ellipsoids are drawn at the 30% probability level; C-bound H atoms are omitted for clarity and selected atoms are labeled. In both cases, the Si atom is located on a crystallographically imposed center of inversion, and therefore the asymmetric unit consists of a half molecule and the symmetry related sites (e.g., O1 and O1* at Si1) are trans to each other. Selected bond lengths (Å) and angles (deg) for 5 are as follows: Pd1–Cl1 2.290(1), Pd1–Cl2 2.303(1), Pd1–N1 2.028(2), Pd1–N2 2.036(2), Si1–O1 1.794(2), Si1–O2 1.760(2), Si1–O3 1.770(2), N1–Pd1–Cl1 175.93(7), N2–Pd1–Cl2 176.15(7), Cl1–Pd1–Cl2 91.60(3), Cl1–Pd1–N2 88.91(7), Cl2–Pd1–N1 87.65(7), and N1–Pd1–N2 92.10(9); for 6: Cu1–Cl1 2.238(1), Cu1–Cl2 2.256(2), Cu1–N1 2.008(4), Cu1–N2 1.990(4), Si1–O1 1.790(3), Si1–O2 1.770(3), Si1–O3 1.755(3), N1–Cu1–Cl1 153.97(12), N2–Cu1–Cl2 162.50(12), Cl1–Cu1–Cl2 93.69(5), Cl1–Cu1–N2 90.82(11), Cl2–Cu1–N1 90.31(12), and N1–Cu1–N2 93.03(16).
Figure 6.
Molecular structures of 5 and 6 in the crystal (5 in the structure of solvate 5 · 6 CHCl3); thermal displacement ellipsoids are drawn at the 30% probability level; C-bound H atoms are omitted for clarity and selected atoms are labeled. In both cases, the Si atom is located on a crystallographically imposed center of inversion, and therefore the asymmetric unit consists of a half molecule and the symmetry related sites (e.g., O1 and O1* at Si1) are trans to each other. Selected bond lengths (Å) and angles (deg) for 5 are as follows: Pd1–Cl1 2.290(1), Pd1–Cl2 2.303(1), Pd1–N1 2.028(2), Pd1–N2 2.036(2), Si1–O1 1.794(2), Si1–O2 1.760(2), Si1–O3 1.770(2), N1–Pd1–Cl1 175.93(7), N2–Pd1–Cl2 176.15(7), Cl1–Pd1–Cl2 91.60(3), Cl1–Pd1–N2 88.91(7), Cl2–Pd1–N1 87.65(7), and N1–Pd1–N2 92.10(9); for 6: Cu1–Cl1 2.238(1), Cu1–Cl2 2.256(2), Cu1–N1 2.008(4), Cu1–N2 1.990(4), Si1–O1 1.790(3), Si1–O2 1.770(3), Si1–O3 1.755(3), N1–Cu1–Cl1 153.97(12), N2–Cu1–Cl2 162.50(12), Cl1–Cu1–Cl2 93.69(5), Cl1–Cu1–N2 90.82(11), Cl2–Cu1–N1 90.31(12), and N1–Cu1–N2 93.03(16).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
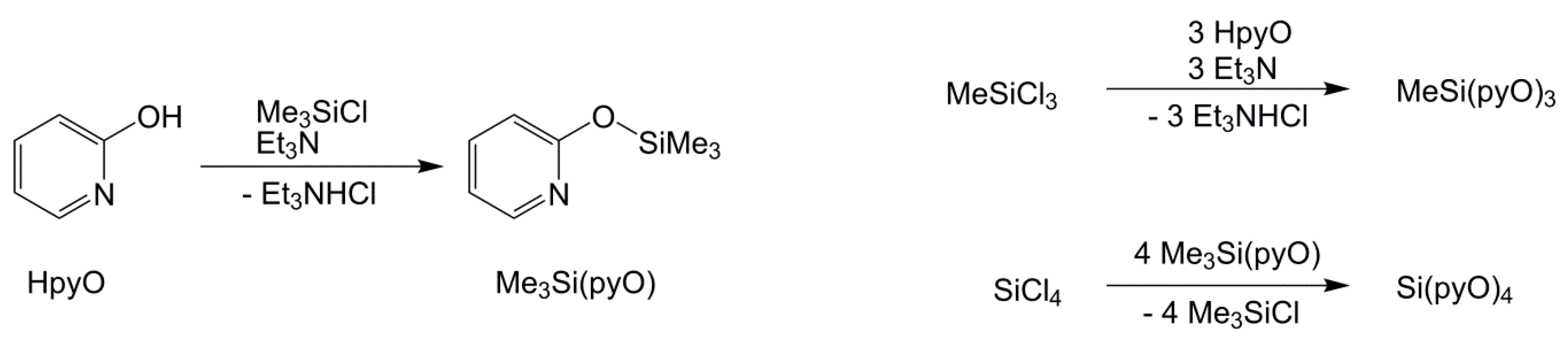{kind=link}
{kind=link}
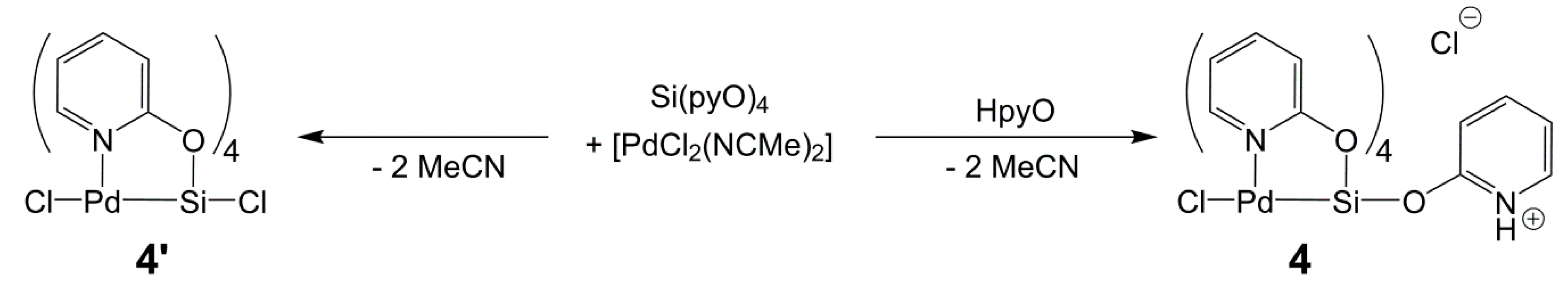{kind=link}
{kind=link}
Table 1.
Crystallographic data from data collection and refinement for MeSi(pyO)3, Si(pyO)4, 1 · 2 CHCl3, and 2.
Table 1.
Crystallographic data from data collection and refinement for MeSi(pyO)3, Si(pyO)4, 1 · 2 CHCl3, and 2.
| Parameter | MeSi(pyO)3 | Si(pyO)4 | 1 · 2 CHCl3 | 2 |
|---|---|---|---|---|
| Formula | C16H15N3O3Si | C20H16N4O4Si | C23H21Cl7N4O4PdSi | C16H15ClCuN3O3Si |
| Mr | 325.40 | 404.46 | 800.08 | 424.39 |
| T(K) | 200(2) | 200(2) | 180(2) | 200(2) |
| λ(Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | triclinic | tetragonal | monoclinic | triclinic |
| Space group | P-1 | I41/a | C2/c | P-1 |
| a(Å) | 9.1581(7) | 9.5163(7) | 14.8719(5) | 8.7497(4) |
| b(Å) | 9.3250(7) | 9.5163(7) | 10.5112(5) | 9.2334(5) |
| c(Å) | 11.4078(9) | 21.824(2) | 39.3020(13) | 23.5781(13) |
| α(°) | 92.440(6) | 90 | 90 | 88.255(4) |
| β(°) | 109.582(6) | 90 | 95.404(3) | 89.283(4) |
| γ(°) | 116.896(6) | 90 | 90 | 68.654(4) |
| V(Å3) | 796.41(12) | 1976.4(3) | 6116.4(4) | 1773.36(17) |
| Z | 2 | 4 | 8 | 4 |
| ρcalc(g·cm−1) | 1.36 | 1.36 | 1.74 | 1.59 |
| µMo Kα (mm−1) | 0.2 | 0.2 | 1.3 | 1.5 |
| F(000) | 340 | 840 | 3184 | 864 |
| θmax(°), Rint | 28.0, 0.0263 | 25.0, 0.0238 | 25.0, 0.0355 | 28.0, 0.0310 |
| Completeness | 99.9% | 99.8% | 99.9% | 99.8% |
| Reflections collected | 12193 | 3724 | 52652 | 28693 |
| Reflns unique | 3836 | 873 | 5379 | 8555 |
| Restraints | 0 | 0 | 18 | 0 |
| Parameters | 209 | 66 | 403 | 453 |
| GoF | 1.066 | 1.137 | 1.147 | 1.073 |
| R1, wR2 [I > 2σ(I)] | 0.0343, 0.0871 | 0.0404, 0.0934 | 0.0339, 0.0765 | 0.0350, 0.0858 |
| R1, wR2 (all data) | 0.0426, 0.0925 | 0.0567, 0.1062 | 0.0423, 0.0818 | 0.0444, 0.0900 |
| Largest peak/hole (e·Å−3) | 0.22, −0.31 | 0.16, −0.28 | 0.67, −0.65 | 0.77, −0.28 |
Table 2.
Crystallographic data from data collection and refinement for 3, 3 · CHCl3, and 4 · 4 CHCl3.
Table 2.
Crystallographic data from data collection and refinement for 3, 3 · CHCl3, and 4 · 4 CHCl3.
| Parameter | 3 | 3 · CHCl3 | 4 · 4 CHCl3 |
|---|---|---|---|
| Formula | C21H19ClCuN4O4Si | C22H20Cl4CuN4O4Si | C29H25Cl14N5O5PdSi |
| Mr | 518.48 | 637.85 | 1154.33 |
| T(K) | 200(2) | 200(2) | 200(2) |
| λ(Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | tetragonal | monoclinic | triclinic |
| Space group | I41/a | P21 | P-1 |
| a(Å) | 18.3774(5) | 9.2616(4) | 11.2079(5) |
| b(Å) | 18.3774(5) | 15.6201(7) | 13.1431(6) |
| c(Å) | 26.4847(8) | 9.2773(5) | 15.1242(7) |
| α(°) | 90 | 90 | 78.113(4) |
| β(°) | 90 | 93.049(4) | 87.403(4) |
| γ(°) | 90 | 90 | 89.899(4) |
| V(Å3) | 8944.6(6) | 1340.22(11) | 2177.81(17) |
| Z | 16 | 2 | 2 |
| ρcalc(g·cm−1) | 1.54 | 1.58 | 1.76 |
| µMo Kα (mm−1) | 1.2 | 1.3 | 1.4 |
| F(000) | 4240 | 646 | 1144 |
| θmax(°), Rint | 28.0, 0.0425 | 28.0, 0.0302 | 27.0, 0.0271 |
| Completeness | 99.9% | 99.9% | 99.9% |
| Reflns collected | 71,378 | 22,760 | 34,910 |
| Reflns unique | 5409 | 6449 | 9502 |
| Restraints | 0 | 1 | 12 |
| Parameters | 290 | 326 | 562 |
| GoF | 1.081 | 1.060 | 1.059 |
| χFlack | −0.008(4) | ||
| R1, wR2 [I > 2σ(I)] | 0.0278, 0.0679 | 0.0269, 0.0624 | 0.0230, 0.0557 |
| R1, wR2 (all data) | 0.0355, 0.0712 | 0.0302, 0.0639 | 0.0275, 0.0577 |
| Largest peak/hole (e·Å−3) | 0.45, −0.25 | 0.44, −0.31 | 0.52, −0.48 |
Table 3.
Selected features of the natural localized molecular orbitals (NLMOs) of the TM→Si interaction in compounds 1, 2, and 3.
Table 3.
Selected features of the natural localized molecular orbitals (NLMOs) of the TM→Si interaction in compounds 1, 2, and 3.
| Feature | 1 | 2 | 3 1 |
|---|---|---|---|
| % contribution TM | 90.7 | 97.9 | 98.6 |
| Hybrid (TM) | 97.6% 4d, 2.2% 5s | 99.6% 3d | 99.5% 3d |
| % contribution Si | 8.3 | 0.8 | 0.8 |
| Hybrid (Si) | 37.7% 3s, 61.7% 3p | 18.2% 3s, 79.5% 3p | 18.0% 3s, 79.7% 3p |
1 Contributions of α-spin and β-spin are essentially identical. TM—transition metal.
Table 4.
Selected features of the bond critical points (BCPs) of the TM–Si interaction in compounds 1, 2, and 3 derived from the topological analyses (AIM) of the wave function.
Table 4.
Selected features of the bond critical points (BCPs) of the TM–Si interaction in compounds 1, 2, and 3 derived from the topological analyses (AIM) of the wave function.
| Feature 1 | 1 | 2 | 3 |
|---|---|---|---|
| ρ(rb) | 0.04461 | 0.01229 | 0.01742 |
| ∇2ρ(rb) | −0.00127 | 0.02835 | 0.02896 |
| G(rb) | 0.01721 | 0.00721 | 0.00958 |
| V(rb) | −0.03475 | −0.00734 | −0.01192 |
| |V(rb)|/G(rb) | 2.018 | 1.017 | 1.244 |
| G(rb)/ρ(rb) | 0.386 | 0.587 | 0.550 |
| H(rb) | −0.01753 | −0.00012 | −0.00234 |
| H(rb)/ρ(rb) | −0.393 | −0.010 | −0.134 |
| Eint | −10.9 | −2.3 | −3.7 |
| WBO | 0.270 | 0.057 | 0.037 |
1 Electron density (ρ(rb) in a.u.), Laplacian of electron density (∇2ρ(rb) in a.u.), Lagrangian kinetic energy density (G(rb) in a.u.), potential energy density (V(rb) in a.u.), ratio |V(rb)|/G(rb), ratio G(rb)/ρ(rb) in a.u., electron energy density (H(rb) in a.u.), ratio H(rb)/ρ(rb) in a.u., estimated interaction energy according to Lepetit et al. [53] and Espinosa et al. [52] Eint = ½ 627.509469 V(rb) in kcal·mol−1. WBO—Wiberg bond order.
Table 5.
Crystallographic data from data collection and refinement for 5 · 2 CHCl3, 5 · 6 CHCl3, 5 · 8 CHCl3, and 6. Rint for the data set of 5 · 2 CHCl3 was not reported in the refinement output because of the HKLF5 format used for twin refinement.
Table 5.
Crystallographic data from data collection and refinement for 5 · 2 CHCl3, 5 · 6 CHCl3, 5 · 8 CHCl3, and 6. Rint for the data set of 5 · 2 CHCl3 was not reported in the refinement output because of the HKLF5 format used for twin refinement.
| Parameter | 5 · 2 CHCl3 | 5 · 6 CHCl3 | 5 · 8 CHCl3 | 6 |
|---|---|---|---|---|
| Formula | C32H28Cl10N6O6Pd2Si | C36H32Cl22N6O6Pd2Si | C38H34Cl28N6O6Pd2Si | C30H26Cl4Cu2N6O6Si |
| Mr | 1187.99 | 1665.46 | 1904.20 | 863.54 |
| T(K) | 200(2) | 200(2) | 200(2) | 200(2) |
| λ(Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | monoclinic | triclinic | monoclinic | monoclinic |
| Space group | C2/c | P-1 | P21/n | P21/n |
| a(Å) | 22.458(2) | 12.1698(6) | 12.3681(5) | 9.1063(8) |
| b(Å) | 11.8102(7) | 12.1951(7) | 13.1356(4) | 11.0475(6) |
| c(Å) | 17.4818(18) | 13.1643(8) | 22.2337(9) | 17.1744(17) |
| α(°) | 90 | 107.904(4) | 90 | 90 |
| β(°) | 111.330(7) | 103.147(4) | 95.990(3) | 104.705(7) |
| γ(°) | 90 | 114.671(4) | 90 | 90 |
| V(Å3) | 4319.1(7) | 1539.81(19) | 3592.4(2) | 1671.2(2) |
| Z | 4 | 1 | 2 | 2 |
| ρcalc(g·cm−1) | 1.83 | 1.80 | 1.76 | 1.72 |
| µMo Kα (mm−1) | 1.5 | 1.6 | 1.6 | 1.7 |
| F(000) | 2344 | 818 | 1868 | 872 |
| θmax(°), Rint | 25.0, / | 27.0, 0.0264 | 27.0, 0.0396 | 25.0, 0.0983 |
| Completeness | 99.8% | 99.9% | 99.9% | 99.7% |
| Reflns collected | 16169 | 18872 | 46707 | 16933 |
| Reflns unique | 3789 | 6728 | 7828 | 2933 |
| Restraints | 9 | 66 | 166 | 0 |
| Parameters | 289 | 410 | 539 | 226 |
| GoF | 1.034 | 1.055 | 1.033 | 0.924 |
| R1, wR2 [I > 2σ(I)] | 0.0474, 0.1009 | 0.0331, 0.0720 | 0.0299, 0.0663 | 0.0441, 0.0776 |
| R1, wR2 (all data) | 0.0958, 0.1166 | 0.0448, 0.0776 | 0.0399, 0.0699 | 0.1057, 0.0907 |
| Largest peak/hole (e·Å−3) | 0.76, −0.85 | 0.56, −0.58 | 0.46, −0.35 | 0.46, −0.72 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ehrlich, L.; Gericke, R.; Brendler, E.; Wagler, J. (2-Pyridyloxy)silanes as Ligands in Transition Metal Coordination Chemistry. Inorganics 2018, 6, 119. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6040119
AMA Style
Ehrlich L, Gericke R, Brendler E, Wagler J. (2-Pyridyloxy)silanes as Ligands in Transition Metal Coordination Chemistry. Inorganics. 2018; 6(4):119. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6040119
Chicago/Turabian StyleEhrlich, Lisa, Robert Gericke, Erica Brendler, and Jörg Wagler. 2018. "(2-Pyridyloxy)silanes as Ligands in Transition Metal Coordination Chemistry" Inorganics 6, no. 4: 119. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics6040119
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.