Bond Forming Reactions Involving Isocyanides at Diiron Complexes



1
Dipartimento di Chimica Industriale “Toso Montanari”, University of Bologna, Viale Risorgimento 4, I-40136 Bologna, Italy
2
Dipartimento di Chimica e Chimica Industriale, University of Pisa, Via G. Moruzzi 13, I-56124 Pisa, Italy
*
Authors to whom correspondence should be addressed.
Inorganics 2019, 7(3), 25; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7030025
Submission received: 28 January 2019
/
Revised: 12 February 2019
/
Accepted: 14 February 2019
/
Published: 26 February 2019
(This article belongs to the Special Issue Binuclear Complexes)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The versatility of isocyanides (CNR) in organic chemistry has been tremendously enhanced by continuous advancement in transition metal catalysis. On the other hand, the urgent need for new and more sustainable synthetic strategies based on abundant and environmental-friendly metals are shifting the focus towards iron-assisted or iron-catalyzed reactions. Diiron complexes, taking advantage of peculiar activation modes and reaction profiles associated with multisite coordination, have the potential to compensate the lower activity of Fe compared to other transition metals, in order to activate CNR ligands. A number of reactions reported in the literature shows that diiron organometallic complexes can effectively assist and promote most of the “classic” isocyanide transformations, including CNR conversion into carbyne and carbene ligands, CNR insertion, and coupling reactions with other active molecular fragments in a cascade sequence. The aim is to evidence the potential offered by diiron coordination of isocyanides for the development of new and more sustainable synthetic strategies for the construction of complex molecular architectures.
1. Introduction
Since the discovery of isocyanide-based multicomponent reactions (MCRs) (e.g., the Passerini and Ugi reactions), isocyanides (CNR) have been recognized as extremely valuable synthetic tools for the construction of complex molecules, particularly heterocycles [1,2,3,4]. Development of MCRs takes advantage of the complex nature of isocyanides, which can act both as nucleophiles and electrophiles. On the other hand, isocyanides are long-known ligands, and their reactivity can be considerably modified upon transition-metal coordination [5,6,7]. Indeed, both the nucleophilicity and the electrophilicity of isocyanides can be controlled and enhanced upon metal coordination. As an example, isocyanide ligands have been successfully used for obtaining both carbene [8,9] and carbyne complexes [10]. In general, interest in isocyanide chemistry involving metal species is still growing [11,12]. One reason for this is a resurgent interest in isocyanides as C1 synthons driven by the development of transition-metal-catalyzed isocyanide insertion reactions [13,14,15,16]. Similarly to CO, isocyanides can be incorporated into catalytic mechanisms based on oxidative addition, isocyanide insertion, and reductive elimination. This emerging strategy, based on CNR insertion into M-element bonds, offers tremendous opportunities for the synthesis of fine chemicals containing a nitrogen functionality, and is largely dominated by Pd-catalyzed or Pd-promoted reactions [17,18,19,20,21,22]. Considerable efforts have been made in order to expand the research field to other transition- metals, given the strong interest in replacing noble and rare metals with more abundant and sustainable transition metals, such as iron. Indeed, the number of Fe-catalyzed reactions is rapidly expanding, but those involving isocyanides are still limited [23,24,25].
This mini-review focuses on reactions involving isocyanides and diiron complexes, aimed at the formation of C–C or C–heteroatom bonds for the construction of more complex molecular architectures. The interest in diiron complexes (instead of complexes containing one Fe atom) is based on two major features: First, two adjacent Fe atoms might provide cooperative effects, particularly effective in redox reactions that involve coordinated ligands. As an example, μ-dithiolate diiron has been very actively investigated as mimics of [FeFe]-H2ases active site and as potential catalysts for hydrogen evolution [26,27,28,29]. The second peculiar feature of diiron complexes is the possibility of observing bridging coordination of a variety of carbon-based ligands (including isocyanides and hydrocarbyl ligands derived from CNR ligands), which potentially provide access to activation modes and reaction profiles not observed when the same ligands are bound to a single metal atom [30,31,32].
The review covers most of the research work done by some of us in this field, in the last two decades.
2. Diiron Complexes with Isocyanide Ligands
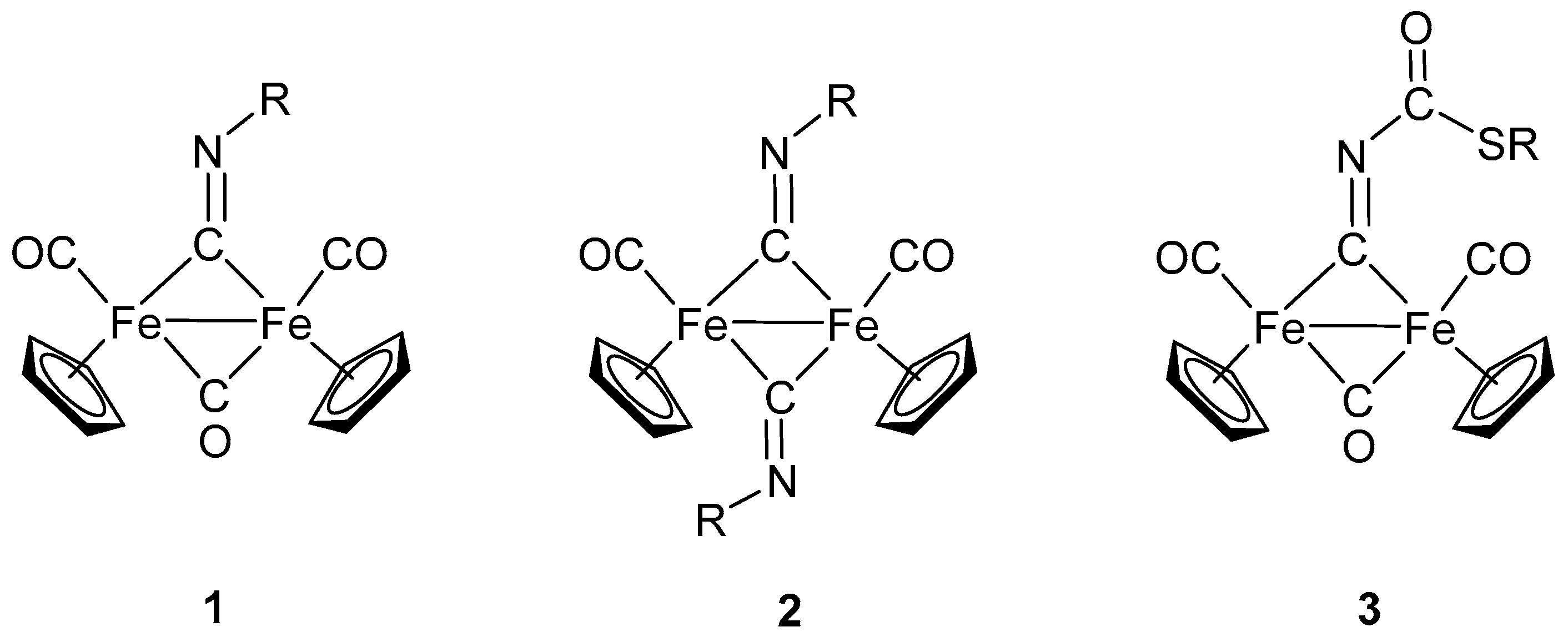
Diiron complexes containing isocyanide ligands have been well known since the 1970s. Examples include the complexes [Fe2(CNEt)9] [33,34] and [Fe2(CNPh)9] [35], formally derived and structurally similar to [Fe2(CO)9]. Beside the homoleptic complexes mentioned above, a prominent role has been assumed by isocyanide complexes derived from the [Fe2(Cp)2(CO)4] frame (Cp = η-C5H5). Direct replacement of CO with CNR in [Fe2(Cp)2(CO)4] generally results in the formation of mixtures of isocyanide complexes [Fe2(Cp)2(CO)4−n(CNR)n] (n = 1–3), with mono-isocyanide and di-isocyanide complexes being prevalent (Scheme 1, compounds 1 [36,37,38] and 2 [39,40], respectively). Diiron complexes of the type [Fe2(Cp)2(CNR)4], containing up to four isocyanide ligands, are also known [41].
Alternative synthetic routes have been designed in order to improve the selectivity [42]; nevertheless, the preparation remains difficult, and separation steps are generally required. The situation is made more complicated by the presence of isomers, in that isocyanides can act both as terminal and bridging ligands. Indeed, CNR shows a higher tendency toward bridging coordination compared to CO, and only with the bulky CNBut ligand does the terminal coordination prevail [43].
Isocyanide ligands, in diiron complexes, might also result from the transformation of other ligands. For example, alkylation (with MeSO3CF3) of the cyanide in the complex [Fe2(Cp)2(CN)(CNMe2)(CO)3] leads to the formation of the isocyanide complex [Fe2(Cp)2(CNMe2)(CO)3(CNMe)]SO3CF3 [44]. A less predictable route to isocyanide complexes is provided by the peculiar reaction of the cyanate (NCO−) with the bridging thiocarbyne (>CSMe) affording the functionalized bridging isocyanide 3 (Scheme 1) [45].
Finally, a resurged interest in isocyanide coordination in diiron complexes has been more recently stimulated by studies concerning [FeFe] hydrogenase model systems. In particular, molecular mimics of the type [Fe2(μ-S2C3H6)(CNR)6−x(CO)x] (x = 2, 3; R = Me, tBu) have been investigated in order to exploit the different donor/acceptor properties of CNR in comparison with CO [46,47,48].
3. Electrophilic Addition at Bridging CNR as Effective Route to Bridging Aminoalkylidyne
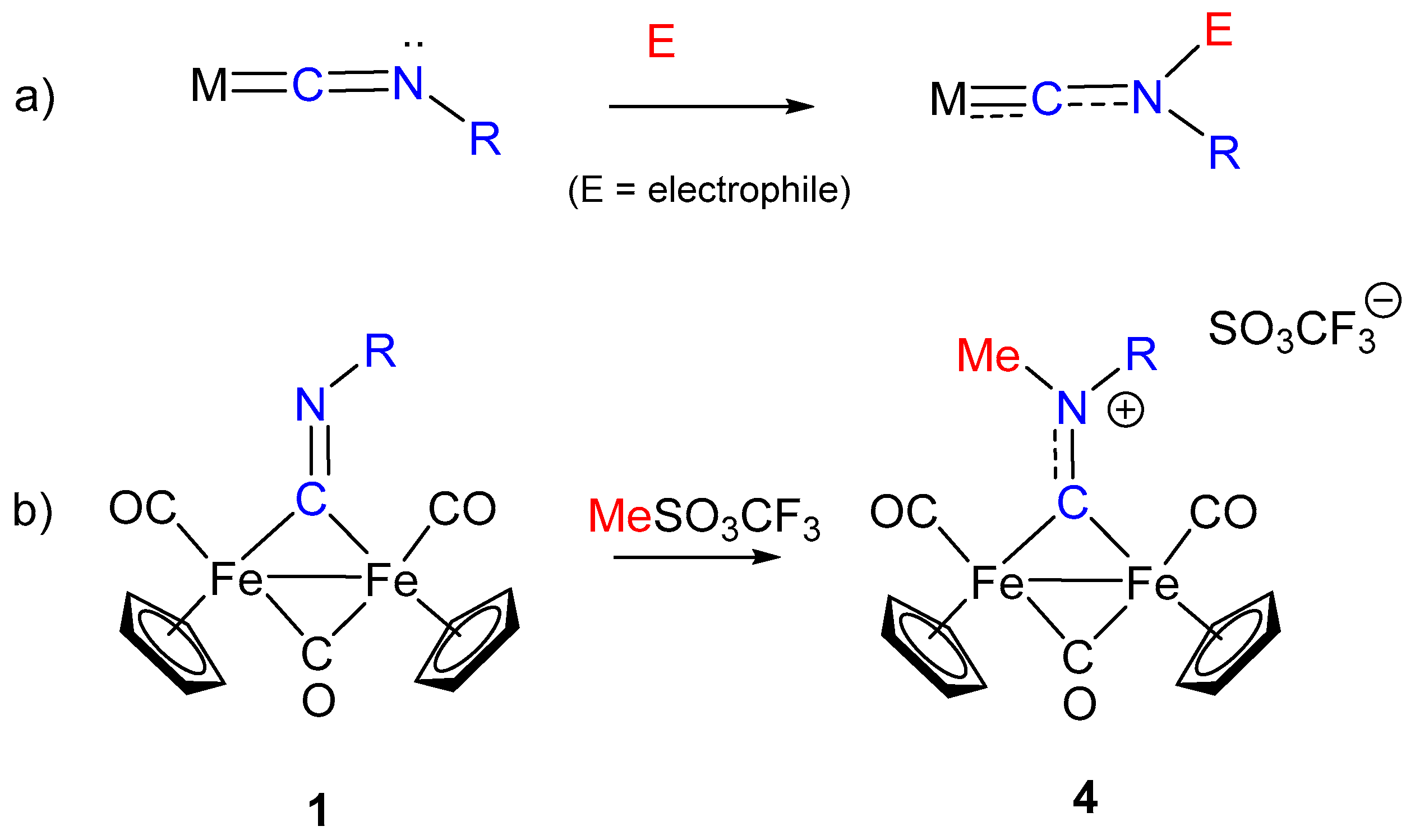
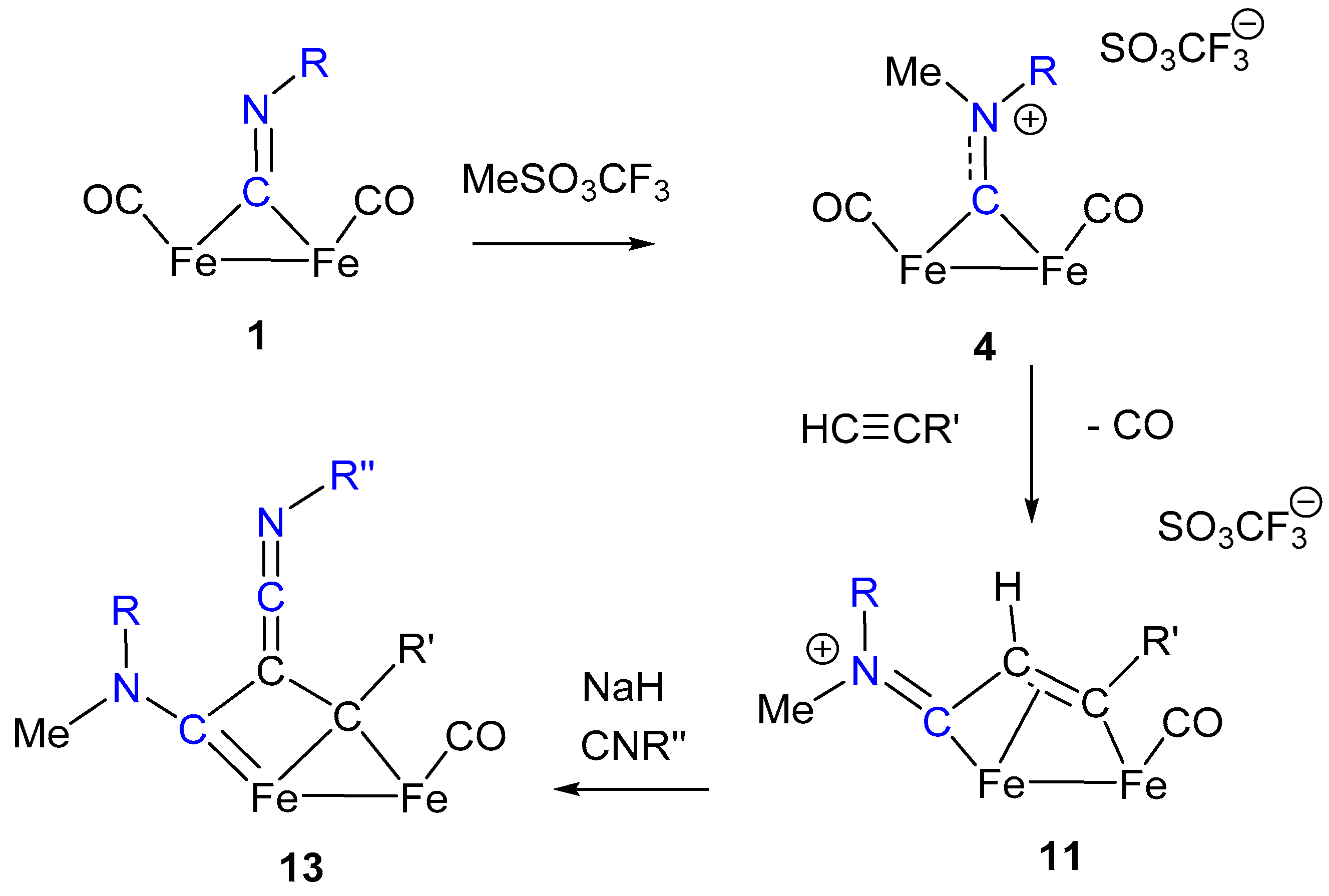
The transformation of isocyanide ligands into carbynes (alkylidynes), upon electrophilic attack at the N atom, is generally observed in mononuclear complexes when the metal is low-valent, and sufficiently electron-rich to activate the CNR ligand towards electrophiles (Scheme 2a) [10]. This requirement usually limits the activation towards electrophiles to low-valent group 6 (Mo or W) or 7 (Re) metal complexes. Conversely, bridging coordination in diiron complexes effectively promotes the reaction of isocyanide ligands toward electrophiles and allows them to extend to iron the transformation of CNR into carbynes. Indeed, treatment with strong alkylating agents (RSO3CF3) of complexes of type 1 (R = Me) transforms isocyanides into the corresponding μ-aminoalkylidyne (aminocarbyne) complexes of the type [Fe2(Cp)2(CO)3(μ-CNRR’)]+, (4) (Scheme 2b) [49,50]. Likewise, di-isocyanide complexes of type 2 are transformed into the corresponding bis-aminoalkylidyne derivatives [Fe2(Cp)2(CO)2(μ-CNRR’)2]2+ [51].
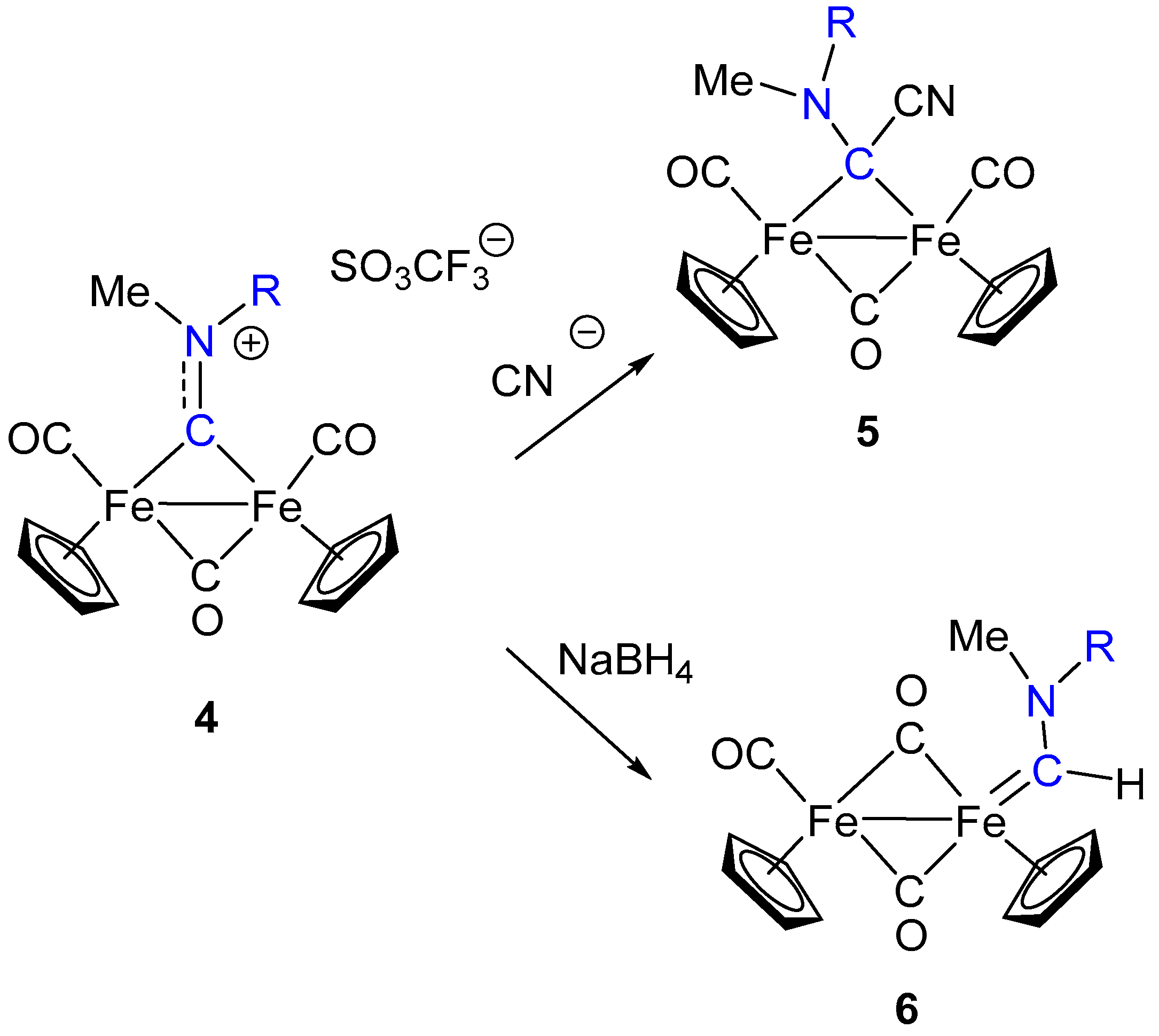
Very recently, the synthesis and isolation of diiron bridging aminoalkylidyne complexes of type 4 have been reinvestigated in order to provide a most convenient access to these complexes [52], which have been proved to be very versatile species and valuable starting materials for obtaining a number of organometallic complexes [52,53]. Although the bridging CNRR’ ligand is usually described as a bridging carbyne (alkylidyne), the >C–N interaction displays some double bond character (C–N bond distances are usually around 130 ppm), and the ligand shows some iminium, or azavinilydene, character. Beside the “formal” description of the >CNRR’ ligand, its reactivity is dominated by the electrophilic character, which is made even stronger by the net positive charge of the diiron complex 4. As a consequence, the bridging amino alkylidyne ligand in 4 can undergo nucleophilic addition at the bridging carbon to form the corresponding aminocarbene complexes 5 and 6 (Scheme 3) [54].
Indeed, the reactivity of complexes 4 with nucleophiles is rather complex: Nucleophilic additions can be selectively directed to bridging carbyne carbon or to other sites (Cp or CO ligands), depending on the nature of the nucleophiles and of the substituent R at the N atom [52,55,56]. Description of the reactivity of 4 with nucleophiles goes beyond the scope of this mini-review. However, it has to be remarked that the bridging aminocarbyne ligand (>CNRR’) is a sort of masked isocyanide (>CNR), in which alkylation of the N atom has produced a considerable enhancement of the electrophilic character of the bridging carbon. Thus, using an appropriate sequence of electrophilic and nucleophilic addition steps, the isocyanide ligand can be transformed into aminocarbyne (compound 4) and aminocarbene ligands (compounds 5 and 6), and the resulting ligands display a molecular architecture in which the parent isocyanide moiety has been totally incorporated (evidenced in blue in Scheme 3). Interestingly, the reaction sequence described above (i.e., electrophilic addition at the N atom and nucleophilic addition at the C atom), required to accomplish the transformation of CNR into aminocarbene, is opposite to that normally observed in the conversion of isocyanides into carbenes, which is based on nucleophilic addition at the isocyanide ligand, followed by electrophilic addition at the N atom [8,9].
4. Insertion Reactions Involving Isocyanides in Diiron Complexes
Diiron complexes offer a limited number of examples of isocyanide insertion despite the fact that CNR insertion into the Fe–C of mononuclear complexes is a known possibility, well investigated also in its mechanistic aspects [57,58]. Floriani et al. reported on the reaction of the diiron complex [Fe2Mes4] (Mes = 2,4,6-Me3C6H2) with CNtBu, in THF at 0 °C, leading to the formation of the bridging iminoacyl complex 7 as a result of isocyanide insertion into the Fe–aryl bond (Scheme 4) [59,60].
Isocyanide insertion into iron bridging hydride bond is observed in the dinuclear complex [Fe2(μ-H)(μ-PCy2)(CO)4(μ-CO)(dppm)] (8), leading to the formation of the bridging formimidoyl complex 9 (Scheme 4) [61]. This reaction is consistent with other similar isocyanide insertions into metal μ-H bond, which are more frequently observed in dinuclear group 6 and 9 metal complexes [62,63,64,65], and provides a unique synthetic route to the formation of bridging formimidoyl ligands.
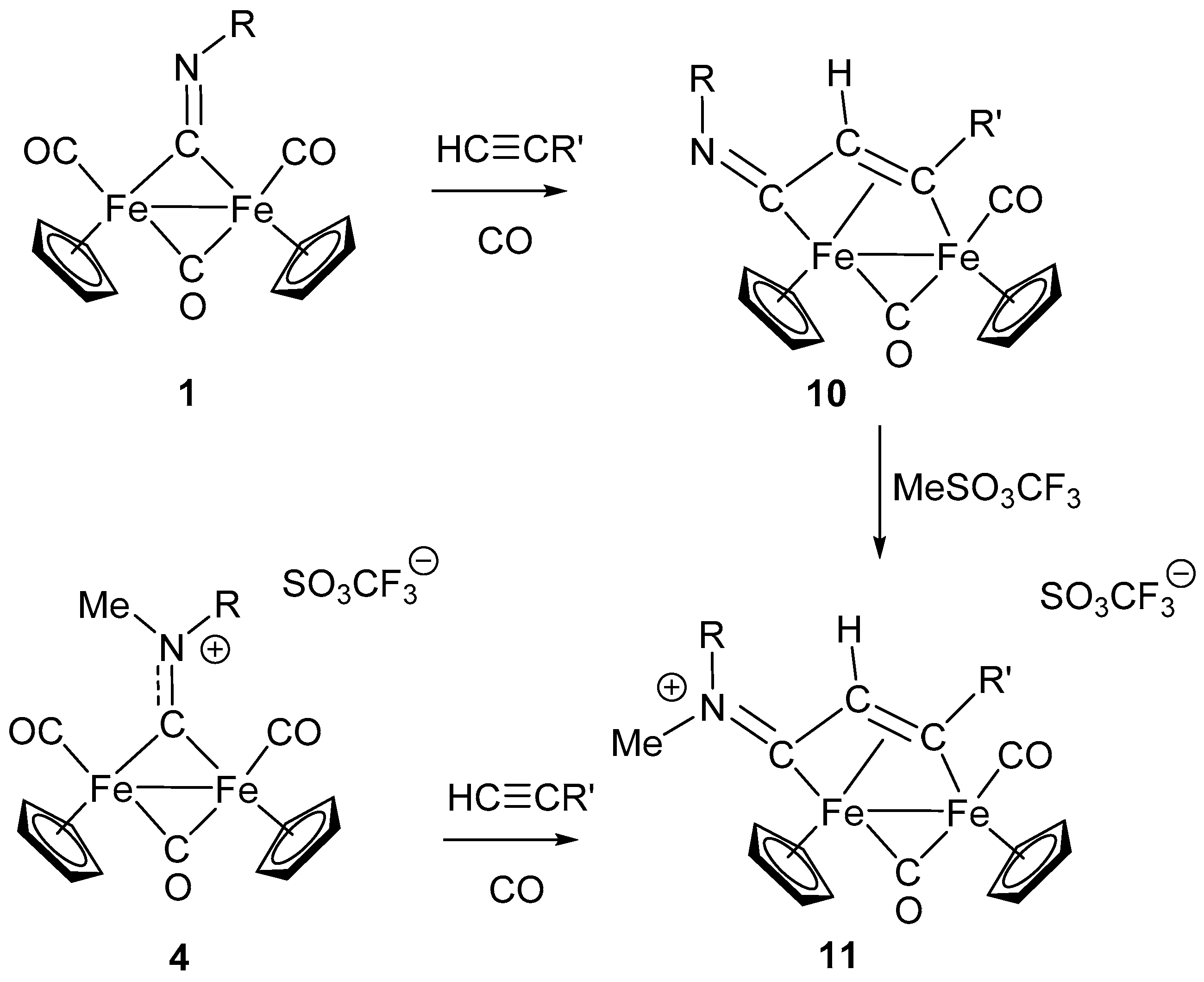
In addition to the examples above reported, in which isocyanide inserts into the Fe–C or Fe–H bond, a somewhat related reaction is that reported in Scheme 5, where alkynes insert into the Fe–CNR bond. The reaction is not properly an isocyanide insertion, but rather the insertion of alkyne into an Fe-bridging isocyanide bond. Nevertheless, it provides a route to the assembly of isocyanides with alkynes, assisted and promoted by diiron coordination. The frame resulting from alkyne insertion into the Fe-isocyanide can be described as enimino or azabutadienyl, and remains coordinated as a bridging ligand in a η1:η2 coordination mode (compound 10, Scheme 5) [66]. The reaction requires photolytic activation in order to remove one CO, and the insertion of monosubstituted alkynes into the Fe–C bond is regioselective, with the less hindered alkyne carbon bound to the imine carbon (as shown in Scheme 5). The alkyne insertion described above is consistent with analogous reactions observed in diiron complexes of the type [Fe2(Cp)2(CO)3(L)] in which alkynes insert into the Fe-bridging L ligands (L = CO [67,68], CS [69]).
Interestingly, the bridging enimine ligand in 10 can be readily alkylated to form the corresponding cationic bridging vinyliminium complexes in 11 (Scheme 5). These latter can be alternatively obtained by alkyne insertion into Fe-bridging aminocarbyne ligands of complex 4, as shown in Scheme 5 [70,71]. This second synthetic route affords a more convenient and general access to diiron vinyliminium complexes (compounds 11) in that the net positive charge of 4 makes CO elimination viable via treatment with Me3NO, rather than with the less selective photochemical method. The need for CO removal has been explained by density functional theory (DFT) mechanistic investigation on the alkyne insertion reaction [72]. In both synthetic routes leading to 11, the overall remarkable result is the coupling of an alkyne with a bridging isocyanide, accompanied (assisted) by N alkylation. Finally, it is worth noting that the convenient access to 11, by alkyne–isocyanide assembly, has made it possible to fully investigate the huge potential of bridging vinyliminium ligands as versatile and extremely valuable precursors of a large variety of new organometallic species [30,53,73,74,75].
5. Addition of Isocyanides to Coordinated Ligands
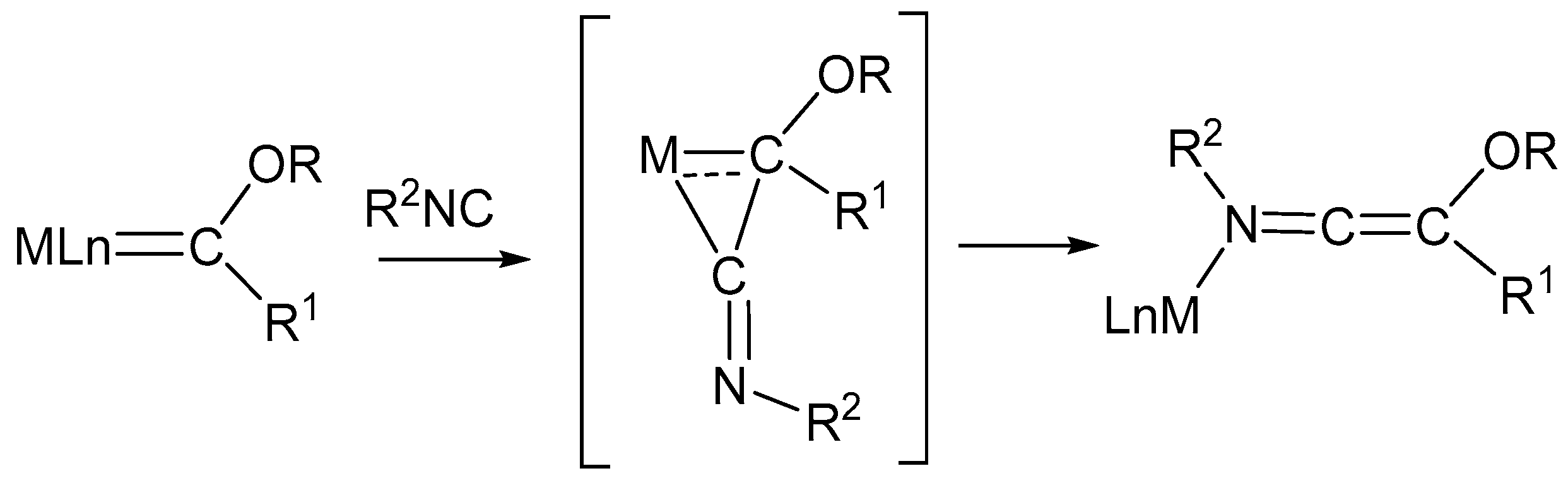
In addition to the insertion reactions described above, isocyanides are potentially able to react with Fe-coordinated ligands due to their remarkable carbene-like reactivity. In particular, the reactions of isocyanides with metal carbene complexes are known to form ketenimine complexes, which, in turn, are valuable intermediates and powerful synthetic tools [76]. The mechanism of the reaction of Fischer-type carbenes with methylisocyanide was studied theoretically by DFT method [77,78]. The reaction is believed to proceed via metallacyclopropanimine species which are generated by nucleophilic attack of the isocyanide carbon atom on the electrophilic carbene carbon (Scheme 6).
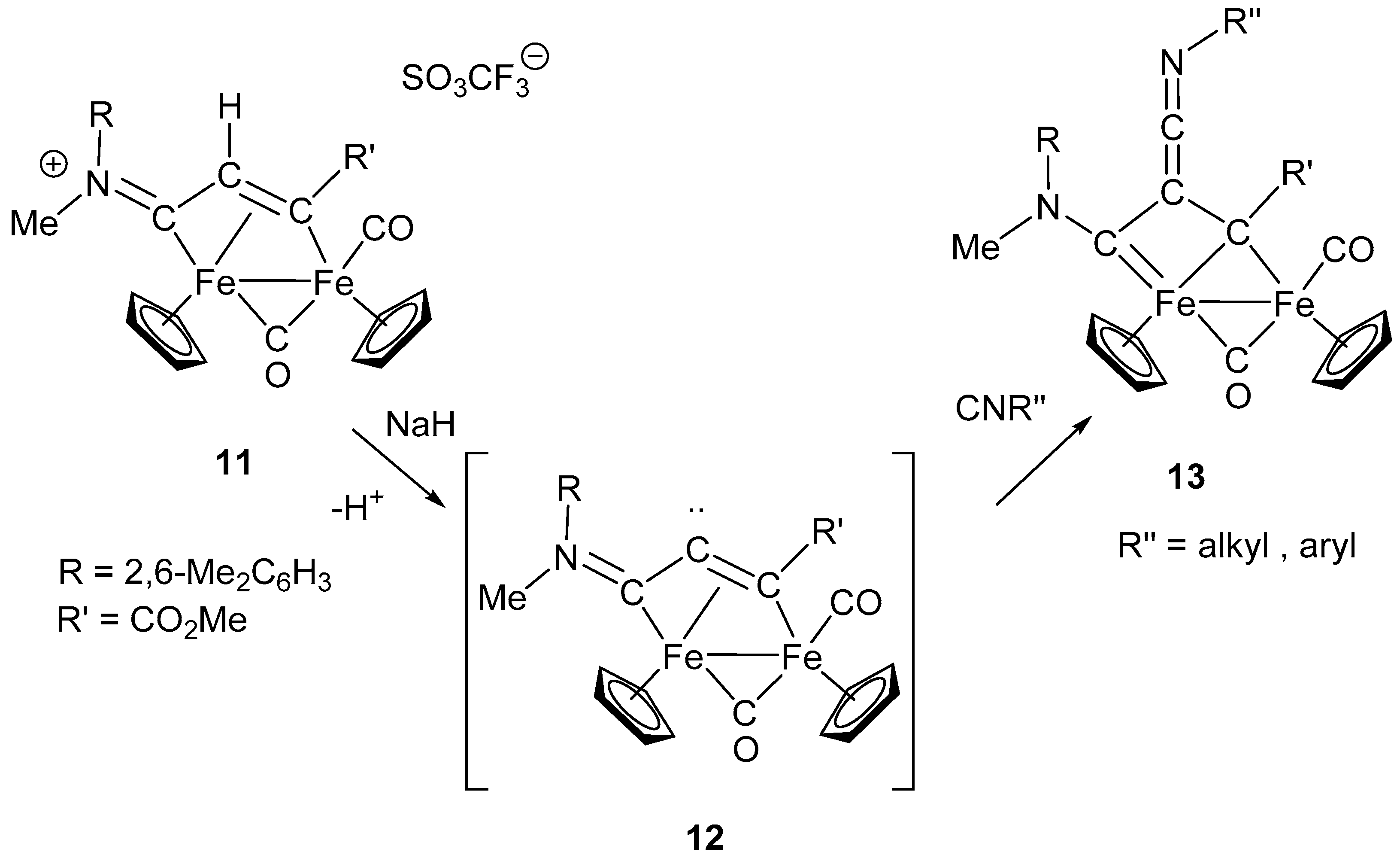
Examples include Fe–carbene complexes but not diiron complexes [79]. However, a similar reaction, producing a ketenimine moiety, has been reported for the diiron bridging ligand shown in Scheme 7 [80].
Removal of the α-hydrogen from the bridging vinyliminium 11 is believed to generate the intermediate 12, which shows some carbene character and reacts with isocyanides to form the ketenimine complex 13 (Scheme 7). The reaction does not have a general character and is limited to some specific combinations of substituents on the vinyliminium ligand (R = xylyl and R’= COOMe). Nevertheless, the formation of the ketenimine is peculiar for many aspects. One is that the observed addition of isocyanide with formation of a C–C double bond is made possible by a rearrangement of the bridging ligand, which assumes the nature of bis-alkylidene in the complex 13. A second consideration concerns the architecture of the bridging frame, which is the result of the assembly, step by step, of several molecular fragments, including two isocyanides. The overall sequence starting from the bridging isocyanide ligand in 1, followed by addition and insertion steps (i.e., N alkylation, alkyne insertion, proton removal and isocyanide addition) is summarized in Scheme 8, to evidence the role of the isocyanides in the molecular assembly of the bridging frame.
6. Nucleophilic Addition at Isocyanide Ligands
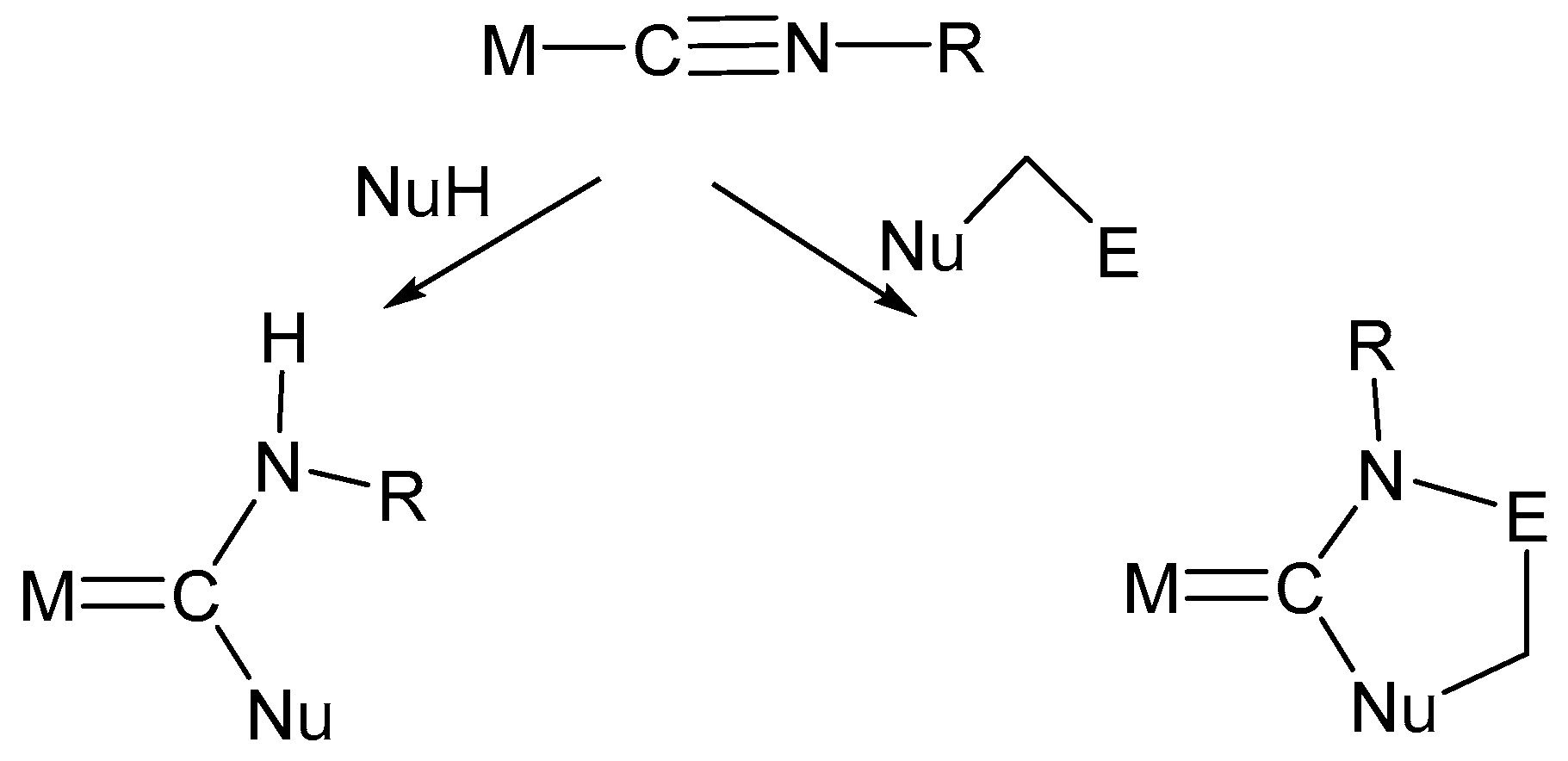
Upon coordination to a transition metal center, isocyanides can be activated toward nucleophilic addition. In particular, the use of protic nucleophiles (NuH) or nucleophiles also containing electrophilic centers (Nu–(CH2)nE), such as haloalcohols and haloamines, has provided a valuable synthetic route for the synthesis of aminocarbene ligands and of N heterocyclic carbene ligands (NHCs), respectively (Scheme 9) [8,9].
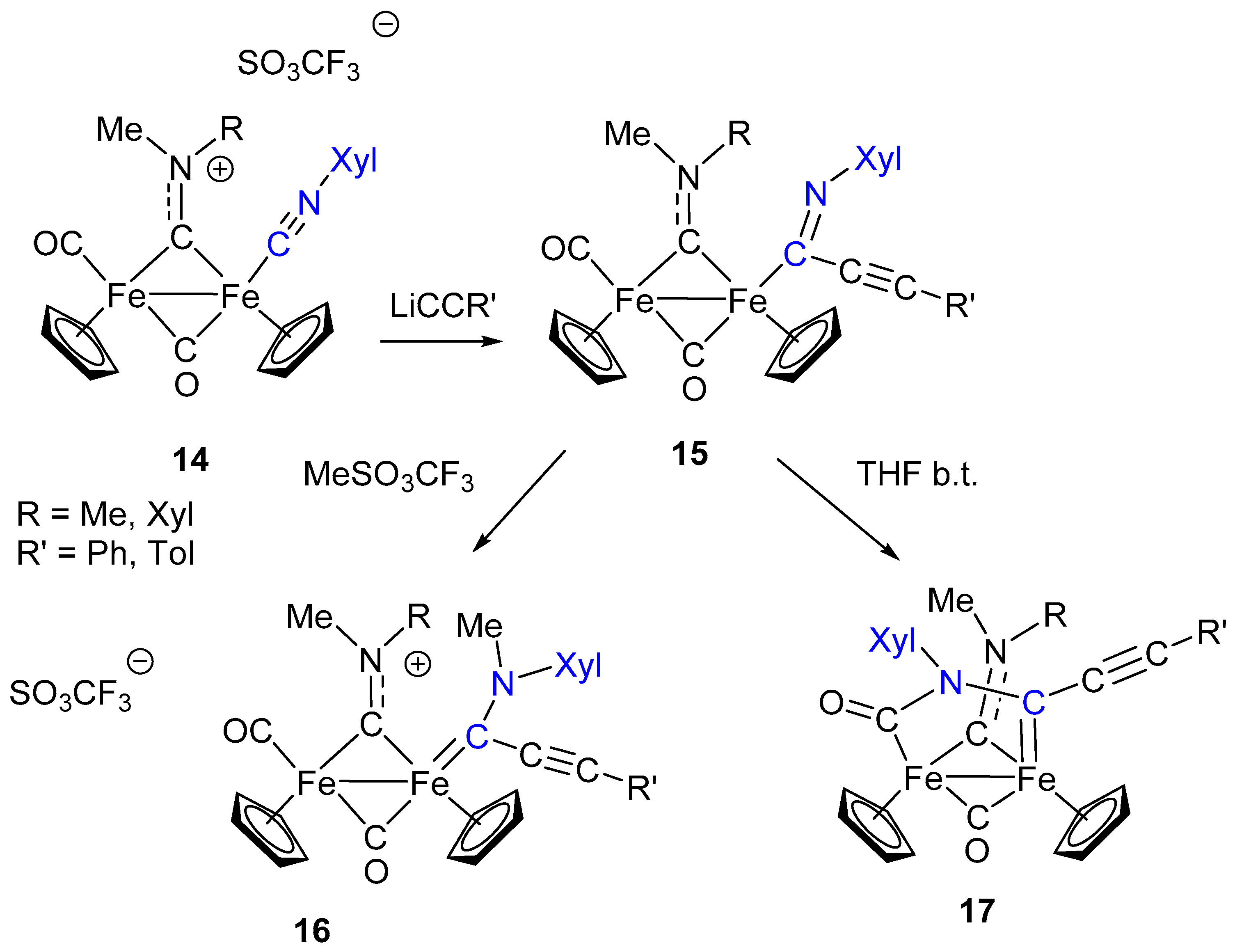
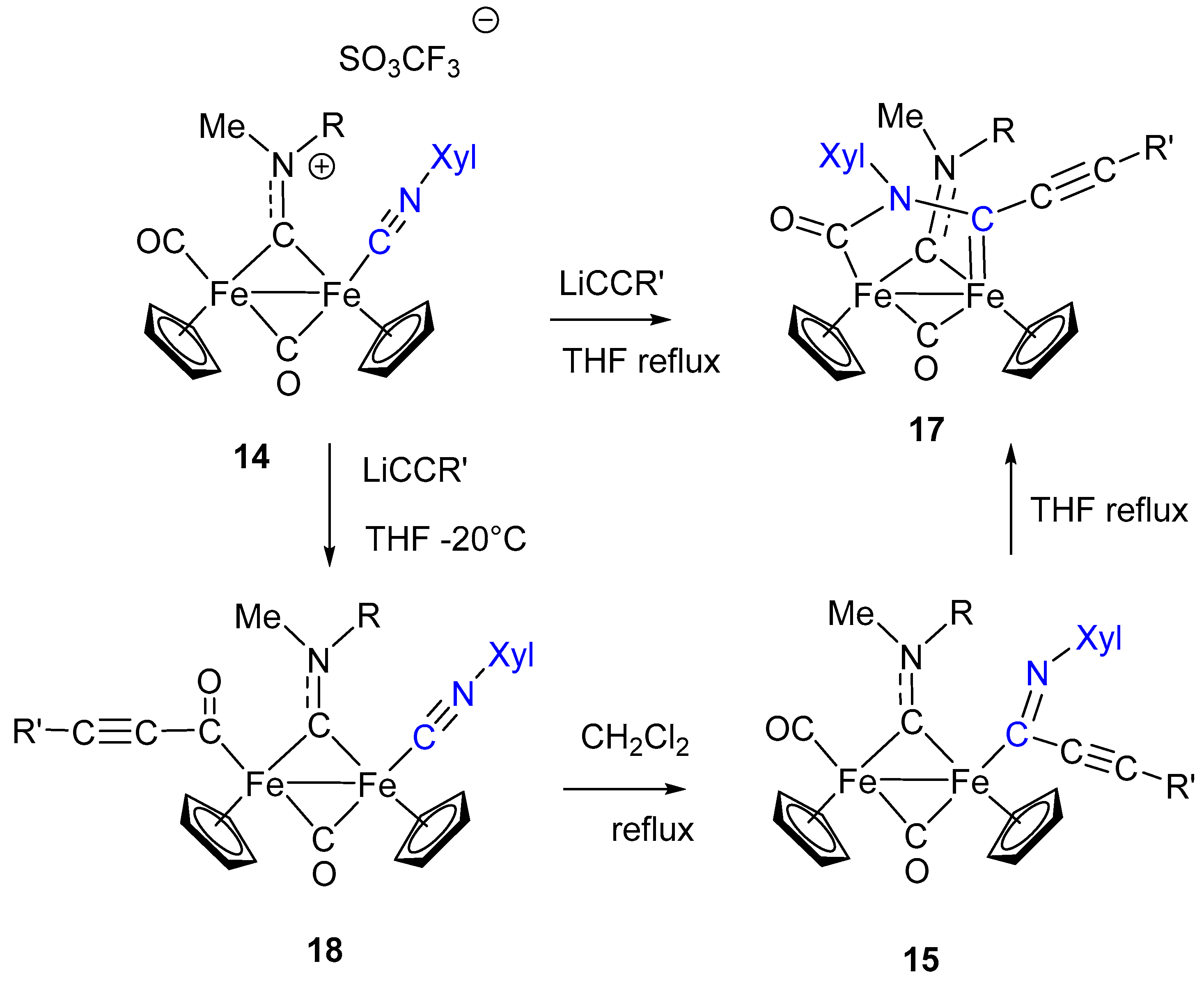
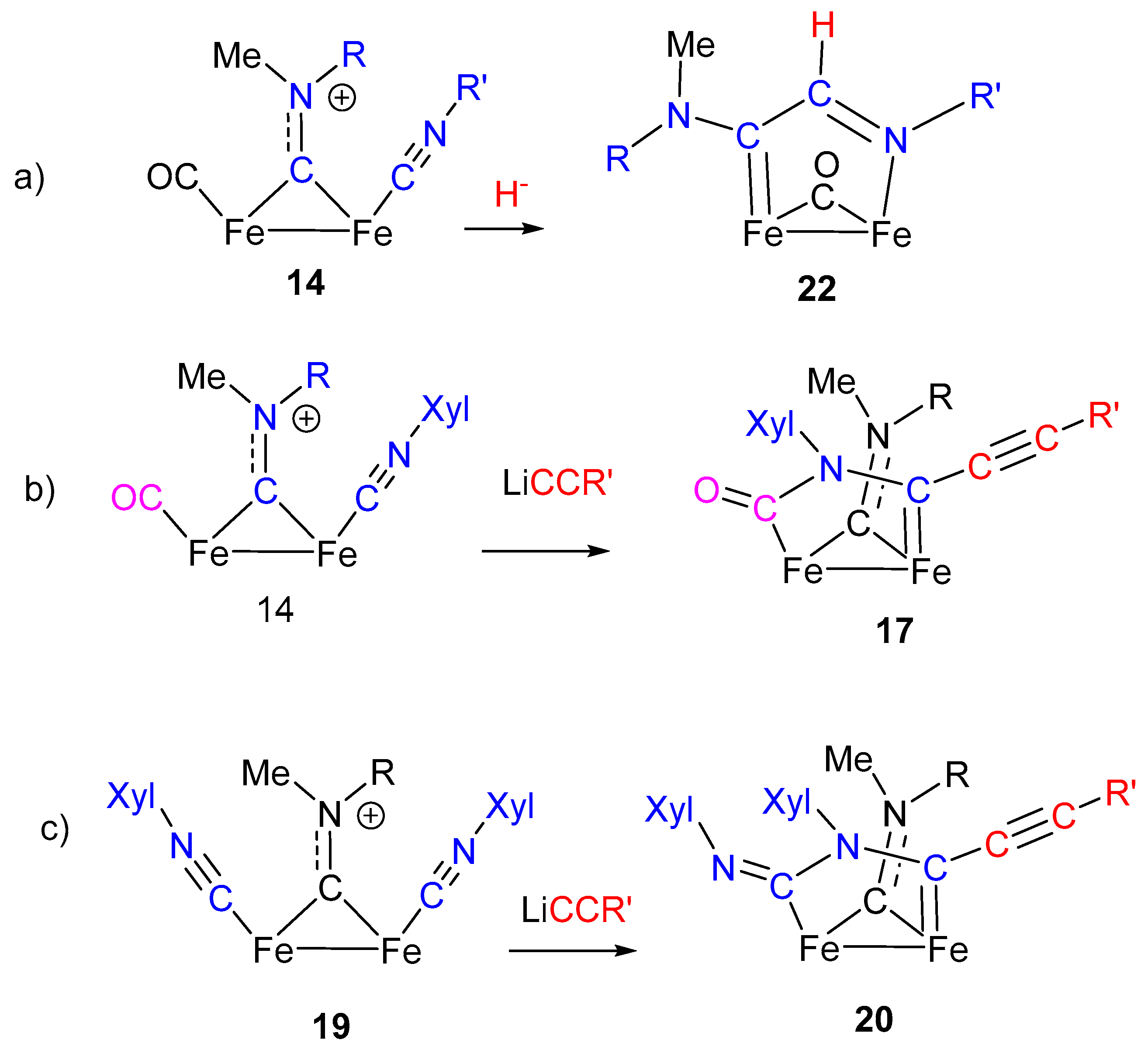
Isocyanide activation toward nucleophilic addition generally requires “electron-poor” metal centers, with reduced electron density (usually, metal ions in the higher oxidation state), to enhance the donor/acceptor properties of the CNR ligand. The nature of the other ligands can also affect the electron density on the metal and consequently the σ/π ratio in the M–CNR bond. Indeed, the most general and successful approach to the transformation of isocyanides into carbenes has been found on palladium and platinum (noble metals in general), but very few examples involving iron have been reported, and these are essentially limited to mononuclear complexes [81,82]. An example of nucleophilic addition at coordinated CNR in diiron complexes is shown in Scheme 10. The reaction of the aminocarbyne complex 14, containing a CNR ligand terminally bound to one Fe, with lithium acetylide leads to the formation of the imidoyl complexes 15 which, in turn, can be methylated at the N atom generating the corresponding aminoalkylidene species 16 [83].
By contrast with the isocyanide transformation to aminocarbene shown in Scheme 3, based on isocyanide methylation followed by nucleophilic addition, the analogous transformation of 14 into the aminocarbene 16 follows the reverse and most classic approach used for this scope, consisting in the nucleophilic addition to CNR, followed by N methylation, as described in Scheme 9. A significant difference is that CNR activation by Fe is not strong enough to allow the addition of nucleophilic species based on heteroatoms (O or N), which are generally used in this type of reaction. Conversely, the observed addition requires a stronger, carbon-based nucleophile: The acetylide. On the other hand, the imidoyl product 15 is stable enough to be isolated and well characterized, which is not so usual [84]. Nonetheless, aminoalkyilidenes (aminocarbenes) are certainly more stable compared to the imidoyl [85]. Therefore, the imidoyl complex 15, upon treatment with MeSO3CF3, readily undergoes methylation with formation of the aminocarbene 16. Interestingly, in the absence of a specific electrophilic reagent able to attack the N atom, the imidoyl ligand in 15 gives rise (upon heating in THF) to an intramolecular rearrangement, consisting in the coupling with the terminally coordinated CO. In other words, even in the absence of a specific alkylation step, the imidoyl can be transformed into aminocarbene by intramolecular addition to CO. A quite remarkable feature is that both the acetylide attack (to form 15) and the reaction at the imidoyl N atom (to form 17) do not involve the bridging aminocarbyne ligand, which simply acts as a spectator one. This is quite surprising, in view of the electrophilic character of bridging aminocarbyne, well remarked in the previous paragraphs. An additional observation is that 17 is the result of a stepwise construction of a rather complex bridging organic frame based on the assembly of different molecular units (CNR, acetylide and CO), with the isocyanide ligand having a pivotal role in the observed transformations.
7. Cascade and Coupling Reactions Triggered by Nucleophilic Addition at Isocyanide Ligands
The formation of complexes 17 evidences a peculiar feature of the diiron complexes described above: Nucleophilic addition at CNR can promote rearrangements involving other ligands in a sort of “cascade” sequence. In particular, the formation of 17, via assembly of acetylide, isocyanide and a CO, is certainly assisted and made possible by the presence of the diiron frame for two major reasons: (a) The presence of more ligands, compared to mononuclear complexes, at conveniently close distance in order to promote intramolecular reactions and rearrangements; (b) the possibility of stabilizing the organic frame resulting from intramolecular coupling by bridging coordination.
A detailed investigation has shown that, under more controlled reaction conditions, the sequence leading from 14 to 17 consists of three steps: (i) Acetylide attack on CO, (ii) acetylide migration to the CNR ligand, (iii) coupling of CO and iminoacyl ligands (Scheme 11) [86].
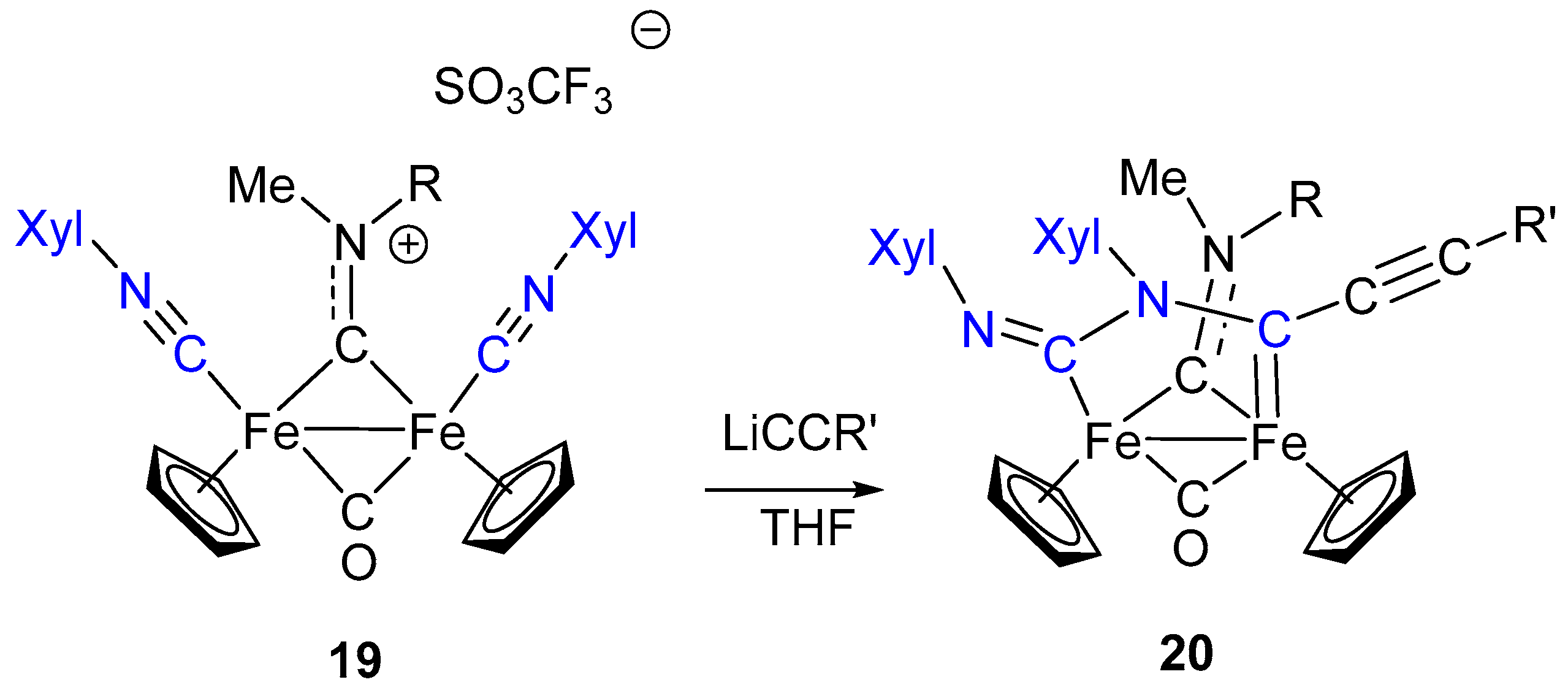
The example provided by the synthesis of 17 is not unique in that a similar reaction is reported for the diiron complex 19, containing two terminally bound isocyanides (Scheme 12) [87]. Likewise the reaction described in Scheme 11, the nucleophilic attack of lithium acetylide to the terminally bound CNXyl ligand generates an imidoyl intermediate which, in turn, reacts with another ligand. In this case, the intramolecular rearrangement involves the other CNR ligand, leading to the formation of the complex 20. The overall result is the coupling of two isocyanide ligands, initiated by a nucleophilic addition, with formation of a C–N bond. The resulting frame, displaying a C–N–C–N atom chain sequence, remains anchored to the iron atoms through bridging coordination in the form of aminocarbene-N aminoacyl ligand. Compared to the reaction sequence described in Scheme 11, the formation of 20 occurs in one single step, and intermediate species have been not isolated in this case.
Reductive coupling of two isocyanide ligands is a long-known reaction [88], and examples include a number of mononuclear isocyanide complexes of group 4 [89], group 5 [88,90,91] and group 6 [92] transition metals. Coupling of isocyanide ligands involving dinuclear complexes is far less common [93,94,95]. Thus, the CNR coupling shown in Scheme 12 has a distinctive character for two reasons: It involves a diiron complex, which is rather uncommon [96], and it provides an example of isocyanide coupling taking place via C–N bond formation, which is rarely observed [62,95], with respect to the predominant C–C coupling mode.
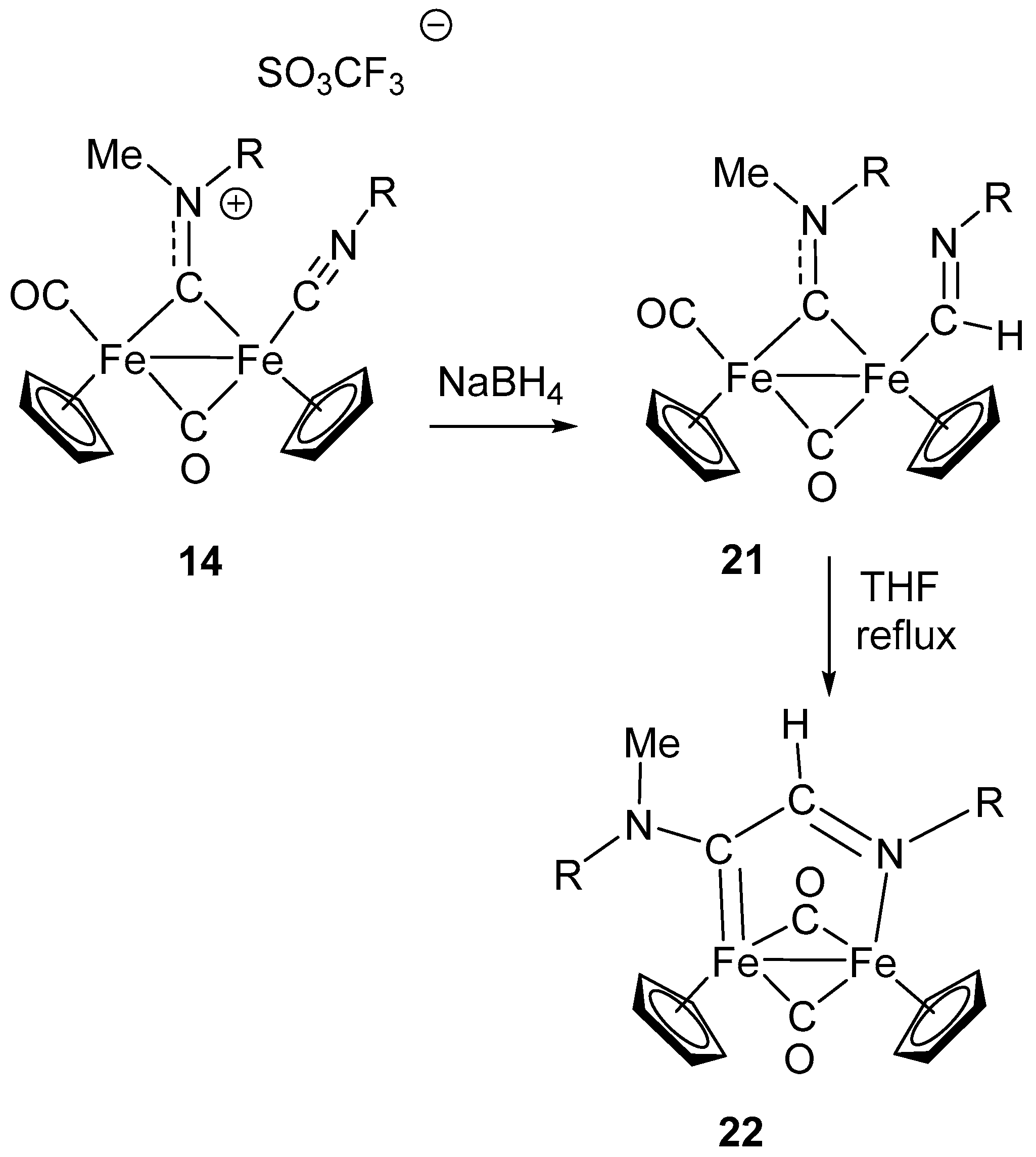
A further example of nucleophilic addition at coordinated CNR ligand giving rise to a sequence of intramolecular rearrangements is shown in Scheme 13. Hydride addition to the isocyanide complex 14 generates the formimidoyl intermediate 21, which further rearranges, after prolonged heating in THF, to form the aminocarbene-aldimine complex 22 [97]. The reaction, providing an uncommon example of Fe-formimidoyl formation by hydride addition at a CNR ligand [98], further demonstrates the effectiveness of diiron complexes in stabilizing unusual intermediate species (e.g., the formimidoyl function). Upon thermal treatment, the formimidoyl 21 rearranges, involving the bridging aminocarbyne ligand, with formation of complex 22, containing a bridging aminocarbene-aldimine ligand, coordinated to the two Fe atoms through the imine nitrogen and the carbene carbon.
By contrast with the previously described intramolecular rearrangements and coupling reactions triggered by nucleophilic attack at CNR (Scheme 11 and Scheme 12), in this case the bridging aminocarbyne ligand is involved. Considering that the μ-aminocarbyne is a “methylated” isocyanide ligand, the aminocarbyne–formimidoyl coupling, leading to 22, can be considered as the assembly of two isocyanide ligands, via C–C coupling. Interestingly, one isocyanide has been activated by electrophilic addition (aminocarbyne), and the other one has been activated by nucleophilic addition (formimidoyl).
A common feature of the reactions described in this paragraph is that nucleophilic addition at a terminally coordinated CNR gives rise to a sequence of coupling reactions: In many cases intermediate species are stable enough to be isolated. The overall result, summarized in Scheme 14, shows strong analogies with multicomponent reactions involving isocyanides.
The unusual activation mode and unique C–N and C–C bond forming reactions, promoted by diiron coordination, lead to the construction of functionalized molecular fragments, containing nitrogen atoms and multiple bonds, which remain bridging through a variety of different multisite coordination modes.
8. Conclusions
Diiron complexes offer a variety of rather distinct reaction patterns and activation modes for CNR ligands, resulting in transformations and rearrangements that should be exploited for synthetic use. Interestingly, in spite of the fact that Fe is considered to be less effective, compared to other transition metals, in promoting most relevant activation patterns for CNR ligands (e.g., enhancement of the nucleophilicity/electrophilicity of CNR, insertion of CNR and multicomponent reactions), all of these reaction profiles can indeed be accessible in a diiron complex. This is likely due to a number of reasons: (a) Diiron coordination of CNR can be both bridging or terminal, the former being more effective in enhancing the electrophilicity of CNR, whereas the terminal coordination seems to favor nucleophilic attack; (b) multisite coordination provides reaction paths otherwise unavailable in mononuclear complexes; this allows the isolatation and observation of a number of intermediate species and functionalities rarely reported in the literature; (c) diiron frames allow for the assembly and putting together, at a convenient distance, of a number of a ligands and reactive molecular fragments (including isocyanides), and promote intramolecular rearrangements and coupling reactions. A number of examples involving CNR ligands in diiron complexes and resembling isocyanide-based multicomponent reactions (MCRs) have been reviewed.
Almost all of the reactions shown here are to be considered Fe-assisted, rather than Fe-catalyzed, in that reactions are stoichiometric and not catalytic. Nevertheless, the quite large amount of examples of transformations, couplings of terminal and bridging ligands, often observed in a cascade sequence and leading to the construction of molecular fragments of higher complexity, are a clear indication of the potential offered by diiron coordination for the development of new and more sustainable synthetic strategies.
Funding
This research was funded by MIUR (Ministero dell’Università e della rRicerca Scientifica) grant: PRIN2015, 20154X9ATP_003.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Van Berkel, S.S.; Bögels, B.G.M.; Wijdeven, M.A.; Westermann, B.; Rutjes, F.P.J.T. Recent advances in asymmetric isocyanide-based multicomponent reactions. Eur. J. Org. Chem. 2012, 3543–3559. [Google Scholar] [CrossRef]
- Lygin, A.V.; de Meijere, A. Isocyanides in the synthesis of nitrogen heterocycles. Angew. Chem. Int. Ed. 2010, 49, 9094–9124. [Google Scholar] [CrossRef] [PubMed]
- Kaim, L.E.; Grimaud, L. Beyond the Ugi reaction: Less conventional interactions between isocyanides and iminium species. Tetrahedron 2009, 65, 2153–2171. [Google Scholar] [CrossRef]
- Dömling, A. Recent developments in isocyanide based multicomponent reactions in applied chemistry. Chem. Rev. 2006, 106, 17–89. [Google Scholar] [CrossRef] [PubMed]
- Cotton, F.A. Progress in Inorganic Chemistry; Interscience: New York, NY, USA, 1959; Volume 1, pp. 284–379. [Google Scholar]
- Malatesta, L.; Bonati, F. Isocyanide Complexes of Transition Metals; Wiley: New York, NY, USA, 1969. [Google Scholar]
- Yamamoto, Y. Zerovalent transition metal complexes of organic isocyanides. Coord. Chem. Rev. 1980, 32, 193–233. [Google Scholar] [CrossRef]
- Michelin, R.A.; Pombeiro, A.J.L.; da Silva, M.F.C.G. Aminocarbene complexes derived from nucleophilic addition to isocyanide ligands. Coord. Chem. Rev. 2001, 218, 75–112. [Google Scholar] [CrossRef]
- Basato, M.; Michelin, R.A.; Mozzon, M.; Sgarbossa, P.; Tassan, A. N-heterocyclic carbenes from transition metal coordinated functional isocyanides of the type o-(CH2Y)C6H4N≡C (Y = OSiMe3, OH; N3; AsR+3 ). J. Organomet. Chem. 2005, 690, 5414–5420. [Google Scholar] [CrossRef]
- Pombeiro, A.J.L.; Guedes da Silva, M.F.C. Aminocarbyne complexes derived from isocyanides activated towards electrophilic addition. Coord. Chem. Rev. 2001, 218, 43–74. [Google Scholar] [CrossRef]
- Boyarskiy, V.P.; Bokach, N.A.; Luzyanin, K.V.; Kukushkin, V.Y. Metal-mediated and metal-catalyzed reactions of isocyanides. Chem. Rev. 2015, 115, 2698–2779. [Google Scholar] [CrossRef] [PubMed]
- Mahmudov, K.T.; Kukushkin, V.Y.; Gurbanov, A.V.; Kinzhalov, M.A.; Boyarskiy, V.P.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Isocyanide metal complexes in catalysis. Coord. Chem. Rev. 2019, 384, 65–89. [Google Scholar] [CrossRef]
- Vlaar, T.; Ruijter, E.; Maes, B.U.W.; Orru, R.V.A. Palladium-catalyzed migratory insertion of isocyanides: An emerging platform in cross-coupling chemistry. Angew. Chem. Int. Ed. 2013, 52, 7084–7097. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.; Ding, Q.; Wu, J. Recent advances in isocyanide insertion chemistry. Chem. Soc. Rev. 2013, 42, 5257–5269. [Google Scholar] [CrossRef] [PubMed]
- Lang, S. Unravelling the labyrinth of palladium-catalysed reactions involving isocyanides. Chem. Soc. Rev. 2013, 42, 4867–4880. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Xu, B. Metal-catalyzed C–H functionalization involving isocyanides. Chem. Soc. Rev. 2017, 46, 1103–1123. [Google Scholar] [CrossRef] [PubMed]
- Abellán-López, A.; Chicote, M.T.; Bautista, D.; Vicente, J. Cyclopalladated complexes derived from phenylacetone oxime. Insertion reactions of carbon monoxide, isocyanides, and alkynes. Novel amidines of the isoquinoline series. Organometallics 2013, 32, 7612–7624. [Google Scholar] [CrossRef]
- Frutos-Pedreño, R.; González-Herrero, P.; Vicente, J.; Jones, P.G. Sequential insertion of alkynes and CO or isocyanides into the Pd–C bond of cyclopalladated phenylacetamides. Synthesis of eight-membered palladacycles, benzo[d]azocine-2,4(1H,3H)-diones, and highly functionalized acrylonitrile and acrylamide derivatives. Organometallics 2012, 31, 3361–3372. [Google Scholar] [CrossRef]
- Vicente, J.; Saura-Lamas, I.; García-López, J.A.; Bautista, D. Eight-membered palladacycles derived from the insertion of olefins into the Pd–C Bond of ortho-palladated pharmaceuticals phenethylamine and phentermine. Synthesis of stable Heck-type intermediates containing accessible β-hydrogens and its use in the synthesis of 2-styrylphenethylamines, tetrahydroisoquinolines, and eight-membered cyclic amidines. Organometallics 2010, 29, 4320–4338. [Google Scholar]
- Vicente, J.; Abad, J.A.; Förtsch, W.; López-Sáez, M.J.; Jones, P.G. Reactivity of Ortho-Palladated Phenol Derivatives with Unsaturated Molecules. 1. Insertion of CO, isocyanides, alkenes, and alkynes. CO/alkene, alkyne/isocyanide, and isocyanide/alkene sequential insertion reactions. Organometallics 2004, 23, 4414–4429. [Google Scholar] [CrossRef]
- Vahabi, A.H.; Alizadeh, A.; Khavasi, H.R.; Bazgir, A. Palladium-catalyzed migratory insertion of isocyanides into C(thiophene)–SMe bonds: Access to atom-transfer reactions. Eur. J. Org. Chem. 2017, 2017, 5347–5356. [Google Scholar] [CrossRef]
- Tereniak, S.J.; Stahl, S.S. Mechanistic basis for efficient, site-selective, aerobic catalytic turnover in Pd-catalyzed C–H imidoylation of heterocycle-containing molecules. J. Am. Chem. Soc. 2017, 139, 14533–14541. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, I.; San Segundo, M.; Correa, A. Iron-catalyzed C(sp3)–H functionalization of N,N-dimethylanilines with isocyanides. Chem. Commun. 2018, 54, 1627–1630. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chen, Y.; Li, W.; Xie, Q.; Shao, L. Microwave-assisted synthesis of phenanthridines by radical insertion/cyclization of biphenyl isocyanides. J. Org. Chem. 2016, 81, 8426–8435. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.D.; Foster, G.P.; Putinas, J.M. The catalytic activation and functionalization of C–H bonds. Aldimine formation by the insertion of isonitriles into aromatic C–H Bonds. J. Am. Chem. Soc. 1987, 109, 5047–5048. [Google Scholar] [CrossRef]
- Li, Y.; Rauchfuss, T.B. Synthesis of Diiron(I) dithiolato carbonyl complexes. Chem. Rev. 2016, 116, 7043–7077. [Google Scholar] [CrossRef] [PubMed]
- Capon, J.F.; Gloaguen, F.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J. Electron and proton transfers at diiron dithiolate sites relevant to the catalysis of proton reduction by the [FeFe]-hydrogenases. Coord. Chem. Rev. 2009, 253, 1476–1494. [Google Scholar] [CrossRef]
- Mejia-Rodriguez, R.; Chong, D.; Reibenspies, J.H.; Soriaga, M.P.; Darensbourg, M.Y. The hydrophilic phosphatriazaadamantane ligand in the development of H2 production electrocatalysts: Iron hydrogenase Model Complexes. J. Am. Chem. Soc. 2004, 126, 12004–12014. [Google Scholar] [CrossRef] [PubMed]
- Cheah, M.H.; Tard, C.; Borg, S.J.; Liu, X.; Ibrahim, S.K.; Pickett, C.J.; Best, S.P. Modeling [Fe–Fe] Hydrogenase: Evidence for Bridging Carbonyl and Distal Iron Coordination Vacancy in an Electrocatalytically Competent Proton Reduction by an Iron Thiolate Assembly That Operates through Fe(0)–Fe(II) Levels. J. Am. Chem. Soc. 2007, 129, 11085–11092. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, R.; Salmi, M.; Zanotti, V. C–C bond formation in diiron complexes. Chem. Eur. J. 2012, 18, 10174–10194. [Google Scholar] [CrossRef] [PubMed]
- Busetto, L.; Maitlis, P.M.; Zanotti, V. Bridging vinylalkylidene transition metal complexes. Coord. Chem. Rev. 2010, 254, 470–486. [Google Scholar] [CrossRef]
- Busetto, L.; Zanotti, V. Carbene ligands in diiron complexes. J. Organomet. Chem. 2005, 690, 5430–5440. [Google Scholar] [CrossRef]
- Basset, J.M.; Green, M.; Howard, J.A.K.; Stone, F.G.A. Formation of nona(ethyl isocyanide)di-iron from penta(ethyl isocyanide)iron and reaction of penta(t-butyl isocyanide)iron with diphenylacetylene; X-ray crystal structures of nona(ethyl isocyanide)di-iron and tris(t-butyl isocyanide){1,4-bis-(t-butylimino)-2,3-diphenylbuta-1,3-diene}iron. J. Chem. Soc. Chem. Commun. 1978, 22, 1000–1001. [Google Scholar]
- Basset, J.M.; Barker, G.K.; Green, M.; Howard, J.A.K.; Stone, F.G.A.; Wolsey, W.C. Chemistry of low-valent metal isocyanide complexes. Part 3. The synthesis, structure, and dynamic behaviour of nonakis(ethyl and isopropyl isocyanide)di-iron and -ruthenium complexes. Crystal structure of [Fe2(µ-CNEt)3(CNEt)6]. J. Chem. Soc. Dalton Trans. 1981, 219–227. [Google Scholar] [CrossRef]
- Ruiz, J.; Riera, V.; Vivanco, M.; García-Granda, S.; Pertierra, P. A homoleptic (aryl isocyanlde)iron(0) dimer. X-ray structure determination of nonakis(phenyl isocyanide)diiron. Organometallics 1992, 11, 2734–2736. [Google Scholar] [CrossRef]
- Bellerby, J.; Boylan, M.J.; Ennis, M.; Manning, A.R. An infrared spectroscopic study of the tautomeric equilibria in solutions of tricarbonylbis(η-dienyl)isocyanidedi-iron complexes. J. Chem. Soc. Dalton Trans. 1978, 9, 1185–1189. [Google Scholar] [CrossRef]
- Joshi, K.K.; Mills, O.S.; Pauson, P.L.; Shaw, B.W.; Stubbs, W.H. An iron complex with a bridging isonitrile group. Chem. Commun. 1965, 10, 181–182. [Google Scholar] [CrossRef]
- Treiche, P.M.; Stenson, J.P.; Benedict, J.J. Reactions of several cationic organometallic isocyanide complexes with sodium borohydride. Inorg. Chem. 1971, 10, 1183–1187. [Google Scholar] [CrossRef]
- Cotton, F.A.; Frenz, B.A. Low-valent metal isocyanide complexes. IV.1 Crystal and molecular structures of cis-anti-Bis(penthaptocyclopentadienyl)dicarbonylbis(μ-methyl isocyanide)-diiron(Fe–Fe), (η5-C5H5)2Fe2(CO)2(μ-CNCH3)2. Inorg. Chem. 1974, 13, 253–256. [Google Scholar] [CrossRef]
- Hunt, I.D.; Mills, O.S. Carbon compounds of the transition metals. XXlX. Crystal structure of trans-anti-Bis(pentahapto-cyclopentadienyl)dicarbonyl-bis(μ-phenylisonitrile)-diiron(Fe–Fe), (h5-C5H5)2Fe2(CO)2(μ-C NPh)2. Acta Cryst. 1977, B33, 2432–2435. [Google Scholar] [CrossRef]
- Fehlhammer, W.P.; Mayr, A.; Kehr, W. Isocyanid-und heteroallen-verbrückte metallkomplexe iv. die krjstall- und molekolstruktur von trans-anti-bis[η-cyclopentadienyl(μ-phenylisocyanid)-(phenylisocyanid)eisen](Fe–Fe),[Fe(η-C5H5)(μ-CNC6H5)CNC6H5]2. direkter stereochemischer vergleich. J. Organomet. Chem. 1980, 197, 327–334. [Google Scholar] [CrossRef]
- Coville, N.J.; Albers, M.O.; Singleton, E. An investigation of the reaction between [{Fe(η5-C5H5)(CO)2}2] and aryl isonitriles. J. Chem. Soc. Dalton Trans. 1982, 1389–1391. [Google Scholar] [CrossRef]
- Campbell, I.L.C.; Stephens, F.S. Crystal and molecular structure of cis-(isobutyl isocyanide)di-µ-carbonylcarbonylbis(π-cyclopentadienyl)di-iron. J. Chem. Soc. Dalton Trans. 1975, 982–985. [Google Scholar] [CrossRef]
- Boss, K.; Dowling, C.; Manning, A.R. Preparation, spectra and structure of [Fe2(η-C5H5)2(L)(CN)(μ-CO){ μ-CN(R’)R}],[Fe2(η-C5H5)2(CO)(CN){μ-CNMe2}2]+ and [Fe2(η-C5H5)2(CN)2(μ-CNMe2}2] zwitterions (L = CO or organoisocyanide) and their reactions with alkyl and protic electrophiles. J. Organomet. Chem. 1996, 509, 197–207. [Google Scholar] [CrossRef]
- Busetto, L.; Carlucci, L.; Zanotti, V.; Albano, V.G.; Braga, D. Synthesis, reactions, and X-ray structures of the functionalized isocyanide complexes [Fe2{µ-CNC(O)SR}(µ-CO)(CO)2(cp)2](cp = η-C5H5, R = Me or Et) and of their carbyne and carbene derivatives. J. Chem. Soc. Dalton Trans. 1990, 243–250. [Google Scholar] [CrossRef]
- Nehring, J.L.; Heinekey, D.M. Dinuclear iron isonitrile complexes: Models for the iron hydrogenase active site. Inorg. Chem. 2003, 42, 4288–4292. [Google Scholar] [CrossRef] [PubMed]
- Boyke, C.A.; Rauchfuss, T.B.; Wilson, S.R.; Rohmer, M.-M.; Bénard, M. [Fe2(SR)2(µ-CO)(CNMe)6]2+ and Analogues: A new class of diiron dithiolates as structural models for the HoxAir state of the Fe-only hydrogenase. J. Am. Chem. Soc. 2004, 126, 15151–15160. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Barton, B.E.; Chambers, G.M.; Rauchfuss, T.B.; Arrigoni, F.; Zampella, G. Preparation and Protonation of Fe2(pdt)(CNR)6, Electron-rich analogues of Fe2(pdt)(CO)6. Inorg. Chem. 2016, 55, 3401–3412. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.; Manning, A.R.; Stephens, F.S. Reactions of [Fe2(η-dienyl)2(CO)4–n(CNR)n] with strong acids. The structure of cis-µ-carbonyl-µ-methyliminiomethylene-bis[carbonyl(η-cyclopentadienyl)iron] tetrafluoroborate. J. Chem. Soc., Dalton Trans. 1979, 23–27. [Google Scholar] [CrossRef]
- Willis, S.; Manning, A.R.; Stephens, F.S. Reactions of [Fe2(η-dienyl)2(CO)4–n(CNR)n] complexes (dienyl = C5H5, C5H4Me, or C9H7; R = alkyl or benzyl; n= 1 or 2) with alkyl halides and other alkylating agents. The crystal structure of cis-[Fe2(η-C5H4Me)2(CO)3{C(NMe2)µ}]I. J. Chem. Soc. Dalton Trans. 1980, 186–191. [Google Scholar] [CrossRef]
- Cox, G.; Dowling, C.; Manning, A.R.; McArdle, P.; Cunningham, D. A reinvestigation of the reaction of [Fe,(η-C5H5)2(CO)4−n(CNR)n](n = 1 or 2) with strong alkylating agents. J. Organomet. Chem. 1992, 438, 143–158. [Google Scholar] [CrossRef]
- Agonigi, G.; Bortoluzzi, M.; Marchetti, F.; Pampaloni, G.; Zacchini, S.; Zanotti, V. Regioselective Nucleophilic Additions to Diiron Carbonyl Complexes Containing a Bridging Aminocarbyne Ligand: A Synthetic, Crystallographic and DFT Study. Eur. J. Inorg. Chem. 2018, 960–971. [Google Scholar] [CrossRef]
- Marchetti, F. Constructing organometallic architectures from aminoalkylidyne diiron complexes. Eur. J. Inorg. Chem. 2018, 2018, 3987–4003. [Google Scholar] [CrossRef]
- Zanotti, V.; Bordoni, S.; Busetto, L.; Carlucci, L.; Palazzi, A.; Serra, R.; Albano, V.G.; Monari, M.; Prestopino, F.; Laschi, F.; et al. Diiron aminoalkylidene complexes. Organometallics 1995, 14, 5232–5241. [Google Scholar] [CrossRef]
- Albano, V.G.; Busetto, L.; Camiletti, C.; Castellari, C.; Monari, M.; Zanotti, V. Selective C–C bond formation at diiron µ-aminocarbyne complexes. J. Chem. Soc. Dalton Trans. 1997, 4671–4676. [Google Scholar] [CrossRef]
- Albano, V.G.; Bordoni, S.; Busetto, L.; Marchetti, F.; Monari, M.; Zanotti, V. Carbon monoxide–isocyanide coupling promoted by acetylide addition to a diiron complex. J. Organomet. Chem. 2003, 684, 37–43. [Google Scholar] [CrossRef]
- Bellachioma, G.; Cardaci, G.; Macchioni, A.; Reichenbach, G. Mechanism of isocyanide insertion into the methyl–iron bond of [Fe(CO)2L2(CNR)CH3]BPh4. Inorg. Chem. 1992, 31, 63–66. [Google Scholar] [CrossRef]
- Bellachioma, G.; Cardaci, G.; Macchioni, A.; Zuccaccia, C. Effect of anions on the isocyanide insertion reaction in cationic alkyl complexes of iron(II): Kinetic, thermodynamic and solution interionic structural studies. J. Organomet. Chem. 2000, 593, 119–126. [Google Scholar] [CrossRef]
- Klose, A.; Solari, E.; Ferguson, R.; Floriani, C. Insertion reactions of isocyanides and nitriles into unsupported iron–aryl bonds: The synthesis of a dimeric iron(II) homoleptic iminoacyl complex. Organometallics 1993, 12, 2414–2416. [Google Scholar] [CrossRef]
- Klose, A.; Solari, E.; Floriani, C.; Chiesi-Villa, A.; Rizzoli, C.; Re, N. Magnetic properties diagnostic for the existence of Iron(II)–Iron(II) bonds in dinuclear complexes which derive from stepwise insertion reactions on unsupported iron–aryl bonds. J. Am. Chem. Soc. 1994, 116, 9123–9135. [Google Scholar] [CrossRef]
- Hogarth, G.; Lavender, M.H.; Shukri, K. Diiron–hydride complexes: synthesis, structure, and reactivity of trans-[Fe2(CO)4(µ-H)(µ-CO)(µ-PCy2)(µ-Ph2PCH2PPh2)]. Organometallics 1995, 14, 2325–2341. [Google Scholar] [CrossRef]
- Alvarez, M.A.; García, M.E.; García-Vivó, D.; Ruiz, M.A.; Vega, M.F. Insertion, rearrangement, and coupling processes in the reactions of the unsaturated hydride complex [W2(η5-C5H5)2(H)(μ-PCy2)(CO)2] with isocyanides. Organometallics 2013, 32, 4543–4555. [Google Scholar] [CrossRef]
- Alvarez, M.A.; García, M.E.; Ramos, A.; Ruiz, M.A. Reactivity of the unsaturated hydride [Mo2(η5-C5H5)2(μ-H)(μ-PCy2)(CO)2] toward P-donor bidentate ligands and unsaturated N-containing organic molecules. Organometallics 2007, 26, 1461–1472. [Google Scholar] [CrossRef]
- Beringhelli, T.; D’Alfonso, G.; Minoja, A.; Ciani, G.; Moret, M.; Sironi, A. Insertion of isocyanides into metal-hydrogen bonds in triangular hydrido-carbonyl clusters of rhenium. X-ray crystal structures of [Re3(μ-H)3(μ-η-CHNR)(CO)10]− (R = p-Tolyl) and of [Re3(μ-H)3(μ3-η2-CHNR)(CO)9]− (R = cyclohexyl). Organometallics 1991, 10, 3131–3138. [Google Scholar] [CrossRef]
- Fujita, K.; Nakaguma, H.; Hanasaka, F.; Yamaguchi, R. Synthesis of a DMPM and hydrido-bridged diiridium complex, [(Cp*Ir)2(μ-dmpm)(μ-H)2][OTf]2, and its reactivity toward alkynes and isocyanides. Organometallics 2002, 21, 3749–3757. [Google Scholar] [CrossRef]
- Albano, V.G.; Busetto, L.; Marchetti, F.; Monari, M.; Zacchini, S.; Zanotti, V. Alkyne-Isocyanide Coupling in [Fe2(CNMe)(CO)3(Cp)2]: A new route to diiron μ-vinyliminium complexes. Organometallics 2007, 26, 3448–3455. [Google Scholar] [CrossRef]
- Dyke, A.F.; Knox, S.A.R.; Naish, P.J.; Taylor, G.E. Organic chemistry of dinuclear metal centres. Part 1. Combination of alkynes with carbon monoxide at di-iron and diruthenium centres: Crystal structure of [Ru2(CO)(µ-CO){µ-σ:η3-C(O)C2Ph2}(η-C5H5)2]. J. Chem. Soc. Dalton Trans. 1982, 1297–1307. [Google Scholar] [CrossRef]
- Boni, A.; Funaioli, T.; Marchetti, F.; Pampaloni, G.; Pinzino, C.; Zacchini, S. Electrochemical, EPR and computational results on [Fe2Cp2(CO)2]-based complexes with a bridging hydrocarbyl ligand. J. Organomet. Chem. 2011, 696, 3551–3556. [Google Scholar] [CrossRef]
- Marchetti, F.; Zacchini, S.; Zanotti, V. Photochemical alkyne insertions into the iron–thiocarbonyl bond of [Fe2(CS)(CO)3(Cp)2]. Organometallics 2016, 35, 2630–2637. [Google Scholar] [CrossRef]
- Albano, V.G.; Busetto, L.; Marchetti, F.; Monari, M.; Zacchini, S.; Zanotti, V. Diiron µ-vinyliminium complexes from acetylene insertion into a metal–aminocarbyne bond. Organometallics 2003, 22, 1326–1331. [Google Scholar] [CrossRef]
- Albano, V.G.; Busetto, L.; Marchetti, F.; Monari, M.; Zacchini, S.; Zanotti, V. Stereochemistry of the insertion of disubstituted alkynes into the metal aminocarbyne bond in diiron complexes. J. Organomet. Chem. 2004, 689, 528–538. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Zacchini, S.; Zanotti, V.; Marchetti, F. DFT mechanistic insights into the alkyne insertion reaction affording diiron μ-vinyliminium complexes and new functionalization pathways. Organometallics 2018, 37, 3718–3731. [Google Scholar] [CrossRef]
- Zanotti, V. Reactions of bridging C3 ligands in diiron complexes: Unconventional routes to new functionalized organic frames. Pure Appl. Chem. 2010, 82, 1555–1568. [Google Scholar] [CrossRef]
- Albano, V.G.; Busetto, L.; Marchetti, F.; Monari, M.; Zacchini, S.; Zanotti, V. C–C bond formation by cyanide addition to dinuclear vinyliminium complexes. J. Organomet. Chem. 2006, 691, 4234–4243. [Google Scholar] [CrossRef]
- Busetto, L.; Marchetti, F.; Zacchini, S.; Zanotti, V. Reactions of diazo compounds at μ-vinyliminium ligands: synthesis of novel dinuclear azine–bis(alkylidene) complexes. Organometallics 2007, 26, 3577–3584. [Google Scholar] [CrossRef]
- Aumann, R. Ketenimine complexes from carbene complexes and isocyanides: Versatile building blocks for carbocycles and N-heterocycles [New Synthetic Methods (74)]. Angew. Chem. Int. ed. Engl. 1988, 27, 1456–1467. [Google Scholar] [CrossRef]
- Fernández, I.; Cossío, F.P.; Sierra, M.A. Mechanism of the Generation of Ketenimine–M(CO)n complexes (M = Cr, W, Fe) from Fischer carbenes and isocyanides. Organometallics 2007, 26, 3010–3017. [Google Scholar] [CrossRef]
- Cadierno, V.; García-Álvarez, J.; Gimeno, J.; Rubio-García, J. Reaction of isocyanides with iminophosphorane-based carbene ligands: Synthesis of unprecedented ketenimine–ruthenium complexes. J. Organomet. Chem. 2005, 690, 5856–5862. [Google Scholar] [CrossRef]
- Mitsudo, T.; Watanabe, H.; Komiya, Y.; Watanabe, Y.; Takaegami, Y.; Nakatsu, K.; Kinoshita, K.; Miyagawa, Y. Synthesis, crystal structure and reactivities of the first η4-vinlylketen metal complexes. J. Organomet. Chem. 1980, 190, C39–C42. [Google Scholar] [CrossRef]
- Busetto, L.; Marchetti, F.; Zacchini, S.; Zanotti, V. Addition of Isocyanides at Diiron μ-Vinyliminium Complexes: Synthesis of novel ketenimine-bis(alkylidene) complexes. Organometallics 2008, 27, 5058–5066. [Google Scholar] [CrossRef]
- Ruiz, J.; García, L.; Mejuto, C.; Vivanco, M.; Díaz, M.R.; García-Granda, S. Strong electron-donating metalla-N-heterocyclic carbenes. Chem. Commun. 2014, 50, 2129–2132. [Google Scholar] [CrossRef] [PubMed]
- Yu, I.; Wallis, C.J.; Patrick, B.O.; Diaconescu, P.L.; Mehrkhodavandi, P. Phosphine-tethered carbene ligands: Template synthesis and reactivity of cyclic and acyclic functionalized carbenes. Organometallics 2010, 29, 6065–6076. [Google Scholar] [CrossRef]
- Marchetti, F.; Zacchini, S.; Zanotti, V. Growing the molecular architecture at alkynyl(amino)carbene ligands in diiron µ-aminocarbyne complexes. Eur. J. Inorg. Chem. 2016, 2016, 4820–4828. [Google Scholar] [CrossRef]
- Wang, D.-L.; Hwang, W.-S.; Lee, L.; Chiang, M.Y. Reaction of N-(3-methyl-2-thienylmethylidene)aniline with diiron nonacarbonyl: Cyclometalation induced methyl migration, imidoyl complex formation, and hydrogenation. J. Organomet. Chem. 1999, 579, 211–216. [Google Scholar] [CrossRef]
- Lappert, M.F. Contributions to the chemistry of carbenemetal chemistry. J. Organomet. Chem. 2005, 690, 5467–5473. [Google Scholar] [CrossRef]
- Marchetti, F.; Zacchini, S.; Zanotti, V. Carbon monoxide–isocyanide coupling promoted by acetylide addition to a diiron complex. Chem. Commun. 2015, 51, 8101–8104. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, F.; Zacchini, S.; Zanotti, V. C–N coupling of isocyanide ligands promoted by acetylide addition to diiron aminocarbyne complexes. Organometallics 2015, 34, 3658–3664. [Google Scholar] [CrossRef]
- Carnahan, E.M.; Protasiewicz, J.D.; Lippard, S.J. The 15 years of reductive coupling: What have we learned? Acc. Chem. Res. 1993, 26, 90–97. [Google Scholar] [CrossRef]
- Bashall, A.; Collier, P.E.; Gade, L.H.; McPartlin, M.; Mountford, P.; Pugh, S.M.; Radojevic, S.; Schubart, M.; Scowen, I.J.; Trösch, D.J.M. C–C and C–N coupling reactions of an imidotitanium complex with isocyanides. Organometallics 2000, 19, 4784–4794. [Google Scholar] [CrossRef]
- Rehder, D.; Bottcher, C.; Collazo, C.; Hedelt, R.; Schmidt, H. Isocyanide–Group 5 complexes and metal-centred C–C coupling. J. Organomet. Chem. 1999, 585, 294–307. [Google Scholar] [CrossRef]
- Collazo, C.; Rodewald, D.; Schmidt, H.; Rehder, D. Niobium-Centered C–C Coupling of Isonitriles. Organometallics 1996, 15, 4884–4887. [Google Scholar] [CrossRef]
- Acho, J.A.; Lippard, S.J. Synthesis and Structural Characterization of [Cr(t-C4H9HNC≡CNH-t-C4H9)(CN-t-C4H9)4I]I. Reductive coupling of isocyanide ligands in a first row transition metal complex. Organometallics 1994, 13, 1294–1299. [Google Scholar] [CrossRef]
- Shen, J.; Yap, G.P.A.; Theopold, K.H. Chromium mediated reductive coupling of isonitrile forms unusual heterocycles. J. Am. Chem. Soc. 2014, 136, 3382–3384. [Google Scholar] [CrossRef] [PubMed]
- Ojo, W.S.; Petillon, F.Y.; Schollhammer, P.; Talarmin, J. C–C, C–S, and C–N coupling versus dealkylation processes in the cationic tris(thiolato)dimolybdenum(III) complexes [Mo2Cp2(μ-SMe)3L2]+ (L = xylNC, t-BuNC, CO, MeCN). Organometallics 2008, 27, 4207–4222. [Google Scholar] [CrossRef]
- Cabon, N.; Paugam, E.; Petillon, F.Y.; Schollhammer, F.; Talarmin, J. Unexpected Coupling of Cp and two RNC ligands at a {Mo2(μ-SMe)3} nucleus. Organometallics 2003, 22, 4178–4180. [Google Scholar] [CrossRef]
- Okazaki, M.; Suto, K.; Kudo, N.; Takano, M.; Ozawa, F. Synthesis and structure of cubane-type tetrairon clusters possessing μ3-isonitrile ligands. Reductive coupling of two isonitriles on redox-responsive tetrairon reaction sites. Organometallics 2012, 31, 4110–4113. [Google Scholar] [CrossRef]
- Marchetti, F.; Zacchini, S.; Zanotti, V. Coupling of isocyanide and μ-aminocarbyne ligands in diiron complexes promoted by hydride addition. Organometallics 2014, 33, 3990–3997. [Google Scholar] [CrossRef]
- Cabon, N.; Petillon, F.Y.; Orain, P.Y.; Schollhammer, F.; Talarmin, J.; Muir, K.W. Controlled nucleophilic activation of different sites in [Mo2Cp2L2(μ-SMe)2(μ-L’)]+ cations (L = ButNC, xylNC, CO; L’ = SMe or PPh2). J. Organomet. Chem. 2005, 690, 4583–4601. [Google Scholar] [CrossRef]
Scheme 1.
Selected examples of diiron complexes with isocyanide ligands.

Scheme 2.
Electrophilic addition at isocyanide ligands: (a) addition at terminally bound CNR, (b) addition at bridging CNR.
Scheme 2.
Electrophilic addition at isocyanide ligands: (a) addition at terminally bound CNR, (b) addition at bridging CNR.

Scheme 3.
Carbyne to carbene conversion via nucleophilic addition.

Scheme 4.
Examples of CNR insertion in diiron complexes.

Scheme 5.
Alkyne insertion into Fe-bridging CNR.

Scheme 6.
Carbene–isocyanide coupling.

Scheme 7.
Isocyanide addition to a bridging iminium ligand.

Scheme 8.
Stepwise assembly of molecular fragments, including two isocyanides.

Scheme 9.
Aminocarbenes from nucleophilic addition to CNR ligands.

Scheme 10.
Isocyanide to aminocarbene conversion via nucleophilic addition.

Scheme 11.
Acetylide addition at CNR and subsequent rearrangements.

Scheme 12.
Coupling of two CNR ligands triggered by acetylide addition.

Scheme 13.
Isocyanide–aminocarbyne coupling initiated by hydride addition to the isocyanide.

Scheme 14.
“Cascade” reactions triggered by nucleophilic addition: (a) Hydride + CNR + aminocarbyne; (b) acetilyde + CNR + CO; (c) acetilyde + CNR + CNR.
Scheme 14.
“Cascade” reactions triggered by nucleophilic addition: (a) Hydride + CNR + aminocarbyne; (b) acetilyde + CNR + CO; (c) acetilyde + CNR + CNR.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mazzoni, R.; Marchetti, F.; Cingolani, A.; Zanotti, V. Bond Forming Reactions Involving Isocyanides at Diiron Complexes. Inorganics 2019, 7, 25. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7030025
AMA Style
Mazzoni R, Marchetti F, Cingolani A, Zanotti V. Bond Forming Reactions Involving Isocyanides at Diiron Complexes. Inorganics. 2019; 7(3):25. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7030025
Chicago/Turabian StyleMazzoni, Rita, Fabio Marchetti, Andrea Cingolani, and Valerio Zanotti. 2019. "Bond Forming Reactions Involving Isocyanides at Diiron Complexes" Inorganics 7, no. 3: 25. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7030025
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.