Metal Coordination Complexes as Redox Mediators in Regenerative Dye-Sensitized Solar Cells

 , , ,
, , ,
Abstract
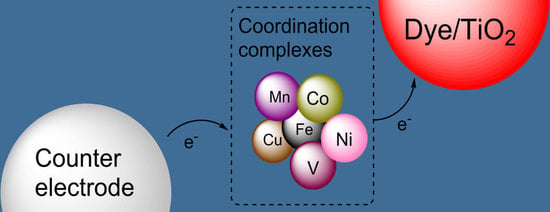
:
1. Introduction
2. Principles of DSSC Operation
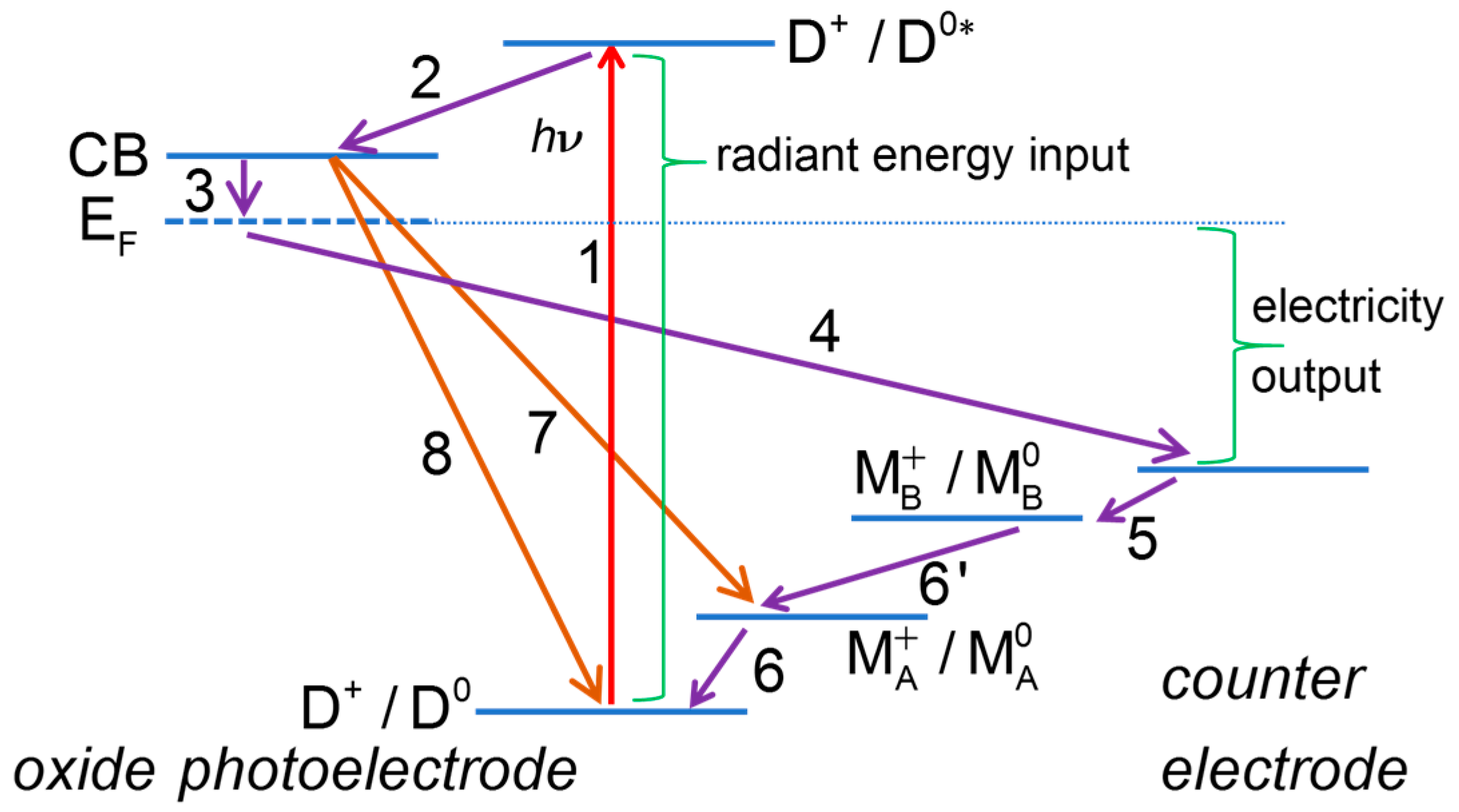
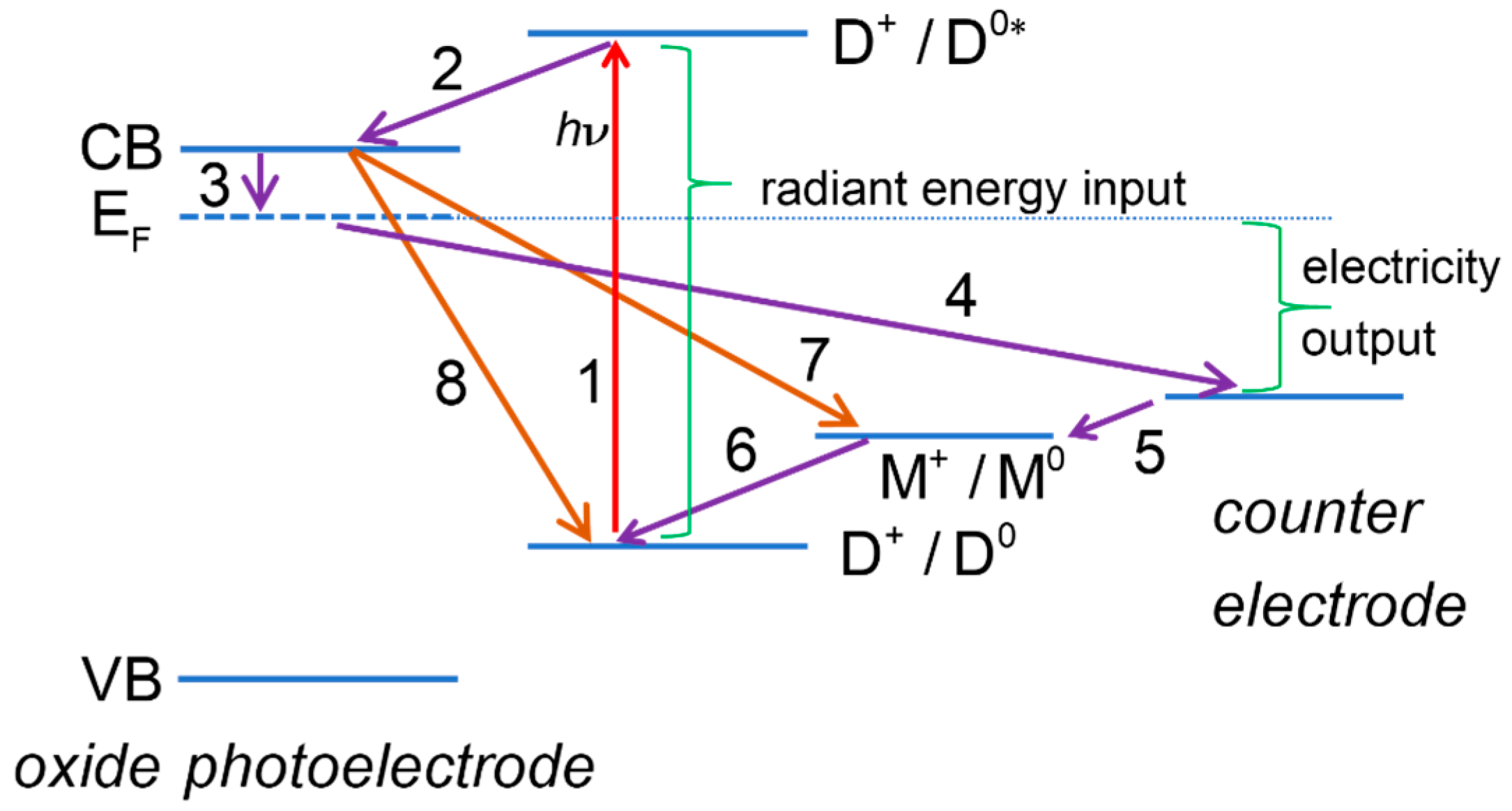
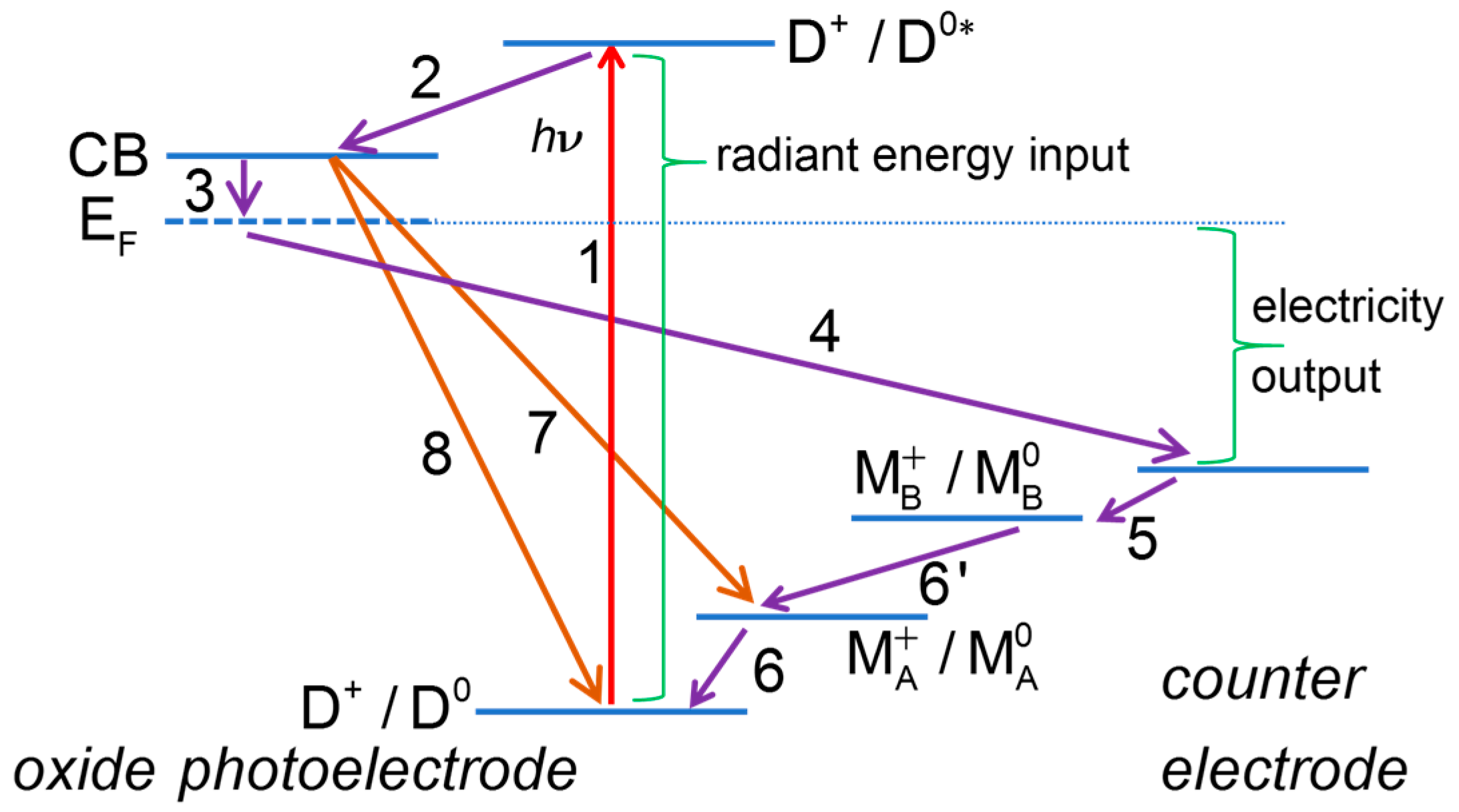
2.1. Useful Processes
2.1.1. Dye Photoexcitation
2.1.2. Electron Injection into the Conduction Band of the Semiconductor Oxide
2.1.3. Electron Collection
2.1.4. Dye Regeneration
2.1.5. Oxidized Mediator Transport from Photoelectrode to CE
2.1.6. Electron Flow through the External Circuit
2.1.7. Electron Transfer at the Counter Electrode
2.1.8. Reduced Mediator Transport from Counter Electrode to Photoelectrode
2.2. Deleterious Processes
2.2.1. Deactivation of Excited Dye D0* with Evolution of Heat or Light
2.2.2. Deactivation Excited State by Reaction with a Species in Solution
2.2.3. Recombination Reactions of Semiconductor Electrons
2.2.4. Recombination of TCO Contact Electrons with the Oxidized Dye or the Oxidized Redox Mediator
3. Incident Photon-To-Current Efficiency and Power Conversion Efficiency
4. Requirements for Efficient Redox Mediators
4.1. Redox Potential Less Positive but Close to That of the Dye
4.2. Long-Term Stability of Both the Reduced and the Oxidized Form
4.3. Solubility
4.4. Fast Diffusion
4.5. Low Light Absorption
4.6. Electrode Kinetics at the Counter Electrode
4.7. Advantages and Drawbacks of Iodide–Triiodide
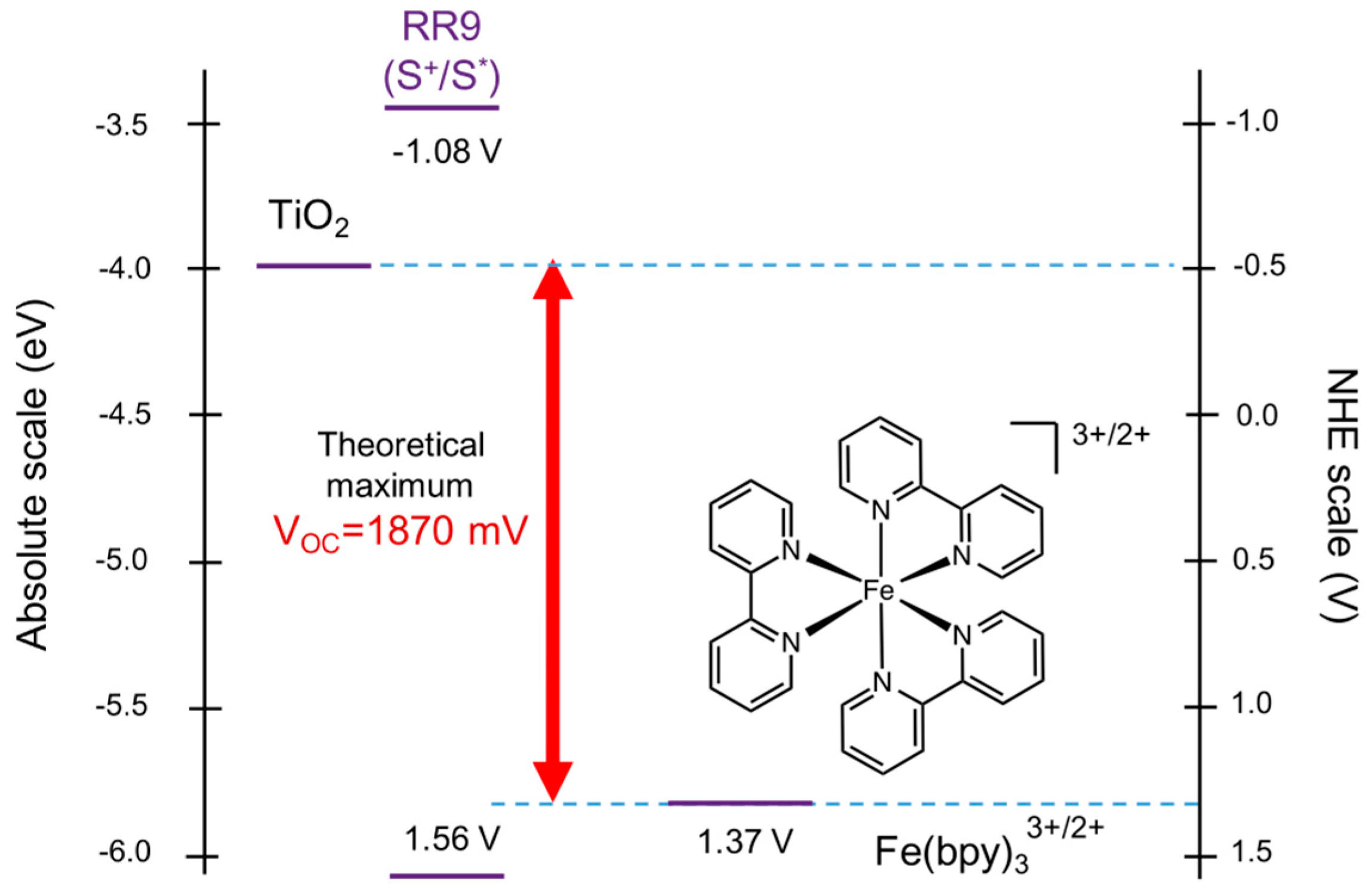
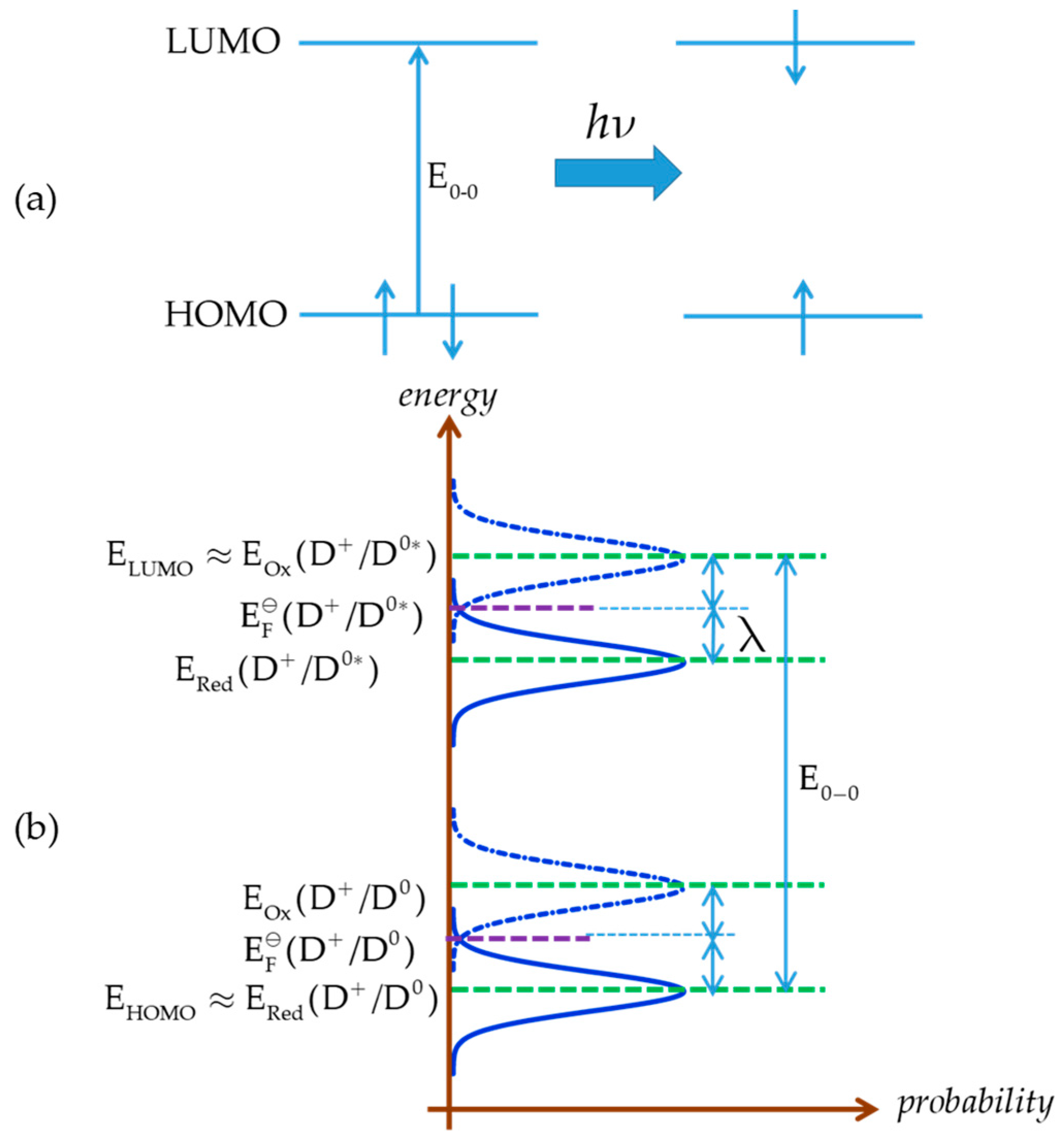
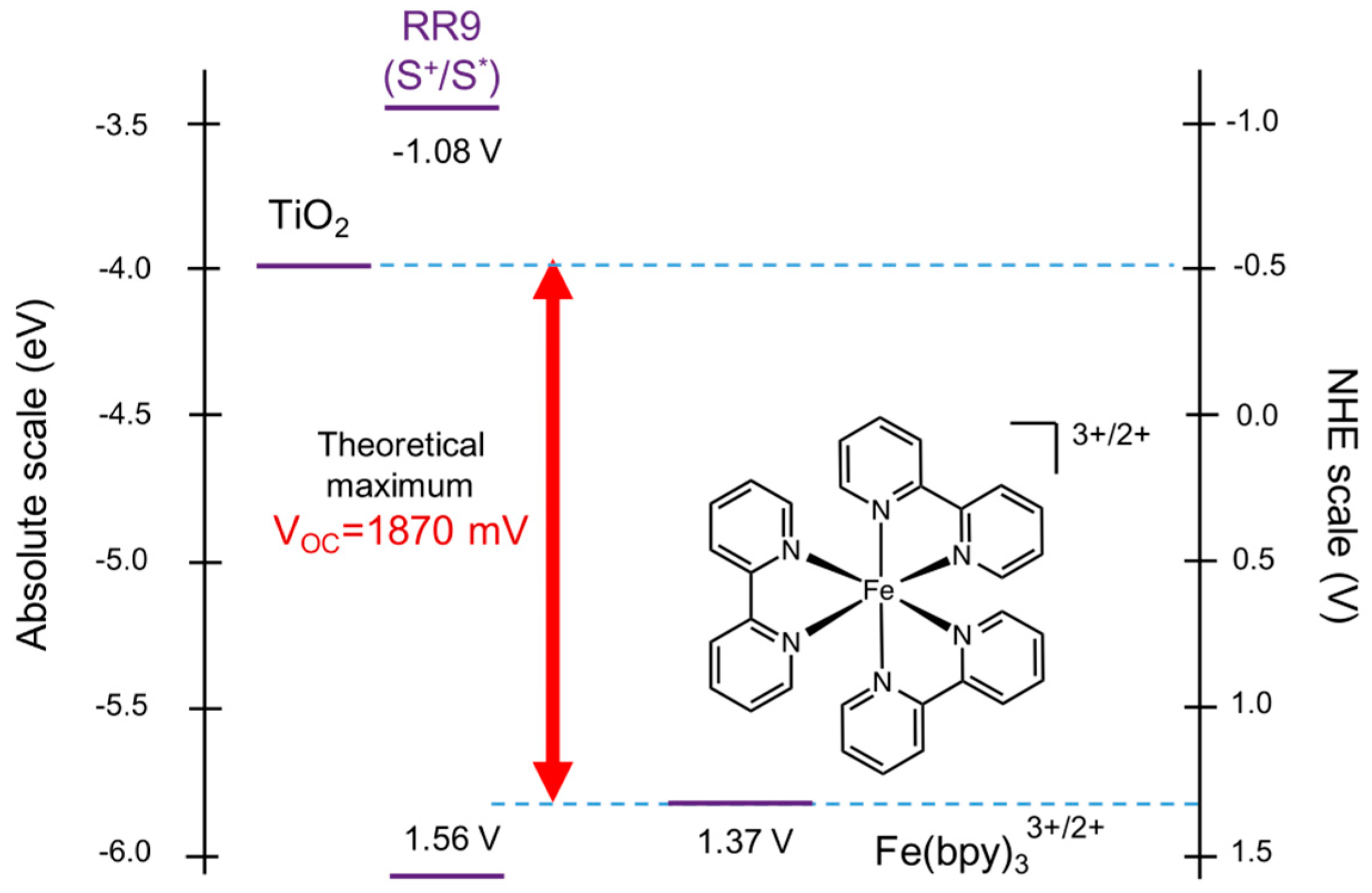
5. Redox Potential Definitions and Measurements
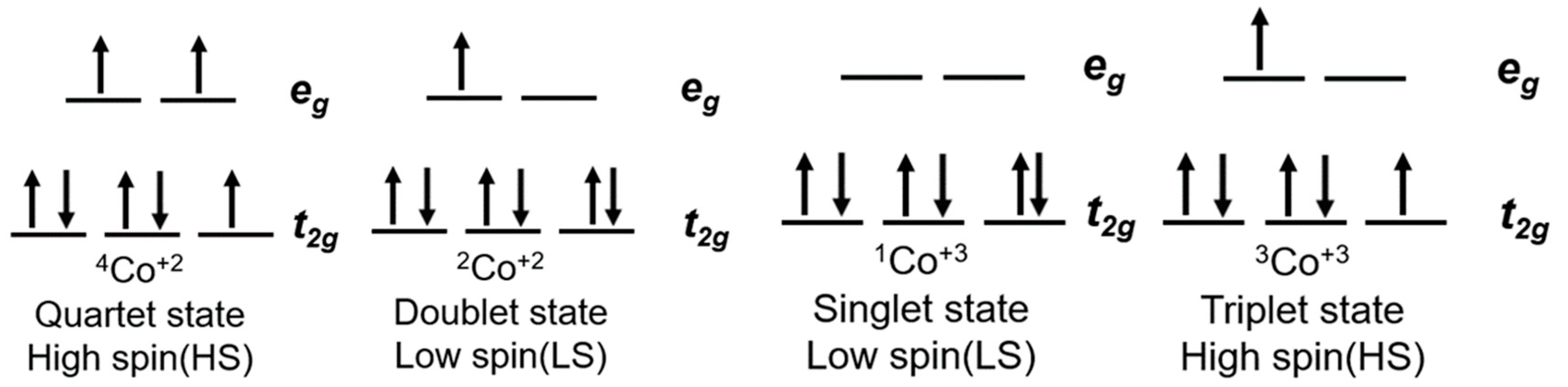
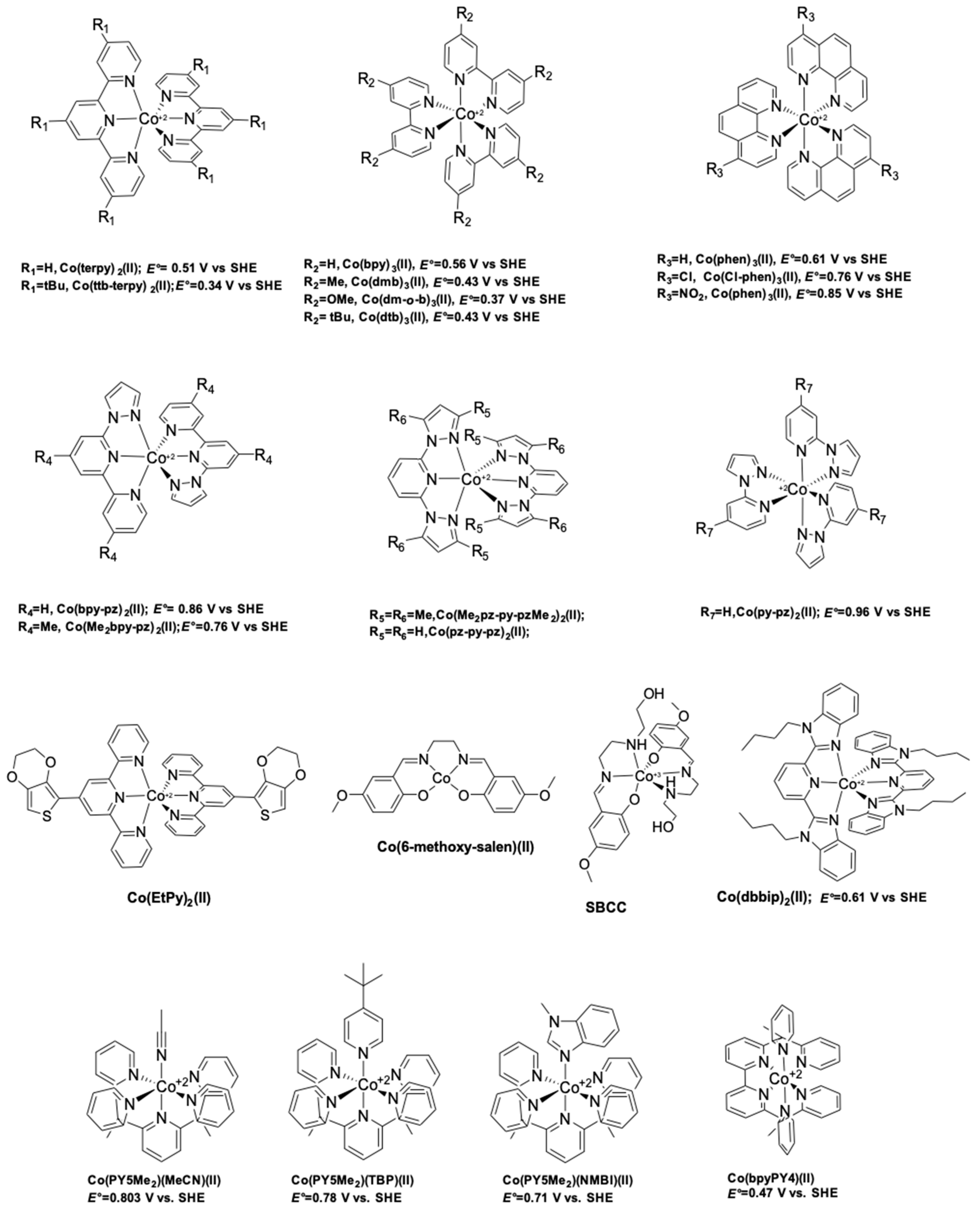
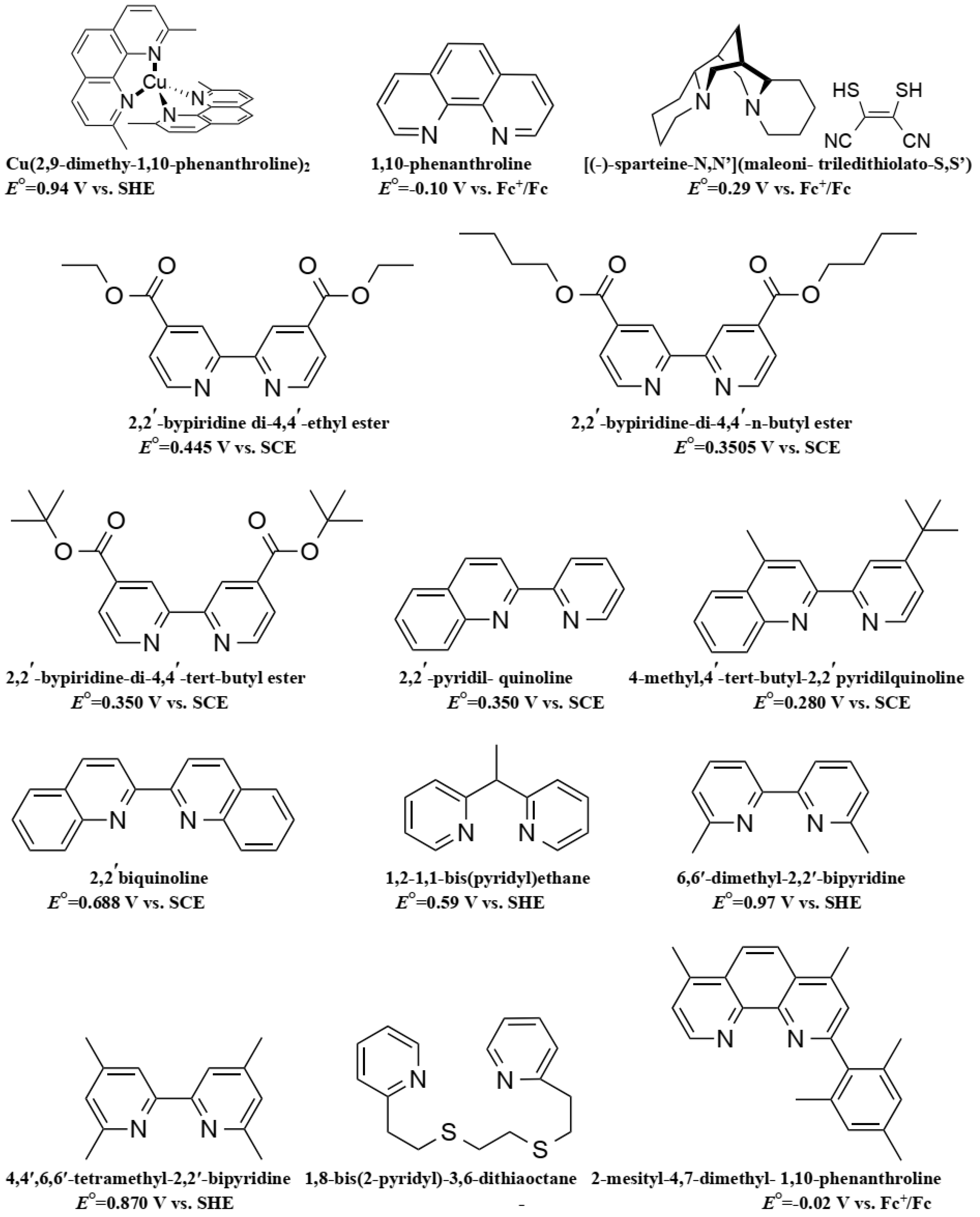
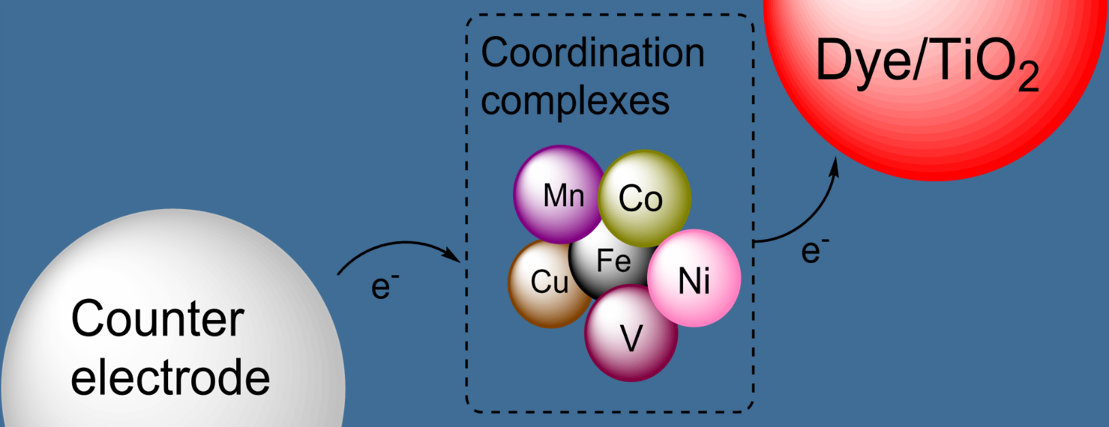
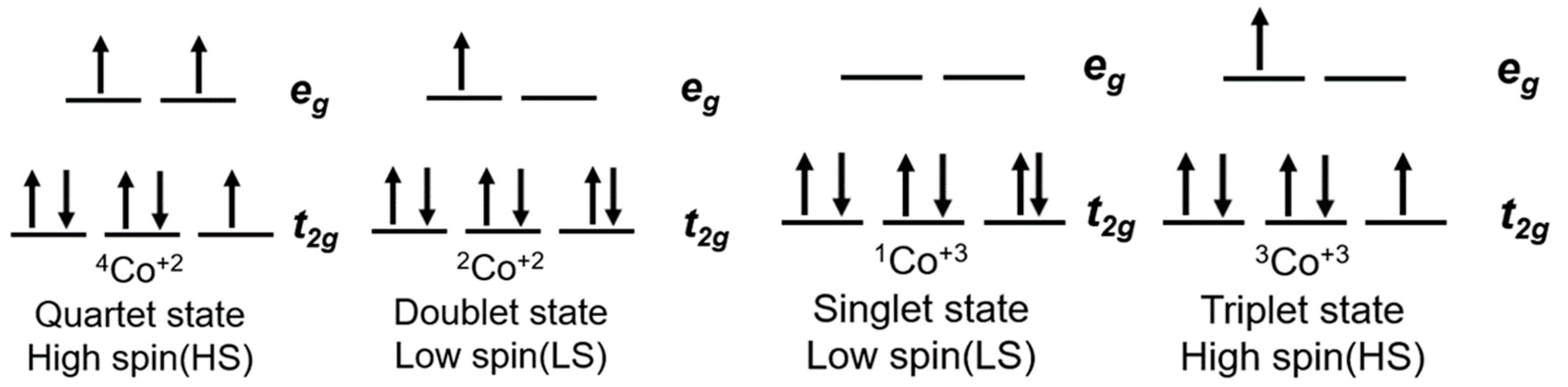
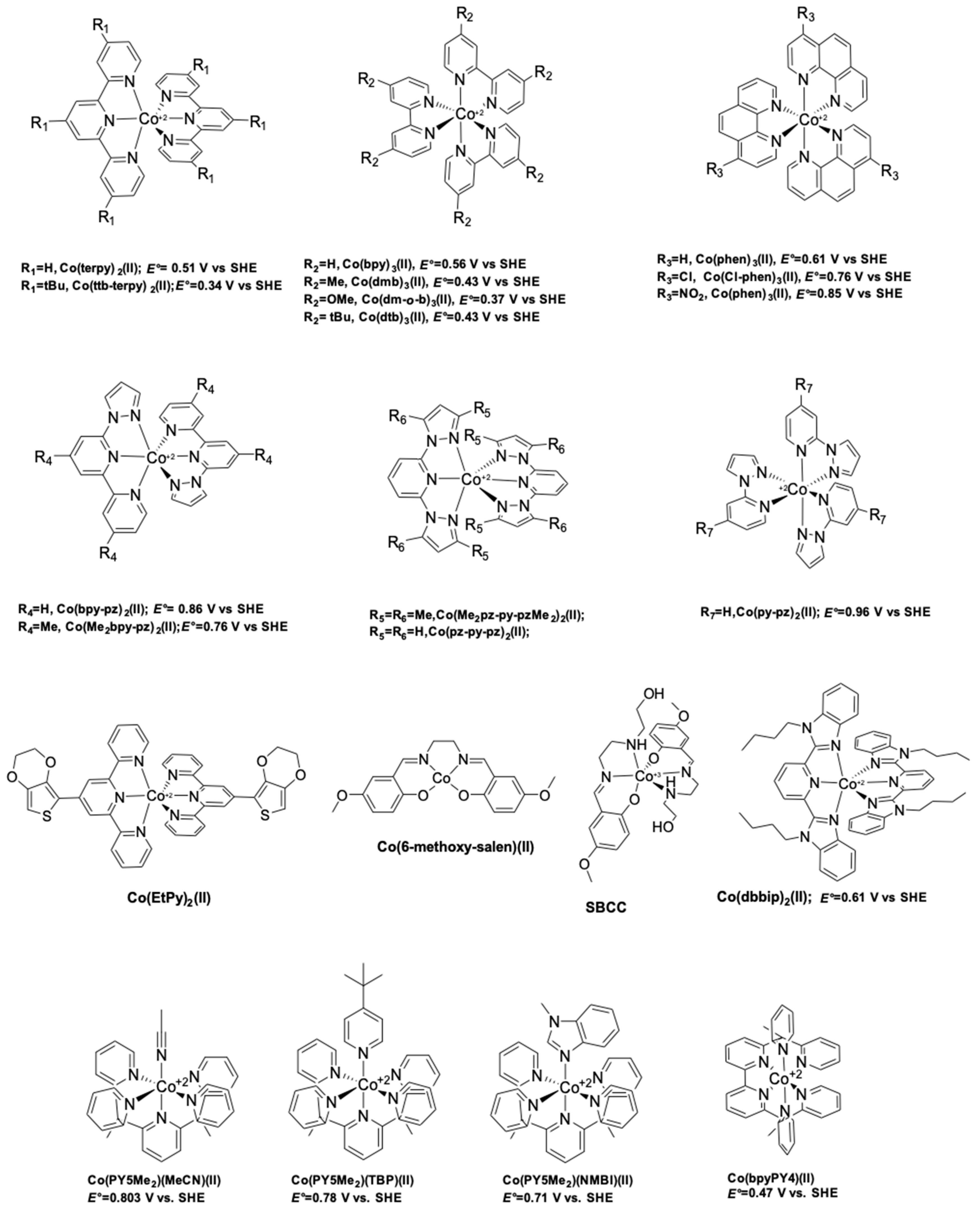
6. Cobalt Mediators
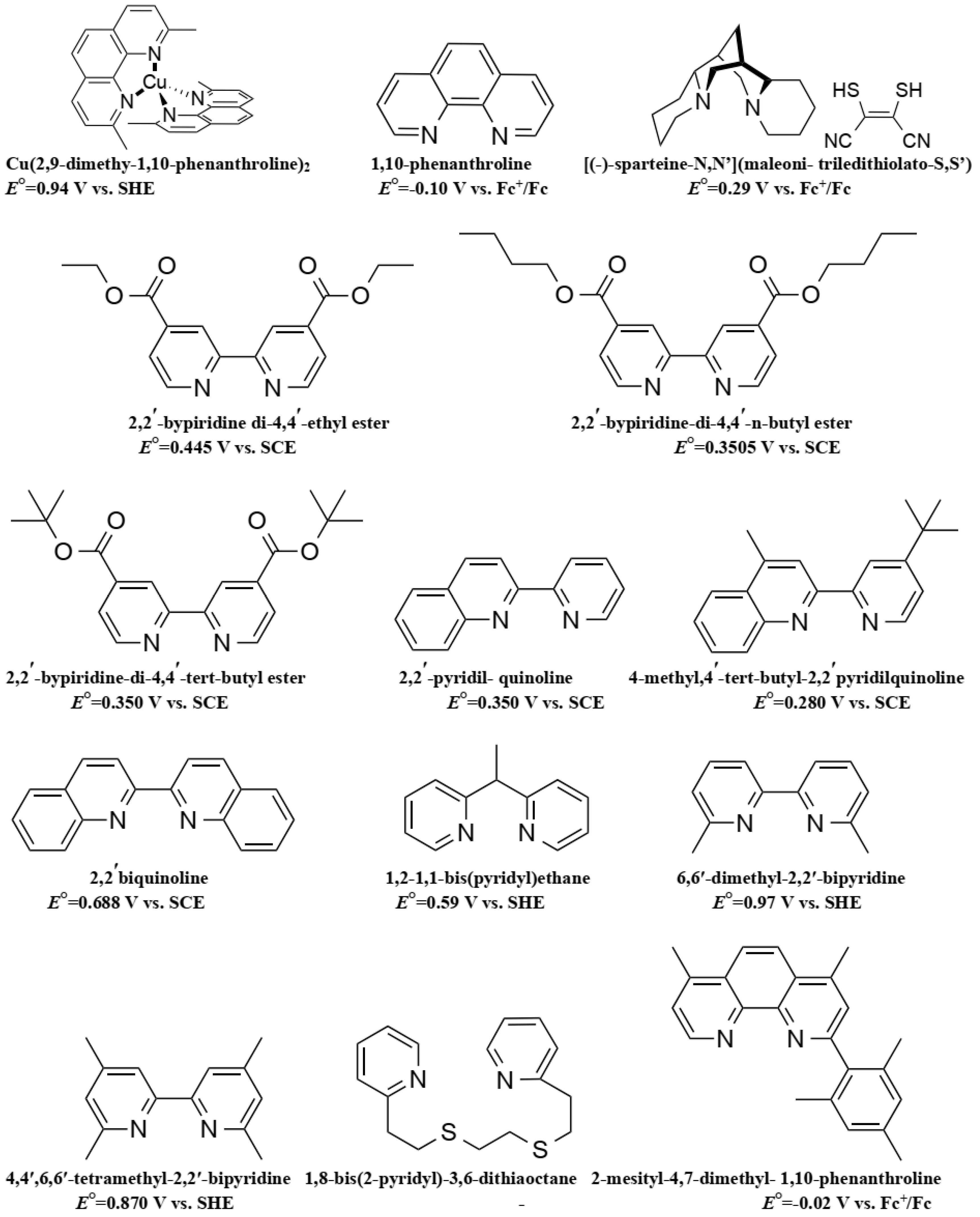
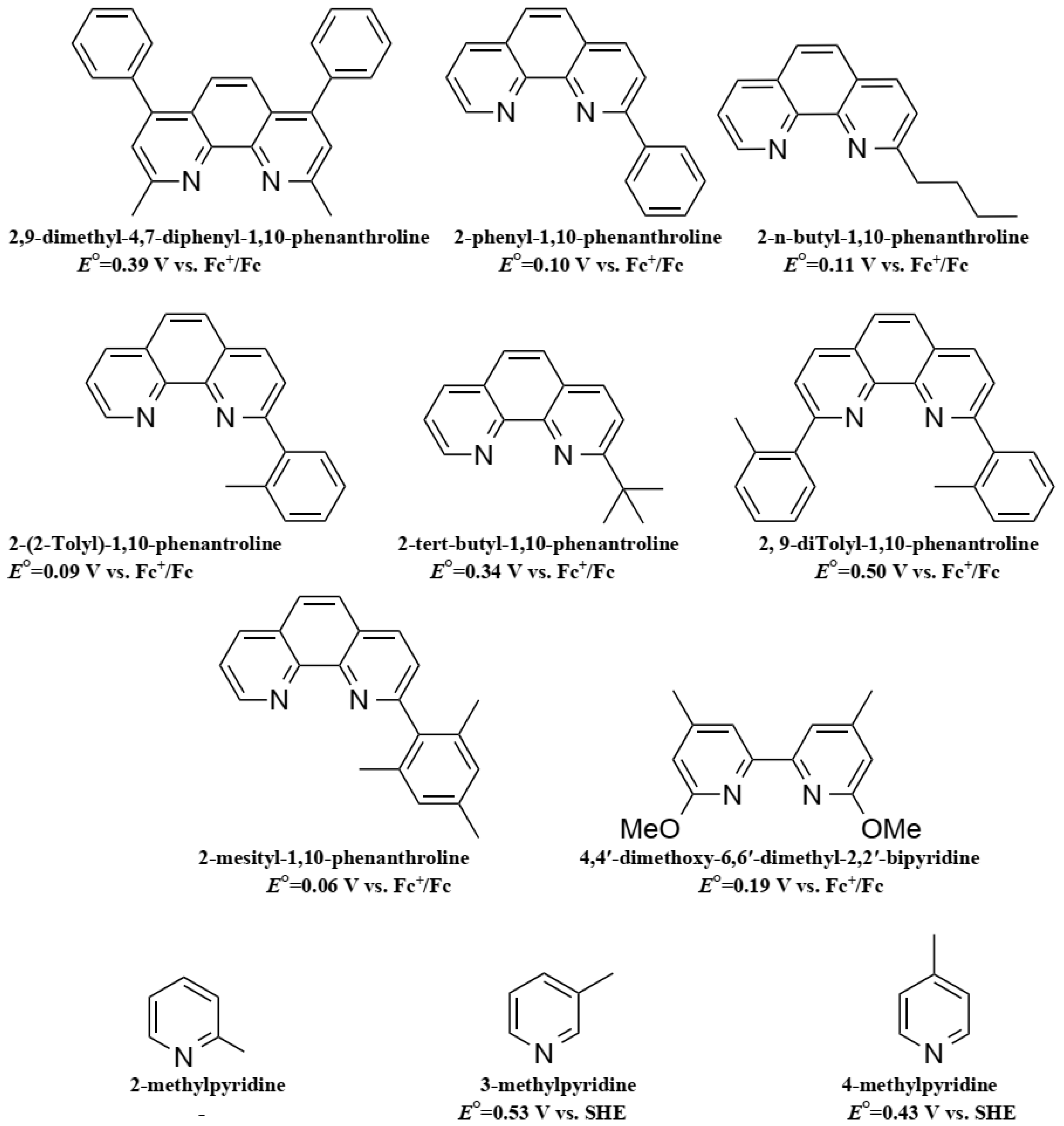
7. Copper Mediators
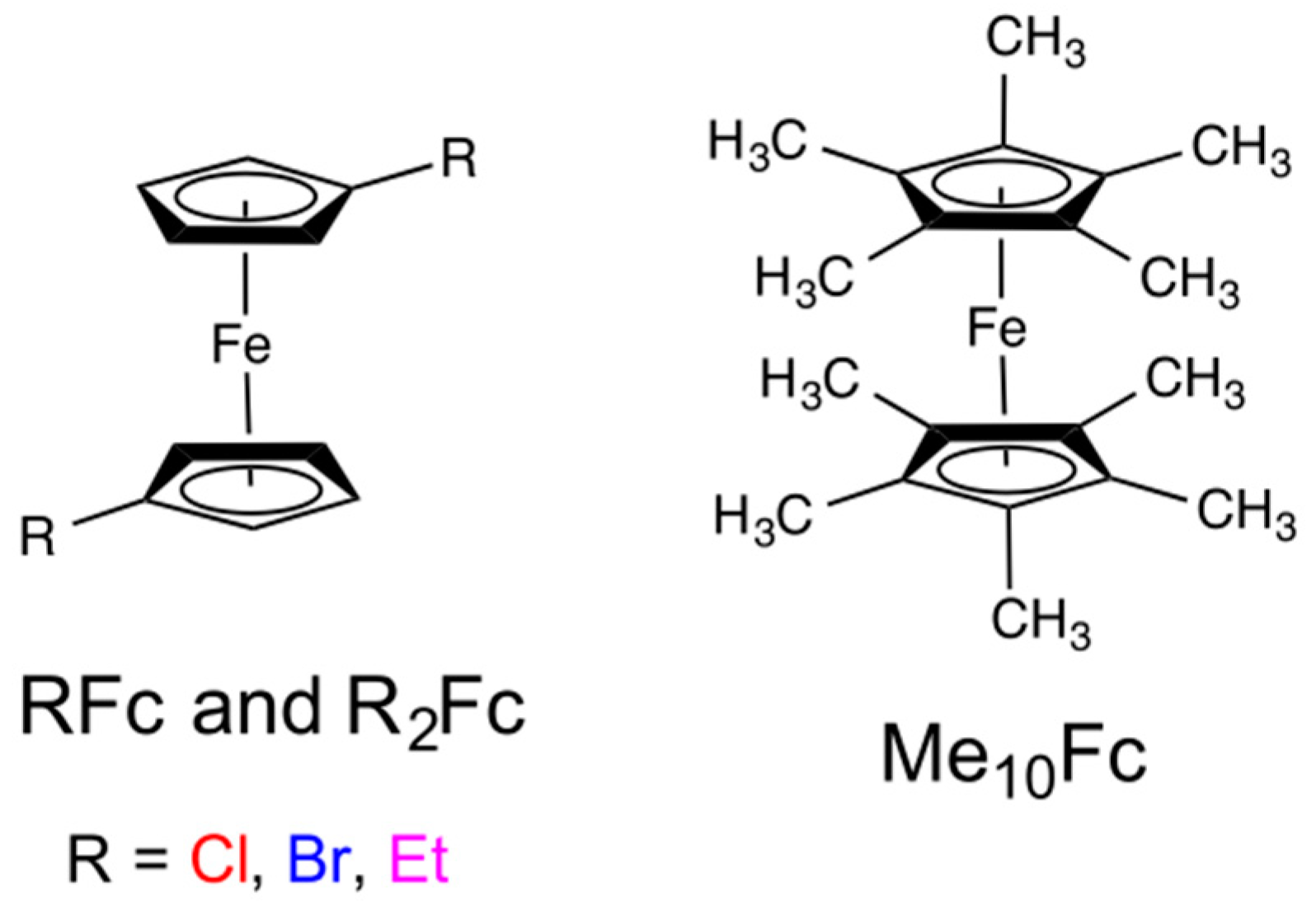
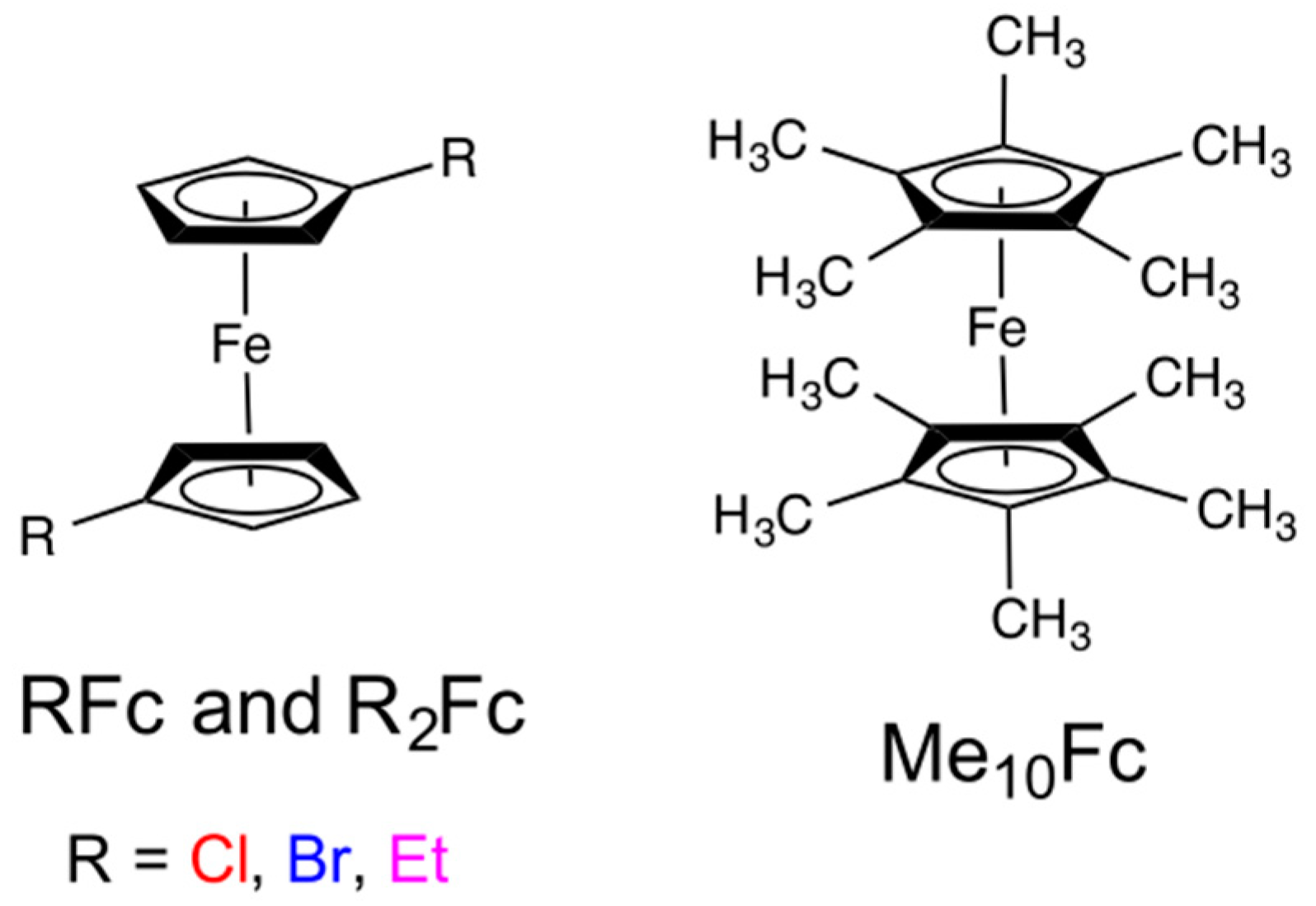
8. Iron Mediators
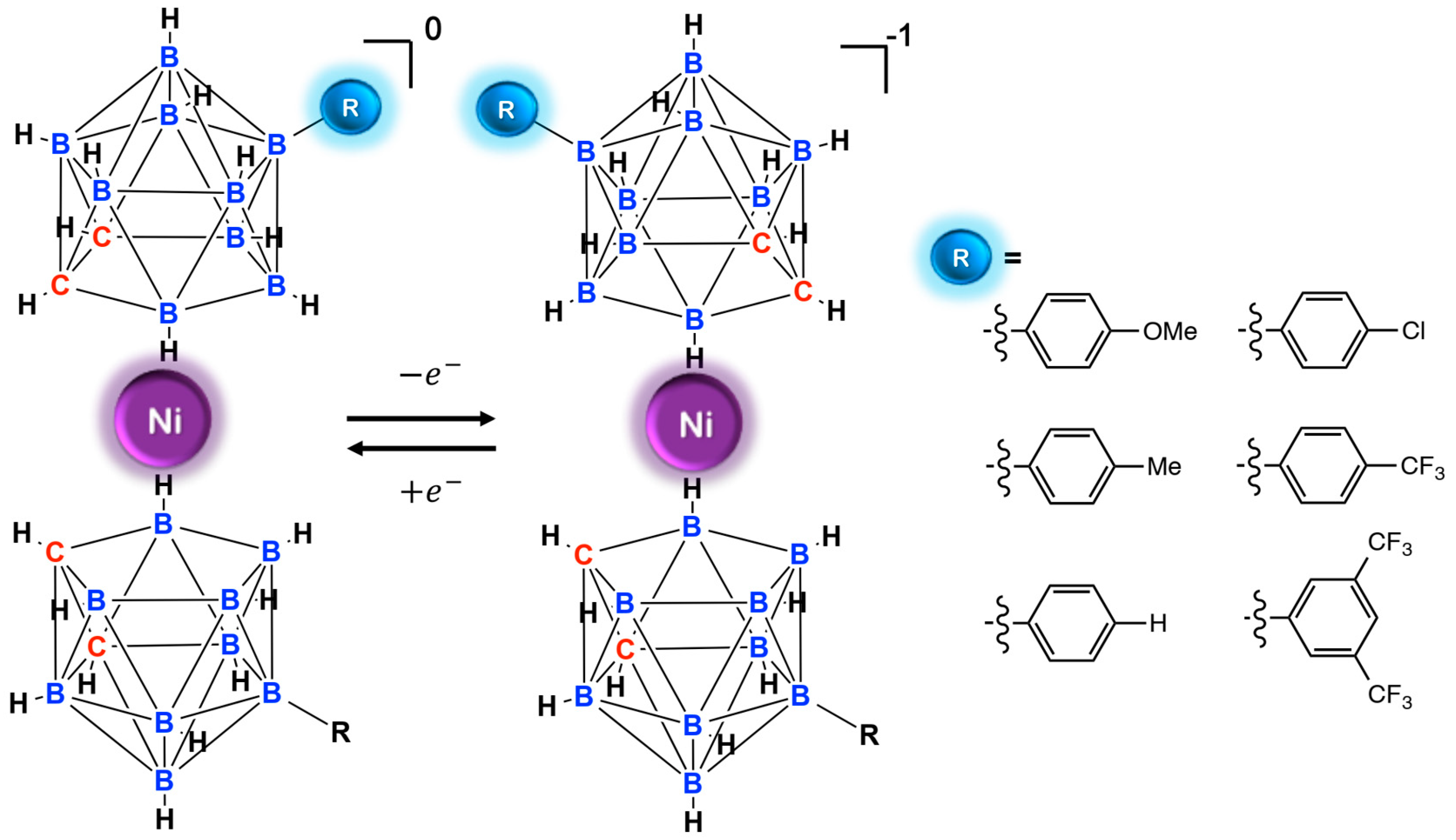
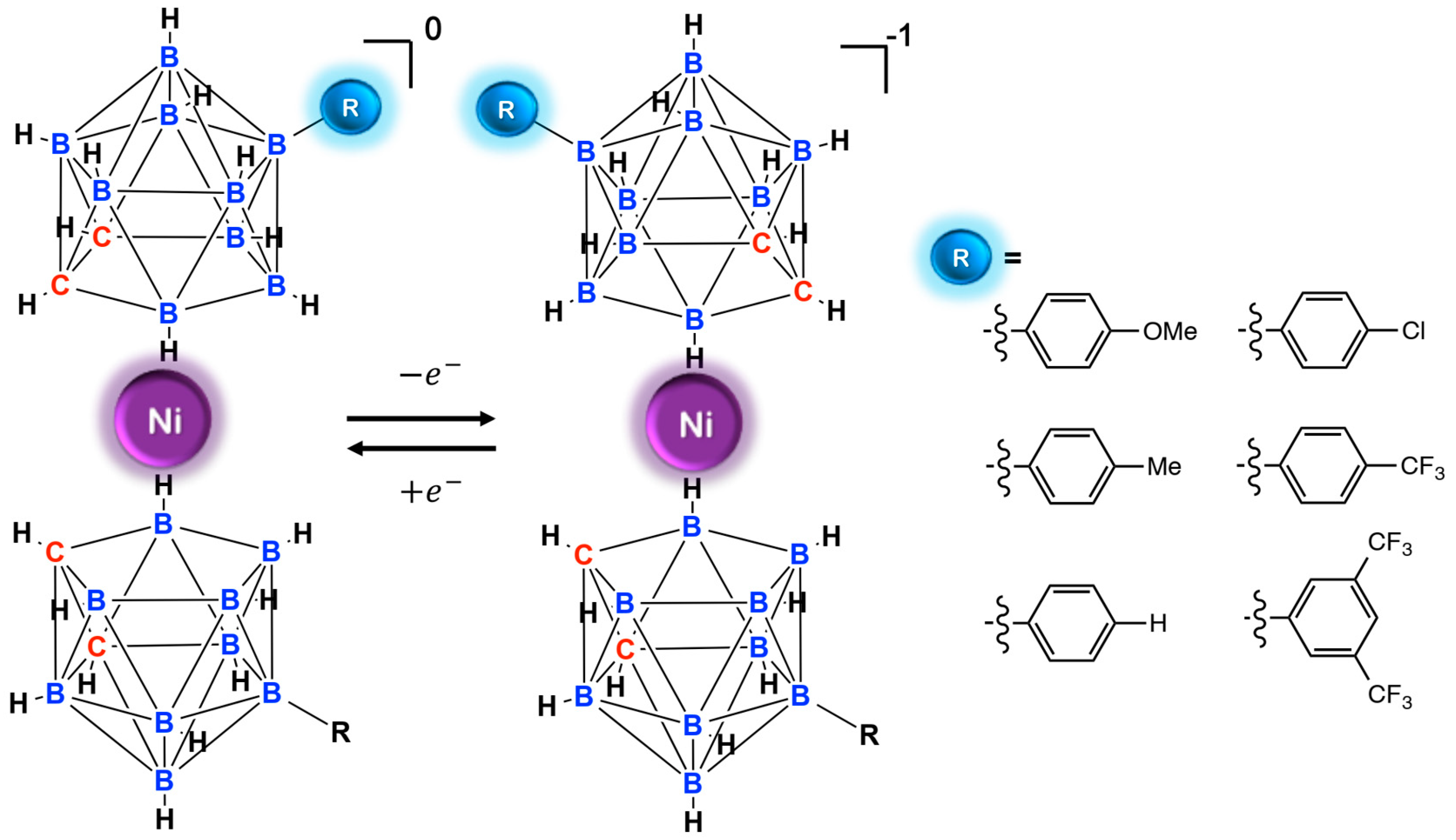
9. Nickel Mediators
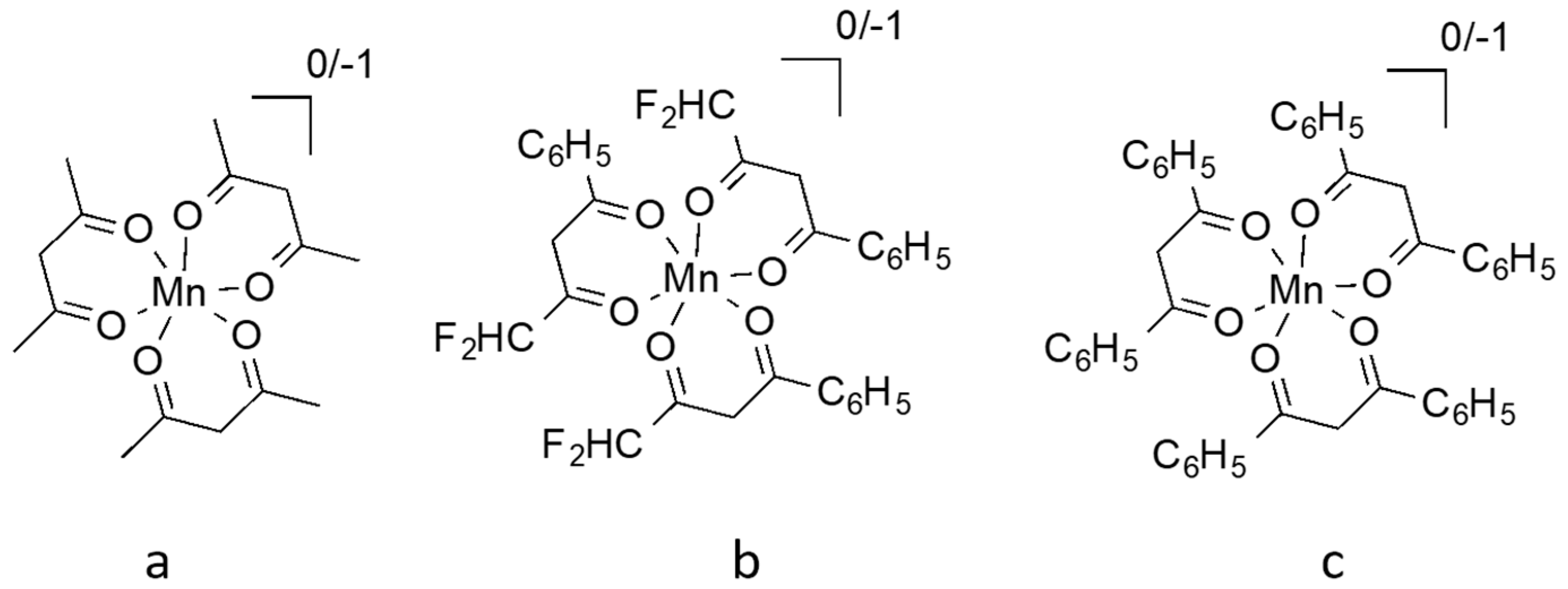
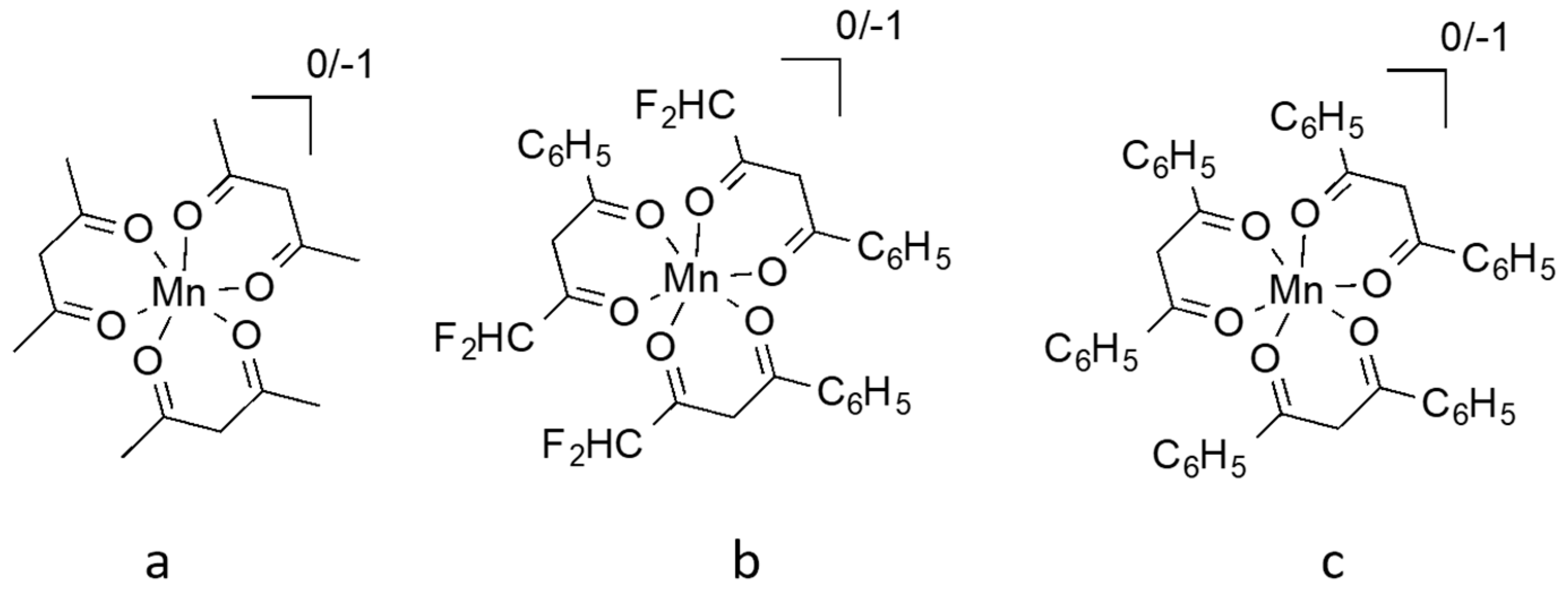
10. Manganese Mediators
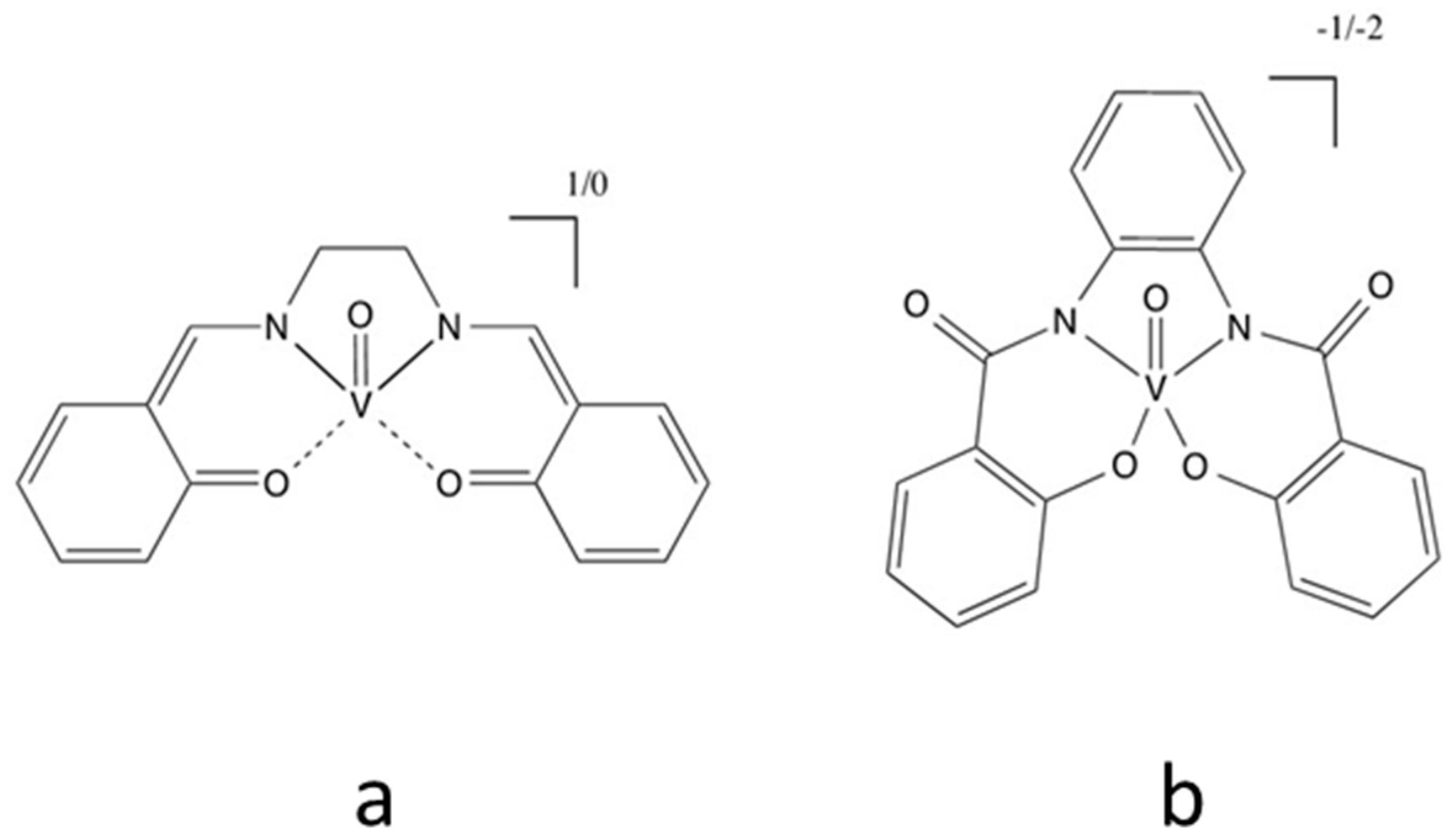
11. Vanadium Mediators
12. Dual Redox Mediator Systems
12.1. Phenothiazine or Ferrocene as Mediator and Co Complex as Co-Mediator
12.2. Iodide as Mediator and Co Complex as Co-Mediator
12.3. Co Complex as Mediator and TEMPO and Co-Mediator
12.4. Co Complex as Mediator and p-Anisylamine as Co-Mediator
12.5. Fe Complex as Mediator and Cu Complex as Co-Mediator
12.6. Co Complexes as Mediator and Co-Mediator
13. Concluding Remarks
Funding
Conflicts of Interest
Abbreviations
| ACN | acetonitrile |
| APCE | absorbed photon-to-current efficiency |
| CB | conduction band |
| CE | counter electrode |
| CTSC | contact phase of the mesoporous oxide |
| CV | cyclic voltammetry |
| DSSC | dye-sensitized solar cell |
| D0, D0* | ground and excited dye of the dye in the reduced form |
| D+ | oxidized dye |
| EF, EFӨ | Fermi level, standard Fermi level |
| e− | electron |
| E | electrode potential |
| FF | fill factor |
| IPCE | incident photon-to-current efficiency |
| Jph | photon number flux |
| Jel | electron number flux |
| jSC | short-circuit current density of DSSC |
| jMPP | current density of DSSC at the maximum power point |
| L | Avogadro constant |
| LHE | light-harvesting efficiency |
| M0, M+ | reduced and oxidized form of redox mediator |
| MEV | microelectrode voltammetry |
| P0 | incident light intensity |
| PCE | solar-to-electrical energy conversion efficiency |
| PE | photoelectrode |
| Q0 | magnitude of electronic charge |
| RDEV | rotating disk electrode voltammetry |
| SC | mesoporous semiconductor phase |
| TAS | transient absorption spectroscopy |
| UOC | open-circuit photovoltage |
| Г | molar surface concentration (based on geometric surface area) |
| ∆GӨR EC | recombination standard free energy |
| ∆GӨR EG | regeneration standard free energy |
| ελ | molar extinction (attenuation) coefficient of a dye |
| Rλ | fraction of reflected light |
| σ | molecule area of a dye |
| σλ | light-absorption molecular cross-section of a dye |
| φCOLL | collection efficiency |
| φINJ | injection efficiency |
References
- Moser, J. Notiz über Verstärkung photoelektrischer Ströme durch optische Sensibilisirung. Monatshefte für Chemie 1887, 8, 373. [Google Scholar] [CrossRef]
- Gerischer, H.; Tributsch, H. Elektrochemische Untersuchungen zur spektralen Sensibilisierung von ZnO-Einkristallen. Berichte der Bunsengesellschaft für Phys. Chemie 1968, 72, 437–445. [Google Scholar] [CrossRef]
- Tributsch, H.; Gerischer, H. Elektrochemische Untersuchungen über den Mechanismus der Sensibilisierung und übersensibilisierung an ZnO-Einkristallen. Berichte der Bunsengesellschaft für Phys. Chemie 1969, 73, 251–260. [Google Scholar] [CrossRef]
- Memming, R. Electron transfer process with excited molöecules at semiconductor electrodes. Prog. Surf. Sci. 2000, 17, 7–73. [Google Scholar] [CrossRef]
- Matsumura, M.; Matsudaira, S.; Tsubomura, H.; Takata, M.; Yanagida, H. Dye Sensitization and Surface Structures of Semiconductor Electrodes. Ind. Eng. Chem. Prod. Res. Dev. 1980, 19, 415–421. [Google Scholar] [CrossRef]
- Tsubomura, H.; Matsumura, M.; Nomzra, Y.; Amamiya, T. Dye sensitised zinc oxide: Aqueous electrolyte: Platinum photocell. Nature 1976, 261, 402–403. [Google Scholar] [CrossRef]
- Anderson, S.; Constable, E.C.; Dare-Edwards, M.P.; Goodenouigh, J.B.; Hamnett, A.; Seddon, K.R.; Wright, R.D. Chemical modification of a titanium (IV) oxide electrode to give stable dye sensitisation without a supersensitiser. Nature 1979, 280, 571–573. [Google Scholar] [CrossRef]
- Dare-Edwards, M.P.; Goodenough, J.B.; Hamnett, A.; Seddon, K.R.; Wright, R.D. Sensitisation of semiconducting electrodes with ruthenium-based dyes. Faraday Discuss. Chem. Soc. 1980, 285. [Google Scholar] [CrossRef]
- Moser, J.; Grätzel, M. Photosensitized electron injection in colloidal semiconductors. J. Am. Chem. Soc. 1984, 106, 6557–6564. [Google Scholar] [CrossRef]
- Desilvestro, J.; Grätzel, M.; Kavan, L.; Moser, J.; Augustynski, J. Highly efficient sensitization of titanium dioxide. J. Am. Chem. Soc. 1985, 107, 2988–2990. [Google Scholar] [CrossRef]
- Vlachopoulos, N.; Liska, P.; Augustynski, J.; Grätzel, M. Very efficient visible light energy harvesting and conversion by spectral sensitization of high surface area polycrystalline titanium dioxide films. J. Am. Chem. Soc. 1988, 110, 1216–1220. [Google Scholar] [CrossRef]
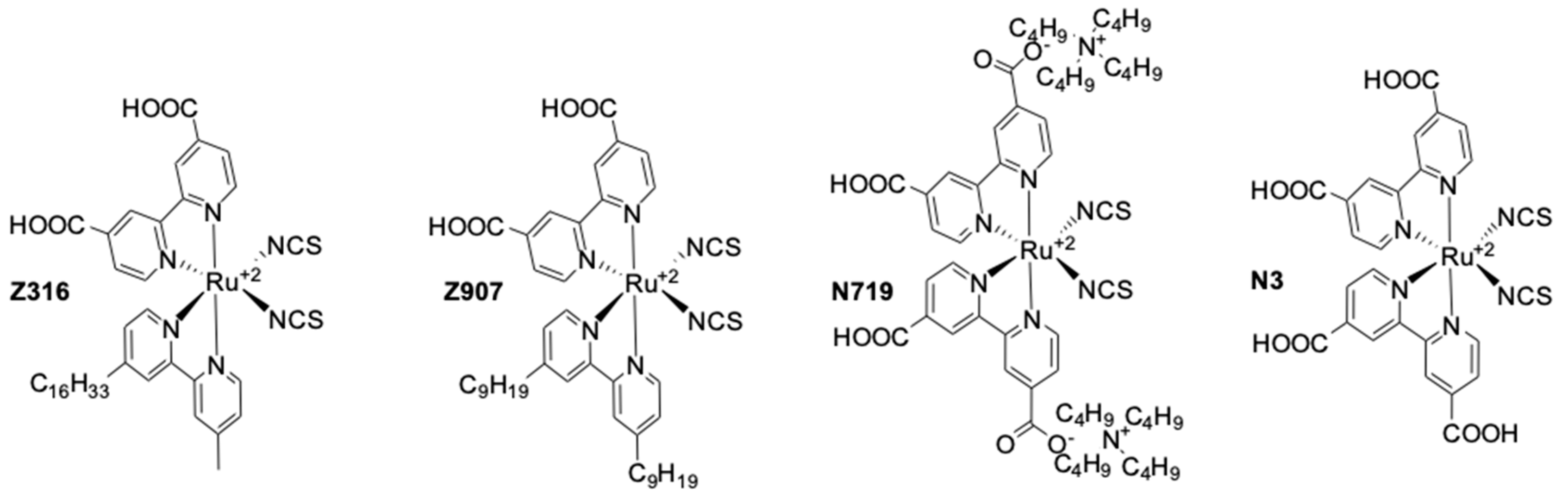
- Liska, P.; Vlachopoulos, N.; Nazeeruddin, M.K.; Comte, P.; Grätzel, M. cis-diaquabis(2,2′-bipyridyl-4,4′-dicarboxylate)-ruthenium(II) sensitizes wide band gap oxide semiconductors very efficiently over a broad spectral range in the visible. J. Am. Chem. Soc. 1988, 110, 3686–3687. [Google Scholar] [CrossRef]
- Gratzel, M.; Liska, P. Photoelectrochemical Cells. U.S. Patent US4927721, 1990. [Google Scholar]
- Dabestani, R.; Bard, A.J.; Campion, A.; Fox, M.A.; Mallouk, T.E.; Webber, S.E.; White, J.M. Sensitization of Titanium Dioxide and Strontium Titanate Electrodes by Ruthenium(II) An Evaluation of Sensitization Efficiency for Component Photoelectrodes in a Muitipanel Device. J. Phys. Chem. 1988, 1872–1878. [Google Scholar] [CrossRef]
- O’Regan, B.; Grätzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar] [CrossRef]
- Nazeeruddin, M.K.; Kay, A.; Rodicio, I.; Humphry-Baker, R.; Mueller, E.; Liska, P.; Vlachopoulos, N.; Grätzel, M. Conversion of light to electricity by cis-X2bis(2,2′-bipyridyl-4,4′-dicarboxylate)ruthenium(II) charge-transfer sensitizers (X = Cl−, Br−, I−, CN−, and SCN−) on nanocrystalline titanium dioxide electrodes. J. Am. Chem. Soc. 1993, 115, 6382–6390. [Google Scholar] [CrossRef]
- Hagfeldt, A.; Didriksson, B.; Palmqvist, T.; Lindström, H.; Södergren, S.; Rensmo, H.; Lindquist, S.-E. Verification of high efficiencies for the Grätzel-cell. A 7% efficient solar cell based on dye-sensitized colloidal TiO2 films. Sol. Energy Mater. Sol. Cells 1994, 31. [Google Scholar] [CrossRef]
- Ahmad, S.; Guillén, E.; Kavan, L.; Grätzel, M.; Nazeeruddin, M.K. Metal free sensitizer and catalyst for dye sensitized solar cells. Energy Environ. Sci. 2013, 6, 3439. [Google Scholar] [CrossRef]
- Asghar, M.I.; Miettunen, K.; Halme, J.; Vahermaa, P.; Toivola, M.; Aitola, K.; Lund, P. Review of stability for advanced dye solar cells. Energy Environ. Sci. 2010, 3, 418. [Google Scholar] [CrossRef]
- Grätzel, M. Photoelectrochemical cells. Nature 2001, 414. [Google Scholar] [CrossRef] [PubMed]
- Grätzel, M. Solar Energy Conversion by Dye-Sensitized Photovoltaic Cells. Inorg. Chem. 2005, 44, 6841–6851. [Google Scholar] [CrossRef] [PubMed]
- Grätzel, M. Recent advances in sensitized mesoscopic solar cells. Acc. Chem. Res. 2009, 42. [Google Scholar] [CrossRef] [PubMed]
- Hagfeld, A.; Grätzel, M. Light-induced redox reactions in nanocrystalline systems. Chem. Rev. 1995, 95, 49–68. [Google Scholar] [CrossRef]
- Hagfeldt, A.; Grätzel, M. Molecular photovoltaics. Acc. Chem. Res. 2000, 33. [Google Scholar] [CrossRef]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-Sensitized Solar Cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef] [PubMed]
- Hamann, T.W.; Ondersma, J.W. Dye-sensitized solar cell redox shuttles. Energy Environ. Sci. 2011, 4, 370–381. [Google Scholar] [CrossRef]
- Kavan, L. Electrochemistry and dye-sensitized solar cells. Curr. Opin. Electrochem. 2017, 2, 88–96. [Google Scholar] [CrossRef]
- Li, C.T.; Lin, R.Y.; Lin, J.T. Sensitizers for aqueous-based solar cells. Chem. Asian J. 2017, 12, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Pashaei, B.; Shahroosvand, H.; Abbasi, P. Transition metal complex redox shuttles for dye-sensitized solar cells. RSC Adv. 2015, 5, 94814–94848. [Google Scholar] [CrossRef]
- Bella, F.; Gerbaldi, C.; Barolo, C.; Grätzel, M. Aqueous dye-sensitized solar cells. Chem. Soc. Rev. 2015, 44, 3431–3473. [Google Scholar] [CrossRef] [PubMed]
- Peter, L.M. The Grätzel Cell: Where Next? J. Phys. Chem. Lett. 2011, 2, 1861–1867. [Google Scholar] [CrossRef]
- Peter, L.M. Dye-sensitized nanocrystalline solar cells. Phys. Chem. Chem. Phys. 2007, 9, 2630–2642. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, S.; Yu, Y.; Manseki, K. Iodine/iodide-free dye-sensitized solar cells. Acc. Chem. Res. 2009, 42, 1827–1838. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Vlachopoulos, N.; Gorlov, M.; Kloo, L. Liquid electrolytes for dye-sensitized solar cells. Dalton Trans. 2011, 40, 10289–10303. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lan, Z.; Lin, J.; Huang, M.; Huang, Y.; Fan, L.; Luo, G. Electrolytes in dye-sensitized solar cells. Chem. Rev. 2015, 115, 2136–2173. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N. CuI versus RuII: Dye-sensitized solar cells and beyond. ChemSusChem 2008, 1, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Housecroft, C.E.; Constable, E.C. The emergence of copper(I)-based dye sensitized solar cells. Chem. Soc. Rev. 2015, 44, 8386–8398. [Google Scholar] [CrossRef] [PubMed]
- Bella, F.; Galliano, S.; Gerbaldi, C.; Viscardi, G. Cobalt-based electrolytes for dye-sensitized solar cells: Recent advances towards stable devices. Energies 2016, 9, 384. [Google Scholar] [CrossRef]
- Bignozzi, C.A.; Argazzi, R.; Boaretto, R.; Busatto, E.; Carli, S.; Ronconi, F.; Caramori, S. The role of transition metal complexes in dye sensitized solar devices. Coord. Chem. Rev. 2013, 257, 1472–1492. [Google Scholar] [CrossRef]
- Caramori, S.; Cristino, V.; Boaretto, R.; Argazzi, R.; Di Carlo, A.; Bignozzi, C.A. New Components for Dye-Sensitized Solar Cells. Int. J. Photoenergy 2010, 458614. [Google Scholar] [CrossRef]
- Cong, J.; Yang, X.; Kloo, L.; Sun, L. Iodine/iodide-free redox shuttles for liquid electrolyte-based dye-sensitized solar cells. Energy Environ. Sci. 2012, 5, 9180. [Google Scholar] [CrossRef]
- Johansson, E.M.J.; Sandell, A.; Siegbahn, H.; Rensmo, H.; Mahrov, B.; Boschloo, G.; Figgemeier, E.; Hagfeldt, A. Interfacial properties of photovoltaic TiO2/dye/PEDOT–PSS heterojunctions. Synth. Met. 2005, 149, 157–167. [Google Scholar] [CrossRef]
- Freitag, M.; Boschloo, G. The revival of dye-sensitized solar cells. Curr. Opin. Electrochem. 2017, 2, 111–119. [Google Scholar] [CrossRef]
- Giribabu, L.; Bolligarla, R.; Panigrahi, M. Recent Advances of Cobalt(II/III) Redox Couples for Dye-Sensitized Solar Cell Applications. Chem. Rec. 2015, 15, 760–788. [Google Scholar] [CrossRef] [PubMed]
- Kalyanasundaram, K. Dye-Sensitized Solar Cells; EPFL Press: Lausanne, Switzerland, 1990; ISBN 9781439808665. [Google Scholar]
- Odobel, F.; Pellegrin, Y.; Gibson, E.A.; Hagfeldt, A.; Smeigh, A.L.; Hammarström, L. Recent advances and future directions to optimize the performances of p-type dye-sensitized solar cells. Coord. Chem. Rev. 2012, 256, 2414–2423. [Google Scholar] [CrossRef]
- Wood, C.J.; Summers, G.H.; Clark, C.A.; Kaeffer, N.; Braeutigam, M.; Carbone, L.R.; D’Amario, L.; Fan, K.K.; Farré, Y.; Narbey, S.; et al. Comprehensive comparison of dye-sensitized NiO photocathodes for solar energy conversion. Phys. Chem. Chem. Phys. 2016, 18, 10727–10738. [Google Scholar] [CrossRef] [PubMed]
- Strong, F.C. Theoretical Basis of Bouguer-Beer Law of Radiation Absorption. Anal. Chem. 1952, 24, 338–342. [Google Scholar] [CrossRef]
- Skoog, D.A.; Leary, J.J. Principles of Instrumental Analysis; Saunders College Pub: Fort Worth, TX, USA, 1992; ISBN 9780030233432. [Google Scholar]
- Trasatti, S. International Union of Pure and Applied Chemistry Commission on Electrochemistry* the Absolute Electrode Potential: An Explanatory Note. Pure Appl. Chem. 1986, 58, 955–966. [Google Scholar] [CrossRef]
- Van de Lagemaat, J.; Park, N.-G.; Frank, A.J. Influence of Electrical Potential Distribution, Charge Transport, and Recombination on the Photopotential and Photocurrent Conversion Efficiency of Dye-Sensitized Nanocrystalline TiO2 Solar Cells: A Study by Electrical Impedance and Optical Modulation Techniques. J. Phys. Chem. B 2000, 104, 2044–2052. [Google Scholar] [CrossRef]
- Bertoluzzi, L.; Ma, S. On the methods of calculation of the charge collection efficiency of dye sensitized solar cells. Phys. Chem. Chem. Phys. 2013, 15, 4283–4285. [Google Scholar] [CrossRef] [PubMed]
- Bonhôte, P.; Gogniat, E.; Tingry, S.; Barbé, C.; Vlachopoulos, N.; Lenzmann, F.; Comte, P.; Grätzel, M. Efficient Lateral Electron Transport inside a Monolayer of Aromatic Amines Anchored on Nanocrystalline Metal Oxide Films. J. Phys. Chem. B 1998, 102, 1498–1507. [Google Scholar] [CrossRef] [PubMed]
- Blauch, D.N.; Saveant, J.M. Dynamics of electron hopping in assemblies of redox centers. Percolation and diffusion. J. Am. Chem. Soc. 1992, 114, 3323–3332. [Google Scholar] [CrossRef]
- Thorsmølle, V.K.; Rothenberger, G.; Topgaard, D.; Brauer, J.C.; Kuang, D.-B.; Zakeeruddin, S.M.; Lindman, B.; Grätzel, M.; Moser, J.-E. Extraordinarily Efficient Conduction in a Redox-Active Ionic Liquid. ChemPhysChem 2011, 12, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Saygili, Y.; Söderberg, M.; Pellet, N.; Giordano, F.; Cao, Y.; Munoz-García, A.B.; Zakeeruddin, S.M.; Vlachopoulos, N.; Pavone, M.; Boschloo, G.; et al. Copper Bipyridyl Redox Mediators for Dye-Sensitized Solar Cells with High Photovoltage. J. Am. Chem. Soc. 2016, 138, 15087–15096. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Wang, Y.; Yi, Z.; Zu, N.; Zhang, J.; Zhang, M.; Wang, P. High-efficiency dye-sensitized solar cells: The influence of lithium ions on exciton dissociation, charge recombination, and surface states. ACS Nano 2010, 4, 6032–6038. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Zhou, D.; Shi, Y.; Si, X.; Wang, Y.; Wang, P. Stable and efficient dye-sensitized solar cells: Photophysical and electrical characterizations. Energy Environ. Sci. 2010, 3, 1722–1725. [Google Scholar] [CrossRef]
- Marszalek, M.; Arendse, F.D.; Decoppet, J.-D.; Babkair, S.S.; Ansari, A.A.; Habib, S.S.; Wang, M.; Zakeeruddin, S.M.; Grätzel, M. Ionic liquid-sulfolane composite electrolytes for high-performance and stable dye-sensitized solar cells. Adv. Energy Mater. 2014, 4, 1301235. [Google Scholar] [CrossRef]
- Boschloo, G.; Hagfeldt, A. Characteristics of the iodide/triiodide redox mediator in dye-sensitized solar cells. Acc. Chem. Res. 2009, 42, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Gardner, J.M.; Giaimuccio, J.M.; Meyer, G.J. Evidence for iodine atoms as intermediates in the dye sensitized formation of I−I bonds. J. Am. Chem. Soc. 2008, 130, 17252–17253. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.G.; Farnum, B.H.; Ardo, S.; Meyer, G.J. Iodide Chemistry in Dye-Sensitized Solar Cells: Making and Breaking I−I Bonds for Solar Energy Conversion. J. Phys. Chem. Lett. 2010, 1, 3132–3140. [Google Scholar] [CrossRef]
- Olsen, E.; Hagen, G.; Eric Lindquist, S. Dissolution of platinum in methoxy propionitrile containing LiI/I2. Sol. Energy Mater. Sol. Cells 2000, 63, 267–273. [Google Scholar] [CrossRef]
- Miettunen, K.; Etula, J.; Saukkonen, T.; Jouttijärvi, S.; Halme, J.; Romu, J.; Lund, P. Insights into corrosion in dye solar cells. Prog. Photovoltaics Res. Appl. 2014, 23, 1045–1056. [Google Scholar] [CrossRef]
- Miettunen, K.; Halme, J.; Lund, P. Metallic and plastic dye solar cells. Wiley Interdiscip. Rev. Energy Environ. 2013, 2, 104–120. [Google Scholar] [CrossRef]
- Gennett, T.; Milner, D.F.; Weaver, M.J. Role of solvent reorganization dynamics in electron-transfer processes. Theory-experiment comparisons for electrochemical and homogeneous electron exchange involving metallocene redox couples. J. Phys. Chem. 1985, 89, 2787–2794. [Google Scholar] [CrossRef]
- Feldt, S.M.; Cappel, U.B.; Johansson, E.M.J.; Boschloo, G.; Hagfeldt, A. Characterization of Surface Passivation by Poly(methylsiloxane) for Dye-Sensitized Solar Cells Employing the Ferrocene Redox Couple. J. Phys. Chem. C 2010, 114, 10551–10558. [Google Scholar] [CrossRef]
- Stergiopoulos, T.; Falaras, P. Minimizing Energy Losses in Dye-Sensitized Solar Cells Using Coordination Compounds as Alternative Redox Mediators Coupled with Appropriate Organic Dyes. Adv. Energy Mater. 2012, 2, 616–627. [Google Scholar] [CrossRef]
- Bard, A.J.L.; Faulkner, L.R. Electrochemical Methods, Fundamentals and Applications, 2nd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2001; ISBN 04710433729. [Google Scholar]
- Bagotsky, V.S. Fundamentals of Electrochemistry, 2nd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2005; ISBN 9780471741992. [Google Scholar]
- Koryta, J.J.; Dvořák, J.; Kavan, L.; Dvorak, J.; Kavan, L. Principles of Electrochemistry; John Wiley & Sons: Chichester, UK, 1993; ISBN 9781119966845. [Google Scholar]
- Oldham, K.B.; Myland, J.C.; Bond, A.M. Electrochemical Science and Technology: Fundamentals and Applications; John Wiley & Sons: Chichester, UK, 2011; ISBN 9781119966845. [Google Scholar]
- Gritzner, G.; Kuta, J. Recommendations on reporting electrode potentials in nonaqueous solvents (Recommendations 1983). Pure Appl. Chem. 1984, 56, 461–466. [Google Scholar] [CrossRef]
- Pavlishchuk, V.V.; Addison, A.W. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 °C. Inorganica Chim. Acta 2000, 298, 97–102. [Google Scholar] [CrossRef]
- Bentley, C.L.; Bond, A.M.; Hollenkamp, A.F.; Mahon, P.J.; Zhang, J. Voltammetric Determination of the Iodide/Iodine Formal Potential and Triiodide Stability Constant in Conventional and Ionic Liquid Media. J. Phys. Chem. C 2015, 119, 22392–22403. [Google Scholar] [CrossRef]
- Zhang, J.; Bond, A.M. Practical considerations associated with voltammetric studies in room temperature ionic liquids. Analyst 2005, 130, 1132–1147. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, F. Notizen: Fermi-Niveau und Flachbandpotential von Molekülkristallen aromatischer Kohlenwasserstoffe. Zeitschrift für Naturforsch. A 1967, 22, 843–844. [Google Scholar] [CrossRef]
- Isse, A.A.; Gennaro, A. Absolute Potential of the Standard Hydrogen Electrode and the Problem of Interconversion of Potentials in Different Solvents. J. Phys. Chem. B 2010, 114, 7894–7899. [Google Scholar] [CrossRef] [PubMed]
- Namazian, M.; Lin, C.Y.; Coote, M.L. Benchmark Calculations of Absolute Reduction Potential of Ferricinium/Ferrocene Couple in Nonaqueous Solutions. J. Chem. Theory Comput. 2010, 6, 2721–2725. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.R. Electrochemistry at Semiconductor and Oxidized Metal Electrodes; Plenum Press: New York, NY, USA, 1980; ISBN 0-306-40524-5. [Google Scholar]
- Willig, F.; Gerischer, H. Reaction of Excited Dye Molecules at Electrodes. Top. Appl. Chem. 1976, 61, 31–84. [Google Scholar] [CrossRef]
- Swierk, J.R.; Mallouk, T.E. Design and development of photoanodes for water-splitting dye-sensitized photoelectrochemical cells. Chem. Soc. Rev. 2013, 42, 2357–2387. [Google Scholar] [CrossRef] [PubMed]
- Forster, R.J.; Keyes, T.E. Redox Properties of Ground and Electronically Excited States: [Ru(bpy)2Qbpy]2+ Monolayers. J. Phys. Chem. B 1998, 102, 10004–10012. [Google Scholar] [CrossRef]
- Tilly, D.; Dayaker, G.; Bachu, P. Cobalt mediated C–H bond functionalization: Emerging tools for organic synthesis. Catal. Sci. Technol. 2014, 4, 2756–2777. [Google Scholar] [CrossRef]
- Gosmini, C.; Moncomble, A. Cobalt-catalyzed cross-coupling reactions of aryl halides. Isr. J. Chem. 2010, 50, 568–576. [Google Scholar] [CrossRef]
- Yamazaki, H.; Shouji, A.; Kajita, M.; Yagi, M. Electrocatalytic and photocatalytic water oxidation to dioxygen based on metal complexes. Coord. Chem. Rev. 2010, 254, 2483–2491. [Google Scholar] [CrossRef]
- Losse, S.; Vos, J.G.; Rau, S. Catalytic hydrogen production at cobalt centres. Coord. Chem. Rev. 2010, 254, 2492–2504. [Google Scholar] [CrossRef]
- Bura, T.; Blaskovits, J.T.; Leclerc, M. Direct (Hetero)arylation Polymerization: Trends and Perspectives. J. Am. Chem. Soc. 2016, 138, 10056–10071. [Google Scholar] [CrossRef] [PubMed]
- Josephsen, J.; Schaffer, C. The Position of 2,2-Bipyridine and 1,10-Phenanthroline in the Spectrochemical Series. Acta Chem. Scand. A 1977, 31, 813–824. [Google Scholar] [CrossRef]
- Krivokapic, I.; Zerara, M.; Daku, M.L.; Vargas, A.; Enachescu, C.; Ambrus, C.; Tregenna-Piggott, P.; Amstutz, N.; Krausz, E.; Hauser, A. Spin-crossover in cobalt(II) imine complexes. Coord. Chem. Rev. 2007, 251, 364–378. [Google Scholar] [CrossRef]
- Kremer, S.; Henke, W.; Reinen, D.; Kremer, S.; Henke, W.; Reinen, D. High-Spin-Low-Spin Equilibria of Cobalt2+ in the Terpyridine Complexes Co(terpy)2X2·nH2O. Inorg. Chem. 1982, 21, 3013–3022. [Google Scholar] [CrossRef]
- Hayami, S.; Komatsu, Y.; Shimizu, T.; Kamihata, H.; Lee, Y.H. Spin-crossover in cobalt(II) compounds containing terpyridine and its derivatives. Coord. Chem. Rev. 2011, 255, 1981–1990. [Google Scholar] [CrossRef]
- Marcus, R.A. Chemical and Electrochemical Electron-Transfer Theory. Annu. Rev. Phys. Chem. 1964, 15, 155–196. [Google Scholar] [CrossRef]
- Nusbaumer, H.; Moser, J.E.; Zakeeruddin, S.M.; Nazeeruddin, M.K.; Grätzel, M. CoII(dbbip))22+ complex rivals tri-iodide/iodide redox mediator in dye-sensitized photovoltaic cells. J. Phys. Chem. B 2001, 105, 10461–10464. [Google Scholar] [CrossRef]
- Hodgkin, D.C.; Kamper, J.; MacKay, M.; Pickworth, J.; Trueblood, K.N.; White, J.G. Structure of vitamin B12. Nature 1956, 178, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Nusbaumer, H.; Zakeeruddin, S.M.; Moser, J.-E.; Grätzel, M. An alternative efficient redox couple for the dye-sensitized solar cell system. Chem. Eur. J. 2003, 9, 3756–3763. [Google Scholar] [CrossRef] [PubMed]
- Cameron, P.J.; Peter, L.M.; Zakeeruddin, S.M.; Grätzel, M. Electrochemical studies of the Co(III)/Co(II)(dbbip)2 redox couple as a mediator for dye-sensitized nanocrystalline solar cells. Coord. Chem. Rev. 2004, 248, 1447–1453. [Google Scholar] [CrossRef]
- Sapp, S.A.; Elliott, C.M.M.; Contado, C.; Caramori, S.; Bignozzi, C.A. Substituted Polypyridine Complexes of Cobalt(II/III) as Efficient Electron-Transfer Mediators in Dye-Sensitized Solar Cells. J. Am. Chem. Soc. 2002, 124, 11215–11222. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, E.; Yum, J.-H.; Kessler, F.; Gómez García, C.J.; Zuccaccia, C.; Cinti, A.; Nazeeruddin, M.K.; Grätzel, M.; De Angelis, F. Cobalt electrolyte/dye interactions in dye-sensitized solar cells: A combined computational and experimental study. J. Am. Chem. Soc. 2012, 19438–19453. [Google Scholar] [CrossRef] [PubMed]
- Nazeeruddin, K.; Amirnasr, M.; Comte, P.; Mackay, J.R.; McQuillan, A.J.; Houriet, R.; Grätzel, M. Adsorption studies of counterions carried by the sensitizer cis-dithiocyanato(2,2′-bipyridyl-4,4′-dicarboxylate) ruthenium(II) on nanocrystalline TiO2 films. Langmuir 2000, 16, 8525–8528. [Google Scholar] [CrossRef]
- Buscaino, R.; Baiocchi, C.; Barolo, C.; Medana, C.; Grätzel, M.; Nazeeruddin, M.K.K.; Viscardi, G. A mass spectrometric analysis of sensitizer solution used for dye-sensitized solar cell. Inorg. Chim. Acta 2008, 361, 798–805. [Google Scholar] [CrossRef]
- Liu, Y.; Jennings, J.R.; Huang, Y.; Wang, Q.; Zakeeruddin, S.M.; Grätze, M. Cobalt redox mediators for ruthenium-based dye-sensitized solar cells: A combined impedance spectroscopy and near-IR transmittance study. J. Phys. Chem. C 2011, 115, 18847–18855. [Google Scholar] [CrossRef]
- Feldt, S.M.; Gibson, E.A.; Gabrielsson, E.; Sun, L.; Boschloo, G.; Hagfeldt, A. Design of Organic Dyes and Cobalt Polypyridine Redox Mediators for High-Efficiency Dye-Sensitized Solar Cells. J. Am. Chem. Soc. 2010, 132, 16714–16724. [Google Scholar] [CrossRef] [PubMed]
- Tsao, H.N.; Yi, C.; Moehl, T.; Yum, J.H.; Zakeeruddin, S.M.; Nazeeruddin, M.K.; Grätzel, M. Cyclopentadithiophene bridged donor–acceptor dyes achieve high power conversion efficiencies in dye-sensitized solar cells based on the tris-cobalt bipyridine redox couple. ChemSusChem 2011, 4, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Feldt, S.M.; Wang, G.; Boschloo, G.; Hagfeldt, A. Effects of driving forces for recombination and regeneration on the photovoltaic performance of dye-sensitized solar cells using cobalt polypyridine redox couples. J. Phys. Chem. C 2011, 115, 21500–21507. [Google Scholar] [CrossRef]
- Feldt, S.M.; Lohse, P.W.; Kessler, F.; Nazeeruddin, M.K.; Grätzel, M.; Boschloo, G.; Hagfeldt, A. Regeneration and recombination kinetics in cobalt polypyridine based dye-sensitized solar cells, explained using Marcus theory. Phys. Chem. Chem. Phys. 2013, 15, 7087–7097. [Google Scholar] [CrossRef] [PubMed]
- Kavan, L.; Yum, J.H.; Nazeeruddin, M.K.; Grätzel, M. Graphene nanoplatelet cathode for Co(III)/(II) mediated dye-sensitized solar cells. ACS Nano 2011, 5, 9171–9178. [Google Scholar] [CrossRef] [PubMed]
- Roy-Mayhew, J.D.; Boschloo, G.; Hagfeldt, A.; Aksay, I.A. Functionalized graphene sheets as a versatile replacement for platinum in dye-sensitized solar cells. ACS Appl. Mater. Interfaces 2012, 4, 2794–2800. [Google Scholar] [CrossRef] [PubMed]
- Carli, S.; Casarin, L.; Syrgiannis, Z.; Boaretto, R.; Benazzi, E.; Caramori, S.; Prato, M.; Bignozzi, C.A. Single Walled Carbon Nanohorns as Catalytic Counter Electrodes for Co(III)/(II) Electron Mediators in Dye Sensitized Cells. ACS Appl. Mater. Interfaces 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H.; Vlachopoulos, N.; Häggman, L.; Perruchot, C.; Jouini, M.; Boschloo, G.; Hagfeldt, A. PEDOT counter electrodes for dye-sensitized solar cells prepared by aqueous micellar electrodeposition. Electrochim. Acta 2013, 107, 45–51. [Google Scholar] [CrossRef]
- Pringle, J.M.; Armel, V.; MacFarlane, D.R. Electrodeposited PEDOT-on-plastic cathodes for dye-sensitized solar cells. Chem. Commun. 2010, 46, 5367–5369. [Google Scholar] [CrossRef] [PubMed]
- Carli, S.; Casarin, L.; Bergamini, G.; Caramori, S.; Bignozzi, C.A. Conductive PEDOT Covalently Bound to Transparent FTO Electrodes. J. Phys. Chem. C 2014, 118, 16782–16790. [Google Scholar] [CrossRef]
- Tsao, H.N.; Burschka, J.; Yi, C.; Kessler, F.; Nazeeruddin, M.K.; Grätzel, M. Influence of the interfacial charge-transfer resistance at the counter electrode in dye-sensitized solar cells employing cobalt redox shuttles. Energy Environ. Sci. 2011, 4, 4921. [Google Scholar] [CrossRef]
- Yum, J.-H.; Baranoff, E.; Kessler, F.; Moehl, T.; Ahmad, S.; Bessho, T.; Marchioro, A.; Ghadiri, E.; Moser, J.-E.; Yi, C.; et al. A cobalt complex redox shuttle for dye-sensitized solar cells with high open-circuit potentials. Nat. Commun. 2012, 3, 631. [Google Scholar] [CrossRef] [PubMed]
- Bessho, T.; Zakeeruddin, S.M.; Yeh, C.Y.; Diau, E.W.G.; Grätzel, M. Highly efficient mesoscopic dye-sensitized solar cells based on donor–acceptor-substituted porphyrins. Angew. Chemie Int. Ed. 2010, 49, 6646–6649. [Google Scholar] [CrossRef] [PubMed]
- Yella, A.; Lee, H.-W.; Tsao, H.N.; Yi, C.; Chandiran, A.K.; Nazeeruddin, M.K.; Diau, E.W.-G.; Yeh, C.-Y.; Zakeeruddin, S.M.; Grätzel, M. Porphyrin-sensitized solar cells with cobalt (II/III)-based redox electrolyte exceed 12 percent efficiency. Science 2011, 334, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Yella, A.; Gao, P.; Humphry-Baker, R.; Curchod, B.F.E.; Ashari-Astani, N.; Tavernelli, I.; Rothlisberger, U.; Nazeeruddin, M.K.; Grätzel, M. Dye-sensitized solar cells with 13% efficiency achieved through the molecular engineering of porphyrin sensitizers. Nat. Chem. 2014, 6, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Kakiage, K.; Aoyama, Y.; Yano, T.; Oya, K.; Fujisawa, J.-I.J.; Hanaya, M. Highly-efficient dye-sensitized solar cells with collaborative sensitization by silyl-anchor and carboxy-anchor dyes. Chem. Commun. 2015, 51, 15894–15897. [Google Scholar] [CrossRef] [PubMed]
- Yum, J.-H.; Holcombe, T.W.; Kim, Y.; Rakstys, K.; Moehl, T.; Teuscher, J.; Delcamp, J.H.; Nazeeruddin, M.K.; Grätzel, M. Blue-coloured highly efficient dye-sensitized solar cells by implementing the diketopyrrolopyrrole chromophore. Sci. Rep. 2013, 3, 2446. [Google Scholar] [CrossRef] [PubMed]
- Grzybowski, M.; Gryko, D.T. Diketopyrrolopyrroles: Synthesis, Reactivity, and Optical Properties. Adv. Opt. Mater. 2015, 3, 280–320. [Google Scholar] [CrossRef]
- Yao, Z.; Zhang, M.; Wu, H.; Yang, L.; Li, R.; Wang, P. Donor/Acceptor Indenoperylene Dye for Highly Efficient Organic Dye-Sensitized Solar Cells. J. Am. Chem. Soc. 2015, 137, 3799–3802. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Zhang, M.; Li, R.; Yang, L.; Qiao, Y.; Wang, P. A metal-free n-annulated thienocyclopentaperylene dye: Power conversion efficiency of 12% for dye-sensitized solar cells. Angew. Chemie Int. Ed. 2015, 54, 5994–5998. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Sun, D.; Cao, Y.; Tsao, H.N.; Yuan, Y.; Zakeeruddin, S.M.; Wang, P.; Grätzel, M. A Stable Blue Photosensitizer for Color Palette of Dye-Sensitized Solar Cells Reaching 12.6% Efficiency. J. Am. Chem. Soc. 2018, 140, 2405–2408. [Google Scholar] [CrossRef] [PubMed]
- Koussi-Daoud, S.; Schaming, D.; Fillaud, L.; Trippé-Allard, G.; Lafolet, F.; Polanski, E.; Nonomura, K.; Vlachopoulos, N.; Hagfeldt, A.; Lacroix, J.-C. 3,4-Ethylenedioxythiophene-based cobalt complex: An efficient co-mediator in dye-sensitized solar cells with poly(3,4-ethylene dioxythiophene) counter-electrode. Electrochim. Acta 2015, 179, 237–240. [Google Scholar] [CrossRef]
- Nasr-Esfahani, M.; Zendehdel, M.; Yaghoobi Nia, N.; Jafari, B.; Khosravi Babadi, M. Fabrication and characterization of a new dye sensitized solar cell with a new Schiff base cobalt complex as a redox mediator. RSC Adv. 2014, 4, 15961–15967. [Google Scholar] [CrossRef]
- Appleton, T.G. Natural oxygen-carrier and storage proteins contain a transition metal to which the oxygen reversibly coordinates. J. Chem. Educ. 1977, 54, 443. [Google Scholar] [CrossRef]
- Kashif, M.K.; Axelson, J.C.; Du, N.W.; Forsyth, C.M.; Chang, C.J.; Long, R.; Spiccia, L.; Bach, U. A New Direction in Dye-Sensitized Solar Cells Redox Mediator Development: In Situ Fine-Tuning of the Cobalt(II)/(III) Redox Potential through Lewis Base Interactions. J. Am. Chem. Soc. 2012, 134, 16646–16653. [Google Scholar] [CrossRef] [PubMed]
- Kashif, M.K.; Nippe, M.; Duffy, N.W.; Forsyth, C.M.; Chang, C.J.; Long, J.R.; Spiccia, L.; Bach, U. Stable dye-sensitized solar cell electrolytes based on cobalt(II)/(III) complexes of a hexadentate pyridyl ligand. Angew. Chem. Int. Ed. Engl. 2013, 52, 5527–5531. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Huang, F.; Cheng, Y.-B.; Bach, U.; Spiccia, L. Aqueous dye-sensitized solar cell electrolytes based on the cobalt(II)/(III) tris(bipyridine) redox couple. Energy Environ. Sci. 2013, 6, 121–127. [Google Scholar] [CrossRef]
- Dong, C.; Xiang, W.; Huang, F.; Fu, D.; Huang, W.; Bach, U.; Cheng, Y.B.; Li, X.; Spiccia, L. Controlling interfacial recombination in aqueous dye-sensitized solar cells by octadecyltrichlorosilane surface treatment. Angew. Chemie Int. Ed. 2014, 53, 6933–6937. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H.; Jiang, R.; Ye, S.; Hagfeldt, A.; Boschloo, G. Development of high efficiency 100% aqueous cobalt electrolyte dye-sensitised solar cells. Phys. Chem. Chem. Phys. 2016, 18, 8419–8427. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Anderson, A.; Barnes, P.R.F.; Xiaoe, L.; Law, C.; O’Regan, B.C. 2000 hours photostability testing of dye sensitised solar cells using a cobalt bipyridine electrolyte. J. Mater. Chem. A 2014, 2, 4751–4757. [Google Scholar] [CrossRef]
- Bella, F.; Vlachopoulos, N.; Nonomura, K.; Zakeeruddin, S.M.; Grätzel, M.; Gerbaldi, C.; Hagfeldt, A. Direct light-induced polymerization of cobalt-based redox shuttles: An ultrafast way towards stable dye-sensitized solar cells. Chem. Commun. 2015, 51, 16308–16311. [Google Scholar] [CrossRef] [PubMed]
- Freitag, M.; Yang, W.; Fredin, L.A.; D’Amario, L.; Karlsson, K.M.; Hagfeldt, A.; Boschloo, G. Supramolecular Hemicage Cobalt Mediators for Dye-Sensitized Solar Cells. ChemPhysChem 2016, 17, 3845–3852. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Yang, W.; Pazoki, M.; Boschloo, G.; Kloo, L. Cation-Dependent Photostability of Co(II/III)-Mediated Dye-Sensitized Solar Cells. J. Phys. Chem. C 2015, 119, 24704–24713. [Google Scholar] [CrossRef]
- Hathaway, B.J. Copper. Coord. Chem. Rev. 1981, 35, 211–252. [Google Scholar] [CrossRef]
- Hathaway, B.J.; Billing, D.E. The electronic properties and stereochemistry of mono-nuclear complexes of the copper(II) ion. Coord. Chem. Rev. 1970, 5, 143–207. [Google Scholar] [CrossRef]
- Karlin, K.D.; Yandell, J.K. Redox behavior of blue copper model complexes. Redox potentials and electron-transfer kinetics of some copper(II)–copper(I) complexes with nitrogen and thioether donors. Inorg. Chem. 1984, 23, 1184–1188. [Google Scholar] [CrossRef]
- Gross, E.L. Plastocyanin: Structure and function. Photosynth. Res. 1993, 37, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Burke, P.J.; Henrick, K.; McMillin, D.R. Crystal and molecular structures of bis(4,4′,6,6′-tetramethyl-2,2′-bipyridyl)copper(I) perchlorate, bis(4,4′,6,6′-tetramethyl-2,2′-bipyridyl) copper(II) diperchlorate, and bis(4,4′,6,6′-tetramethyl-2,2′-bipyridyl)copper(II) diperchlorate dihydrate. Inorg. Chem. 1982, 21, 1881–1886. [Google Scholar] [CrossRef]
- Rorabacher, D.B. Electron Transfer by Copper Centers. Chem. Rev. 2004, 104, 651–698. [Google Scholar] [CrossRef] [PubMed]
- Armaroli, N. Photoactive mono- and polynuclear Cu(I)–phenanthrolines. A viable alternative to Ru(II)–polypyridines? Chem. Soc. Rev. 2001, 30, 113–124. [Google Scholar] [CrossRef]
- Hattori, S.; Wada, Y.; Yanagida, S.; Fukuzumi, S. Blue copper model complexes with distorted tetragonal geometry acting as effective electron-transfer mediators in dye-sensitized solar cells. J. Am. Chem. Soc. 2005, 127, 9648–9654. [Google Scholar] [CrossRef] [PubMed]
- Kubota, L.T.; Gushikem, Y. Cyclic voltammetry studies of copper and nickel hexacyanoferrate immobilized on a silica gel surface coated with titanium(IV) oxide. J. Electroanal. Chem. 1993, 362, 219–225. [Google Scholar] [CrossRef]
- Brugnati, M.; Caramori, S.; Cazzanti, S.; Marchini, L.; Argazzi, R.; Bignozzi, C.A. Electron Transfer Mediators for Photoelectrochemical Cells Based on Cu(I) Metal Complexes. Int. J. Photoenergy 2007, 2007, 1–10. [Google Scholar] [CrossRef]
- Bai, Y.; Yu, Q.; Cai, N.; Wang, Y.; Zhang, M.; Wang, P. High-efficiency organic dye-sensitized mesoscopic solar cells with a copper redox shuttle. Chem. Commun. 2011, 47, 4376–4378. [Google Scholar] [CrossRef] [PubMed]
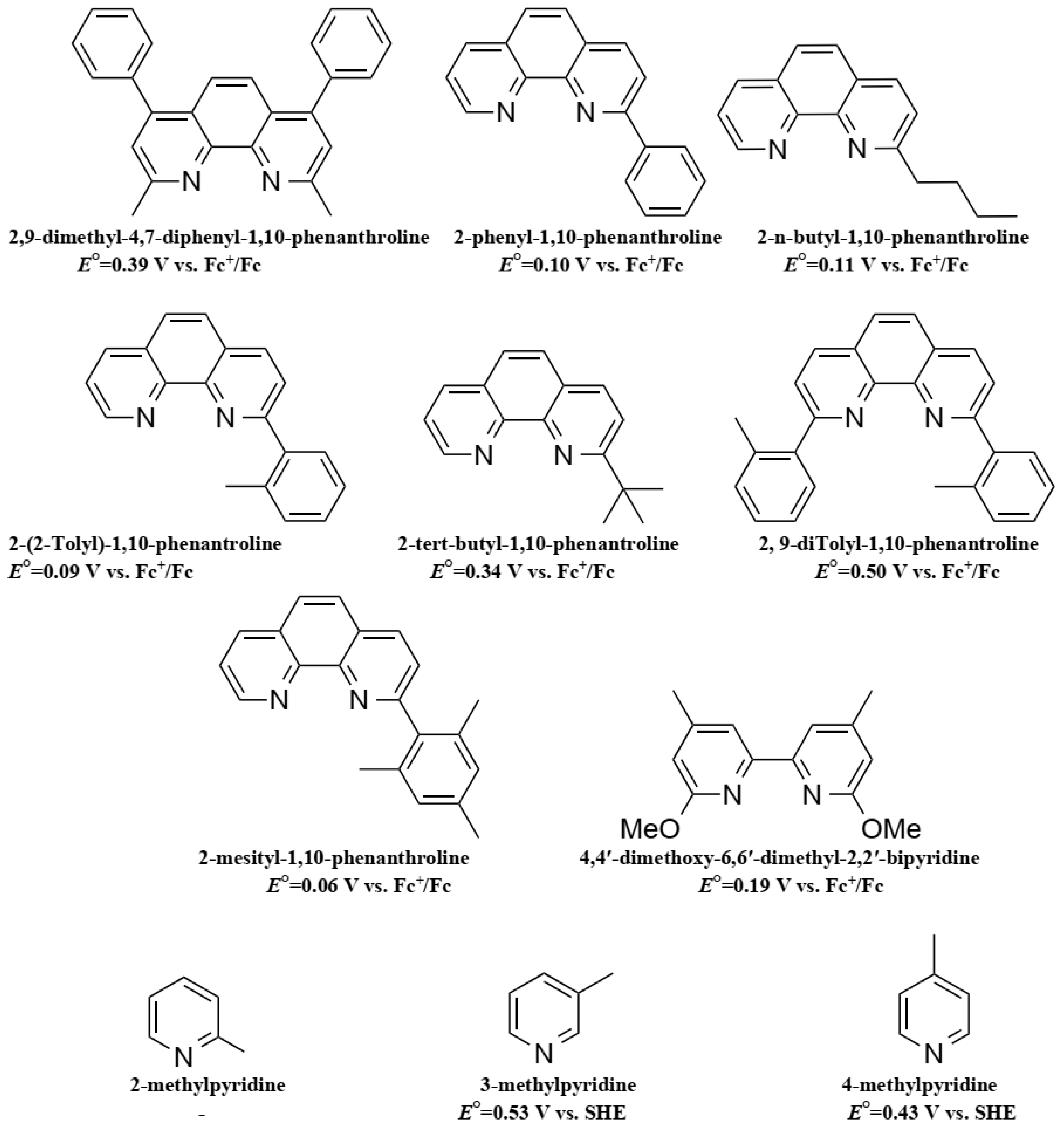
- Freitag, M.; Giordano, F.; Yang, W.; Pazoki, M.; Hao, Y.; Zietz, B.; Grätzel, M.; Hagfeldt, A.; Boschloo, G. Copper phenanthroline as a fast and high-performance redox mediator for dye-sensitized solar cells. J. Phys. Chem. C 2016, 120, 9595–9603. [Google Scholar] [CrossRef]
- Pradhan, S.C.; Hagfeldt, A.; Soman, S. Resurgence of DSCs with copper electrolyte: A detailed investigation of interfacial charge dynamics with cobalt and iodine based electrolytes. J. Mater. Chem. A 2018, 6, 22204. [Google Scholar] [CrossRef]
- Cong, J.; Kinschel, D.; Daniel, Q.; Safdari, M.; Gabrielsson, E.; Chen, H.; Svensson, P.H.; Sun, L.; Kloo, L. Bis(1,1-bis(2-pyridyl)ethane)copper(I/II) as an efficient redox couple for liquid dye-sensitized solar cells. J. Mater. Chem. A 2016, 4, 14550–14554. [Google Scholar] [CrossRef]
- Sun, Z.-Z.; Zheng, K.-M.; Li, Q.-S.; Li, Z.-S. Rational design of Co-based redox mediators for dye-sensitized solar cells by density functional theory. RSC Adv. 2014, 4, 31544–31551. [Google Scholar] [CrossRef]
- Kavan, L.; Saygili, Y.; Freitag, M.; Zakeeruddin, S.M.; Hagfeldt, A.; Grätzel, M. Electrochemical Properties of Cu(II/I)-Based Redox Mediators for Dye-Sensitized Solar Cells. Electrochim. Acta 2017, 227, 194–202. [Google Scholar] [CrossRef]
- Magni, M.; Giannuzzi, R.; Colombo, A.; Cipolla, M.P.; Dragonetti, C.; Caramori, S.; Carli, S.; Grisorio, R.; Suranna, G.P.; Bignozzi, C.A.; et al. Tetracoordinated Bis-phenanthroline Copper-Complex Couple as Efficient Redox Mediators for Dye Solar Cells. Inorg. Chem. 2016, 55, 5245–5253. [Google Scholar] [CrossRef] [PubMed]
- Ferdowsi, P.; Saygili, Y.; Zakeeruddin, S.M.; Mokhtari, J.; Grätzel, M.; Hagfeldt, A.; Kavan, L. Alternative bases to 4-tert-butylpyridine for dye-sensitized solar cells employing copper redox mediator. Electrochim. Acta 2018, 265, 194–201. [Google Scholar] [CrossRef]
- Saygili, Y.; Stojanovic, M.; Michaels, H.; Tiepelt, J.; Teuscher, J.; Massaro, A.; Pavone, M.; Giordano, F.; Zakeeruddin, S.M.; Boschloo, G.; et al. Effect of Coordination Sphere Geometry of Copper Redox Mediators on Regeneration and Recombination Behavior in Dye-Sensitized Solar Cell Applications. ACS Appl. Energy Mater. 2018, 1, 4950–4962. [Google Scholar] [CrossRef]
- Li, J.; Yang, X.; Yu, Z.; Gurzadyan, G.G.; Cheng, M.; Zhang, F.; Cong, J.; Wang, W.; Wang, H.; Li, X.; et al. Efficient dye-sensitized solar cells with [copper(6,6[prime or minute]-dimethyl-2,2[prime or minute]-bipyridine)2]2+/1+ redox shuttle. RSC Adv. 2017, 7, 4611–4615. [Google Scholar] [CrossRef]
- Richert, S.A.; Tsang, P.K.S.; Sawyer, D.T. Ligand-centered Redox Processes for MnL3, FeL3, and CoL3 Complexes (L = Acetylacetonate, 8-quinolinate, Picolinate, 2,2′-bipyridyl, 1,10-phenanthroline) and For Their Tetrakis(2,6-dichlorophenyl)porphinato Complexes [M(POR)]. Inorg. Chem. 1989, 28, 2471–2475. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Y.; Zhang, W.; Stojanovic, M.; Dar, M.I.; Pechy, P.; Saygili, Y.; Hagfeldt, A.; Zakeeruddin, S.M.; Grätzel, M. Electron-Affinity-Triggered Variations on the Optical and Electrical Properties of Dye Molecules Enabling Highly Efficient Dye-Sensitized Solar Cells. Angew. Chem. 2018, 130, 14321–14324. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, R.; Jiang, R.; Canto-Aguilar, E.J.; Oskam, G.; Boschloo, G. Improving the mass transport of copper-complex redox mediators in dye-sensitized solar cells by reducing the inter-electrode distance. Phys. Chem. Chem. Phys. 2017, 19, 32132–32142. [Google Scholar] [CrossRef] [PubMed]
- Freitag, M.; Teuscher, J.; Saygili, Y.; Zhan, X.; Giordano, F.; Liska, P.; Hua, J.; Zakeeruddin, S.M.; Moser, J.-E.; Grätzel, M.; et al. Dye-sensitized solar cells for efficient power generation under ambient lighting. Nat. Photonics 2017, 11, 372–378. [Google Scholar] [CrossRef]
- Cao, Y.; Liu, Y.; Zakeeruddin, S.M.; Hagfeldt, A.; Grätzel, M. Direct Contact of Selective Charge Extraction Layers Enables High-Efficiency Molecular Photovoltaics. Joule 2018, 2, 1108–1117. [Google Scholar] [CrossRef]
- Hoffeditz, W.L.; Katz, M.J.; Deria, P.; Cutsail, G.E., III; Pellin, M.J.; Farha, O.K.; Hupp, J.T.; Cutsail, G.E.; Pellin, M.J.; Farha, O.K.; et al. One Electron Changes Everything. A Multispecies Copper Redox Shuttle for Dye-Sensitized Solar Cells. J. Phys. Chem. C 2016, 120, 3731–3740. [Google Scholar] [CrossRef]
- Wang, Y.; Hamann, T.W. Improved performance induced by in situ ligand exchange reactions of copper bipyridyl redox couples in dye-sensitized solar cells. Chem. Commun. 2018, 54, 12361. [Google Scholar] [CrossRef] [PubMed]
- Colombo, A.; Dragonetti, C.; Magni, M.; Roberto, D.; Demartin, F.; Caramori, S.; Bignozzi, C.A. Efficient Copper Mediators Based on Bulky Asymmetric Phenanthrolines for DSSCs. ACS Appl. Mater. Interfaces 2014, 6, 13945–13955. [Google Scholar] [CrossRef] [PubMed]
- Colombo, A.; Ossola, R.; Magni, M.; Roberto, D.; Jacquemin, D.; Castellano, C.; Demartin, F.; Dragonetti, C. Intriguing C–H···Cu interactions in bis-(phenanthroline)Cu(I) redox mediators for dye-sensitized solar cells. Dalt. Trans. 2018, 47, 1018–1022. [Google Scholar] [CrossRef] [PubMed]
- Magni, M.; Colombo, A.; Dragonetti, C.; Mussini, P. Steric vs electronic effects and solvent coordination in the electrochemistry of phenanthroline-based copper complexes. Electrochim. Acta 2014, 141, 324–330. [Google Scholar] [CrossRef]
- Benazzi, E.; Magni, M.; Colombo, A.; Dragonetti, C.; Caramori, S.; Bignozzi, C.A.; Grisorio, R.; Suranna, G.P.; Cipolla, M.P.; Manca, M.; et al. Bis(1,10-phenanthroline) copper complexes with tailored molecular architecture: From electrochemical features to application as redox mediators in dye-sensitized solar cells. Electrochim. Acta 2018, 271, 180–189. [Google Scholar] [CrossRef]
- Colombo, A.; Di Carlo, G.; Dragonetti, C.; Magni, M.; Orbelli Biroli, A.; Pizzotti, M.; Roberto, D.; Tessore, F.; Benazzi, E.; Bignozzi, C.A.; et al. Coupling of Zinc Porphyrin Dyes and Copper Electrolytes: A Springboard for Novel Sustainable Dye-Sensitized Solar Cells. Inorg. Chem. 2017, 56, 14189–14197. [Google Scholar] [CrossRef] [PubMed]
- Dragonetti, C.; Magni, M.; Colombo, A.; Melchiorre, F.; Biagini, P.; Roberto, D. Coupling of a Copper Dye with a Copper Electrolyte: A Fascinating Springboard for Sustainable Dye-Sensitized Solar Cells. ACS Appl. Energy Mater. 2018, 1, 751–756. [Google Scholar] [CrossRef]
- Karpacheva, M.; Malzner, F.J.; Wobill, C.; Büttner, A.; Constable, E.C.; Housecroft, C.E. Cuprophilia: Dye-sensitized solar cells with copper(I) dyes and copper(I)/(II) redox shuttles. Dye. Pigment. 2018, 156, 410–416. [Google Scholar] [CrossRef]
- Leandri, V.; Daniel, Q.; Chen, H.; Sun, L.; Gardner, J.M.; Kloo, L. Electronic and Structural Effects of Inner Sphere Coordination of Chloride to a Homoleptic Copper(II) Diimine Complex. Inorg. Chem. 2018, 57, 4556–4562. [Google Scholar] [CrossRef] [PubMed]
- Pernechele, R. New Copper Redox Couples for Liquid Dye-Sensitized Solar Cells, Degree Project in Chemical Science and Engineering; KTH Royal Institute of Technology: Stockholm, Sweden, 2017. [Google Scholar]
- Michaels, H.; Benesperi, I.; Edvinsson, T.; Muñoz-Garcia, A.B.; Pavone, M.; Boschloo, G.; Freitag, M. Copper Complexes with Tetradentate Ligands for Enhanced Charge Transport in Dye-Sensitized Solar Cells. Inorganics 2018, 6, 53. [Google Scholar] [CrossRef]
- Hu, M.; Shen, J.; Yu, Z.; Liao, R.; Gurzadyan, G.G.; Yang, X.; Hagfeldt, A.; Wang, M.; Sun, L. Efficient and Stable Dye-Sensitized Solar Cells Based on a Tetradentate Copper(II/I) Redox Mediator. ACS Appl. Mater. Interfaces 2018, 10, 30409–30416. [Google Scholar] [CrossRef] [PubMed]
- Freitag, M.; Daniel, Q.; Pazoki, M.; Sveinbjörnsson, K.; Zhang, J.; Sun, L.; Hagfeldt, A.; Boschloo, G. High-efficiency dye-sensitized solar cells with molecular copper phenanthroline as solid hole conductor. Energy Environ. Sci. 2015, 8, 2634–2637. [Google Scholar] [CrossRef]
- Cao, Y.; Saygili, Y.; Ummadisingu, A.; Teuscher, J.; Luo, J.; Pellet, N.; Giordano, F.; Zakeeruddin, S.M.; Moser, J.-E.; Freitag, M.; et al. 11% efficiency solid-state dye-sensitized solar cells with copper(II/I) hole transport materials. Nat. Commun. 2017, 8, 15390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wu, Y.; Bahng, H.W.; Cao, Y.; Yi, C.; Saygili, Y.; Luo, J.; Liu, Y.; Kavan, L.; Moser, J.E.; et al. Comprehensive control of voltage loss enables 11.7% efficient solid-state dye-sensitized solar cells. Energy Environ. Sci. 2018, 11, 1779–1787. [Google Scholar] [CrossRef]
- Ruess, R.; Nguyen, T.H.Q.; Schlettwein, D. Metal Complexes as Redox Shuttles in Dye-Sensitized Solar Cells Based on Electrodeposited ZnO: Tuning Recombination Kinetics and Conduction Band Energy. J. Electrochem. Soc. 2018, 165, H3115–H3121. [Google Scholar] [CrossRef]
- Kavan, L.; Krysova, H.; Janda, P.; Tarabkova, H.; Saygili, Y.; Freitag, M.; Zakeeruddin, S.M.; Hagfeldt, A.; Grätzel, M. Novel highly active Pt/graphene catalyst for cathodes of Cu(II/I)-mediated dye-sensitized solar cells. Electrochim. Acta 2017, 251, 167–175. [Google Scholar] [CrossRef]
- Rodrigues, R.R.; Cheema, H.; Delcamp, J.H. A High-Voltage Molecular-Engineered Organic Sensitizer-Iron Redox Shuttle Pair: 1.4 V DSSC and 3.3 V SSM-DSSC Devices. Angew. Chemie Int. Ed. 2018, 57, 5472–5476. [Google Scholar] [CrossRef] [PubMed]
- Braterman, P.S.; Song, J.I.; Peacock, R.D. Electronic absorption spectra of the iron(II) complexes of 2,2′-bipyridine, 2,2′-bipyrimidine, 1,10-phenanthroline, and 2,2′:6′,2″-terpyridine and their reduction products. Inorg. Chem. 1992, 31, 555–559. [Google Scholar] [CrossRef]
- Daeneke, T.; Uemura, Y.; Duffy, N.W.; Mozer, A.J.; Koumura, N.; Bach, U.; Spiccia, L. Aqueous dye-sensitized solar cell electrolytes based on the ferricyanide-ferrocyanide redox couple. Adv. Mater. 2012, 24, 1222–1225. [Google Scholar] [CrossRef] [PubMed]
- Horvat-Radošević, V.; Kvastek, K.; Križekar, D. Kinetics of the [Fe(CN)6]3−/[Fe(CN)6]4− Redox Couple Reaction on Anodically Passivated Fe80B20. Croat. Chem. Acta 1997, 70, 537–561. [Google Scholar]
- Ameur, Z.O.; Husein, M.M. Electrochemical Behavior of Potassium Ferricyanide in Aqueous and (w/o) Microemulsion Systems in the Presence of Dispersed Nickel Nanoparticles. Sep. Sci. Technol. 2013, 48, 681–689. [Google Scholar] [CrossRef]
- Rutkowska, I.A.; Andrearczyk, A.; Zoladek, S.; Goral, M.; Darowicki, K.; Kulesza, P.J. Electrochemical characterization of Prussian blue type nickel hexacyanoferrate redox mediator for potential application as charge relay in dye-sensitized solar cells. J. Solid State Electrochem. 2011, 15, 2545–2552. [Google Scholar] [CrossRef]
- Hamann, T.W.; Farha, O.K.; Hupp, J.T. Outer-Sphere Redox Couples as Shuttles in Dye-Sensitized Solar Cells. Performance Enhancement Based on Photoelectrode Modification via Atomic Layer Deposition. J. Phys. Chem. C 2008, 112, 19756–19764. [Google Scholar] [CrossRef]
- Penner, R.M.; Heben, M.J.; Longin, T.L.; Lewis, N.S. Fabrication and Use of Nanometer-Sized Electrodes in Electrochemistry. Science 1990, 250, 1118–1121. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Stanbury, D.M. Kinetics and Mechanism of the One-Electron Reduction of Iodine by [RuII(NH3)5 isn]2+ in Aqueous Solution. Inorg. Chem. 1998, 37, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Gregg, B.A.; Pichot, F.; Ferrere, S.; Fields, C.L. Interfacial Recombination Processes in Dye-Sensitized Solar Cells and Methods to Passivate the Interfaces. J. Phys. Chem. B 2001, 105, 1422–1429. [Google Scholar] [CrossRef]
- Klahr, B.M.; Hamann, T.W. Performance Enhancement and Limitations of Cobalt Bipyridyl Redox Shuttles in Dye-Sensitized Solar Cells. J. Phys. Chem. C 2009, 113, 14040–14045. [Google Scholar] [CrossRef]
- Ito, S. Investigation of Dyes for Dye-Sensitized Solar Cells: Ruthenium-Complex Dyes, Metal-Free Dyes, Metal-Complex Porphyrin Dyes and Natural Dyes. In Solar Cells—Dye-Sensitized Devices; Kosyachenko, L.A., Ed.; InTech: Rijeka, Croatia; Shanghai, China, 2011; pp. 19–48. ISBN 978-953-307-735-2. [Google Scholar]
- Ondersma, J.; Hamann, T. Impedance Investigation of Dye-Sensitized Solar Cells Employing Outer-Sphere Redox Shuttles. J. Phys. Chem. C 2010, 638–645. [Google Scholar] [CrossRef]
- Daeneke, T.; Kwon, T.-H.; Holmes, A.B.; Duffy, N.W.; Bach, U.; Spiccia, L. High-efficiency dye-sensitized solar cells with ferrocene-based electrolytes. Nat. Chem. 2011, 3, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Congiu, M.; Nunes-Neto, O.; De Marco, M.L.; Dini, D.; Graeff, C.F.O. Cu2−xS films as counter-electrodes for dye solar cells with ferrocene-based liquid electrolytes. Thin Solid Films 2016, 612, 22–28. [Google Scholar] [CrossRef]
- Daeneke, T.; Mozer, A.J.; Kwon, T.-H.; Duffy, N.W.; Holmes, A.B.; Bach, U.; Spiccia, L. Dye regeneration and charge recombination in dye-sensitized solar cells with ferrocene derivatives as redox mediators. Energy Environ. Sci. 2012, 5, 7090–7099. [Google Scholar] [CrossRef]
- Cariello, M.; Ahn, S.; Park, K.-W.; Chang, S.-K.; Hong, J.; Cooke, G. An investigation of the role increasing π-conjugation has on the efficiency of dye-sensitized solar cells fabricated from ferrocene-based dyes. RSC Adv. 2016, 6, 9132–9138. [Google Scholar] [CrossRef]
- El-Zohry, A.M.; Cong, J.; Karlsson, M.; Kloo, L.; Zietz, B. Ferrocene as a rapid charge regenerator in dye-sensitized solar cells. Dye. Pigment. 2016, 132, 360–368. [Google Scholar] [CrossRef]
- Takaichi, J.; Morimoto, Y.; Ohkubo, K.; Shimokawa, C.; Hojo, T.; Mori, S.; Asahara, H.; Sugimoto, H.; Fujieda, N.; Nishiwaki, N.; et al. Redox chemistry of nickel(II) complexes supported by a series of noninnocent β-diketiminate ligands. Inorg. Chem. 2014, 53, 6159–6169. [Google Scholar] [CrossRef] [PubMed]
- Zilbermann, I.; Maimon, E.; Cohen, H.; Meyerstein, D. Redox chemistry of nickel complexes in aqueous solutions. Chem. Rev. 2005, 105, 2609–2625. [Google Scholar] [CrossRef] [PubMed]
- Li, T.C.; Spokoyny, A.M.; She, C.; Farha, O.K.; Mirkin, C.A.; Marks, T.J.; Hupp, J.T. Ni(III)/(IV)Bis(dicarbollide) as a Fast, Noncorrosive Redox Shuttle for Dye-Sensitized Solar Cells. J. Am. Chem. Soc. 2010, 132, 4580–4582. [Google Scholar] [CrossRef] [PubMed]
- Hawthorne, M.F.; Zink, J.I.; Skelton, J.M.; Bayer, M.J.; Liu, C.; Livshits, E.; Baer, R.; Neuhauser, D. Electrical or Photocontrol of the Rotary Motion of a Metallacarborane. Science 2004, 303, 1849–1851. [Google Scholar] [CrossRef] [PubMed]
- Spokoyny, A.M.; Li, T.C.; Farha, O.K.; Machan, C.W.; She, C.; Stern, C.L.; Marks, T.J.; Hupp, J.T.; Mirkin, C.A. Electronic Tuning of Nickel-Based Bis(dicarbollide) Redox Shuttles in Dye-Sensitized Solar Cells. Angew. Chemie Int. Ed. 2010, 49, 5339–5343. [Google Scholar] [CrossRef] [PubMed]
- Li, T.C.; Fabregat-Santiago, F.; Farha, O.K.; Spokoyny, A.M.; Raga, S.R.; Bisquert, J.; Mirkin, C.A.; Marks, T.J.; Hupp, J.T. SiO2 Aerogel Templated, Porous TiO2 Photoanodes for Enhanced Performance in Dye-Sensitized Solar Cells Containing a Ni(III)/(IV) Bis(dicarbollide) Shuttle. J. Phys. Chem. C 2011, 115, 11257–11264. [Google Scholar] [CrossRef]
- Yamaguchi, K.S.; Sawyer, D.T. The Redox Chemistry of Manganese(III) and -(IV) Complexes. Isr. J. Chem. 1985, 25, 164–176. [Google Scholar] [CrossRef]
- Morrison, M.M.; Sawyer, D.T. Redox chemistry of the polyimine complexes of manganese(II), -(III), and -(IV) in acetonitrile. Inorg. Chem. 1978, 17, 333–337. [Google Scholar] [CrossRef]
- Arora, C.L. Lecture demonstration of the various oxidation states of manganese. J. Chem. Educ. 1977, 54, 302–303. [Google Scholar] [CrossRef]
- Kolb, D. Oxidation states of manganese. J. Chem. Educ. 1988, 65, 1004–1005. [Google Scholar] [CrossRef]
- Krewald, V.; Neese, F.; Pantazis, D.A. Resolving the Manganese Oxidation States in the Oxygen-evolving Catalyst of Natural Photosynthesis. Isr. J. Chem. 2015, 55, 1219–1232. [Google Scholar] [CrossRef]
- Perera, I.R.; Gupta, A.; Xiang, W.; Daeneke, T.; Bach, U.; Evans, R.A.; Ohlin, C.A.; Spiccia, L. Introducing manganese complexes as redox mediators for dye-sensitized solar cells. Phys. Chem. Chem. Phys. 2014, 16, 12021–12028. [Google Scholar] [CrossRef] [PubMed]
- Carli, S.; Benazzi, E.; Casarin, L.; Bernardi, T.; Bertolasi, V.; Argazzi, R.; Caramori, S.; Bignozzi, C.A. On the stability of manganese tris(β-diketonate) complexes as redox mediators in DSSCs. Phys. Chem. Chem. Phys. 2016, 18, 5949–5956. [Google Scholar] [CrossRef] [PubMed]
- Hossain, F.; Rigsby, M.A.; Duncan, C.T.; Milligan, P.L.; Lord, R.L.; Baik, M.; Schultz, F.A. Synthesis, Structure, and Properties of Low-Spin Manganese(III)–Poly(pyrazolyl) borate Complexes. Inorg. Chem. 2007, 46, 2596–2603. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulou, A.; Vlasiou, M.; Tziouris, P.A.; Tsiafoulis, C.; Tsipis, A.C.; Rehder, D.; Kabanos, T.A.; Keramidas, A.D.; Stathatos, E. Oxidovanadium(IV/V) complexes as new redox mediators in dye-sensitized solar cells: A combined experimental and theoretical study. Inorg. Chem. 2015, 54, 3979–3988. [Google Scholar] [CrossRef] [PubMed]
- Oyaizu, K.; Hayo, N.; Sasada, Y.; Kato, F.; Nishide, H. Enhanced bimolecular exchange reaction through programmed coordination of a five-coordinate oxovanadium complex for efficient redox mediation in dye-sensitized solar cells. Dalt. Trans. 2013, 42, 16090–16095. [Google Scholar] [CrossRef] [PubMed]
- Cazzanti, S.; Caramori, S.; Argazzi, R.; Elliott, C.M.; Bignozzi, C.A. Efficient non-corrosive electron-transfer mediator mixtures for dye-sensitized solar cells. J. Am. Chem. Soc. 2006, 128, 9996–9997. [Google Scholar] [CrossRef] [PubMed]
- Cong, J.; Hao, Y.; Sun, L.; Kloo, L. Two redox couples are better than one: Improved current and fill factor from cobalt-based electrolytes in dye-sensitized solar cells. Adv. Energy Mater. 2014, 4, 1301273. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, H.; Chen, X.; Yan, F. Imidazolium functionalized cobalt tris(bipyridyl) complex redox shuttles for high efficiency ionic liquid electrolyte dye-sensitized solar cells. J. Mater. Chem. A 2013, 1, 11933–11941. [Google Scholar] [CrossRef]
- Hao, Y.; Yang, W.; Zhang, L.; Jiang, R.; Mijangos, E.; Saygili, Y.; Hammarström, L.; Hagfeldt, A.; Boschloo, G. A small electron donor in cobalt complex electrolyte significantly improves efficiency in dye-sensitized solar cells. Nat. Commun. 2016, 7, 13934. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
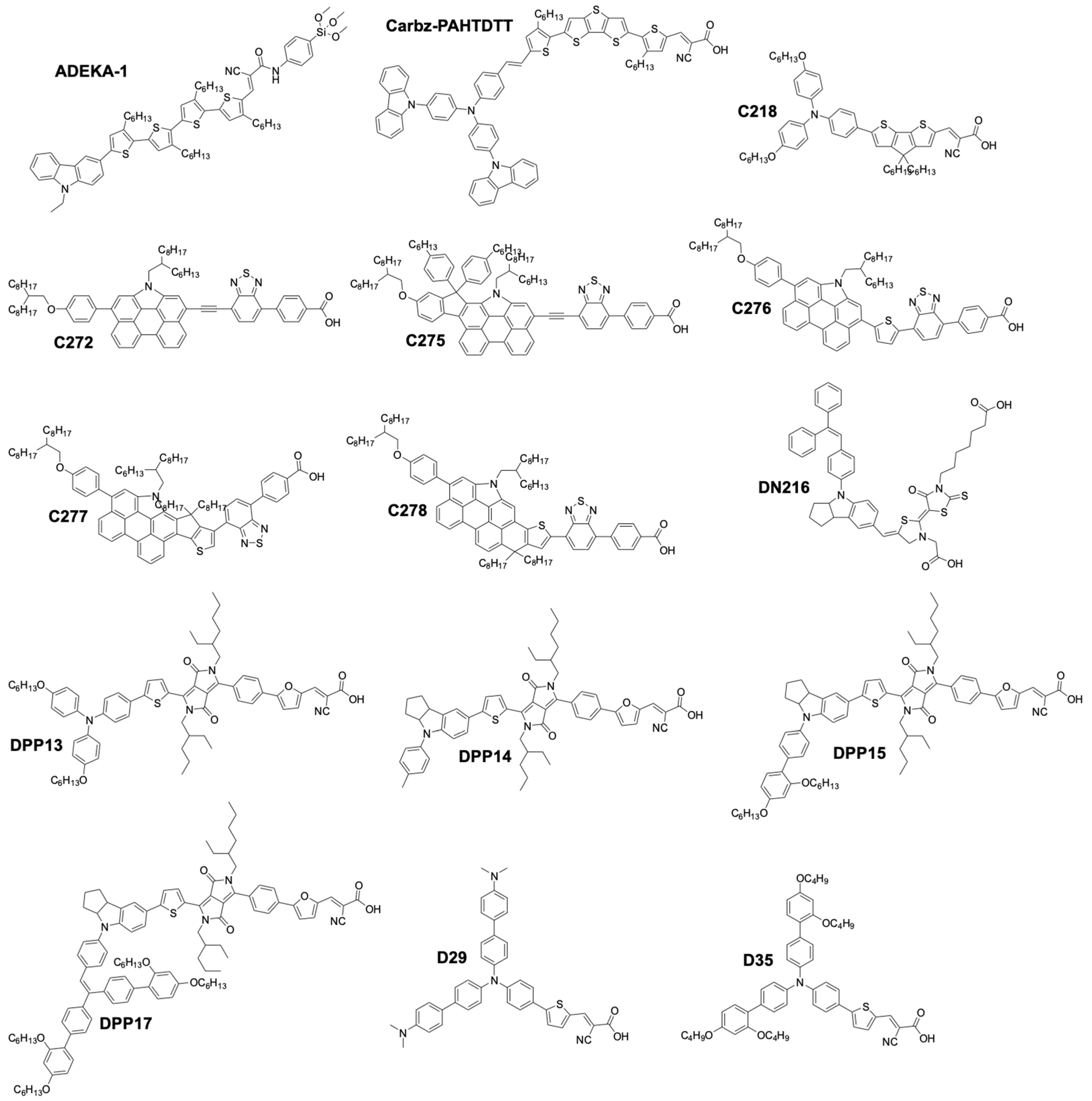
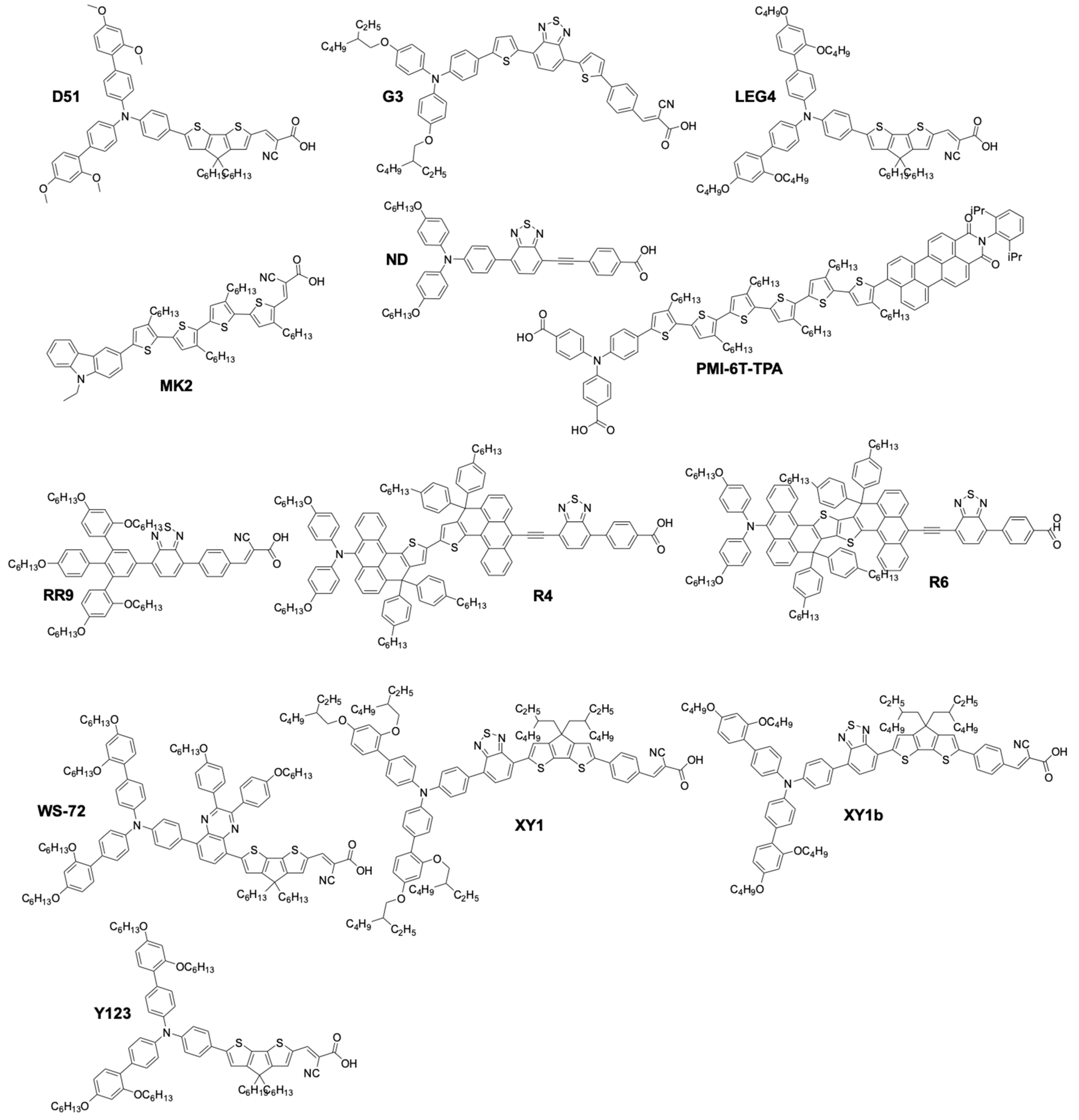
| Dye | Mediator | UOC (V) | jSC (mA·cm−2) | FF | PCE (%) | PCEref (%) | CE | Ref. |
|---|---|---|---|---|---|---|---|---|
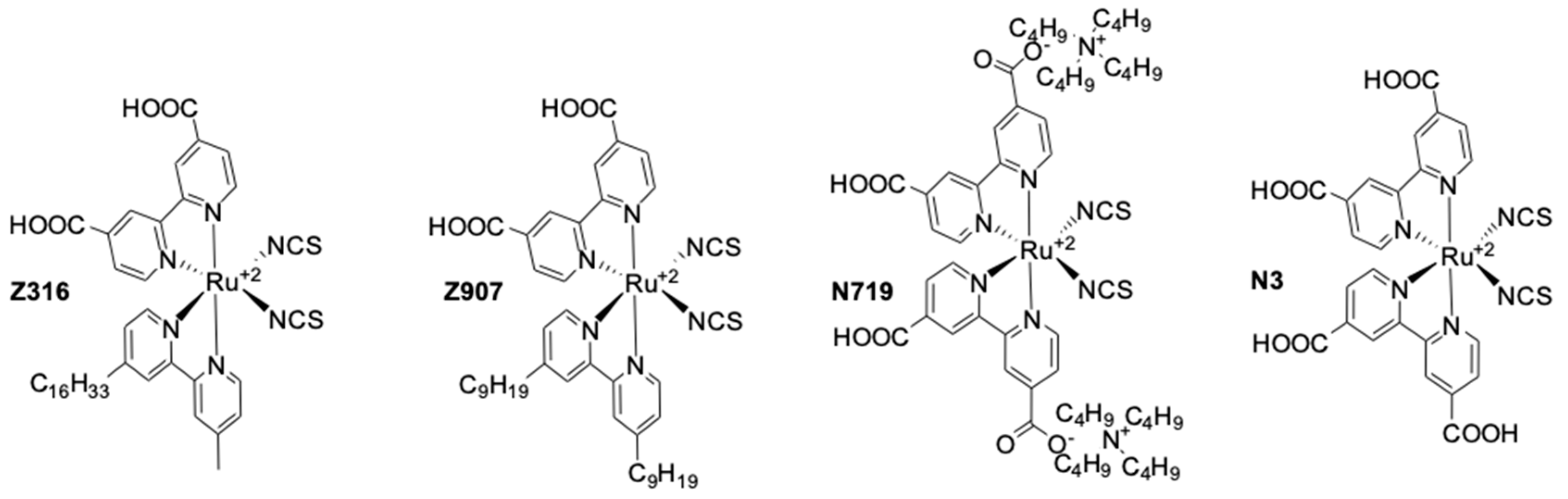
| Z316 | [Co(dbbip)2](III/II) | 0.67 | 6.8 | 0.46 | 2.2 | - | Pt | [94] |
| N3 | [Co(dtb)2](III/II) | 0.44 | 4.8 | 0.62 | 1.3 | 82 1 | Au | [98] |
| N719 | [Co(bpy)3](III/II) | 0.58 | 3.0 | 0.66 | 1.1 | 21 1 | Pt | [99] |
| Z907 | [Co(bpy)3](III/II) | 0.65 | 4.43 | 0.74 | 2.1 | 40 1 | Pt | [99] |
| Z907 | [Co(bpy)3](III/II) | 0.75 | 14 | 0.62 | 6.5 2 | 84 1 | Pt | [102] |
| D35 | [Co(bpy)3](III/II) | 0.9 | 10.7 | 0.68 | 6.7 | 118 1 | Pt | [103] |
| D35 | [Co(Cl-phen)3](III/II) | 0.93 | 7.3 | 0.59 | 4.01 | - | Pt | [105] |
| D35 | [Co(phen)3](III/II) | 1.0 | 6.4 | 0.56 | 3.57 | 89 3 | Pt | [105] |
| D35 | [Co(NO2-phen)3](III/II) | 1.00 | 4.384 | 0.51 | 2.29 | 57 3 | Pt | [105] |
| Y123 | [Co(bpy)3](III/II) | 0.86 | 14.6 | 0.7 | 8.8 | 140 4/122 5 | Pt | [104] |
| Y123 | [Co(phen)3](III/II) | 0.94 | 14.8 | 0.69 | 9.5 | 92 6 | PEDOT | [113] |
| Y123 | [Co(bpy)3](III/II) | 0.91 | 15.9 | 0.71 | 10.6 | - | PEDOT | [113] |
| Y123 | [Co(bpy-pz)3](III/II | 1.00 | 13.06 | 0.77 | 10.8 7 | - | PProDOT | [114] |
| Y123 | [Co(bpy-pz)3](III/II) | 1.02 | 12.54 | 0.69 | 8.87 8 | 135 | PProDOT | [114] |
| YD2-o-C8 | [Co(bpy)3](III/II) | 0.97 | 17.3 | 0.71 | 11.9 | - | Pt | [116] |
| YD2 | [Co(bpy)3](III/II) | 0.83 | 14.9 | 0.69 | 8.4 | - | Pt | [116] |
| YD2-o-C8 | [Co(bpy)3](III/II) | 0.94 | 17.7 | 0.74 | 12.5 9 | - | Pt | [116] |
| SM315 | [Co(bpy)3](III/II) | 0.91 | 18.1 | 0.78 | 13 | - | GNP 10 | [117] |
| SM371 | [Co(bpy)3](III/II) | 0.96 | 15.9 | 0.79 | 12 | - | GNP | [117] |
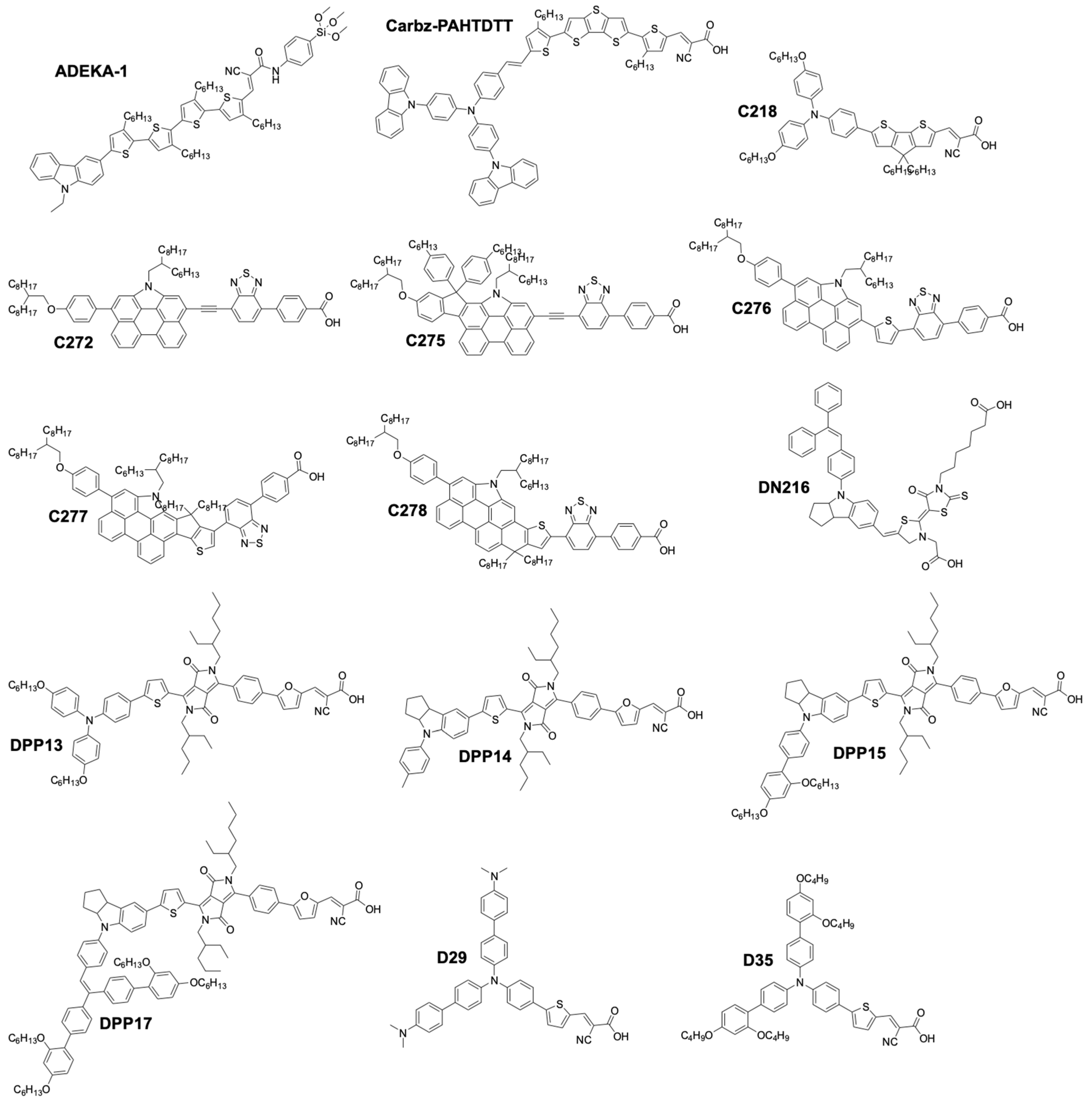
| ADEKA-1 | [Co(phen)3](III/II) | 1.01 | 18.2 | 0.77 | 14.7 11 | 131 1 | Au/GNP | [118] |
| DPP17 | [Co(bpy)3](III/II) | 0.76 | 17.9 | 0.74 | 10.1 | 142 1 | Graphene | [119] |
| DPP13 | [Co(bpy)3](III/II) | 0.743 | 15.6 | 0.78 | 8.97 | 1181 | Graphene | [119] |
| DPP14 | [Co(bpy)3](III/II) | 0.716 | 15.2 | 0.76 | 8.23 | 106 1 | Graphene | [119] |
| DPP15 | [Co(bpy)3](III/II) | 0.745 | 17.6 | 0.75 | 9.81 | 132 1 | Graphene | [119] |
| C272 | [Co(phen)3](III/II) | 0.90 | 15.8 | 0.74 | 10.6 | - | Au/Cr | [121] |
| C275 | [Co(phen)3](III/II) | 0.96 | 17.0 | 0.77 | 12.5 | - | Au/Cr | [122] |
| C276 | [Co(bpy)3](III/II) | 0.82 | 15.5 | 0.74 | 9.4 | - | Au/Cr | [122] |
| C277 | [Co(bpy)3](III/II) | 0.82 | 19.4 | 0.72 | 11.5 | - | Au/Cr | [122] |
| R6 | [Co(bpy)3](III/II) | 0.84 | 19.5 | 0.73 | 12 | - | Au/Cr | [122] |
| R4 | [Co(bpy)3](III/II) | 0.85 | 19.7 | 0.75 | 12.6 | - | Pt | [123] |
| Z907 | [Co(bpy)3](III/II) | 0.852 | 17.3 | 0.75 | 11.1 | - | Pt | [123] |
| Z907 | [Co(EtPy)2](III/II) [Co(phen)3](III/II) | 0.75 | 5.1 | 0.58 | 2.2 | 157 12 | PEDOT | [124] |
| D35 | [Co(EtPy)2](III/II)/ [Co(phen)3](III/II) | 0.92 | 8.4 | 0.67 | 5.1 | 121 | PEDOT | [124] |
| D35 | SBCC | 0.91 | 5.2 | 0.54 | 2.53 | 72 | Pt | [125] |
| MK2 | [Co(PY5Me2)(NMBI)](III/II) | 0.94 | 11.8 | 0.77 | 8.4 | 120 | PEDOT | [127] |
| MK2 | [Co(PY5Me2)(TBP)](III/II) | 0.99 | 8.1 | 0.76 | 6.1 | 82 | PEDOT | [127] |
| MK2 | [Co(bpyPY4)](III/II) | 0.76 | 14.7 | 0.75 | 8.3 | 106 | Pt | [128] |
| MK2 | [Co(bpy)3](III/II) | 0.69 | 9.8 | 0.74 | 5.0 13 | - | ITO/Pt | [129] |
| MK2 | [Co(bpy)3](III/II) | 0.82 | 10.2 | 0.68 | 5.6 | - | Pt | [130] |
| D51 | [Co(phen)3](III/II) | 0.78 | 8.6 | 0.72 | 4.8 | - | PEDOT | [131] |
| D51 | [Co(bpy-pz)3](III/II) | 0.90 | 8.1 | 0.76 | 5.5 | 120 | PEDOT | [131] |
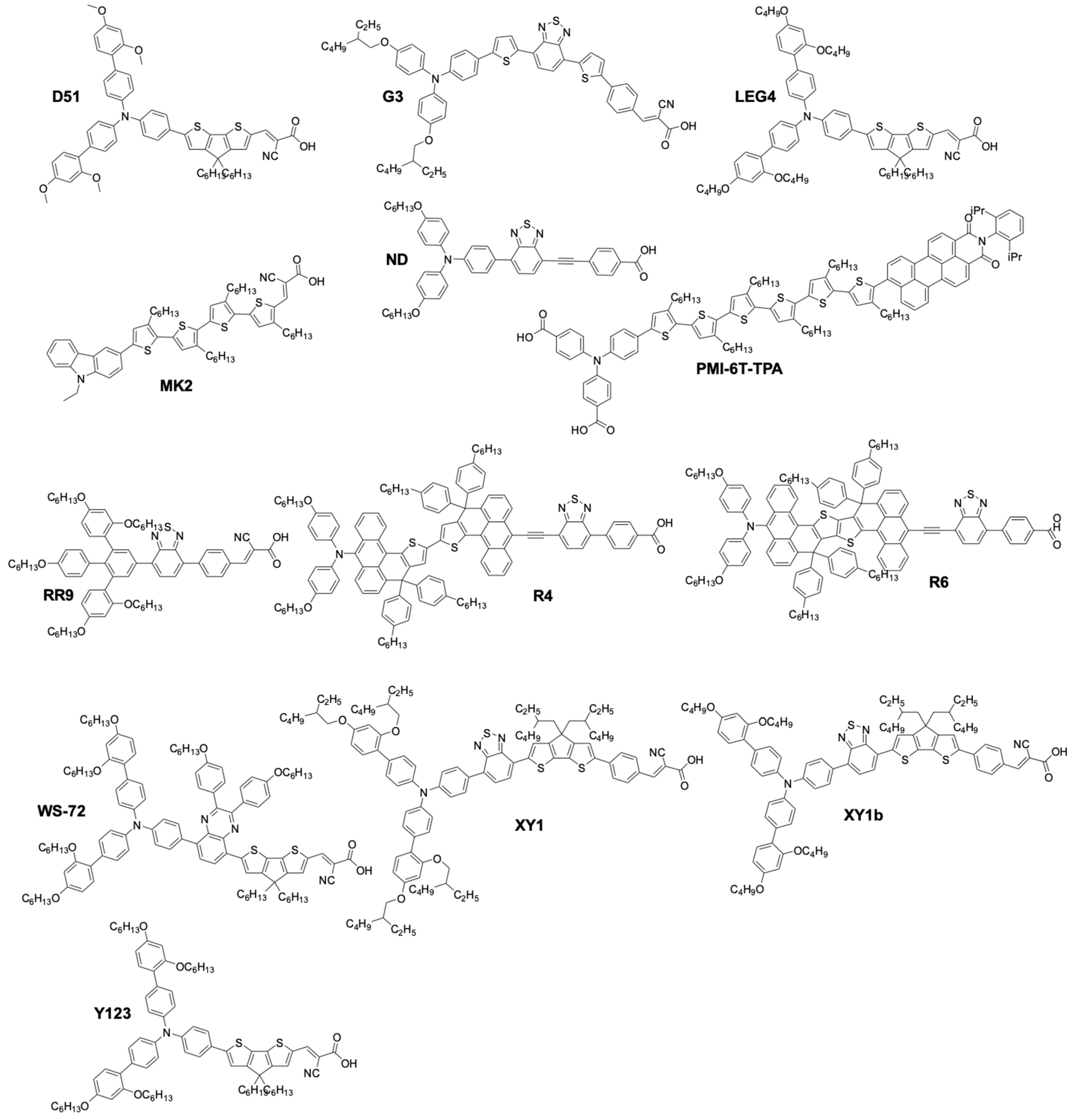
| Complex or Ligand | Irradiance mW·cm−2 or lux | Dye | UOC (V) | jSC (mA·cm−2) | FF | PCE | PCErel | CE | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| [Cu(dmp)2]2+/1+ | 100 # | N719 | 0.79 | 3.2 | 0.55 | 1.4 | - | Pt | [143] |
| [Cu(dmp)2]2+/1+ | 20 # | N719 | - | - | - | 2.2 | a 0.51 | Pt | [143] |
| [Cu(dmp)2]2+/1+ | 100 # | C218 | 0.93 | 11.3 | 0.66 | 7 | a 1.08 | Pt | [146] |
| [Cu(dmp)2]2+/1+ | 23 # | C218 | 0.87 | 2.8 | 0.73 | 7.8 | - | Pt | [146] |
| [Cu(dmp)2]2+/1+ | 100 # | LEG4 | 1.02 | 12.6 | 0.62 | 8.3 | b 1.14 | PEDOT | [159] |
| [Cu(dmp)2]2+/1+ | 50 # | LEG4 | 1.01 | 7.0 | 0.69 | 9.8 | - | PEDOT | [159] |
| [Cu(dmp)2]2+/1+ | 100 # | Y123 | 1.06 | 13.61 | 0.692 | 10.3 | - | PEDOT | [56] |
| [Cu(phen)2]2+/1+ | 100 # | N719 | 0.57 | 0.48 | 0.43 | 1.3 | - | Pt | [143] |
| [Cu(SP)(mmt)]0/−1 | 100 # | N719 | 0.66 | 4.4 | 0.44 | 0.12 | - | Pt | [143] |
| [Cu(SP)(mmt)]0/−1 | 20 # | N719 | - | - | - | 1.9 | a 0.44 | Pt | [143] |
| [Cu(bpye)2]2+/1+ | 100 # | LEG4 | 0.895 | 14.1 | 0.713 | 9 | b 1.18 | PEDOT | [149] |
| [Cu(bpye)2]2+/1+ | 50 # | LEG4 | 0.885 | 7.3 | 0.764 | 9.9 | b 1.19 | PEDOT | [149] |
| [Cu(bpye)2]2+/1+ | 10 # | LEG4 | 0.842 | 1.3 | 0.808 | 8.7 | b 1.24 | PEDOT | [149] |
| [Cu(dmby)2]2+/1+ | 100 # | Y123 | 1.07 | 14.15 | 0.687 | 10 | - | PEDOT | [56] |
| [Cu(dmby)2]2+/1+ | 100 # | LEG4 | 1.048 | 14.4 | 0.681 | 10.3 | a 1.29 b 1.12 | Pt | [155] |
| [Cu(tmby)2]2+/1+ | 100 # | Y123 | 1.04 | 15.53 | 0.640 | 10.3 | - | PEDOT | [56] |
| [Cu(tmby)2]2+/1 | 100 # | ND | 1.02 | 9.26 | 0.750 | 7.15 | a 1.16 b 1.37 | PEDOT | [153] |
| [Cu(tmby)2]2+/1+ | 200 § | D35/XY1 | 0.732 | 0.0272 | 0.79 | 25.5 | c 1.37 | PEDOT | [159] |
| [Cu(tmby)2]2+/1+ | 1000 § | D35/XY1 | 0.797 | 0.138 | 0.80 | 28.9 | c 1.38 | PEDOT | [159] |
| [Cu(tmby)2]2+/1+ | 100 # | D35/XY1 | 1.03 | 16.19 | 0.68 | 11.3 | - | PEDOT | [159] |
| [Cu(tmby)2]2+/1+ | 100 # | Y123/XY1b | 1.05 | 15.74 | 0.79 | 13.1 | - | PEDOT | [160] |
| Cu(PDTO)+ and Cu(TBP)4+x(ACN)y2+ | 100 # | Carbz-PAHTDTT | 0.88 | 4 | 0.52 | 1.83 | - | Inverse-opal Pt | [161] |
| [Cu(tmby)2]2+/1+ | 1000 § | Y123/XY1b | 0.878 | 0.00149 | 0.773 | 31.8 | - | PEDOT | [174] |
| [CuL2]2+/1+ L = 2-mesityl-4,7- dimethyl-1,10- phenanthroline | 100 # | G3 | 0.72 | 9.3 | 0.66 | 4.4 | a 0.60 | Pt | [152] |
| [CuL2]2+/1+ L = 2-mesityl-1,10- phenanthroline | 100 # | G3 | 0.83 | 11.4 | 0.59 | 5.6 | a 1.07 b 1.19 | Pt | [166] |
| [CuL2]2+/1+ L = 2-tolyl-1,10- phenanthroline | 100 # | G3 | 0.87 | 11.1 | 0.62 | 6.0 | a 1.15 b 1.27 | Pt | [166] |
| [CuL2]2+/1+ L = 2-phenyl-1,10- phenanthroline | 100 # | G3 | 0.88 | 8.0 | 0.69 | 4.9 | a 0.94 b 1.04 | Pt | [166] |
| [CuL2]2+/1+ L = 2-n-butyl-1,10- phenanthroline | 100 # | G3 | 0.86 | 10.1 | 0.66 | 5.7 | a 1.10 b 1.21 | Pt | [166] |
| [CuL2]2+/1+ L = 4,4′-dimethoxy- 6,6′-dimethyl-2,2′- Bipyridine | 100 # | Copper dye | 0.684 | 4.01 | 0.75 | 2.06 | a,d 0.38 | Pt | [169] |
| [CuL2]2+/1+ 2-n-butyl-1,10- phenanthroline | 100 # | D | 0.61 | 6.3 | 0.53 | 2 | a,d 0.22 b,d 0.33 | Pt | [168] |
| [Cu(3mpy)-3mpy]2+/+ | 100 # | LEG4 | 0.94 | 12.8 | 0.76 | 9.1 | b 1.37 | PEDOT | [171] |
| Derivative Shuttle | UOC (V) | jSC (mA·cm−2) | FF | (%) | IPCE Max (%) |
|---|---|---|---|---|---|
| Me10Fc+/0 | 0.437 | 6.6 | 0.40 | 1.1 | 79 |
| Et2Fc+/0 | 0.641 | 13.3 | 0.50 | 4.2 | 77 |
| EtFc+/0 | 0.669 | 12.8 | 0.56 | 4.8 | 79 |
| Fc+/Fc | 0.737 | 12.5 | 0.57 | 5.2 | 76 |
| BrFc+/0 | 0.671 | 9.3 | 0.48 | 3.0 | 40 |
| Br2Fc+/0 | 0.599 | 4.4 | 0.46 | 1.2 | 30 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saygili, Y.; Stojanovic, M.; Flores-Díaz, N.; Zakeeruddin, S.M.; Vlachopoulos, N.; Grätzel, M.; Hagfeldt, A. Metal Coordination Complexes as Redox Mediators in Regenerative Dye-Sensitized Solar Cells. Inorganics 2019, 7, 30. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7030030
Saygili Y, Stojanovic M, Flores-Díaz N, Zakeeruddin SM, Vlachopoulos N, Grätzel M, Hagfeldt A. Metal Coordination Complexes as Redox Mediators in Regenerative Dye-Sensitized Solar Cells. Inorganics. 2019; 7(3):30. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7030030
Chicago/Turabian StyleSaygili, Yasemin, Marko Stojanovic, Natalie Flores-Díaz, Shaik M. Zakeeruddin, Nick Vlachopoulos, Michael Grätzel, and Anders Hagfeldt. 2019. "Metal Coordination Complexes as Redox Mediators in Regenerative Dye-Sensitized Solar Cells" Inorganics 7, no. 3: 30. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7030030