Successive Activation of C–H and C–O Bonds of Vinyl Ethers by a Diphosphine and Hydrido-Bridged Diiridium Complex

Abstract
:
{kind=link}
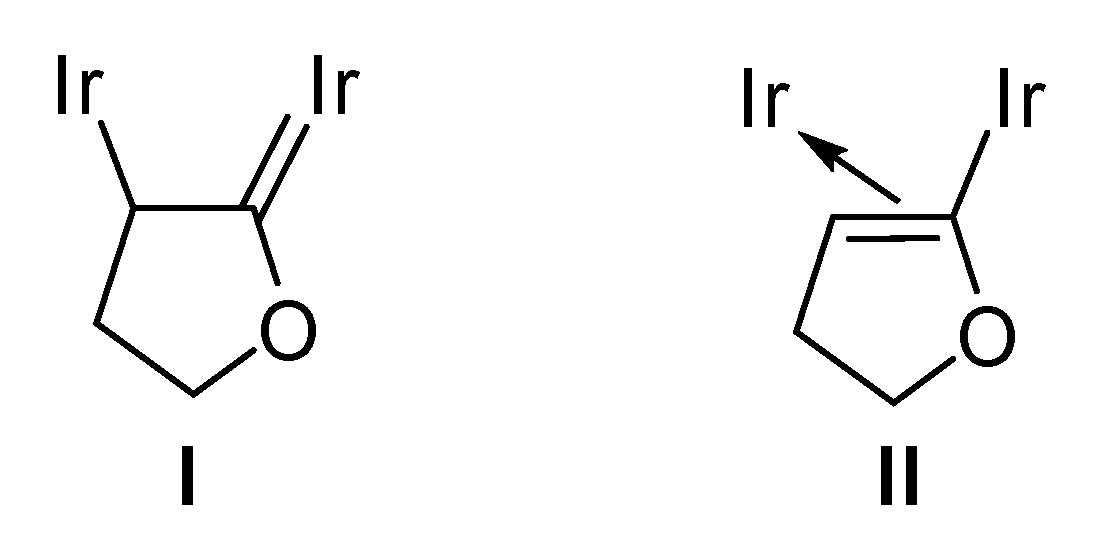{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
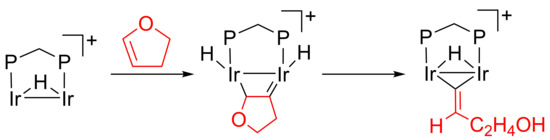
2.1. C–H Activation of 2,3-Dihydrofuran

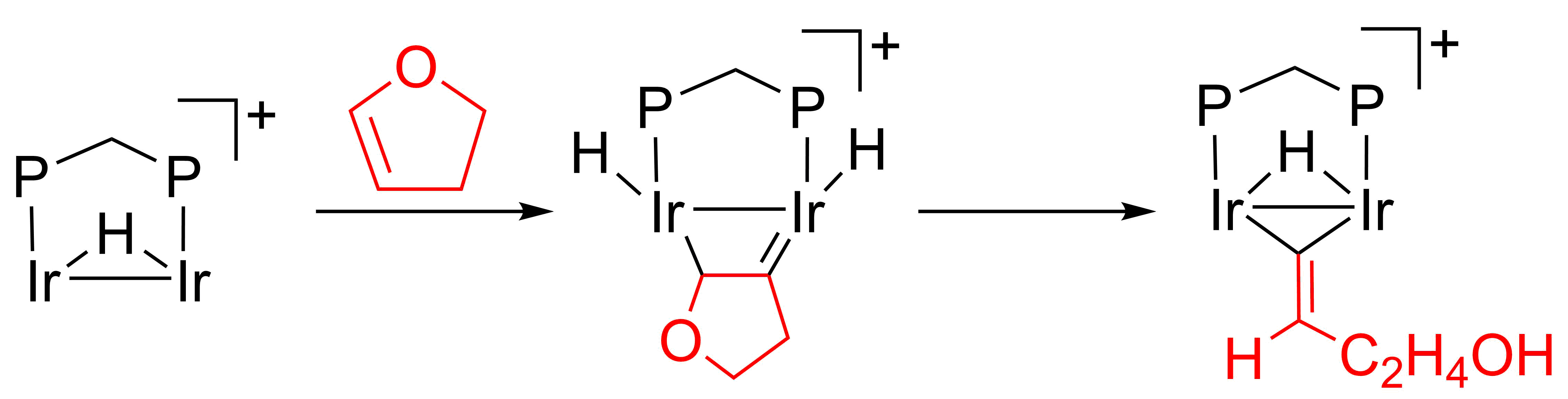
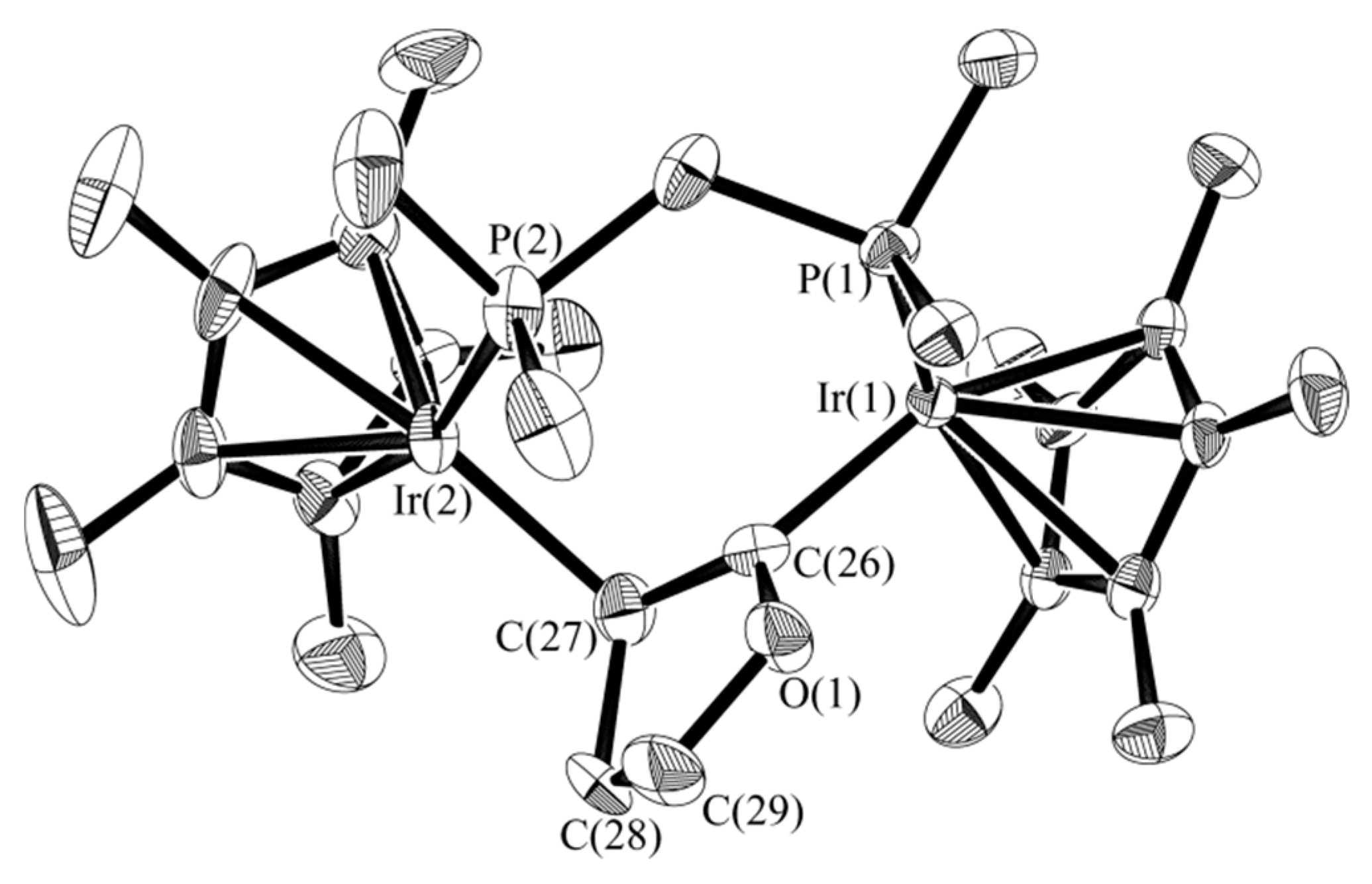
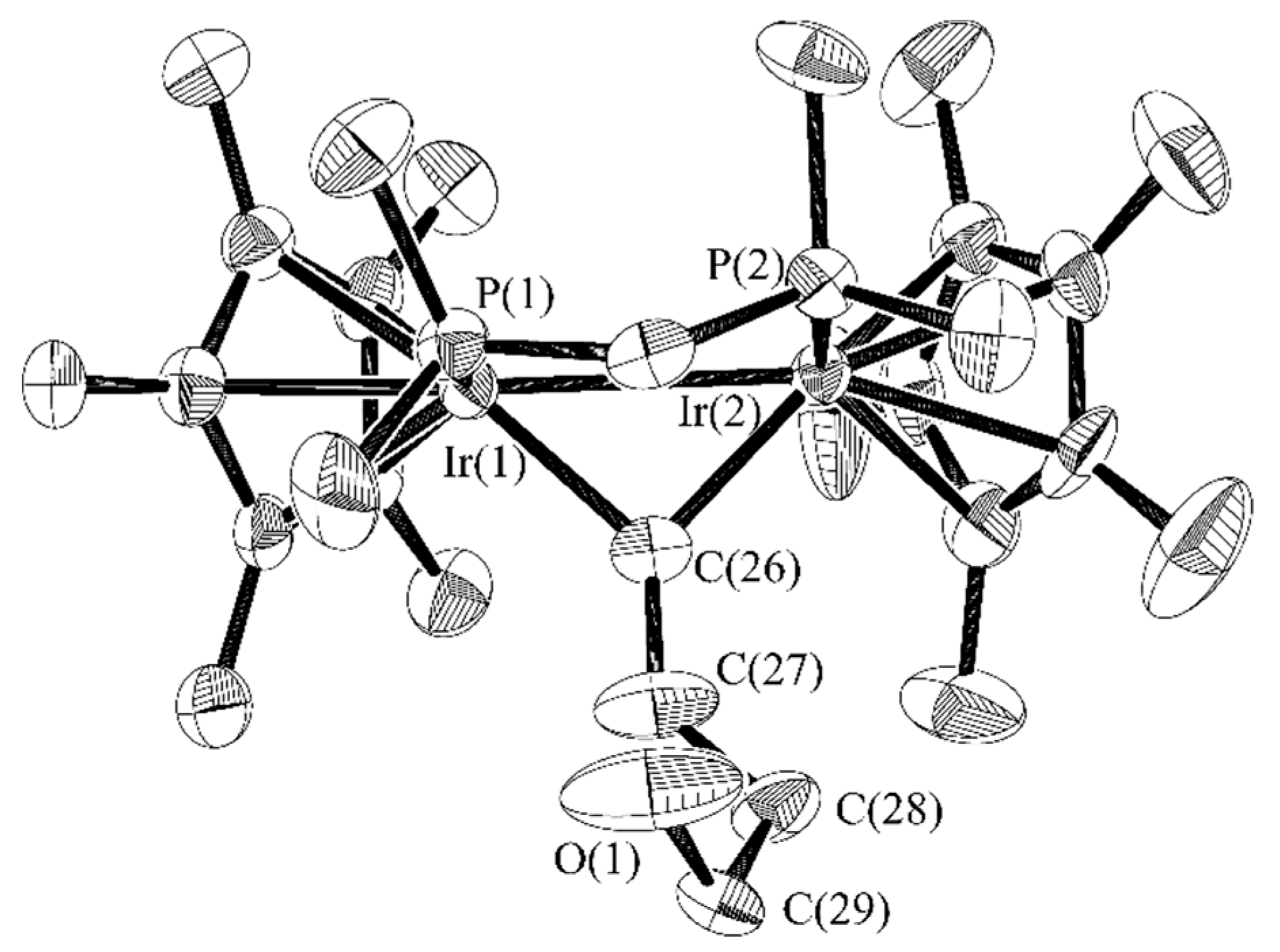
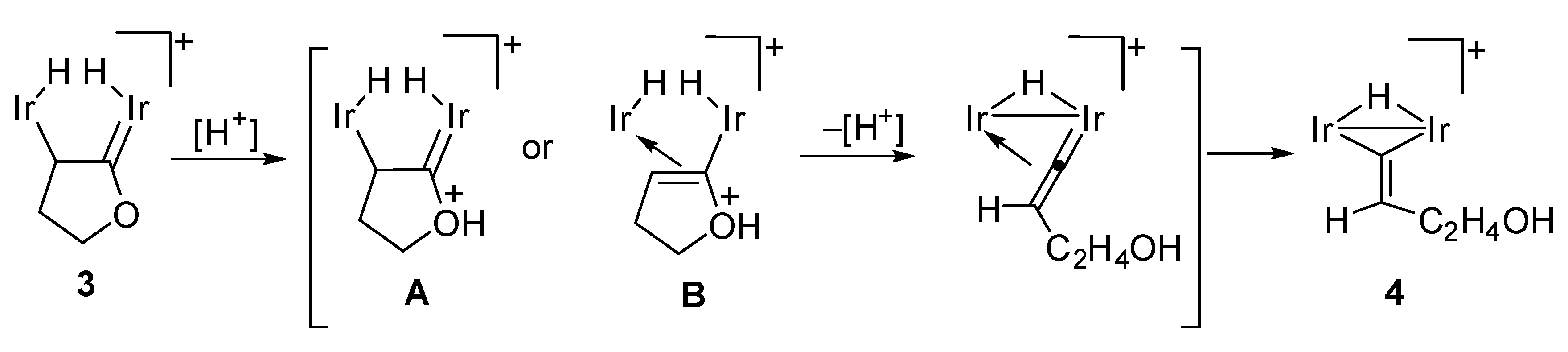
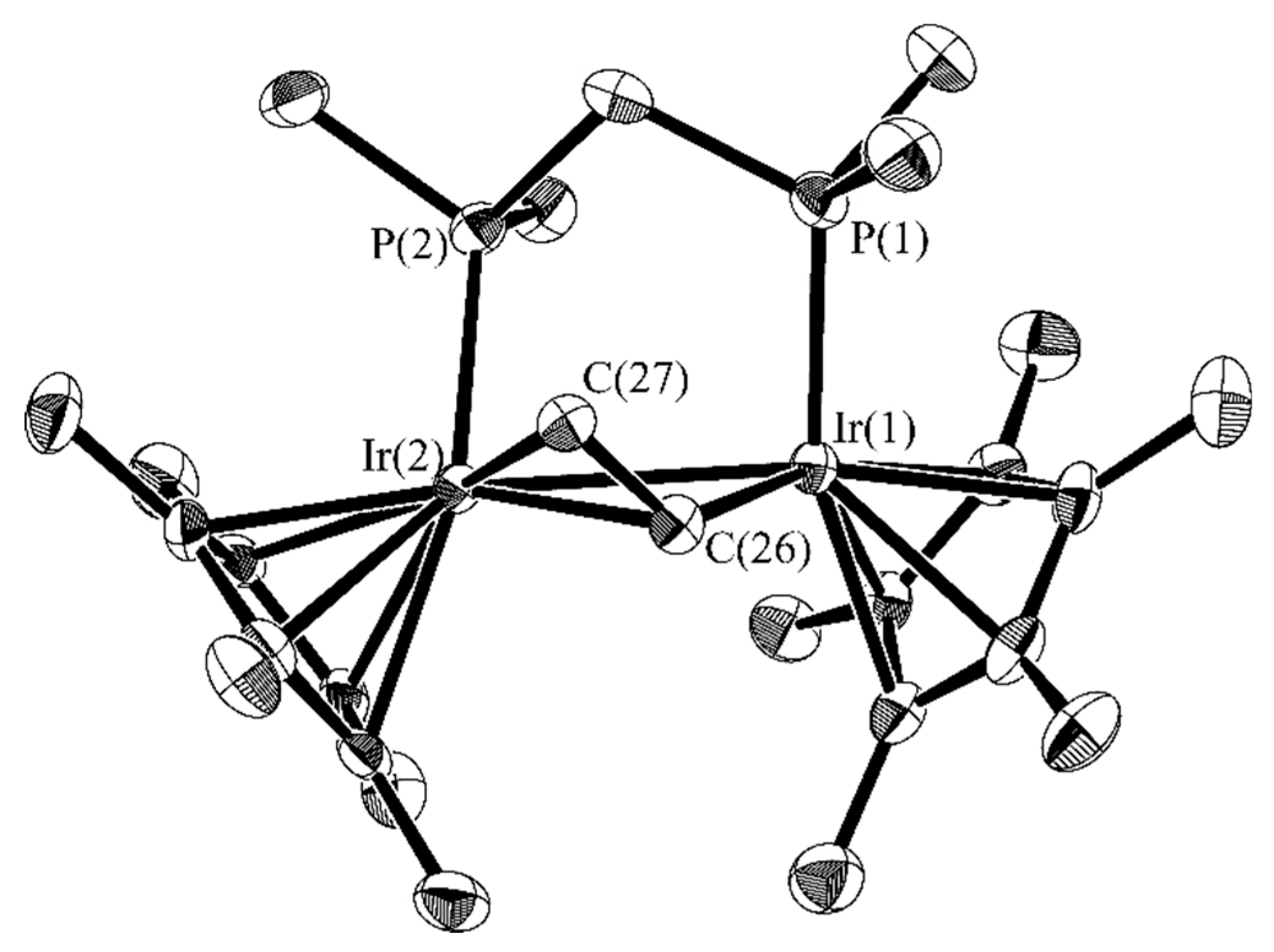
2.2. C–O Bond Activation of 3

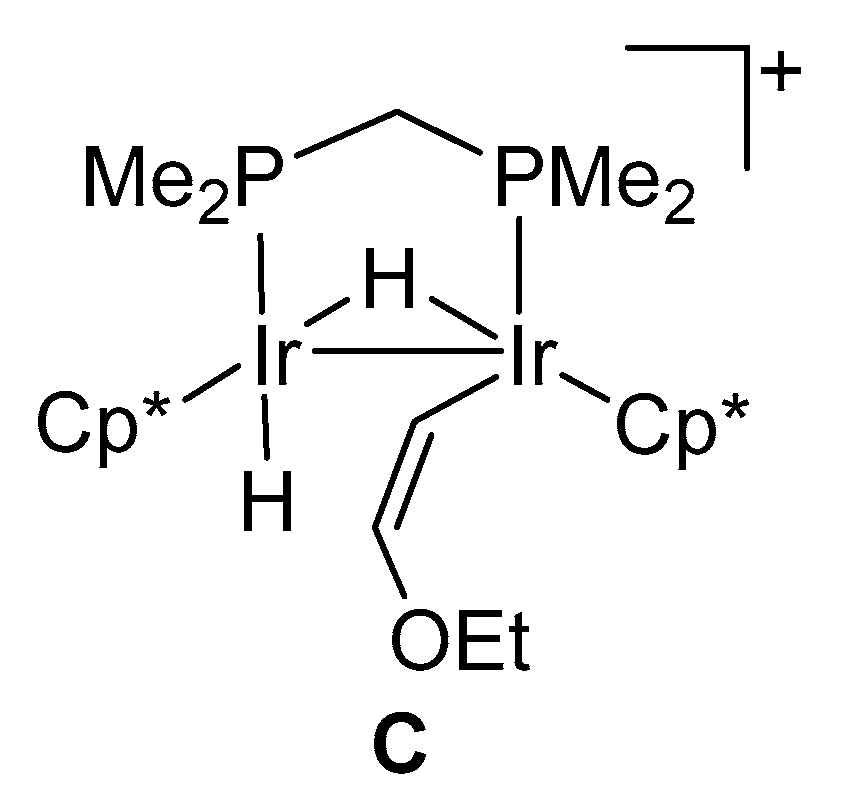
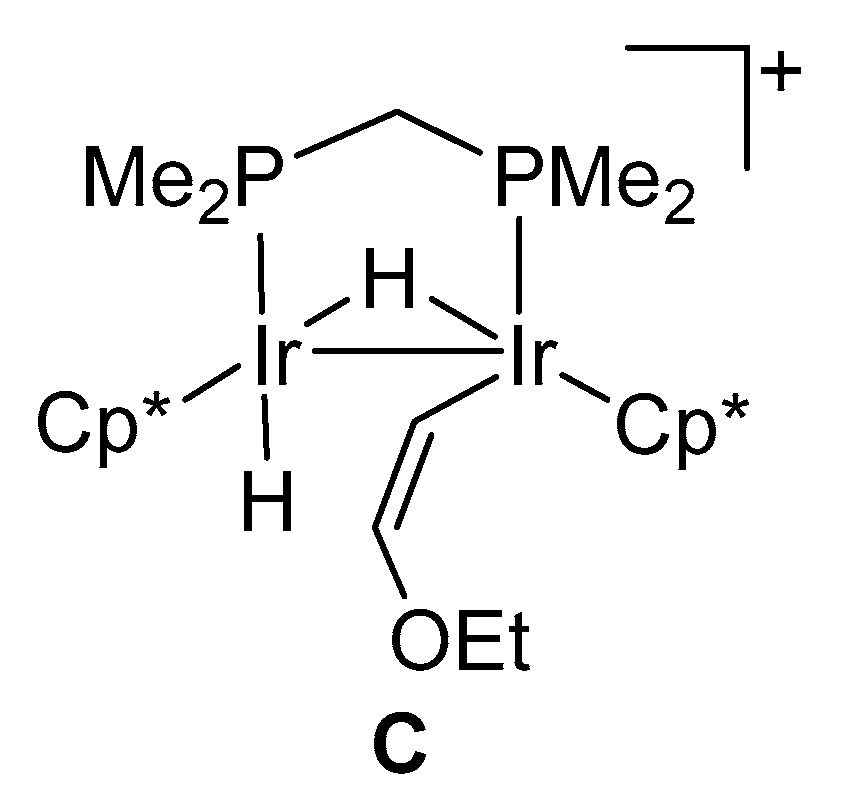
2.3. Successive Activation of C–H and C–O Bonds of Ethyl Vinyl Ether

3. Materials and Methods
3.1. General
3.2. C–H Bond Activation of 2,3-dihydrofuran to Give 3
3.3. Conversion of 3 into Vinylidene Complex 4
3.4. Reaction of Complex 1 with Ethyl Vinyl Ether to Give 5
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References and Notes
- Braun, M. α-Heteroatom-Substituted 1-Alkenyllithium Reagents: Carbanions and Carbenoids for C–C Bond Formation. Angew. Chem. Int. Ed. 1998, 37, 430–451. [Google Scholar] [CrossRef]
- Friesen, R.W. Generation and Reactivity of α-Metalated Vinyl Ethers. J. Chem. Soc. Parkin Trans. 1 2001, 1969–2001. [Google Scholar] [CrossRef]
- Barluenga, J.; Rodríguez, F.; Álvarez-Rodorigo, L.; Fañanás, F.J. Coupling Reactions of Zirconocene Complexes and Heterosubstituted Alkenes. Chem. Soc. Rev. 2005, 34, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, M.; Aoyama, H.; Kiyosu, J.; Shirogane, Y.; Iwasawa, T.; Obora, Y.; Tsuji, Y. Metal Complexes-Catalyzed Hydrolysis and Alcoholysis of Organic Substrates and Their Application to Kinetic Resolution. J. Organomet. Chem. 2007, 692, 472–480. [Google Scholar] [CrossRef]
- Png, Z.M.; Zeng, H.; Ye, Q.; Xu, J. Inverse-Electron-Demand Diels-Alder Reactions: Principles and Applications. Chem. Asian J. 2017, 12, 2142–2159. [Google Scholar] [CrossRef]
- Ozawa, F.; Kubo, A.; Hayashi, T. Catalytic Asymmetric Arylation of 2,3-Dihydrofuran with Aryl Triflates. J. Am. Chem. Soc. 1991, 113, 1417–1419. [Google Scholar] [CrossRef]
- Wu, W.-Q.; Peng, Q.; Dong, D.-X.; Hou, X.-L.; Wu, Y.-D. A Dramatic Switch of Enantioselectivity in Asymmetric Heck Reaction by Benzylic Substituents of Ligands. J. Am. Chem. Soc. 2008, 130, 9717–9725. [Google Scholar] [CrossRef]
- Gøgsig, T.M.; Kleimark, J.; Lill, S.O.N.; Korsager, S.; Lindhardt, A.T.; Norrby, P.-O.; Skrydstrup, T. Mild and Efficient Nickel-Catalyzed Heck Reactions with Electron-Rich Olefins. J. Am. Chem. Soc. 2012, 134, 443–452. [Google Scholar] [CrossRef]
- Wu, C.; Zhou, J.S. Asymmetric Intermolecular Heck Reaction of Aryl Halides. J. Am. Chem. Soc. 2014, 136, 650–652. [Google Scholar] [CrossRef]
- Kazankova, M.A.; Shulyupin, M.O.; Beletskaya, I.P. Catalytic Hydrophosphination of Alkenylalkyl Ethers. Synlett 2003, 2155–2158. [Google Scholar] [CrossRef]
- Pahadi, N.K.; Tunge, J.A. Catalytic Intermolecular Hydroamination of Vinyl Ethers. Synlett 2009, 3135–3138. [Google Scholar] [CrossRef] [PubMed]
- Ebe, Y.; Nishimura, T. Iridium-Catalyzed Branch-Selective Hydroarylation of Vinyl Ethers via C–H Bond Activation. J. Am. Chem. Soc. 2015, 137, 5899–5902. [Google Scholar] [CrossRef] [PubMed]
- Hatano, M.; Ebe, Y.; Nishimura, T.; Yorimitsu, H. Asymmetric Alkylation of N-Sulfonylbenzamides with Vinyl Ethers via C–H Bond Activation Catalyzed by Hydroxoiridium/Chiral Diene Complexes. J. Am. Chem. Soc. 2016, 138, 4010–4013. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.A.; Morken, J.P. Stereoselective Synthesis of Furans by the Pd-Catalyzed Oshima–Utimoto Reaction. Org. Lett. 2005, 7, 3367–3370. [Google Scholar] [CrossRef]
- Deng, L.; Giessert, A.J.; Gerlitz, O.O.; Dai, X.; Diver, S.T.; Davies, H.M.L. Metal Carbene-Promoted Sequential Transformations for the Enantioselective Synthesis of Highly Functionalized Cycloheptadienes. J. Am. Chem. Soc. 2005, 127, 1342–1343. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Kusama, H.; Iwasawa, N. Enantioselective Preparation of 8-Oxabicyclo [3.2.1]octane Derivatives via Asymmetric [3+2]-Cycloaddition of Platinum-Containing Carbonyl Ylides with Vinyl Ethers. J. Am. Chem. Soc. 2010, 132, 8842–8843. [Google Scholar] [CrossRef]
- Sherry, B.D.; Toste, F.D. Gold(I)-Catalyzed Propargyl Claisen Rearrangement. J. Am. Chem. Soc. 2004, 126, 15978–15979. [Google Scholar] [CrossRef] [Green Version]
- Sherry, B.D.; Maus, L.; Laforteza, B.N.; Toste, F.D. Gold(I)-Catalyzed Synthesis of Dihydropyrans. J. Am. Chem. Soc. 2006, 128, 8132–8133. [Google Scholar] [CrossRef]
- Geherty, M.E.; Dura, R.D.; Nelson, S.G. Catalytic Asymmetric Claisen Rearrangement of Unactivated Allyl Vinyl Ethers. J. Am. Chem. Soc. 2010, 132, 11875–11877. [Google Scholar] [CrossRef]
- Aoyama, H.; Tokunaga, M.; Hiraiwa, S.-I.; Shirogane, Y.; Obora, Y.; Tsuji, Y. Hydrolysis of Alkenyl Esters and Ethers Catalyzed by Metal Complexes. Org. Lett. 2004, 6, 509–512. [Google Scholar] [CrossRef]
- Itoh, H.; Yamamoto, E.; Masaoka, S.; Sakai, K.; Tokunaga, M. Kinetic Resolution of P-Chirogenic Compounds by Palladium-Catalyzed Alcoholysis of Vinyl Ethers. Adv. Synth. Catal. 2009, 351, 1796–1800. [Google Scholar] [CrossRef]
- Luo, S.; Vela, J.; Lief, G.R.; Jordan, R.F. Copolymerization of Ethylene and Alkyl Vinyl Ethers by a (Phosphine-Sulfonate)PdMe Catalyst. J. Am. Chem. Soc. 2007, 129, 8946–8947. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Luo, S.; Jordan, R.F. Cationic Polymerization and Insertion Chemistry in the Reactions of Vinyl Ethers with (α-Diimine)PdMe+ Species. J. Am. Chem. Soc. 2010, 132, 5273–5284. [Google Scholar] [CrossRef] [PubMed]
- Carrow, B.P.; Nozaki, K. Synthesis of Functional Polyolefins Using Cationic Bisphosphine Monoxide–Palladium Complexes. J. Am. Chem. Soc. 2012, 134, 8802–8805. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Ren, P. Catalytic Asymmetric C–H Activation of Silyl Enol Ethers as an Equivalent of an Asymmetric Michael Reaction. J. Am. Chem. Soc. 2001, 123, 2070–2071. [Google Scholar] [CrossRef]
- Kikuchi, T.; Takagi, J.; Ishiyama, T.; Miyaura, N. Iridium-catalyzed Vinylic C–H Borylation of Cyclic Vinyl Ethers by Bis(pinacolato)diboron. Chem. Lett. 2008, 37, 664–665. [Google Scholar] [CrossRef]
- Kikuchi, T.; Takagi, J.; Isou, H.; Ishiyama, T.; Miyaura, N. Vinylic C–H Borylation of Cyclic Vinyl Ethers with Bis(pinacolato)diboron Catalyzed by an Iridium(I)-dtbpy Complex. Chem. Asian J. 2008, 3, 2082–2090. [Google Scholar] [CrossRef]
- Pawar, G.G.; Singh, G.; Tiwari, V.K.; Kapur, M. Dehydrogenative Heck Reaction (Fujiwara–Moritani Reaction) of Unactivated Olefins with Simple Dihydropyrans under Aprotic Conditions. Adv. Synth. Catal. 2013, 355, 2185–2190. [Google Scholar] [CrossRef]
- Wenkert, E.; Michelotti, E.L.; Swindell, C.S.; Tingoli, M. Transformation of Carbon–Oxygen into Carbon–Carbon Bonds Mediated by Low-Valent Nickel Species. J. Org. Chem. 1984, 49, 4894–4899. [Google Scholar] [CrossRef]
- Cornella, J.; Martin, R. Ni-Catalyzed Stereoselective Arylation of Inert C–O bonds at Low Temperatures. Org. Lett. 2013, 15, 6298–6301. [Google Scholar] [CrossRef]
- Shilov, A.E.; Shul’pin, G.B. Activation of C–H Bonds by Metal Complexes. Chem. Rev. 1997, 97, 2879–2932. [Google Scholar] [CrossRef] [PubMed]
- Labinger, J.A.; Bercaw, J.E. Understanding and Exploiting C–H Bond Activation. Nature 2002, 417, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.D. Advances in Carbon–Hydrogen Activation. In Comprehensive Organometallic Chemistry III: From Fundamentals to Applications; Crabtree, R.H., Mingos, D.M.P., Eds.; Elsevier Ltd.: Oxford, UK, 2007; Chapter 1.25; pp. 699–723. [Google Scholar] [CrossRef]
- Fernández-Alvarez, F.J.; Iglesias, M.; Oro, L.A.; Passarelli, V. Bond Activation and Catalysis. In Comprehensive Inorganic Chemistry II; Reedijk, J., Poeppelmeier, K., Eds.; Elsevier Ltd.: Oxford, UK, 2013; Chapter 8.9; pp. 399–432. [Google Scholar] [CrossRef]
- Hartwig, J.F. Evolution of C–H Bond Functionalization from Methane to Methodology. J. Am. Chem. Soc. 2016, 138, 2–24. [Google Scholar] [CrossRef]
- Milstein, D. Challenging Metal-Based Transformations. From Single-Bond Activation to Catalysis and Metallaquinonoids. Pure Appl. Chem. 2003, 75, 445–460. [Google Scholar] [CrossRef]
- Oh, M.; Yu, K.; Li, H.; Watson, E.J.; Carpenter, G.B.; Sweigart, D.A. The Remote Activation of Chemical Bonds via Metal Coordination. Adv. Synth. Catal. 2003, 345, 1053–1060. [Google Scholar] [CrossRef]
- Komiya, S.; Hirano, M. Bond Activation by Low Valent Ruthenium Complexes. Dalton Trans. 2003, 1439–1453. [Google Scholar] [CrossRef]
- Yu, D.-G.; Li, B.-J.; Shi, Z.-J. Exploration of New C–O Electrophiles in Cross-Coupling Reactions. Acc. Chem. Res. 2010, 43, 1486–1495. [Google Scholar] [CrossRef]
- Cornella, J.; Zarate, C.; Martin, R. Metal-catalyzed activation of ethers via C–O bond cleavage: A new strategy for molecular diversity. Chem. Soc. Rev. 2014, 43, 8081–8097. [Google Scholar] [CrossRef]
- Tobisu, M.; Chatani, N. Cross-Couplings Using Aryl Ethers via C–O Bond Activation Enabled by Nickel Catalysts. Acc. Chem. Res. 2015, 48, 1717–1726. [Google Scholar] [CrossRef]
- Komiya, S.; Srivastava, R.S.; Yamamoto, A.; Yamamoto, T. Selective Cleavage of C–O and/or Si–O Bonds in Trimethylsilyl Ethers Promoted by Cobalt(I), Rhodium(I), and Ruthenium(II) Hydride Complexes. Organometallics 1985, 4, 1504–1508. [Google Scholar] [CrossRef]
- Deelman, B.-J.; Booij, M.; Meetsma, A.; Teuben, J.H.; Kooijman, H.; Spek, A.L. Activation of Ethers and Sulfides by Organolanthanide Hydrides. Molecular Structures of (Cp*2Y)2(μ-OCH2CH2O)(THF)2 and (Cp*2Ce)2(μ-O)(THF)2. Organometallics 1995, 14, 2306–2317. [Google Scholar] [CrossRef]
- Takaki, K.; Maruo, M.; Kamata, T.; Makioka, Y.; Fujiwara, Y. Selective C–O Bond Cleavage of Vinyl Ethers with Cp*2Sm(thf)n Leading to Vinylsamarium or Enolate Complexes. J. Org. Chem. 1996, 61, 8332–8334. [Google Scholar] [CrossRef] [PubMed]
- Bonanno, J.B.; Henry, T.P.; Neithamer, D.R.; Wolczanski, P.T.; Lobkovsky, E.B. Arylamine C–N Bond Oxidative Addition to (silox)3Ta (silox = tBu3SiO). J. Am. Chem. Soc. 1996, 118, 5132–5133. [Google Scholar] [CrossRef]
- Planas, J.G.; Marumo, T.; Ichikawa, Y.; Hirano, M.; Komiya, S. Carbon–Oxygen and Carbon–Sulfur Bond Activation of Vinyl Esters, Ethers and Sulfides by Low Valent Ruthenium Complexes. J. Chem. Soc. Dalton Trans. 2000, 2613–2625. [Google Scholar] [CrossRef]
- Ferrando, G.; Gérard, H.; Spivak, G.J.; Coalter, J.N., III; Huffman, J.C.; Eisenstein, O.; Caulton, K.G. Facile C(sp2)/OR Bond Cleavage by Ru or Os. Inorg. Chem. 2001, 40, 6610–6621. [Google Scholar] [CrossRef]
- Goj, L.A.; Lail, M.; Pittard, K.A.; Riley, K.C.; Gunnoe, T.B.; Petersen, J.L. Reactions of TpRu(CO)(NCMe)(Ph) with Electron-Rich Olefins: Examples of Stoichiometric C–S, C–O and C–H Bond Cleavage. Chem. Commun. 2006, 982–984. [Google Scholar] [CrossRef]
- Bradley, C.A.; Veiros, L.F.; Pun, D.; Lobkovsky, E.; Keresztes, I.; Chirik, P.J. Carbon–Oxygen Bond Cleavage with η9,η5-Bis(indenyl)zirconium Sandwich Complexes. J. Am. Chem. Soc. 2006, 128, 16600–16612. [Google Scholar] [CrossRef]
- Paneque, M.; Poveda, M.L.; Santos, L.L.; Carmona, E.; Mereiter, K. Generation of Metallacyclic Structures from the Reactions of Vinyl Ethers with a TpMe2IrIII Compound. Organometallics 2008, 27, 6353–6359. [Google Scholar] [CrossRef]
- Trovitch, R.J.; Loblovsky, E.; Bouwkamp, M.W.; Chirik, P.J. Carbon–Oxygen Bond Cleavage by Bis(imino)pyridine Iron Compounds: Catalyst Deactivation Pathways and Observation of Acyl C–O Bond Cleavage in Esters. Organometallics 2008, 27, 6264–6278. [Google Scholar] [CrossRef]
- Kiyota, S.; Kobori, T.; Soeta, H.; Ichikawa, Y.; Komine, N.; Komiya, S.; Hirano, M. Synthesis of and Catalytic Nitrile Hydration by a Cationic Tris(μ-hydroxo)diruthenium(II) Complex Having PMe3 Ligands. Polyhedron 2016, 120, 3–10. [Google Scholar] [CrossRef]
- Shoshani, M.M.; Semeniuchenko, V.; Johnson, S.A. Dismantling of Vinyl Ethers by Pentanuclear [(iPr3P)Ni]5H6: Facile Cooperative C–O, C–C and C–H Activation Pathways. Chem. Eur. J. 2018, 24, 14282–14289. [Google Scholar] [CrossRef] [PubMed]
- Halpern, J. Binuclear Oxidative Addition–Reductive Elimination Reactions. Inorg. Chim. Acta 1982, 62, 31–37. [Google Scholar] [CrossRef]
- Suzuki, H. Activation of Organic Substrates on Multi-Metallic Sites of Transition Metal Polyhydride Clusters Having C5Me5 Groups as Auxiliary Ligands. Eur. J. Inorg. Chem. 2002, 1009–1023. [Google Scholar] [CrossRef]
- Facklar, J.P., Jr. Forty-Five Years of Chemical Discovery Including a Golden Quarter-Century. Inorg. Chem. 2002, 41, 6959–6972. [Google Scholar] [CrossRef]
- Powers, I.G.; Uyeda, C. Metal–Metal Bonds in Catalysis. ACS Catal. 2017, 7, 936–958. [Google Scholar] [CrossRef]
- Rej, S.; Tsurugi, H.; Mashima, K. Multiply-Bonded Dinuclear Complexes of Early-Transition Metals as Minimum Entities of Metal Cluster Catalysts. Coord. Chem. Rev. 2018, 355, 223–239. [Google Scholar] [CrossRef]
- Fujita, K.; Nakaguma, H.; Hamada, T.; Yamaguchi, R. Inter- and Intramolecular Activation of Aromatic C–H Bonds by Diphosphine and Hydrido-Bridged Dinuclear Iridium Complexes. J. Am. Chem. Soc. 2003, 125, 12368–12369. [Google Scholar] [CrossRef]
- Takahashi, Y.; Nonogawa, M.; Fujita, K.; Yamaguchi, R. C–H activation on a Diphosphine and Hydrido-Bridged Diiridium Complex: Generation and Detection of an Active IrII–IrII Species [(Cp*Ir)2(μ-dmpm)(μ-H)]+. Dalton Trans. 2008, 3546–3552. [Google Scholar] [CrossRef]
- Fujita, K.; Takahashi, Y.; Nakaguma, H.; Hamada, T.; Yamaguchi, R. Activation of C–H and H–H Bonds by Dinuclear Iridium Complexes. Oxidative Addition to Highly Active Unsaturated 32e– Diiridium Species. J. Organomet. Chem. 2008, 693, 3375–3382. [Google Scholar] [CrossRef]
- Berry, M.; Howard, J.A.K.; Stone, F.G.A. Chemistry of Di- and Tri-metal Complexes with Bridging Carbene or Carbyne Ligands. Part 2. Formation of Manganese–Platinum, –Palladium, and –Nickel Compounds, and the Crystal Structures of Two Forms of [(OC)4Mn{μ-(1-σ,1—2-η-C=CH–CH2–CH2–O)}Pt(PMe3)2]. J. Chem. Soc. Dalton Trans. 1980, 1601–1608. [Google Scholar] [CrossRef]
- Luecke, H.F.; Arndtsen, B.A.; Burger, P.; Bergman, R.G. Synthesis of Fischer Carbene Complexes of Iridium by C–H Bond Activation of Methyl and Cyclic Ethers: Evidence for Reversible α-Hydrogen Migration. J. Am. Chem. Soc. 1996, 118, 2517–2518. [Google Scholar] [CrossRef]
- Romero, P.E.; Whited, M.T.; Grubbs, R.H. Multiple C–H Activations of Methyl tert-Butyl Ether at Pincer Iridium Complexes: Synthesis and Thermolysis of Ir(I) Fischer Carbenes. Organometallics 2008, 27, 3422–3429. [Google Scholar] [CrossRef]
- Hughes, R.P.; Laritchev, R.B.; Zakharov, L.N.; Rheingold, A.L. Carbon–Fluorine Bond Activation Coupled with Carbon–Carbon Bond Formation at Iridium. Confirmation of Complete Kinetic Diastereoselectivity at the New Carbon Stereocenter by Intramolecular Trapping Using Vinyl as the Migrating Group. J. Am. Chem. Soc. 2005, 127, 6325–6334. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.S.; Lee, H.; Park, H.; Kim, M. Synthesis of Cross-Conjugated Olefins from Alkynes: Regioselective C–C Bond Formation between Alkynes. Organometallics 2002, 21, 3889–3896. [Google Scholar] [CrossRef]
- Wang, L.-S.; Cowie, M. Cationic Vinylidene-Bridged Complexes and Their Reactions with Alkynes to Yield Either Alkyne- and Vinylidene-Bridged or Bis(vinylidene) Products. Facile Interconversion between Terminal and Bridging Vinylidene Bonding Modes. Organometallics 1995, 14, 3040–3057. [Google Scholar] [CrossRef]
- Torkelson, J.R.; McDonald, R.; Cowie, M. C–H Bond Activation and C–C Bond Formation in the Reactions of the Methyl Complex [Ir2(CH3)(CO)2(Ph2PCH2PPh2)2][CF3SO3] with Alkynes. Organometallics 1999, 18, 4134–4146. [Google Scholar] [CrossRef]
- McDonald, F.E.; Connolly, C.B.; Gleason, M.M.; Towne, T.B.; Treiber, K.D. A New Synthesis of 2,3-Dihydrofurans: Cycloisomerization of Alkynyl Alcohols to Endocyclic Enol Ethers. J. Org. Chem. 1993, 58, 6952–6953. [Google Scholar] [CrossRef]
- Fujita, K.; Nakaguma, H.; Hanasaka, F.; Yamaguchi, R. Synthesis of a DMPM and Hydrido-Bridged Diiridium Complex, [(Cp*Ir)2(μ-dmpm)(μ-H)2][OTf]2, and Its Reactivity toward Alkynes and Isocyanides. Organometallics 2002, 21, 3749–3757. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takahashi, Y.; Shimbayashi, T.; Fujita, K.-i. Successive Activation of C–H and C–O Bonds of Vinyl Ethers by a Diphosphine and Hydrido-Bridged Diiridium Complex. Inorganics 2019, 7, 121. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7100121
Takahashi Y, Shimbayashi T, Fujita K-i. Successive Activation of C–H and C–O Bonds of Vinyl Ethers by a Diphosphine and Hydrido-Bridged Diiridium Complex. Inorganics. 2019; 7(10):121. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7100121
Chicago/Turabian StyleTakahashi, Yoshinori, Takuya Shimbayashi, and Ken-ichi Fujita. 2019. "Successive Activation of C–H and C–O Bonds of Vinyl Ethers by a Diphosphine and Hydrido-Bridged Diiridium Complex" Inorganics 7, no. 10: 121. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7100121