Stabilization of Supramolecular Networks of Polyiodides with Protonated Small Tetra-azacyclophanes

 , , , , ,
, , , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
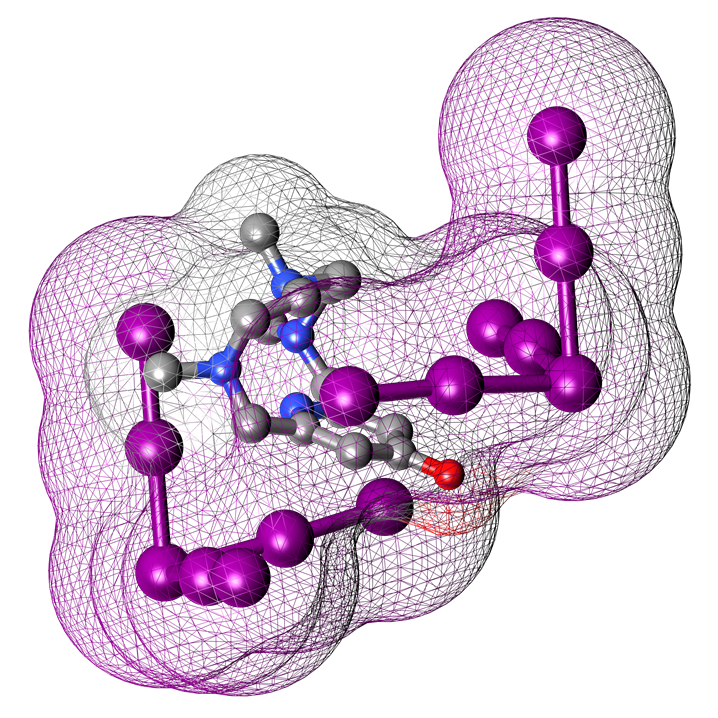
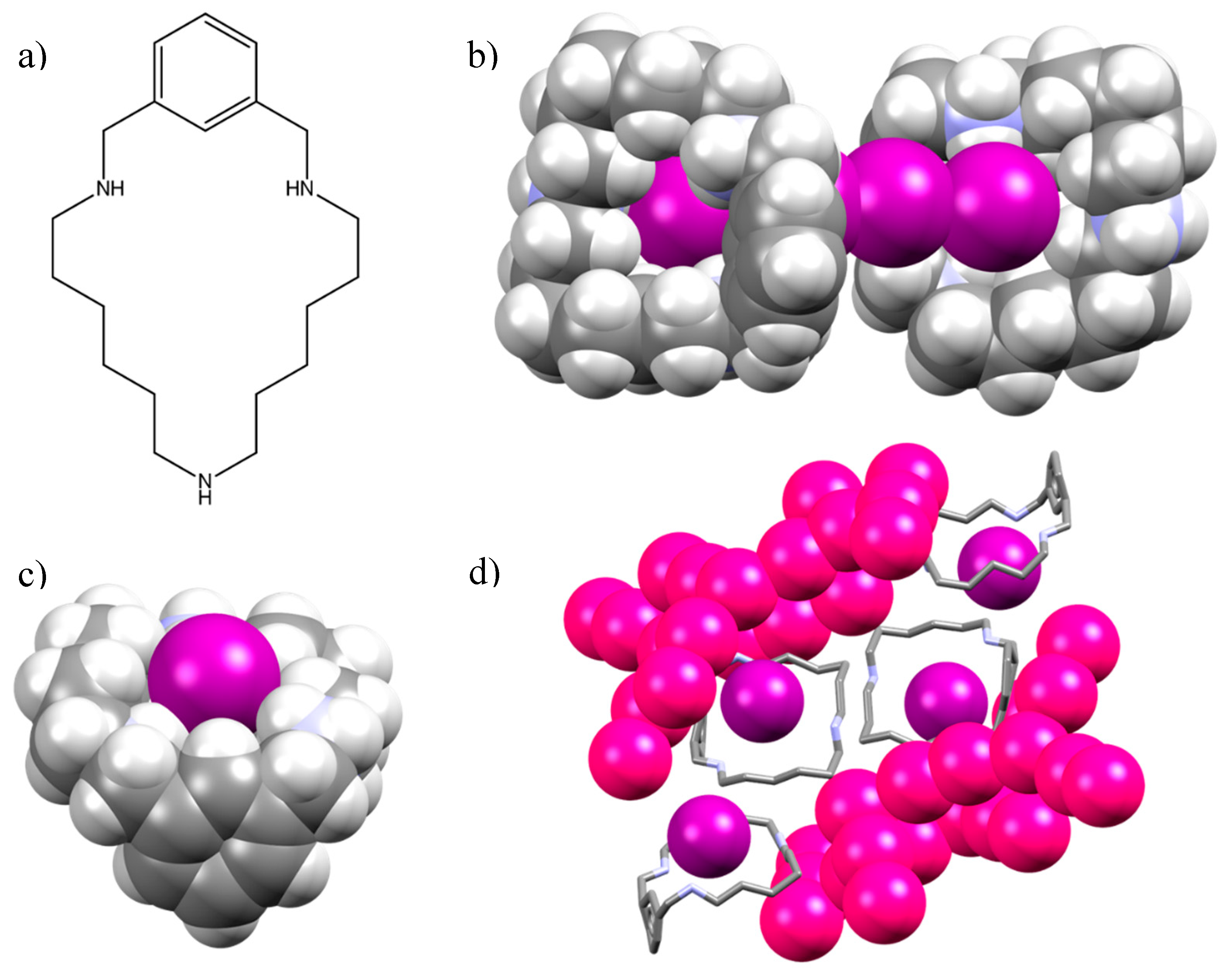
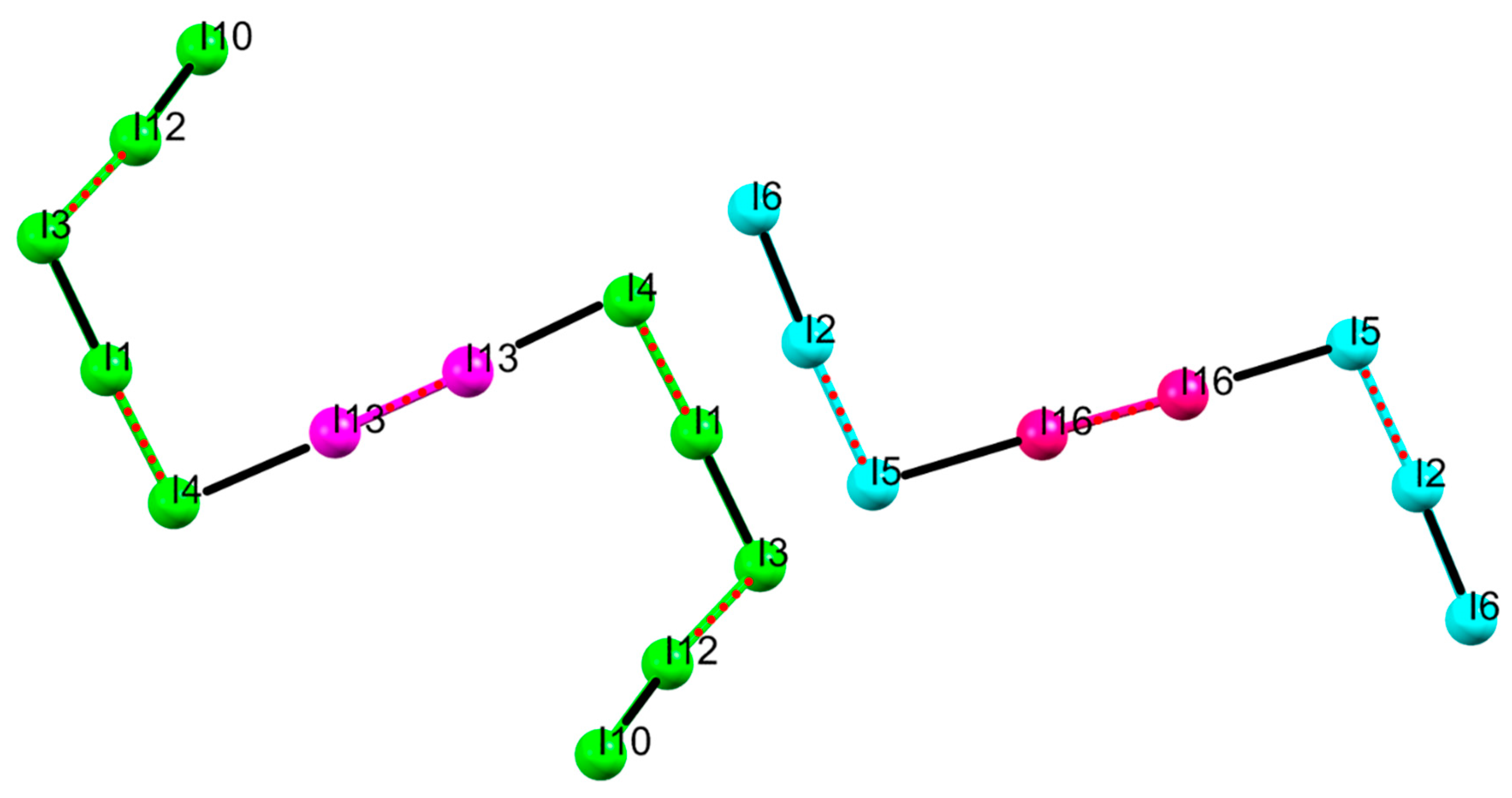
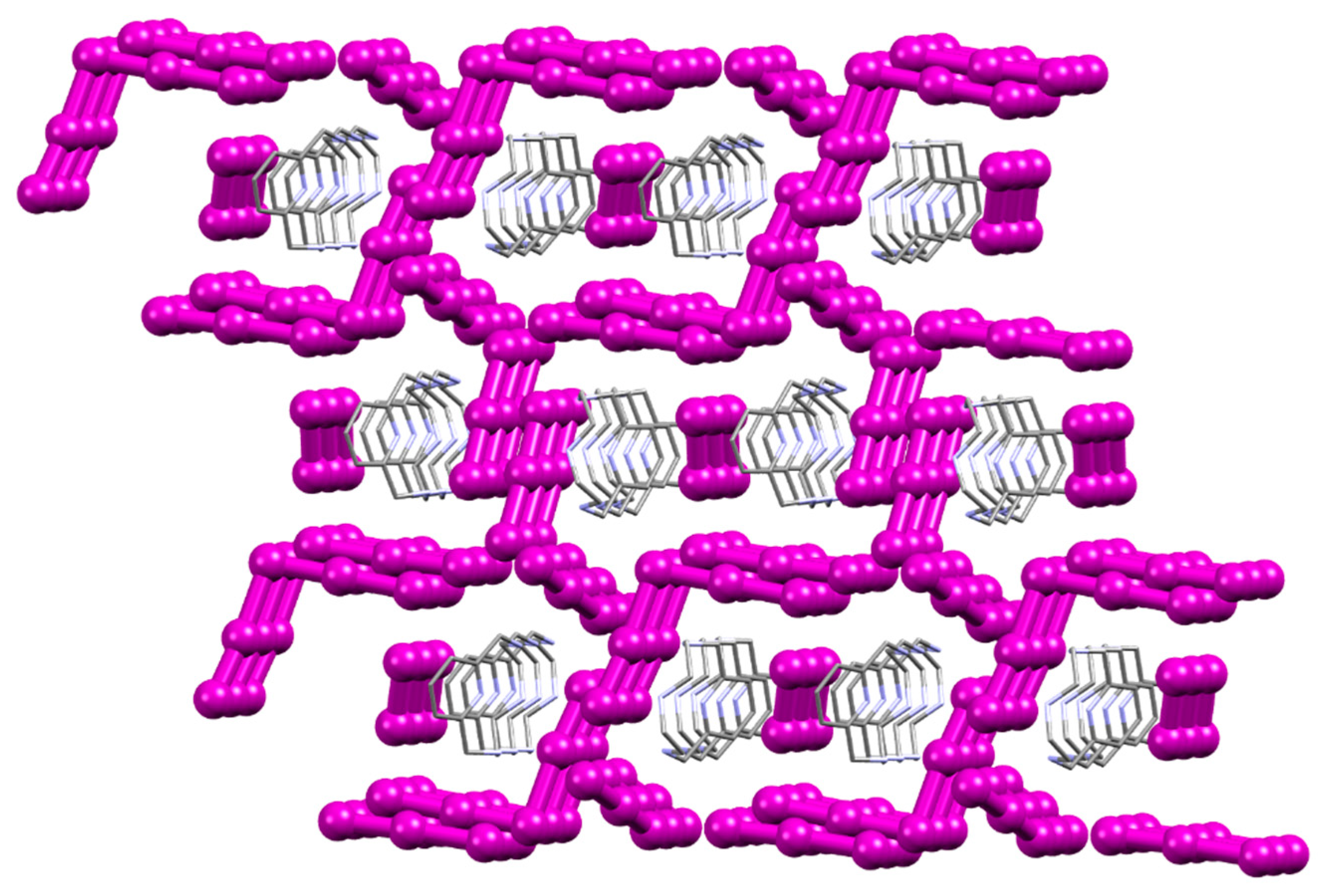
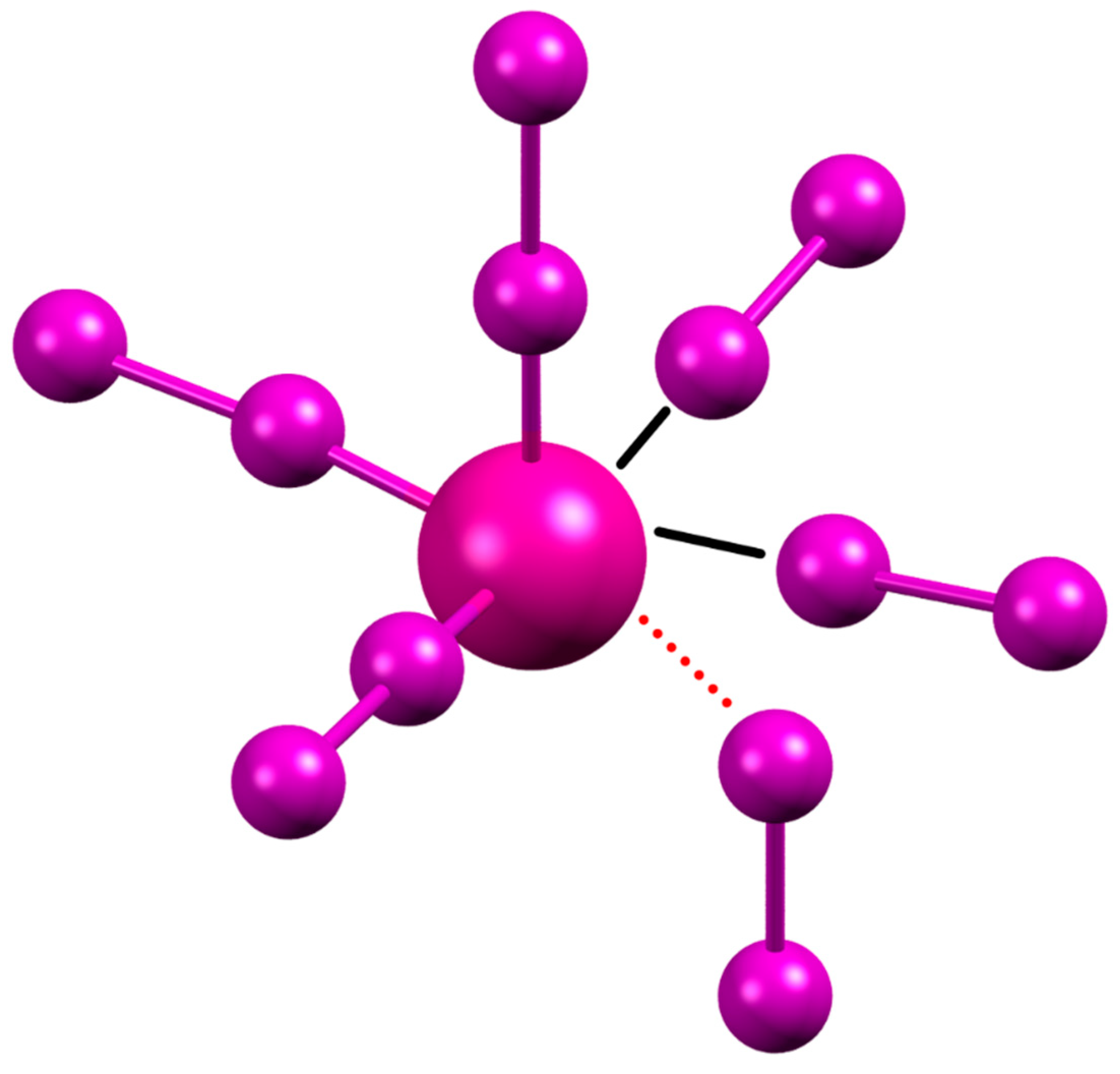
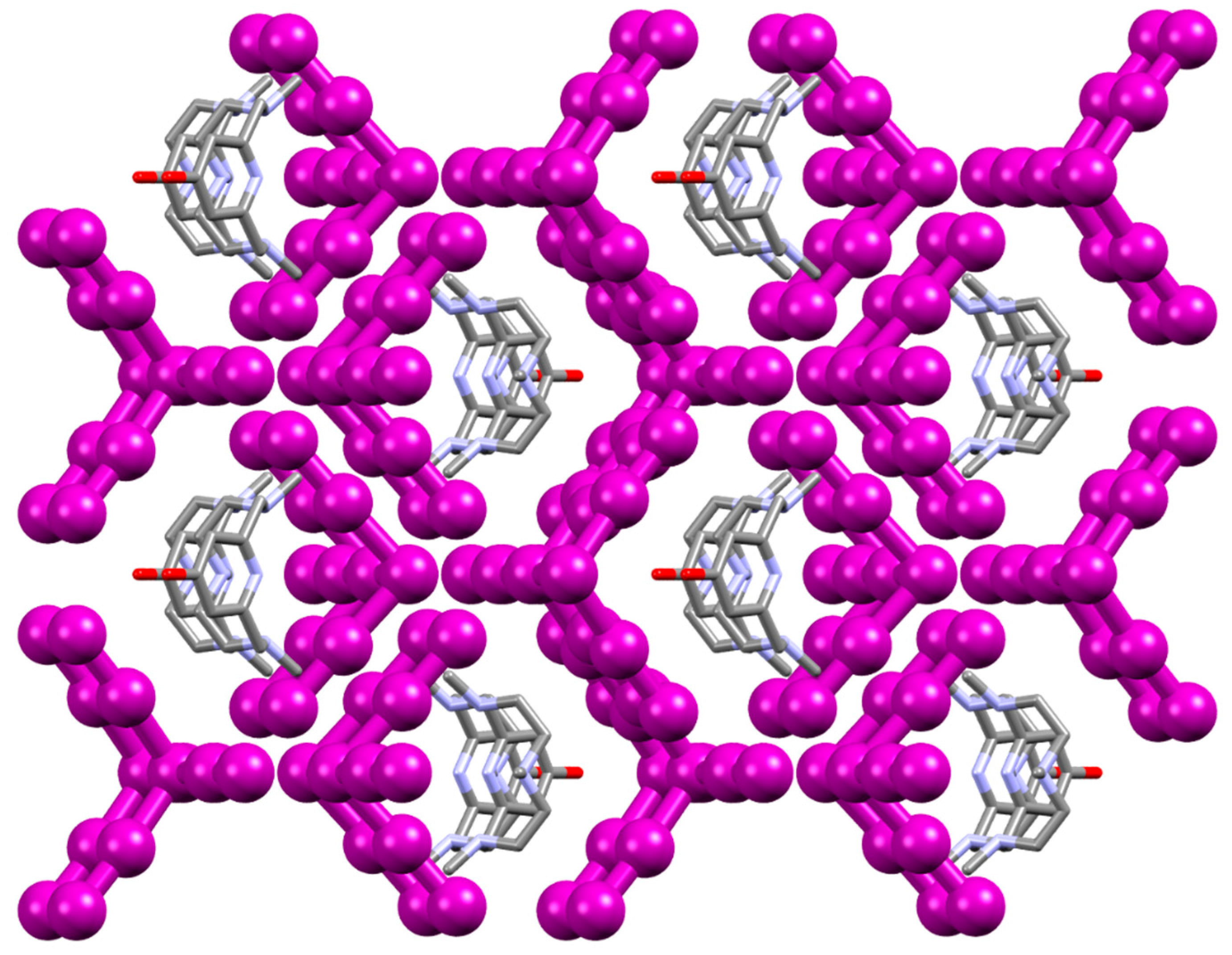
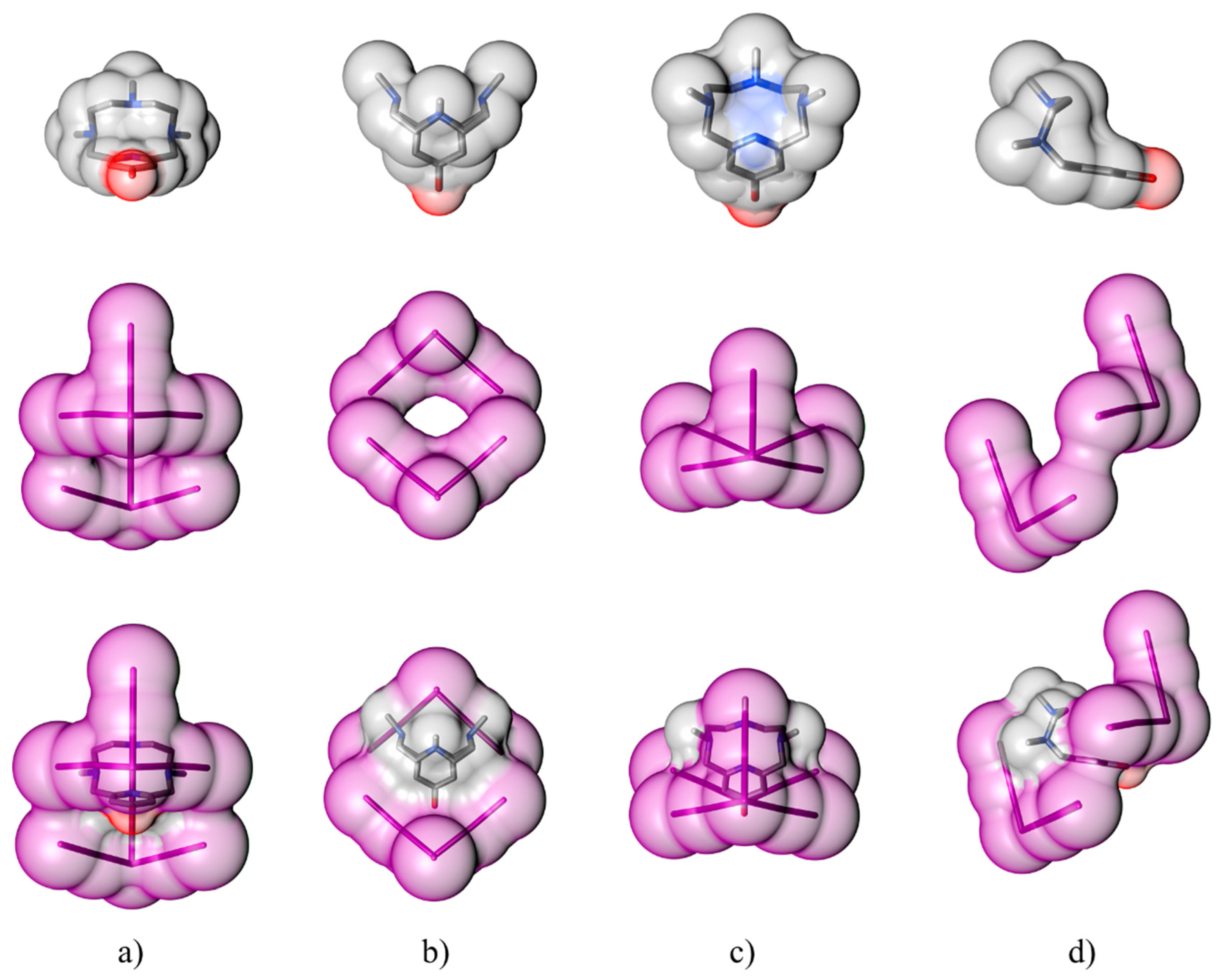
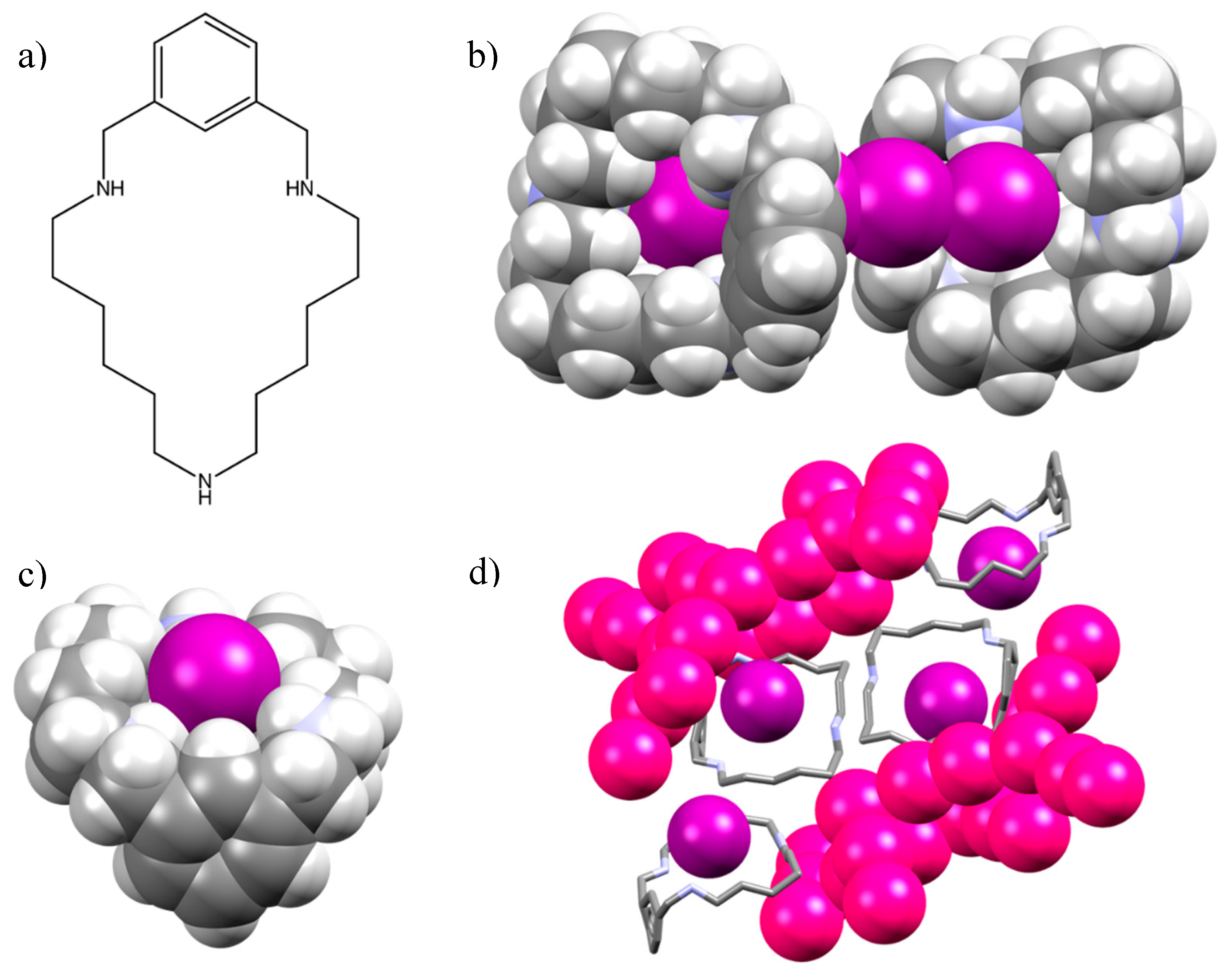
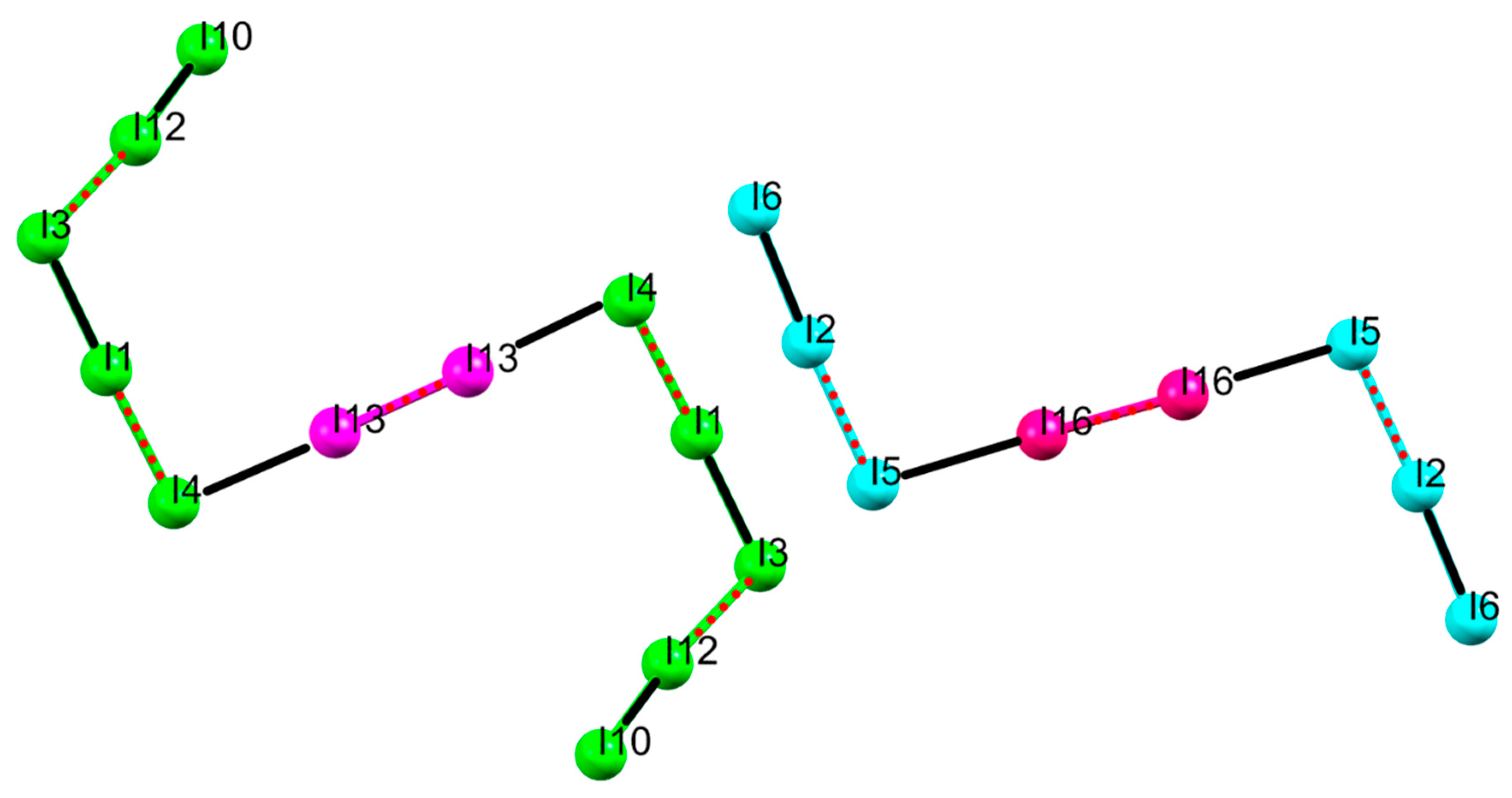
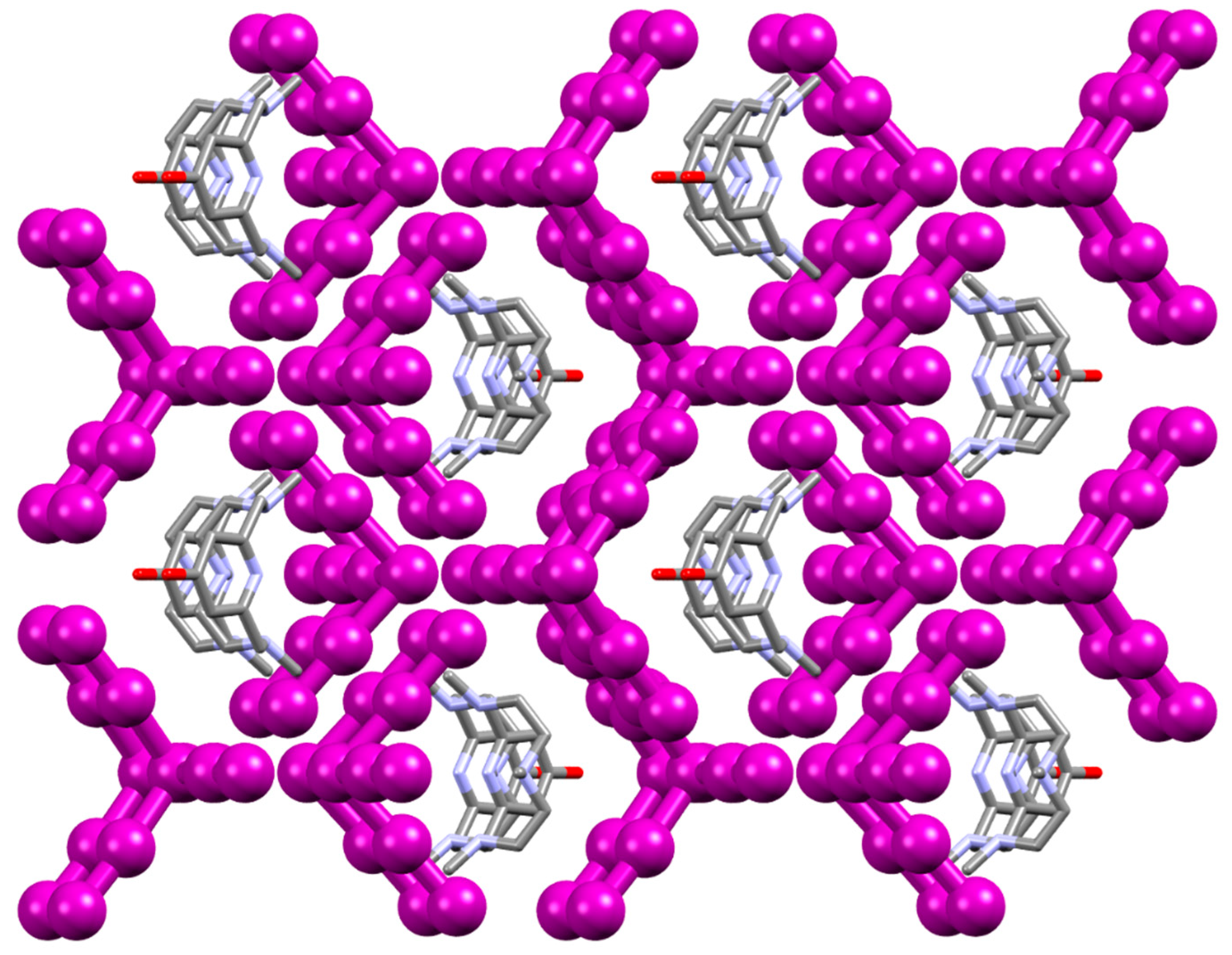
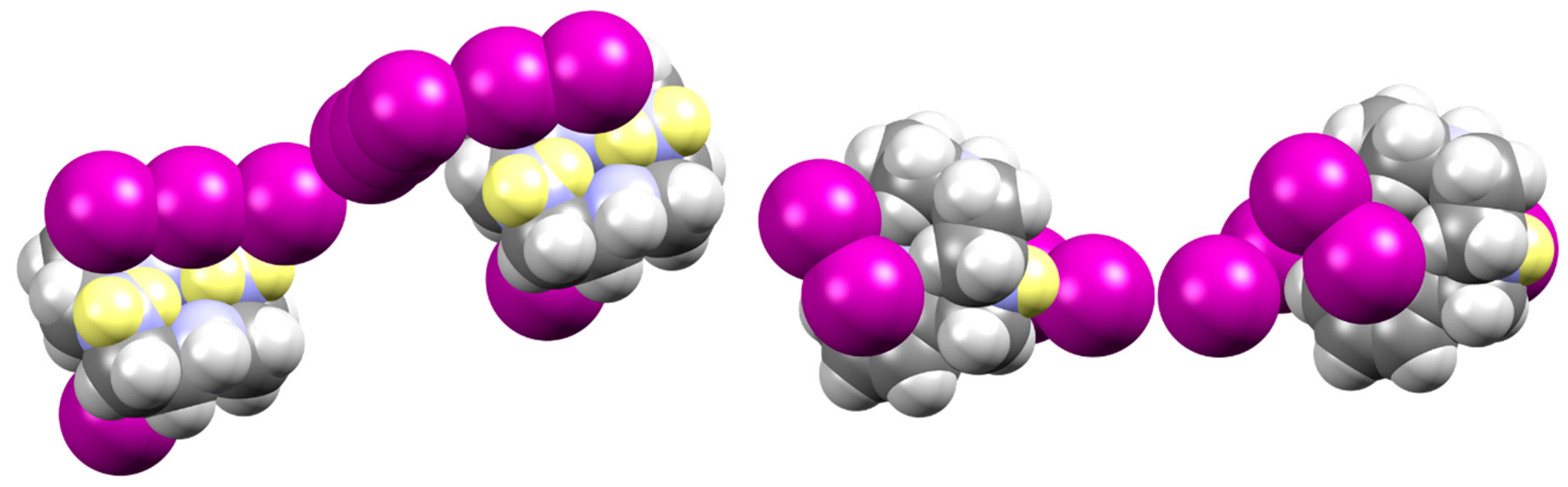
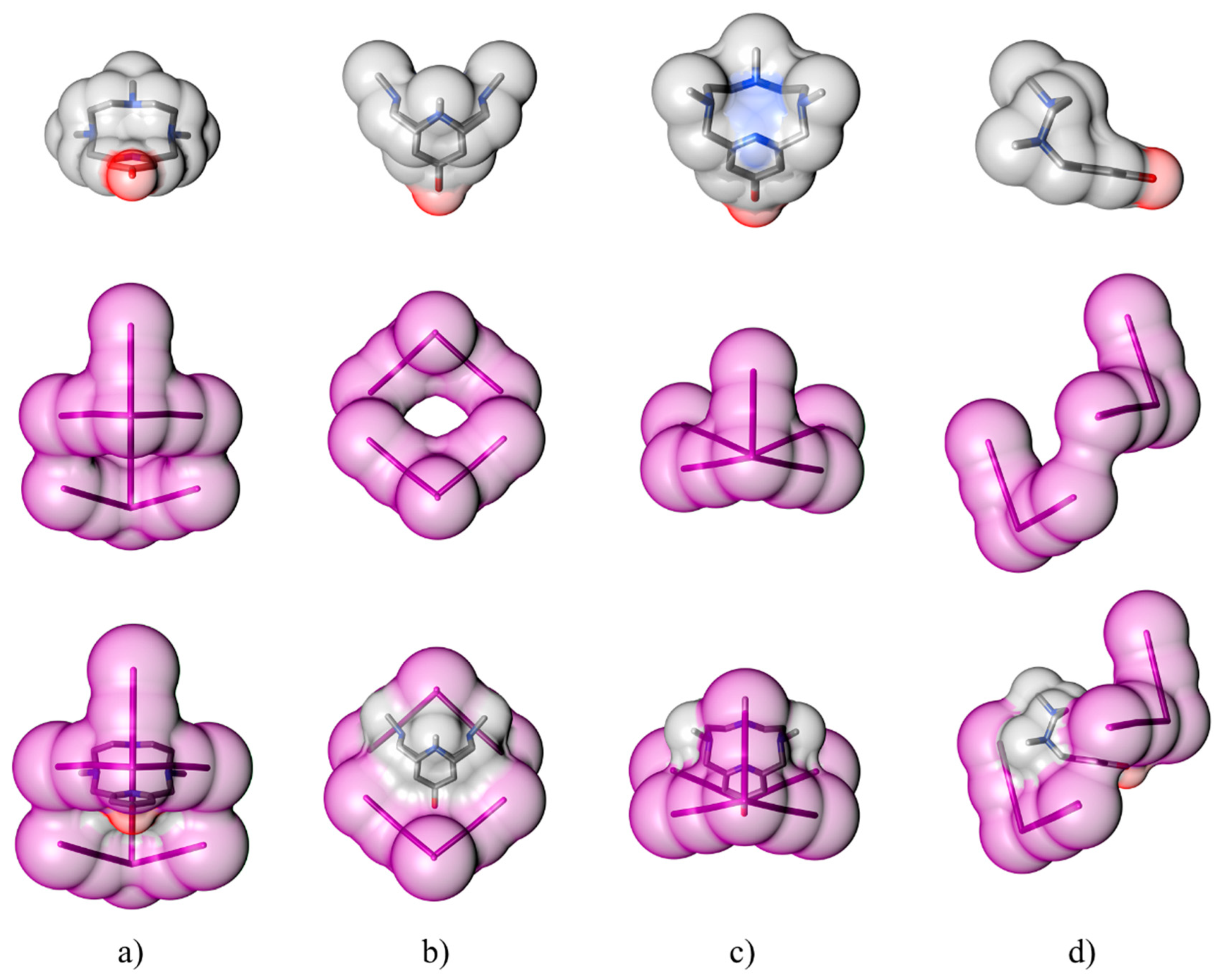
2.1. Crystal Structure of [(H2L1)2I2(I5)(I3)3]
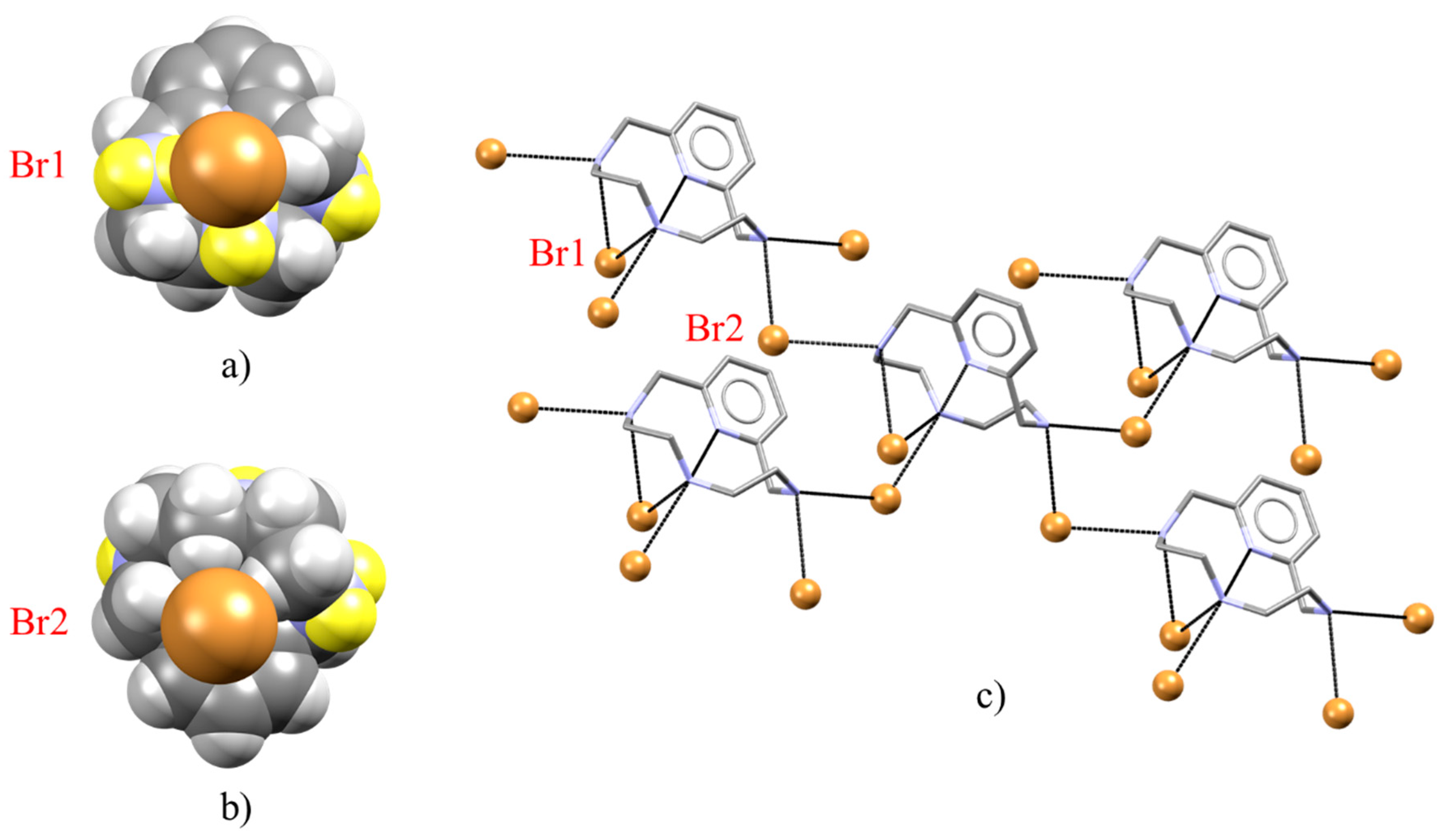
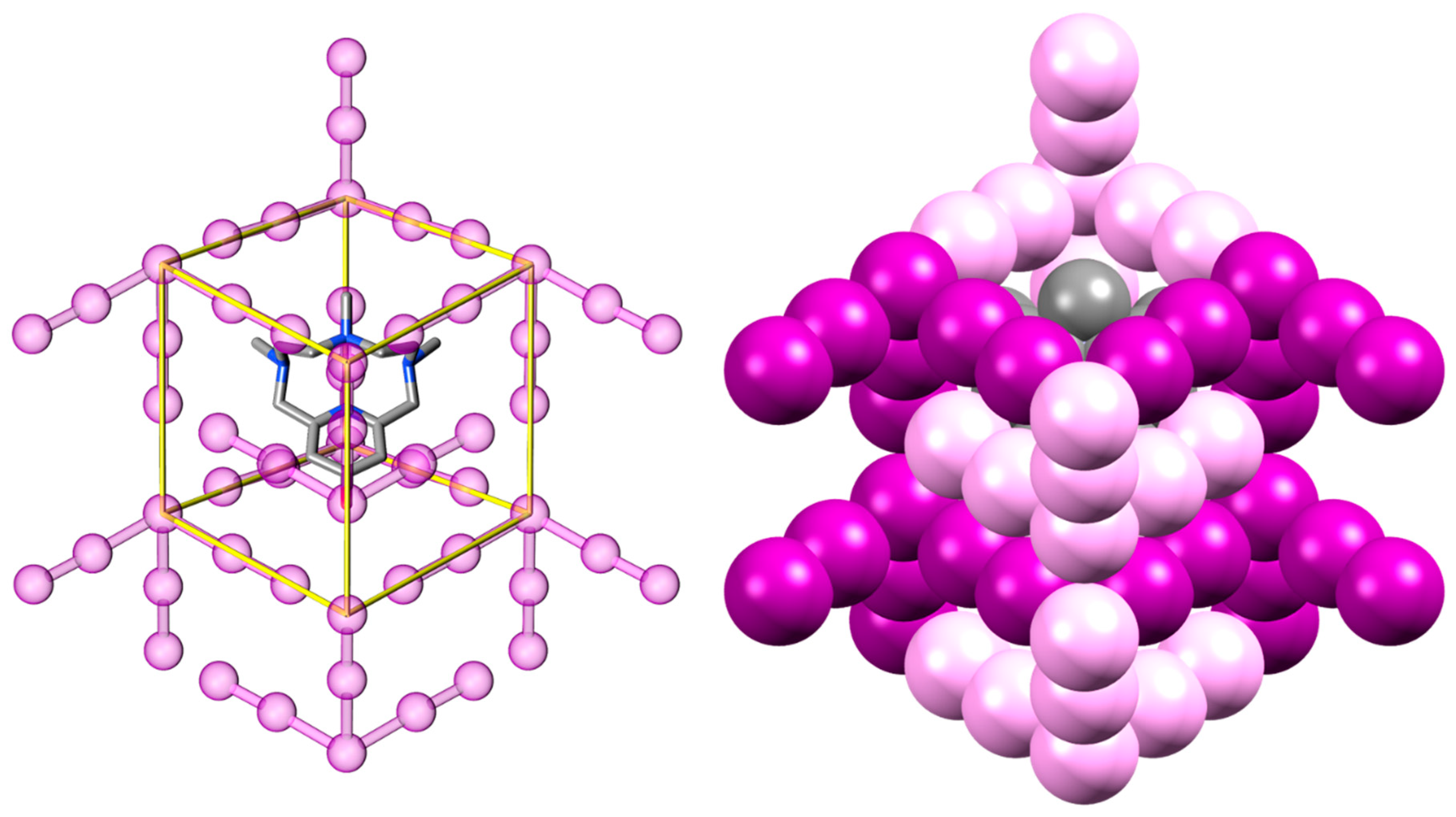
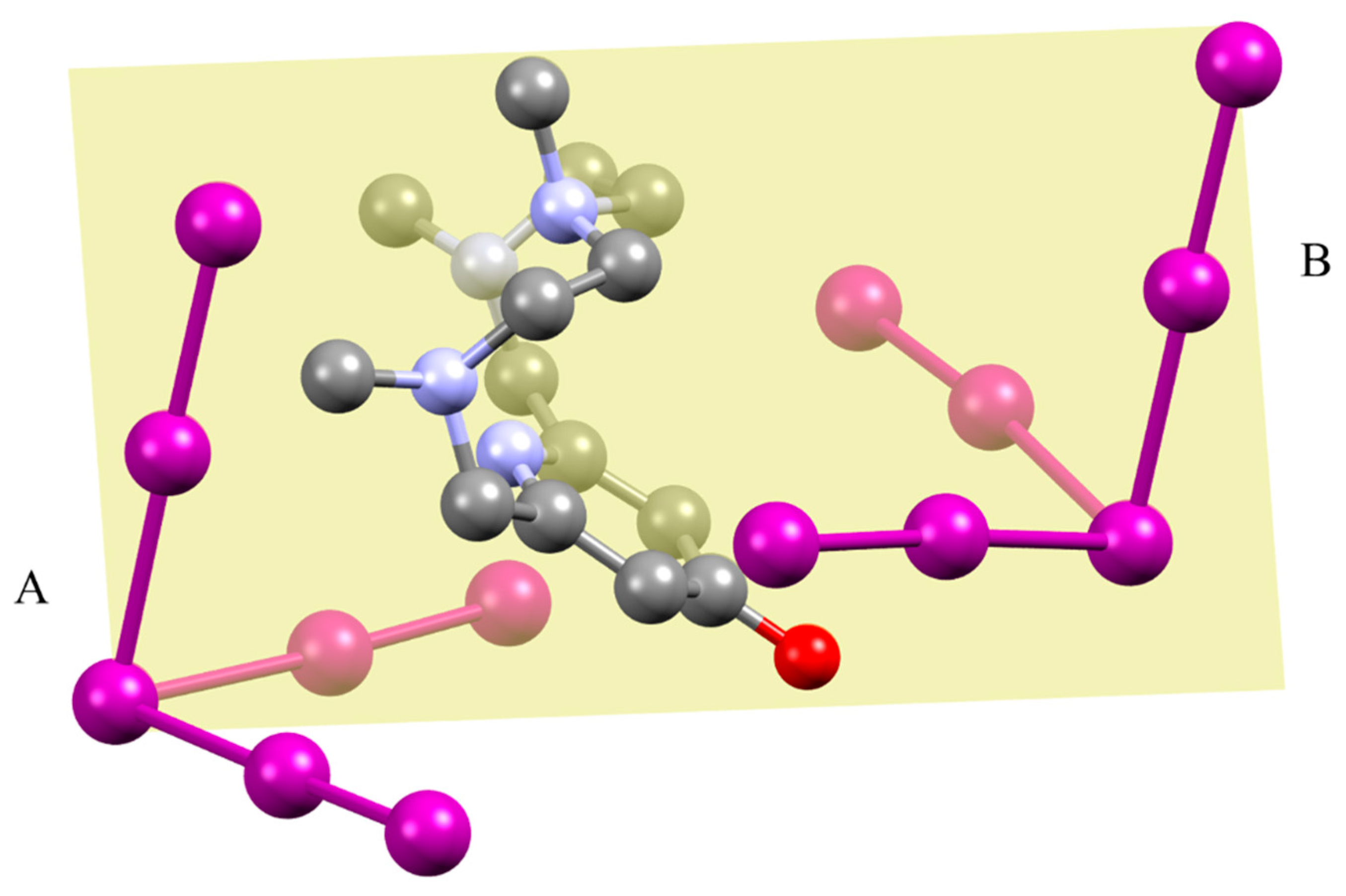
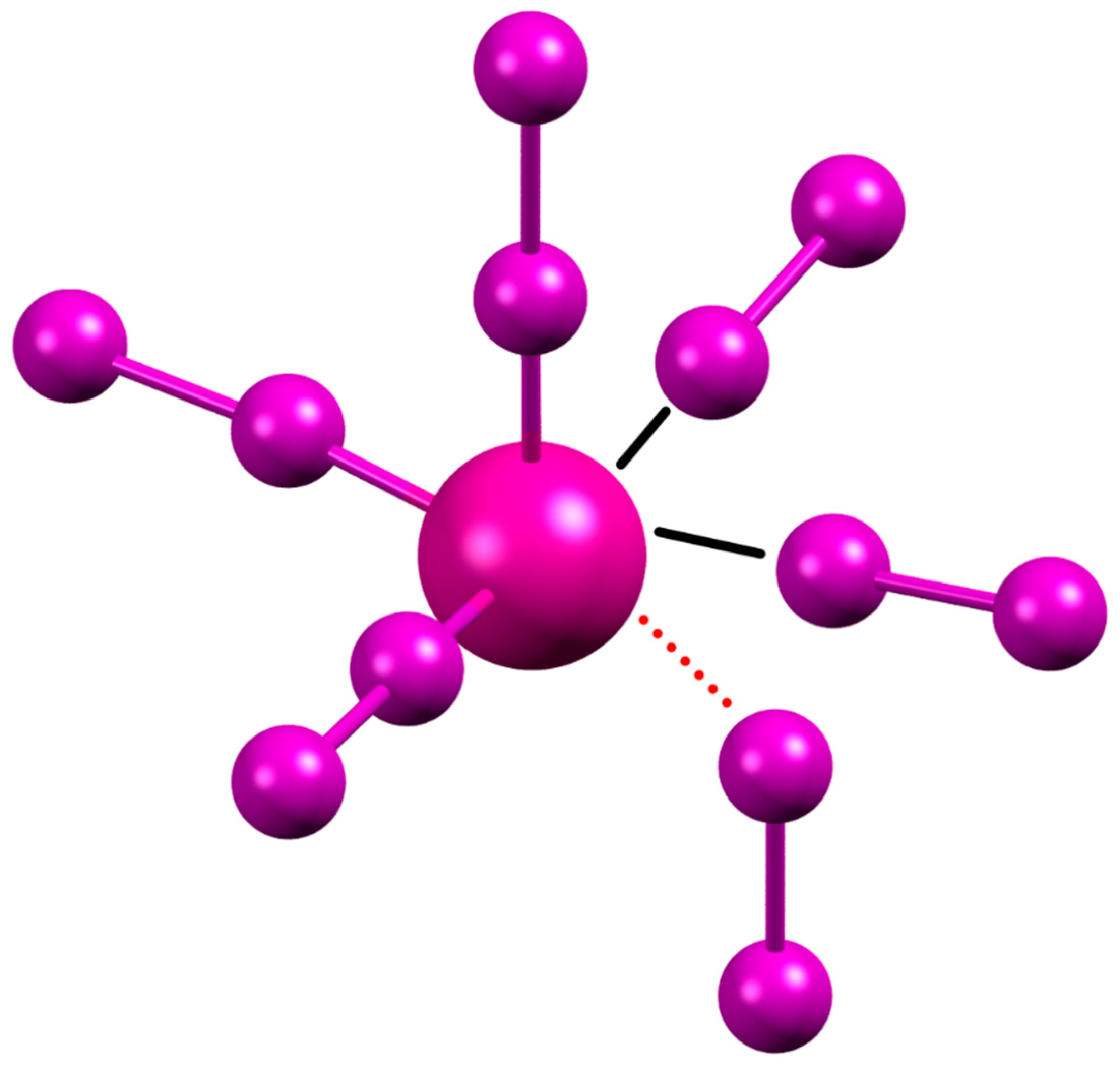
2.2. Crystal Structure of [H2L2-Me3(I7)2]
3. Discussion
4. Materials and Methods
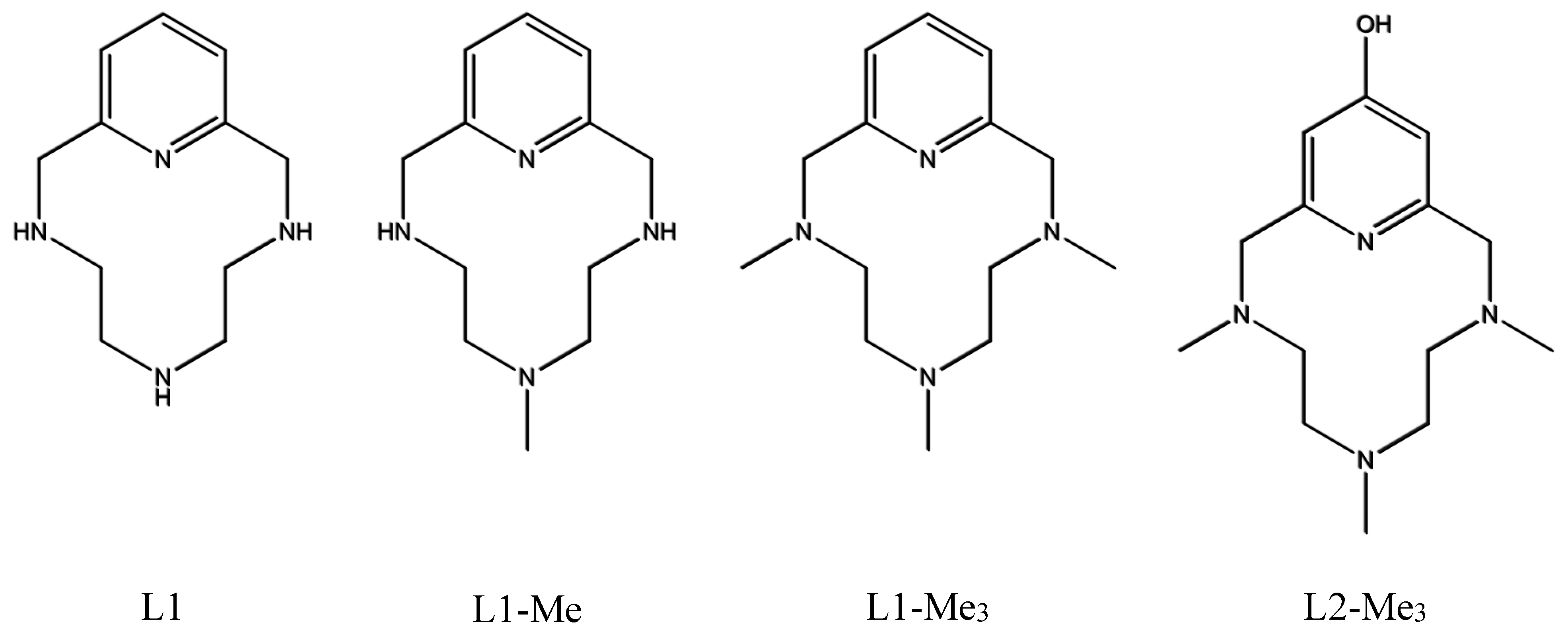
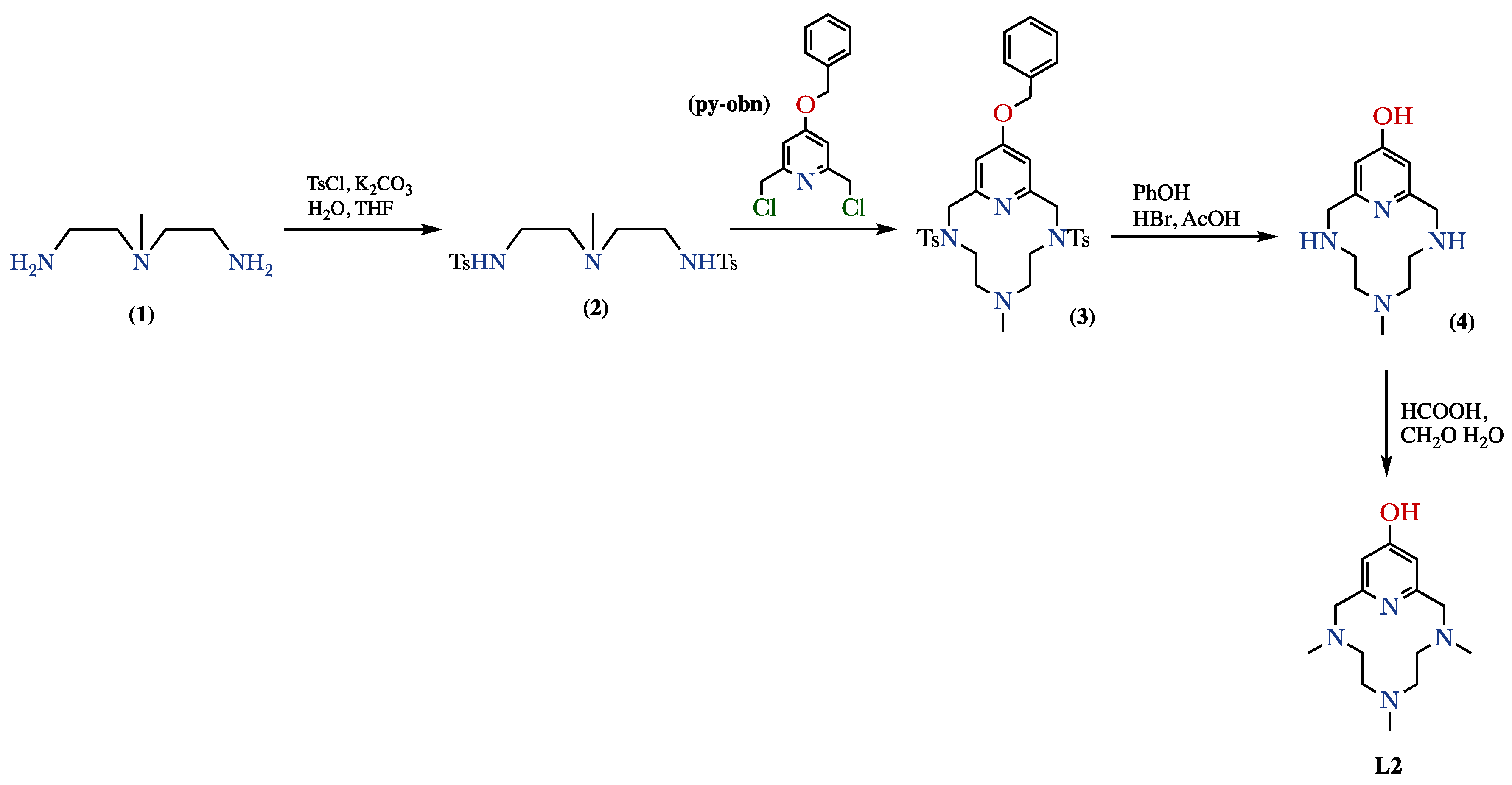
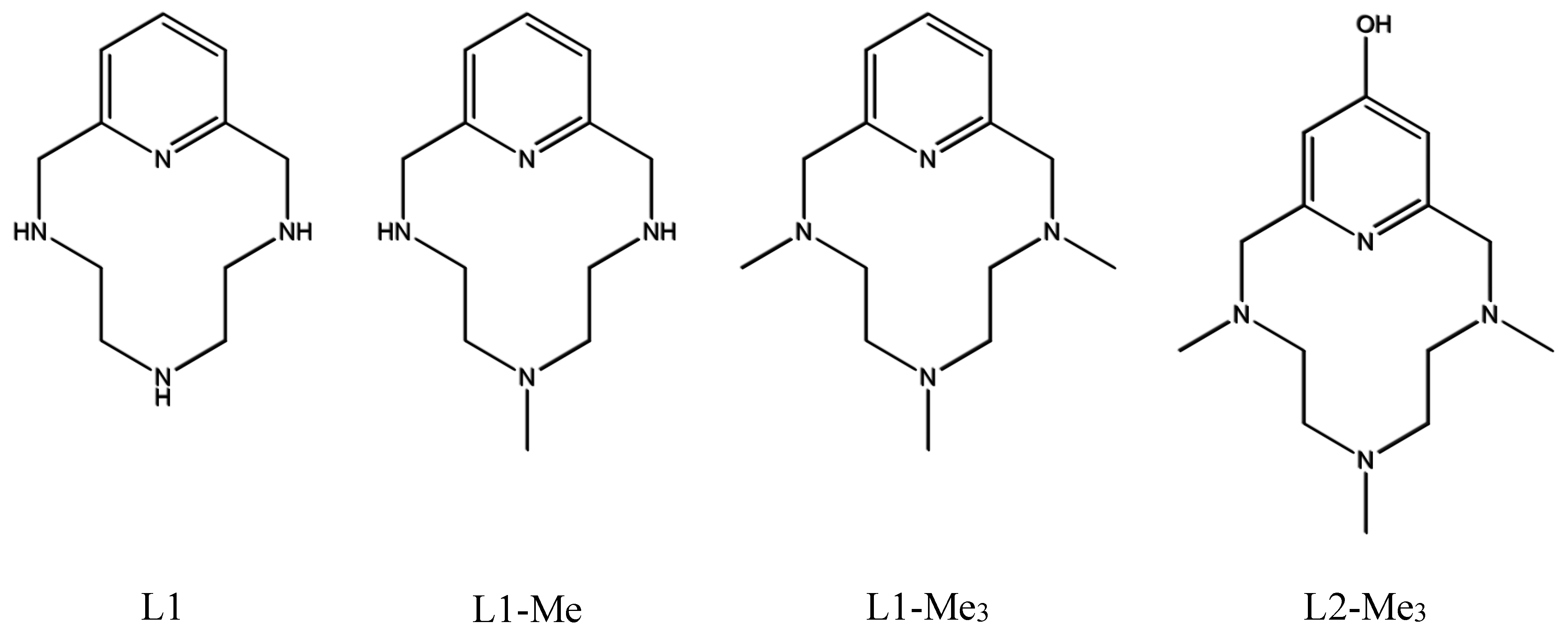
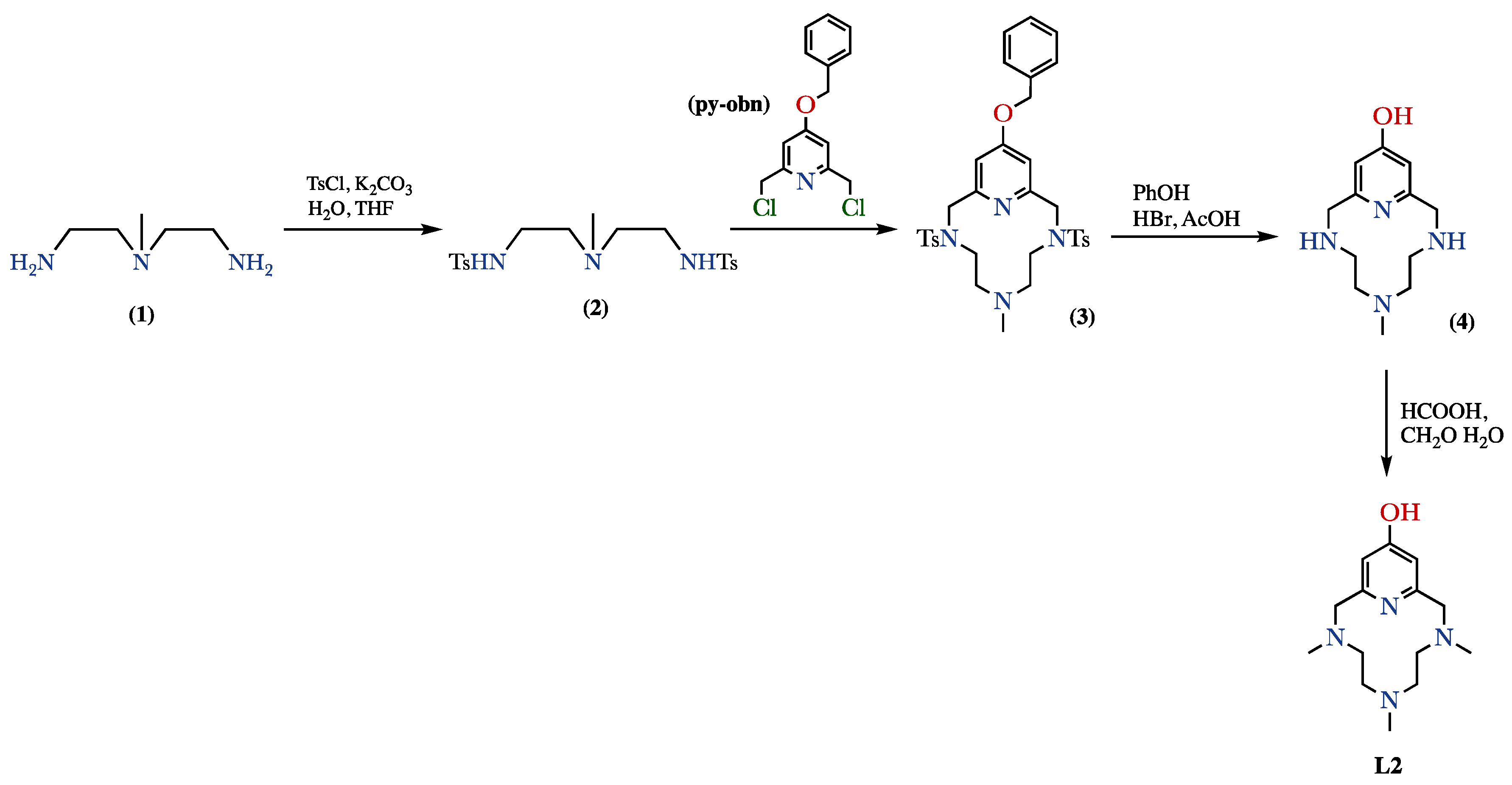
4.1. Ligand Synthesis
4.1.1. Synthesis of 14-Benziloxy-6-(N-methyl)-3,9-bis(p-tolylsulfonyl)-3,6,9-triaza-1(2,6)- pyridinacyclodecaphane (3)
4.1.2. Synthesis of 6-(N-Methyl)-3,6,9-triaza-1(2,6)-pyridinacyclodecaphan-14-ol hydrobromide (4·3HBr)
4.1.3. Synthesis of 3,6,9-Tris(N-methyl)-3,6,9-triaza-1(2,6)-pyridinacyclodecaphan-14-ol (L2–Me3)
4.2. X-ray Structure Analysis
4.2.1. Crystallographic Information for [(H2L1)2I2(I5)(I3)3]
4.2.2. Crystallographic Information for [H2L2–Me3(I7)2]
4.2.3. Software
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gay-Lussac, J.L. Mémoire sur l’iode. Ann. Chimie 1814, 91, 5–160. [Google Scholar]
- Jones, G. On the Existence and Behavior of Complex Polyiodides. J. Phys. Chem. 1930, 34, 673–691. [Google Scholar] [CrossRef]
- Svensson, P.H.; Kloo, L. Synthesis, structure, and bonding in polyiodide and metal iodide-iodine systems. Chem. Rev. 2003, 103, 1649–1684. [Google Scholar] [CrossRef] [PubMed]
- Svensson, P.H.; Gorlov, M.; Kloo, L. Dimensional caging of polyiodides. Inorg. Chem. 2008, 47, 11464–11466. [Google Scholar] [CrossRef] [PubMed]
- Abate, A.; Brischetto, M.; Cavallo, G.; Lahtinen, M.; Metrangolo, P.; Pilati, T.; Radice, S.; Resnati, G.; Rissanen, K.; Terraneo, G. Dimensional encapsulation of I−···I2···I− in an organic salt crystal matrix. Chem. Commun. 2010, 46, 2724–2726. [Google Scholar] [CrossRef] [PubMed]
- García, M.D.; Martí-Rujas, J.; Metrangolo, P.; Peinador, C.; Pilati, T.; Resnati, G.; Terraneo, G.; Ursini, M. Dimensional caging of polyiodides: Cation-templated synthesis using bipyridinium salts. CrystEngComm 2011, 13, 4411–4416. [Google Scholar] [CrossRef]
- Reiss, G.J.; van Megen, M. Two new polyiodides in the 4,4′-bipyridinium diiodide/iodine system. Z. Nat. B 2012, 67, 5–10. [Google Scholar]
- Van Megen, M.; Reiss, G.J. The pseudosymmetric structure of bis(pentane-1,5-diaminium) iodide tris(triiodide). Acta Cryst. E 2012, 68, o1331–o1332. [Google Scholar] [CrossRef]
- Garzón-Tovar, L.; Duarte-Ruiz, Á.; Wurst, K. Non-classical hydrogen bond (CH···I) directed self-assembly formation of a novel 1D supramolecular polymer, based on a copper complex [Cu{(CH3)2SO}6]I4. Inorg. Chem. Commun. 2013, 32, 64–67. [Google Scholar] [CrossRef]
- Müller, M.; Albrecht, M.; Gossen, V.; Peters, T.; Hoffmann, A.; Raabe, G.; Valkonen, A.; Rissanen, K. Anion–π interactions in salts with polyhalide anions: Trapping of I42−. Chem. Eur. J. 2010, 16, 12446–12453. [Google Scholar] [CrossRef]
- Van Megen, M.; Reiss, G.J. I62− Anion Composed of Two Asymmetric Triiodide Moieties: A Competition between Halogen and Hydrogen Bond. Inorganics 2013, 1, 3–13. [Google Scholar] [CrossRef]
- Wu, J.; Lan, Z.; Lin, J.; Huang, M.; Huang, Y.; Fan, L.; Luo, G. Electrolytes in Dye-Sensitized Solar Cells. Chem. Rev. 2015, 115, 2136–2173. [Google Scholar] [CrossRef] [PubMed]
- Bella, F.; Galliano, S.; Falco, M.; Viscardi, G.; Barolo, C.; Gratzel, M.; Gerbaldi, C. Unveiling iodine-based electrolytes chemistry in aqueous dye-sensitized solar cells. Chem. Sci. 2016, 7, 4880–4890. [Google Scholar] [CrossRef]
- De Grotthus, C.J.D. Sur la décomposition de l’eau et des corps qu’elle tient en dissolution à l’aide de l’électricité galvanique. Ann. Chim. 1806, 58, 54–73. [Google Scholar]
- Li, J.; Wang, Z.-S. Lithium-coordinating ionic conductor for solid-state dye-sensitized solar cells. RSC Adv. 2015, 5, 56967–56973. [Google Scholar] [CrossRef]
- Wang, H.; Li, J.; Gong, F.; Zhou, G.; Wang, Z.-S. Ionic Conductor with High Conductivity as Single-Component Electrolyte for Efficient Solid-State Dye-Sensitized Solar Cells. J. Am. Chem. Soc. 2013, 135, 12627–12633. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, H.; Xue, B.; Wang, Z.; Meng, Q.; Chen, L. Solid-State Composite Electrolyte LiI/3-Hydroxypropionitrile/SiO2 for Dye-Sensitized Solar Cells. J. Am. Chem. Soc. 2005, 127, 6394–6401. [Google Scholar] [CrossRef]
- Savastano, M.; Bazzicalupi, C.; Giorgi, C.; García-Gallarín, C.; López de la Torre, M.D.; Pichierri, F.; Bianchi, A.; Melguizo, M. Anion Complexes with Tetrazine-Based Ligands: Formation of Strong Anion–π Interactions in Solution and in the Solid State. Inorg. Chem. 2016, 55, 8013–8024. [Google Scholar] [CrossRef]
- Savastano, M.; Bazzicalupi, C.; García-Gallarín, C.; Gellini, C.; López de la Torre, M.D.; Mariani, P.; Pichierri, F.; Bianchi, A.; Melguizo, M. Iodide and triiodide anion complexes involving anion–π interactions with a tetrazine-based receptor. Dalton Trans. 2017, 46, 4518–4529. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, A.; García-España, E. Azacoronands and Azacyclophanes. In Supramolecular Chemistry: From Molecules to Nanomaterials; Gale, P.A., Steed, J.W., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2012. [Google Scholar] [CrossRef]
- Ilioudis, C.A.; Steed, J.W. Complexation of I42− and I82− by protonated azacyclophanes. CrystEngComm 2004, 6, 239–242. [Google Scholar] [CrossRef]
- Drahos, B.; Kotek, J.; Cisarova, I.; Hermann, P.; Helm, L.; Lukes, I.; Toth, E. Mn2+ Complexes with 12-Membered Pyridine Based Macrocycles Bearing Carboxylate or Phosphonate Pendant Arm: Crystallographic, Thermodynamic, Kinetic, Redox, and 1H/17O Relaxation Studies. Inorg. Chem. 2011, 50, 12785–12801. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Camarena, Á.; Liberato, A.; Delgado-Pinar, E.; Algarra, A.G.; Pitarch-Jarque, J.; Llinares, J.M.; Mañez, M.Á.; Domenech-Carbó, A.; Basallote, M.G.; García-España, E. Coordination Chemistry of Cu2+ Complexes of Small N-Alkylated Tetra-azacyclophanes with SOD Activity. Inorg. Chem. 2018, 57, 10961–10973. [Google Scholar] [CrossRef] [PubMed]
- Verdejo, B.; Ferrer, A.; Blasco, S.; Castillo, C.E.; Gonzalez, J.; Ĺatorre, J.; Máñez, M.A.; Basallote, M.G.; Soriano, C.; García España, E. Hydrogen and Copper Ion-Induced Molecular Reorganizations in Scorpionand-like Ligands. A Potentiometric, Mechanistic, and Solid-State Study. Inorg. Chem. 2007, 46, 5707–5719. [Google Scholar] [CrossRef] [PubMed]
- Alcock, N.W. Secondary Bonding to Nonmetallic Elements. Adv. Inorg. Chem. Radiochem. 1972, 15, 1–58. [Google Scholar] [CrossRef]
- Savastano, M.; Arranz-Mascarós, P.; Bazzicalupi, C.; Clares, M.P.; Godino-Salido, M.L.; Gutiérrez-Valero, M.D.; Inclán, M.; Bianchi, A.; García-España, E.; López-Garzón, R. Construction of green nanostructured heterogeneous catalysts via non-covalent surface decoration of multi-walled carbon nanotubes with Pd(II) complexes of azamacrocycles. J. Catal. 2017, 353, 239–249. [Google Scholar] [CrossRef]
- Bazzicalupi, C.; Bencini, A.; Biagini, S.; Bianchi, A.; Faggi, E.; Giorgi, C.; Marchetta, M.; Totti, F.; Valtancoli, B. Polyamine receptors containing dipyridine or phenanthroline units: Clues for the design of fluorescent chemosensors for metal ions. Chem. Eur. J. 2009, 15, 8049–8063. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef] [PubMed]
- Rimmer, E.L.; Bailey, R.D.; Pennington, W.T.; Hanks, T.W. The reaction of iodine with 9-methylacridine: Formation of polyiodide salts and a charge-transfer complex. J. Chem. Soc. Perkin Trans. 2 1998, 2557–2562. [Google Scholar] [CrossRef]
- Aragoni, M.C.; Arca, M.; Demartin, F.; Devillanova, F.A.; Garau, A.; Isaia, F.; Lippolis, V.; Rizzato, S.; Verani, G. [Ni(L)(MeCN)]2+ complex cation as a template for the assembly of extended I3−···I5− and I5−···I7− polyiodide networks {L = 2,5,8-trithia[9](2,9)-1,10-phenanthrolinophane}. Synthesis and structures of [Ni(L)(MeCN)]I8 and [Ni(L)(MeCN)]I12. Inorg. Chim. Acta 2004, 357, 3803–3809. [Google Scholar] [CrossRef]
- Demartin, F.; Deplano, P.; Devillanova, F.A.; Isaia, F.; Lippolis, V.; Verani, G. Conductivity, FT-Raman spectra, and X-ray crystal structures of two novel [D2I]In (n = 3 and D = N-methylbenzothiazole-2(3H)-selone; n = 7 and D = N-methylbenzothiazole-2(3H)-thione) iodonium salts. First example of I−·3I2 heptaiodide. Inorg. Chem. 1993, 32, 3694–3699. [Google Scholar] [CrossRef]
- Corban, G.J.; Hadjikakou, S.K.; Tsipis, A.C.; Kubicki, M.; Bakas, T.; Hadjiliadis, N. Inhibition of peroxidase-catalyzed iodination by thioamides: Experimental and theoretical study of the antithyroid activity of thioamides. New J. Chem. 2011, 35, 213–224. [Google Scholar] [CrossRef]
- Tebbe, K.; Nagel, K. Untersuchungen an Polyhalogeniden. XXVI [1]. Über N-Propylurotropiniumpolyiodide UrPrIx mit x = 5 und 7: Strukturelle Charakterisierung eines Pentaiodids und eines Heptaiodids. Zeitschrift Anorganische Allgemeine Chemie 1996, 622, 1323–1328. [Google Scholar] [CrossRef]
- Poli, R.; Gordon, J.C.; Khanna, R.K.; Fanwick, P.E. The first discrete structure for the heptaiodide ion. Inorg. Chem. 1992, 31, 3165–3167. [Google Scholar] [CrossRef]
- Renner, M.W.; Barkigia, K.M.; Zhang, Y.; Medforth, C.J.; Smith, K.M.; Fajer, J. Consequences of Oxidation in Nonplanar Porphyrins: Molecular Structure and Diamagnetism of the. pi. Cation Radical of Copper(II) Octaethyltetraphenylporphyrin. J. Am. Chem. Soc. 1994, 116, 8582–8592. [Google Scholar] [CrossRef]
- Svensson, P.H.; Raud, G.; Kloo, L. Metal Iodides in Polyiodide Networks—The Structural Chemistry of Complex Thallium Iodides with Excess Iodine. Eur. J. Inorg. Chem. 2000, 1275–1282. [Google Scholar] [CrossRef]
- Fiolka, C.; Pantenburg, I.; Meyer, G. Transition-Metal(II)–Crown-Ether–Polyiodides. Cryst. Growth Des. 2011, 11, 5159–5165. [Google Scholar] [CrossRef]
- Hendrixson, T.L.; ter Horst, M.A.; Jacobson, R.A. Structure of dipyridinium decaiodide—An infinite chain structure. Acta Cryst. 1991, C47, 2141–2144. [Google Scholar] [CrossRef]
- Stetter, H.; Frank, W.; Mertens, R. Darstellung und komplexbildung von polyazacycloalkan-N-essigsäuren. Tetrahedron 1981, 37, 767–772. [Google Scholar] [CrossRef]
- Wu, C.; He, Y. Synthesis of azacrown containing pyridine ring. Youji Huaxue 1983, 3(6), 437–439. [Google Scholar]
- Costa, J.; Delgado, R. Metal complexes of macrocyclic ligands containing pyridine. Inorg. Chem. 1993, 32, 5257–5265. [Google Scholar] [CrossRef]
- Meijer, A. Manganese chelates and their use as contrast agents in magnetic resonance imaging (MRI). U.S. Patent WO 2011/073371 A1, 23 June 2011. [Google Scholar]
- Eschweile, W. Ersatz von an Stickstoff gebundenen Wasserstoffatomen durch die Methylgruppe mit Hülfe von Formaldehyd. Berichte Deutschen Chemischen Gesellschaft 1905, 38, 880–882. [Google Scholar] [CrossRef]
- Clarke, H.T.; Gillespie, B.H.; Weisshaus, S.Z. The Action of Formaldehyde on Amines and Amino Acids. J. Am. Chem. Soc. 1933, 55, 4571–4587. [Google Scholar] [CrossRef]
- Serrano-Plana, J.; Oloo, W.N.; Acosta-Rueda, L.; Meier, K.K.; Verdejo, B.; García-España, E.; Basallote, M.G.; Münck, E.; Que, L.; Company, A.; et al. Trapping a Highly Reactive Nonheme Iron Intermediate That Oxygenates Strong C–H Bonds with Stereoretention. J. Am. Chem. Soc. 2015, 137, 15833–15842. [Google Scholar] [CrossRef]
- Busto, E.; González-Álvarez, A.; Gotor-Fernández, V.; Alfonso, I.; Gotor, V. Optically active macrocyclic hexaazapyridinophanes decorated at the periphery: Synthesis and applications in the NMR enantiodiscrimination of carboxylic acids. Tetrahedron 2010, 66, 6070–6077. [Google Scholar] [CrossRef]
- Froidevaux, P.; Harrowfield, J.M.; Sobolev, A.N. Calixarenes as Scaffolds: Introduction of Tridentate Rare Earth Metal Binding Units into Calix[4]arene. Inorg. Chem. 2000, 39, 4678–4687. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Camarena, Á.; Delgado-Pinar, E.; Soriano, C.; Alarcón, J.; Llinares, J.M.; Tejero, R.; García-España, E. Enhancement of SOD activity in boehmite supported nanoreceptors. Chem. Commun. 2018, 54, 3871–3874. [Google Scholar] [CrossRef]
- Richman, J.E.; Atkins, T.J. Nitrogen analogs of crown ethers. J. Am. Chem. Soc. 1974, 96, 2268–2270. [Google Scholar] [CrossRef]
- Richman, J.E.; Atkins, T.J.; Oetle, W.T. Organic Synthesis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1988; Volume VI. [Google Scholar]
- Shaw, B.L. Formation of large rings, internal metalation reactions, and internal entropy effects. J. Am. Chem. Soc. 1975, 97, 3856–3857. [Google Scholar] [CrossRef]
- Haskell, B.E.; Bowlus, S.B. New synthesis of L-2-amino-3-oxalylaminopropionic acid, the Lathyrus sativus neurotoxin. J. Org. Chem. 1976, 41, 159–160. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Murray, J.S.; Politzer, P.; Pilati, T.; Ursini, M.; Resnati, G. Halogen bonding in hypervalent iodine and bromine derivatives: Halonium salts. IUCrJ 2017, 4 (Pt 4), 411–419. [Google Scholar] [CrossRef]
- Catalano, L.; Cavallo, G.; Metrangolo, P.; Resnati, G.; Terraneo, G. Halogen Bonding in Hypervalent Iodine Compounds. In Hypervalent Iodine Chemistry; Wirth, T., Ed.; Topics in Current Chemistry; Springer: Cham, Switzerland, 2016; Volume 373. [Google Scholar]
- Terraneo, G.; Resnati, G.; Metrangolo, P. Iodine and Halogen Bonding. In Iodine Chemistry and Applications; Kaiho, T., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015. [Google Scholar]
- Bartashevich, E.V.; Yushina, I.D.; Stash, A.I.; Tsirelson, V.G. Halogen Bonding and Other Iodine Interactions in Crystals of Dihydrothiazolo(oxazino)quinolinium Oligoiodides from the Electron-Density Viewpoint. Cryst. Growth Des. 2014, 14, 5674–5684. [Google Scholar] [CrossRef]
- Arman, H.D.; Gieseking, R.L.; Hanks, T.W.; Pennington, W.T. Complementary halogen and hydrogen bonding: Sulfur···iodine interactions and thioamide ribbons. Chem. Commun. 2010, 46, 1854–1856. [Google Scholar] [CrossRef] [PubMed]
- Arman, H.D.; Rafferty, E.R.; Bayse, C.A.; Pennington, W.T. Complementary Selenium···Iodine Halogen Bonding and Phenyl Embraces: Cocrystals of Triphenylphosphine Selenide with Organoiodides. Cryst. Growth Des. 2012, 12, 4315–4323. [Google Scholar] [CrossRef]
- Bartashevich, E.; Yushina, I.; Kropotina, K.; Muhitdinova, S.; Tsirelson, V. Testing the tools for revealing and characterizing the iodine–iodine halogen bond in crystals. Acta Cryst. B 2017, 73, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Resnati, G.; Pennington, W.T. The halogen bond: A new avenue in recognition and self-assembly. New J. Chem. 2018, 42, 10461–10462. [Google Scholar] [CrossRef]
- Peuronen, A.; Valkonen, A.; Kortelainen, M.; Rissanen, K.; Lahtinen, M. Halogen bonding-based “catch and release”: Reversible solid-state entrapment of elemental iodine with monoalkylated dabco salts. Cryst. Growth Des. 2012, 12, 4157–4169. [Google Scholar] [CrossRef]
- Hu, J.; Wang, D.; Guo, W.; Du, S.; Tang, Z.K. Reversible control of the orientation of iodine molecules inside the AlPO4–11 crystals. J. Phys. Chem. C 2012, 116, 4423–4430. [Google Scholar] [CrossRef]
- Zeng, M.-H.; Wang, Q.-X.; Tan, Y.-X.; Hu, S.; Zhao, H.-X.; Long, L.-S.; Kurmoo, M. Rigid pillars and double walls in a porous metal-organic framework: Single-crystal to single-crystal, controlled uptake and release of iodine and electrical conductivity. J. Am. Chem. Soc. 2010, 132, 2561–2563. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savastano, M.; Martínez-Camarena, Á.; Bazzicalupi, C.; Delgado-Pinar, E.; Llinares, J.M.; Mariani, P.; Verdejo, B.; García-España, E.; Bianchi, A. Stabilization of Supramolecular Networks of Polyiodides with Protonated Small Tetra-azacyclophanes. Inorganics 2019, 7, 48. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7040048
Savastano M, Martínez-Camarena Á, Bazzicalupi C, Delgado-Pinar E, Llinares JM, Mariani P, Verdejo B, García-España E, Bianchi A. Stabilization of Supramolecular Networks of Polyiodides with Protonated Small Tetra-azacyclophanes. Inorganics. 2019; 7(4):48. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7040048
Chicago/Turabian StyleSavastano, Matteo, Álvaro Martínez-Camarena, Carla Bazzicalupi, Estefanía Delgado-Pinar, José M. Llinares, Palma Mariani, Begoña Verdejo, Enrique García-España, and Antonio Bianchi. 2019. "Stabilization of Supramolecular Networks of Polyiodides with Protonated Small Tetra-azacyclophanes" Inorganics 7, no. 4: 48. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7040048