Electrochemical and Computational Insights into the Reduction of [Fe2(CO)6{µ-(SCH2)2GeMe2}] Hydrogenase H-Cluster Mimic


Abstract
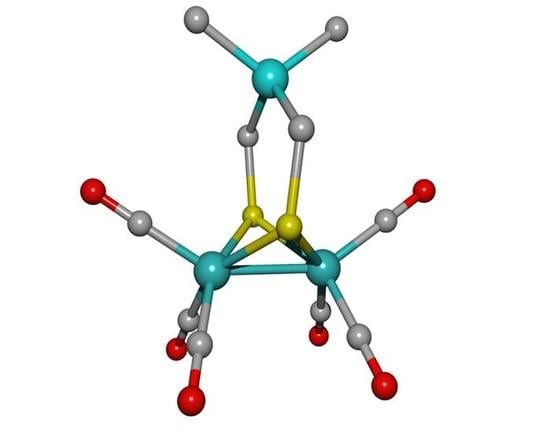
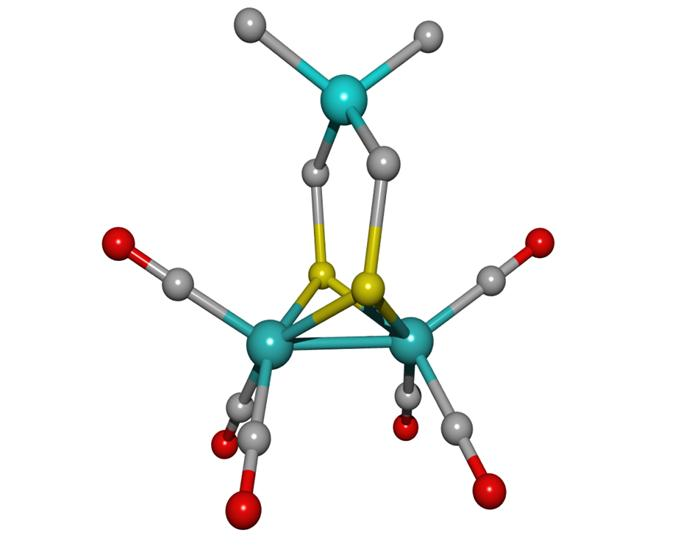
:
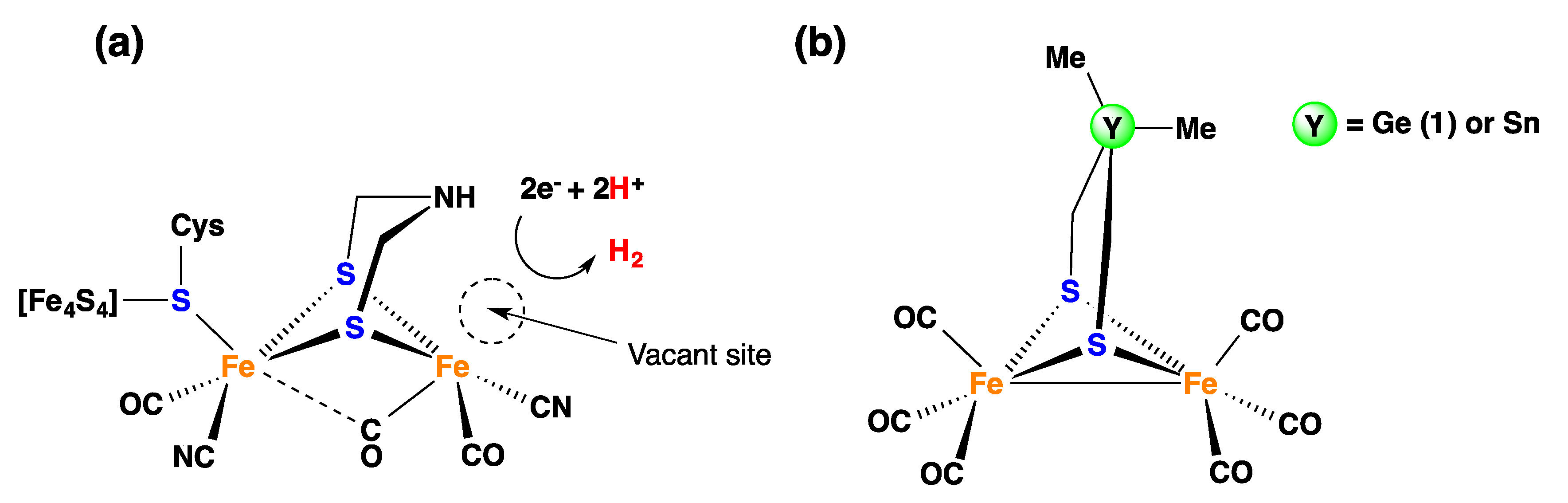
1. Introduction
2. Results and Discussion
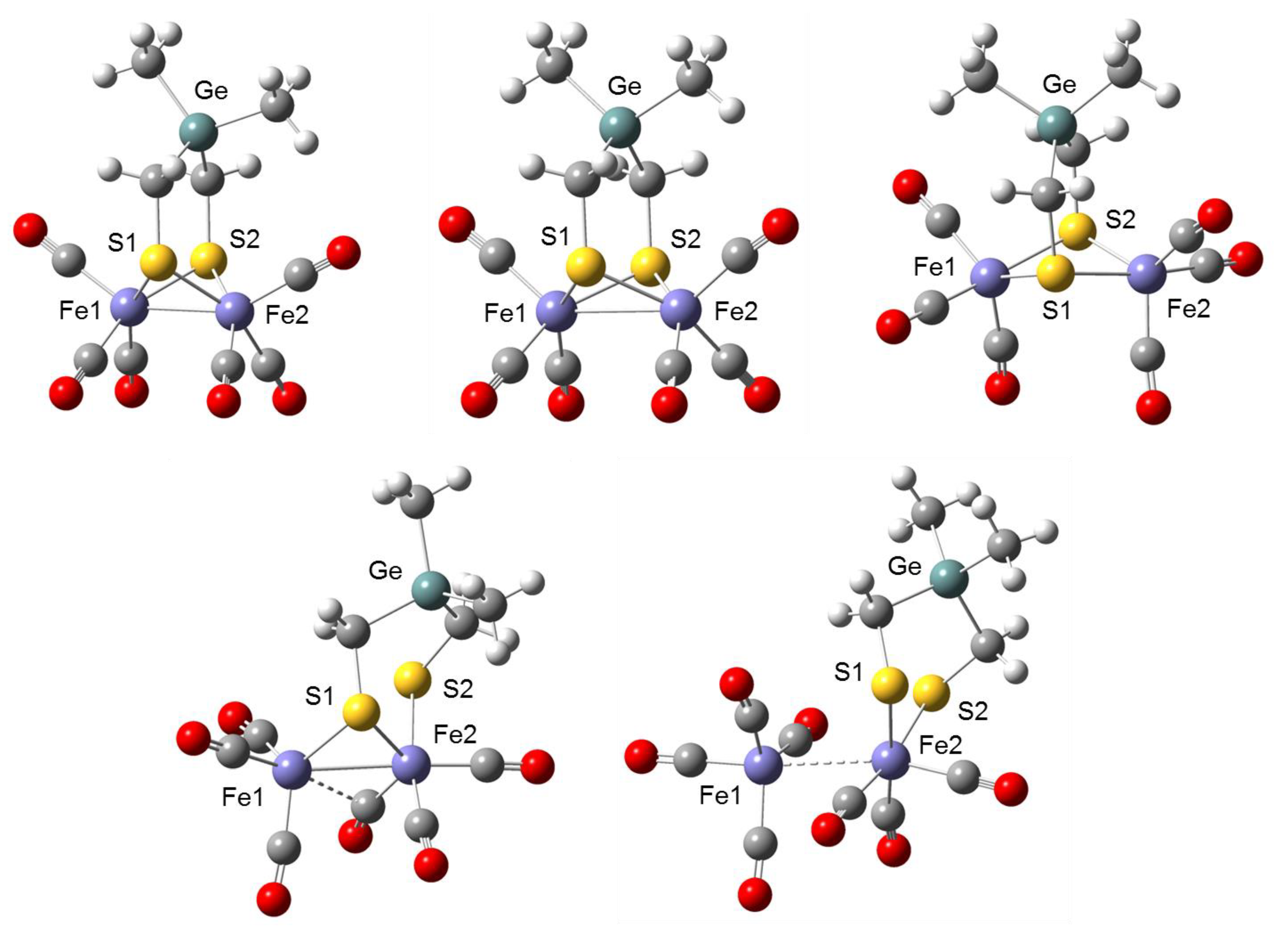
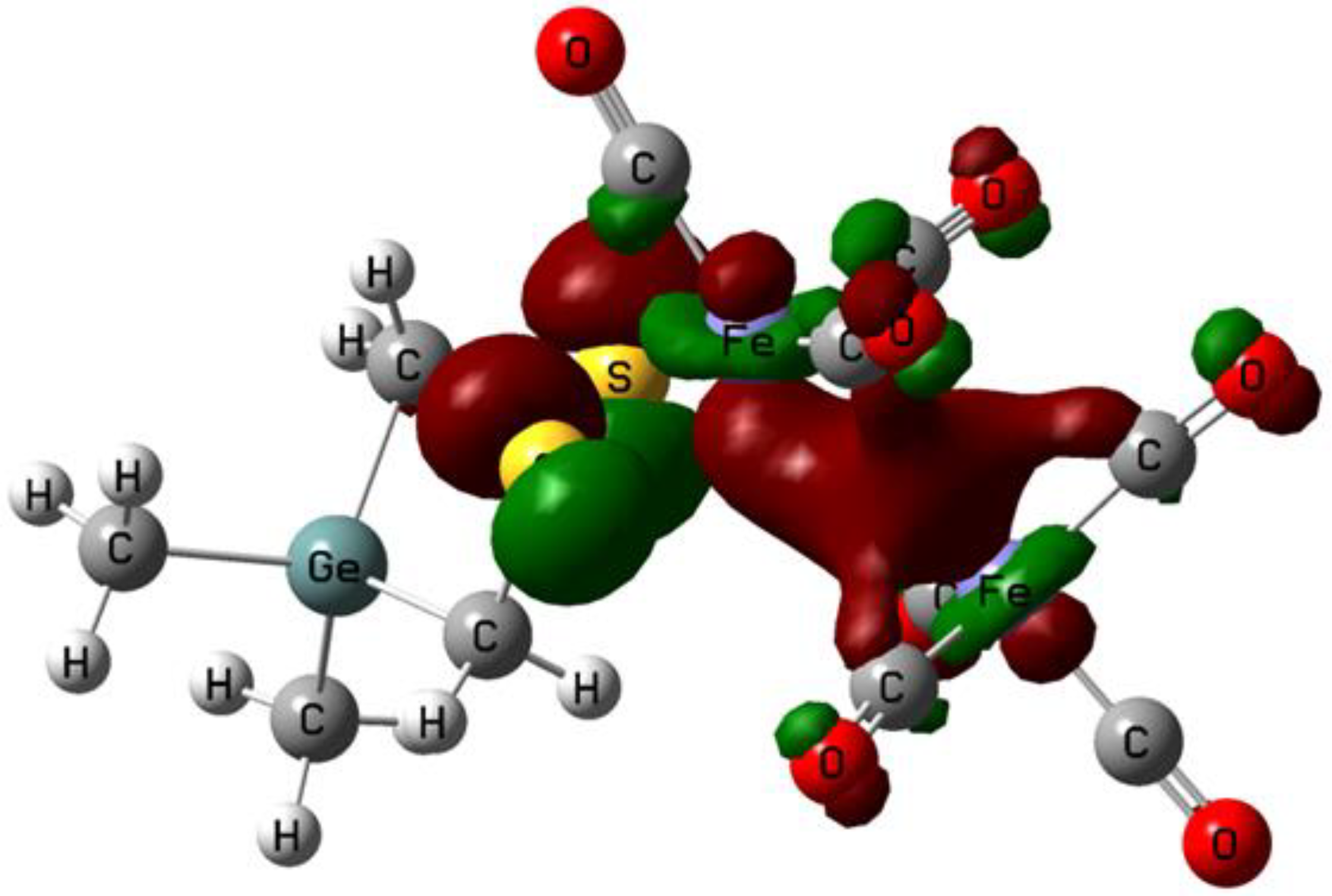
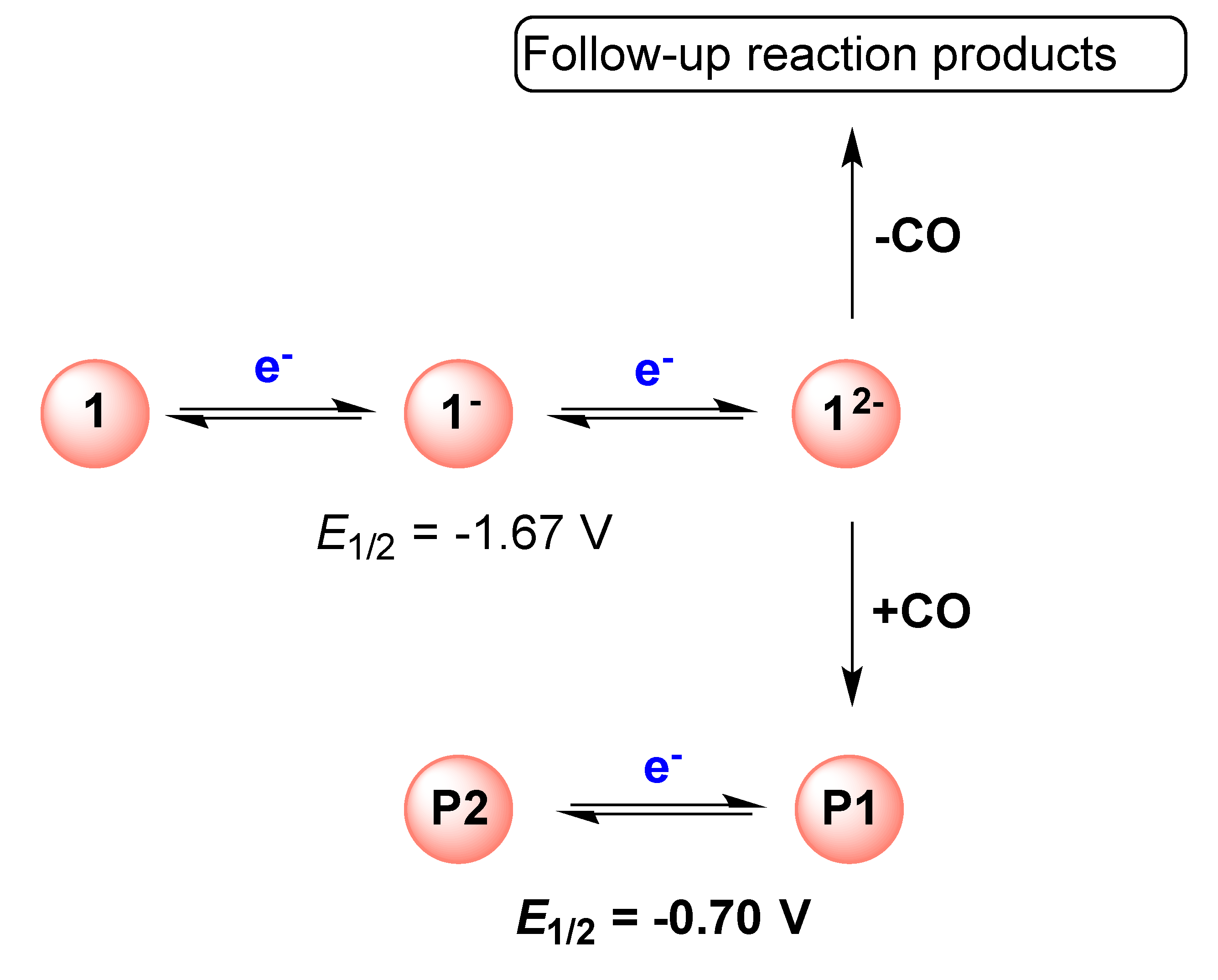
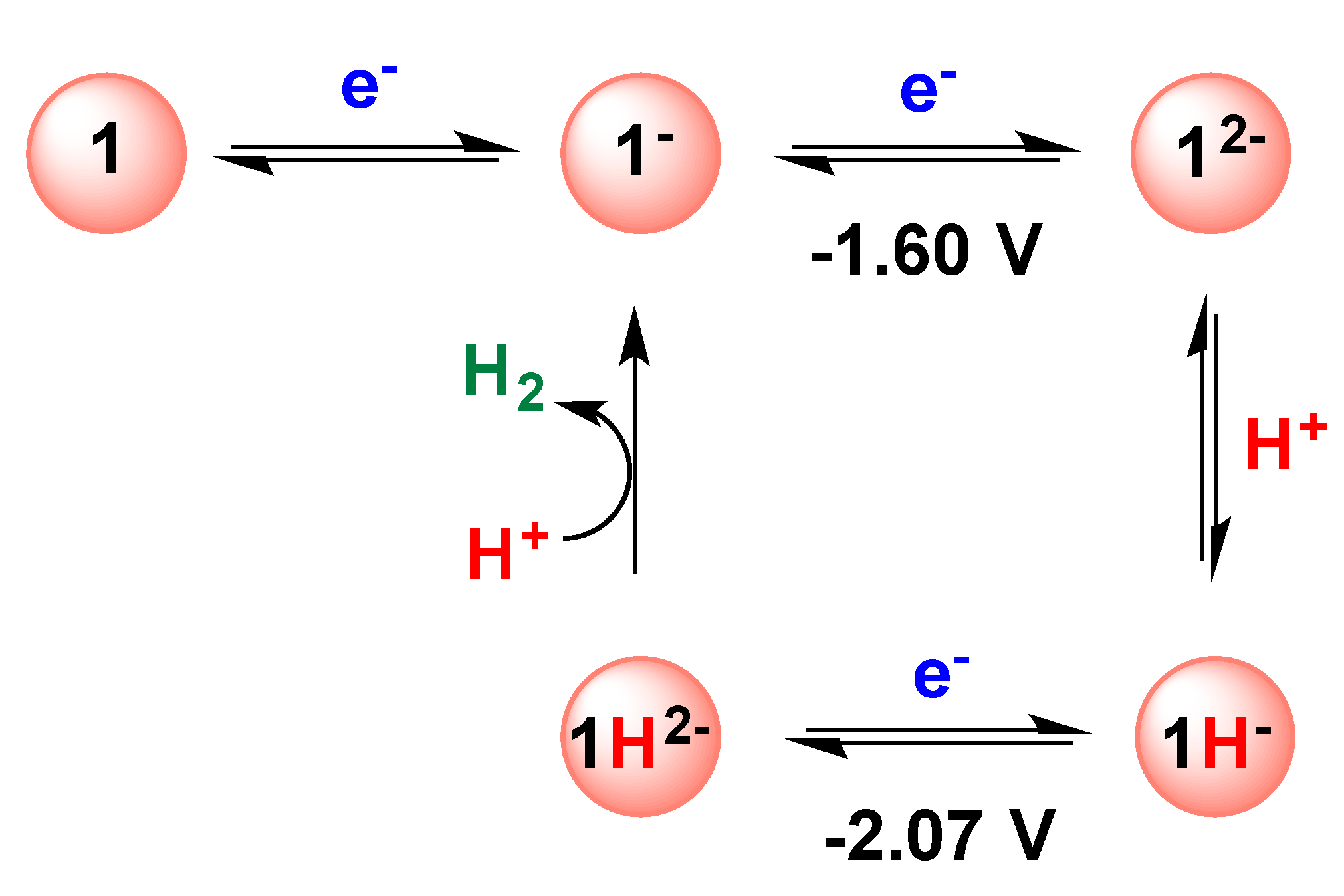
2.1. DFT Calculations on the Reduction of Complex 1
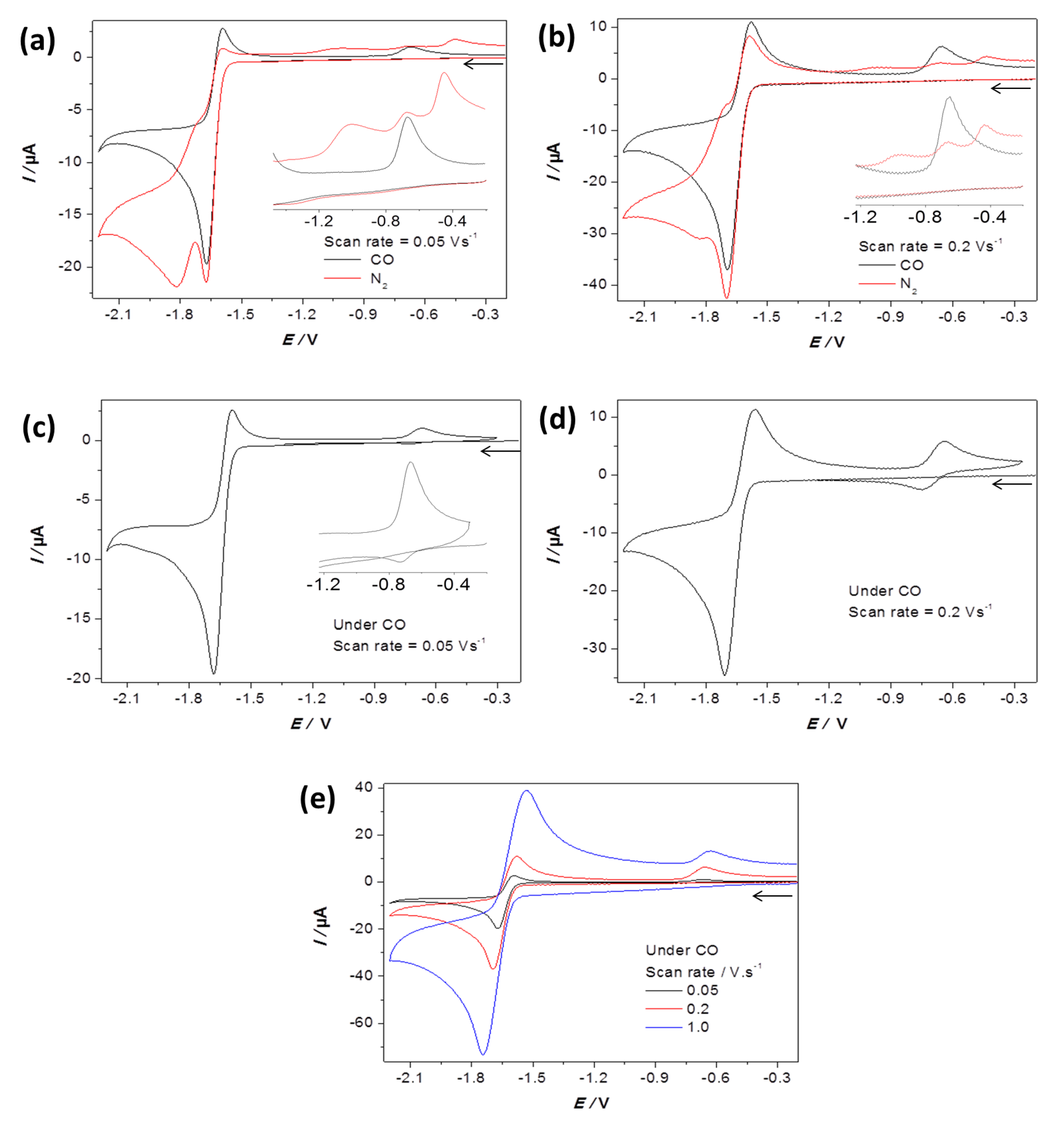
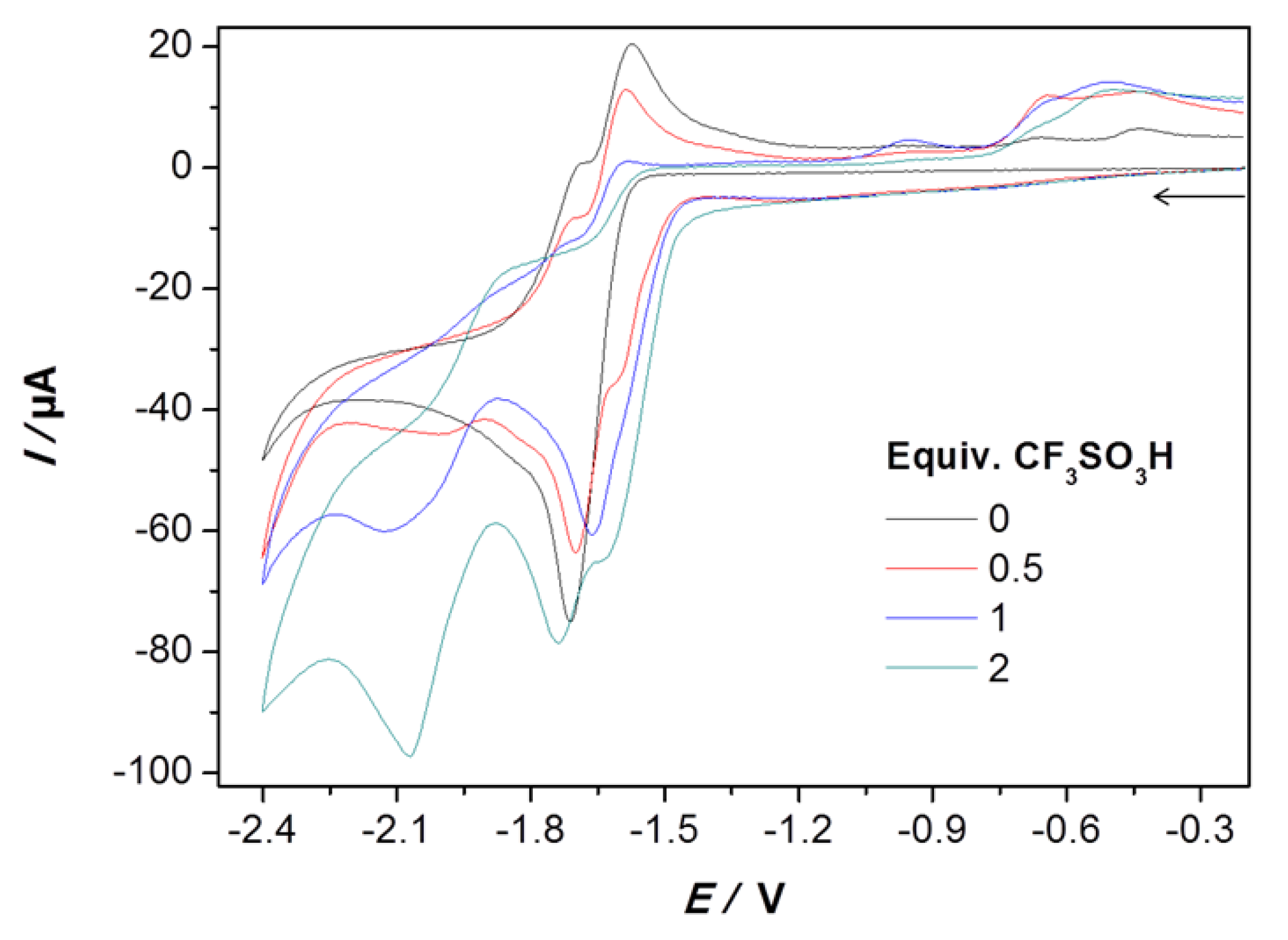
2.2. Electrochemical Reduction of 1 in the Presence of CF3SO3H
3. Experimental Section
3.1. Electrochemistry: Instrumentation and Procedures
3.2. Theoretical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Koroneos, C.; Dompros, A.; Roumbas, G.; Moussiopoulos, N. Life cycle assessment of hydrogen fuel production processes. Int. J. Hydrogen Energy 2004, 29, 1443–1450. [Google Scholar] [CrossRef]
- Rauche, H. Die Kaliindustrie im 21 Jahrhundert; Springer: Berlin, Germany, 2015. [Google Scholar]
- Borup, R.; Meyers, J.; Pivovar, B.; Kim, Y.S.; Mukundan, R.; Garland, N.; Myers, D.; Wilson, M.; Garzon, F.; Wood, D.; et al. Scientific Aspects of Polymer Electrolyte Fuel Cell Durability and Degradation. Chem. Rev. 2007, 107, 3904–3951. [Google Scholar] [CrossRef]
- Ursua, A.; Gandia, L.M.; Sanchis, P. Hydrogen Production from Water Electrolysis: Current Status and Future Trends. Proc. IEEE 2012, 100, 410–426. [Google Scholar] [CrossRef]
- Adams, M.W. The Structure and Mechanism of Iron-Hydrogenases. Biochim. Biophys. Acta Bioenergy 1990, 1020, 115–145. [Google Scholar] [CrossRef]
- Frey, M. Hydrogenases: Hydrogen-Activating Enzyme. ChemBioChem 2002, 3, 153–160. [Google Scholar] [CrossRef]
- Evans, D.J.; Pickett, C.J. Chemistry and the Hydrogenases. Chem. Soc. Rev. 2003, 32, 268–275. [Google Scholar] [CrossRef]
- Volbeda, A.; Fontecilla-Camps, J.C. Structure-Function Relationships of Nickel-Iron Sites in Hydrogenase and a Comparison with the Active Sites of Other Nickel-Iron Enzymes. Coord. Chem. Rev. 2005, 249, 1609–1619. [Google Scholar] [CrossRef]
- Fontecilla-Camps, J.C.; Volbeda, A.; Cavazza, C.; Nicolet, Y. Structure/Function Relationships of [NiFe]- and [FeFe]-Hydrogenases. Chem. Rev. 2007, 107, 4273–4303. [Google Scholar] [CrossRef] [PubMed]
- Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef]
- Adamska-Venkatesh, A.; Roy, S.; Siebel, J.F.; Simmons, T.R.; Fontecave, M.; Artero, V.; Reijerse, E.; Lubitz, W. Spectroscopic Characterization of the Bridging Amine in the Active Site of [FeFe] Hydrogenase Using Isotopologues of the H-Cluster. J. Am. Chem. Soc. 2015, 137, 12744–12747. [Google Scholar] [CrossRef]
- Adamska, A.; Silakov, A.; Lambertz, C.; Rüdiger, O.; Happe, T.; Reijerse, E.; Lubitz, W. Identification and Characterization of the “Super-Reduced” State of the H-Cluster in [FeFe] Hydrogenase: A New Building Block for the Catalytic Cycle? Angew. Chem. Int. Ed. 2012, 51, 11458–11462. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.-J.; Hall, M.B. A Capable Bridging Ligand for Fe-Only Hydrogenase: Density Functional Calculations of a Low-Energy Route for Heterolytic Cleavage and Formation of Dihydrogen. J. Am. Chem. Soc. 2001, 123, 3828–3829. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.W.; Lanzilotta, W.N.; Lemon, B.J.; Seefeldt, L.C. X-ray Crystal Structure of the Fe-Only Hydrogenase (CpI) from Clostridium pasteurianum to 1.8 Angstrom Resolution. Science 1998, 282, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Nicolet, Y.; de Lacey, A.L.; Vernede, X.; Fernandez, V.M.; Hatchikian, E.C.; Fontecilla-Camps, J.C. Crystallographic and FTIR Spectroscopic Evidence of Changes in Fe Coordination Upon Reduction of the Active Site of the Fe-Only Hydrogenase from Desulfovibrio desulfuricans. J. Am. Chem. Soc. 2001, 123, 1596–1601. [Google Scholar] [CrossRef] [PubMed]
- Abul-Futouh, H.; El-khateeb, M.; Görls, H.; Weigand, W. [FeFe]-hydrogenase H-cluster mimics mediated by mixed (S, Se) and (S, Te) bridging moieties: Insight into molecular structures and electrochemical characteristics. Heteroatom Chem. 2018, 29, e21446. [Google Scholar] [CrossRef]
- De Lacey, A.L.; Fernández, V.M.; Rousset, M.; Cammack, R. Activation and Inactivation of Hydrogenase Function and the Catalytic Cycle: Spectroelectrochemical Studies. Chem. Rev. 2007, 107, 4304–4330. [Google Scholar] [CrossRef]
- Sommer, C.; Adamska-Venkatesh, A.; Pawlak, K.; Birrell, J.A.; Rüdiger, O.; Reijerse, E.J.; Lubitz, W. Proton Coupled Electronic Rearrangement within the H-Cluster as an Essential Step in the Catalytic Cycle of [FeFe] Hydrogenases. J. Am. Chem. Soc. 2017, 139, 1440–1443. [Google Scholar] [CrossRef] [PubMed]
- Adamska-Venkatesh, A.; Krawietz, D.; Siebel, J.; Weber, K.; Happe, T.; Reijerse, E.; Lubitz, W. New Redox States Observed in [FeFe] Hydrogenases Reveal Redox Coupling Within the H-Cluster. J. Am. Chem. Soc. 2014, 136, 11339–11346. [Google Scholar] [CrossRef]
- Silakov, A.; Reijerse, E.J.; Albracht, S.P.J.; Hatchikian, E.C.; Lubitz, W. The Electronic Structure of the H-Cluster in the [FeFe]-Hydrogenase from Desulfovibrio desulfuricans: A Q-band 57Fe-ENDOR and HYSCORE Study. J. Am. Chem. Soc. 2007, 129, 11447–11458. [Google Scholar] [CrossRef]
- Li, Y.; Rauchfuss, T.B. Synthesis of Diiron(I) Dithiolato Carbonyl Complexes. Chem. Rev. 2016, 116, 7043–7077. [Google Scholar] [CrossRef] [Green Version]
- Abul-Futouh, H.; Skabeev, A.; Botteri, D.; Zagranyarski, Y.; Görls, H.; Weigand, W.; Peneva, K. Toward a Tunable Synthetic [FeFe]-Hydrogenase H-Cluster Mimic Mediated by Perylene Monoimide Model Complexes: Insight into Molecular Structures and Electrochemical Characteristics. Organometallics 2018, 37, 3278–3285. [Google Scholar] [CrossRef]
- Qian, G.; Zhong, W.; Wei, Z.; Wang, H.; Xiao, Z.; Long, L.; Liu, X. Diiron Hexacarbonyl Complexes Bearing Naphthalene-1,8-Dithiolate Bridge Moiety as Mimics of the Sub-Unit of [FeFe]-Hydrogenase: Synthesis, Characterisation and Electrochemical Investigations. New J. Chem. 2015, 39, 9752–9760. [Google Scholar] [CrossRef]
- Figliola, C.; Male, L.; Horton, P.N.; Pitak, M.B.; Coles, S.J.; Horswell, S.L.; Grainger, R.S. [FeFe]-Hydrogenase Synthetic Mimics Based on Peri-Substituted Dichalcogenides. Organometallics 2014, 33, 4449–4460. [Google Scholar] [CrossRef]
- Figliola, C.; Male, L.; Horswell, S.L.; Grainger, R.S. N-Derivatives of Peri-Substituted Dichalcogenide [FeFe]-Hydrogenase Mimics: Towards Photocatalytic Dyads for Hydrogen Production. Eur. J. Inorg. Chem. 2015, 19, 3146–3156. [Google Scholar] [CrossRef]
- Samuel, A.P.S.; Co, D.T.; Stern, C.L.; Wasielewski, M.R. Ultrafast Photodriven Intramolecular Electron Transfer from a Zinc Porphyrin to a Readily Reduced Diiron Hydrogenase Model Complex. J. Am. Chem. Soc. 2010, 132, 8813–8815. [Google Scholar] [CrossRef]
- Li, P.; Amirjalayer, S.; Hartl, F.; Lutz, M.; Bruin, B.; Becker, R.; Woutersen, S.; Reek, J.N.H. Direct Probing of Photoinduced Electron Transfer in a Self-Assembled Biomimetic [2Fe2S]-Hydrogenase Complex Using Ultrafast Vibrational Spectroscopy. Inorg. Chem. 2014, 53, 5373–5383. [Google Scholar] [CrossRef]
- Gloaguen, F.; Morvan, D.; Capon, J.-F.; Schollhammer, P.; Talarmin, J. Electrochemical Proton Reduction at Mild Potentials by Monosubstituted Diiron Organometallic Complexes Bearing a Benzenedithiolate Bridge. J. Electroanal. Chem. 2007, 603, 15–20. [Google Scholar] [CrossRef]
- Felton, G.A.N.; Vannucci, A.K.; Chen, J.; Lockett, L.T.; Okumura, N.; Petro, B.J.; Zakai, U.I.; Evans, D.H.; Glass, R.S.; Lichtenberger, D.L. Hydrogen Generation from Weak Acids: Electrochemical and Computational Studies of a Diiron Hydrogenase Mimic. J. Am. Chem. Soc. 2007, 129, 12521–12530. [Google Scholar] [CrossRef]
- Schwartz, L.; Singh, P.S.; Eriksson, L.; Lomoth, R.; Ott, S. Tuning the Electronic Properties of Fe2(μ-Arenedithiolate)(CO)6−n(PMe3)n (n = 0, 2) Complexes Related to the [Fe–Fe]-Hydrogenase Active Site. C. R. Chim. 2008, 11, 875–889. [Google Scholar] [CrossRef]
- Capon, J.-F.; Gloaguen, F.; Schollhammer, P.; Talarmin, J. Electrochemical Proton Reduction by Thiolate-Bridged Hexacarbonyl Diiron Clusters. J. Electroanal. Chem. 2004, 566, 241–247. [Google Scholar] [CrossRef]
- Chen, J.; Vannucci, A.K.; Mebi, C.A.; Okumura, N.; Borowski, S.C.; Swenson, M.; Lockett, L.T.; Evans, D.H.; Glass, R.S.; Lichtenberger, D.L. Synthesis of Diiron Hydrogenase Mimics Bearing Hydroquinone and Related Ligands. Electrochemical and Computational Studies of the Mechanism of Hydrogen Production and the Role of O−H···S Hydrogen Bonding. Organometallics 2010, 29, 5330–5340. [Google Scholar] [CrossRef]
- Donovan, E.S.; McCormick, J.J.; Nichol, G.S.; Felton, G.A.N. Cyclic Voltammetric Studies of Chlorine-Substituted Diiron Benzenedithiolato Hexacarbonyl Electrocatalysts Inspired by the [FeFe]-Hydrogenase Active Site. Organometallics 2012, 31, 8067–8070. [Google Scholar] [CrossRef]
- Felton, G.A.N.; Mebi, C.A.; Petro, B.J.; Vannucci, A.K.; Evans, D.H.; Glass, R.S.; Lichtenberger, D.L. Review of Electrochemical Studies of Complexes Containing the [Fe2S2] Core Characteristic of [FeFe]-Hydrogenases Including Catalysis by These Complexes of the Reduction of Acids to Form Dihydrogen. J. Organomet. Chem. 2009, 694, 2681–2699. [Google Scholar] [CrossRef]
- Abul-Futouh, H.; Almazahreh, L.R.; Sakamoto, T.; Stessman, N.Y.T.; Lichtenberger, D.L.; Glass, R.S.; Görls, H.; El-khateeb, M.; Schollhammer, P.; Mloston, G.; et al. [FeFe]-Hydrogenase H-Cluster Mimics with Unique Planar μ-(SCH2)2ER2 Linkers (E = Ge and Sn). Chem. Eur. J. 2017, 23, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Glass, R.S.; Gruhn, N.E.; Lorance, E.; Singh, M.S.; Stessman, N.Y.T.; Zakai, U.I. Synthesis, Gas-Phase Photoelectron Spectroscopic, and Theoretical Studies of Stannylated Dinuclear Iron Dithiolates. Inorg. Chem. 2005, 44, 5728–5737. [Google Scholar] [CrossRef] [PubMed]
- Almazahreh, L.R.; Apfel, U.-P.; Imhof, W.; Rudolph, M.; Görls, H.; Talarmin, J.; Schollhammer, P.; El-khateeb, M.; Weigand, W. A Novel [FeFe] Hydrogenase Model with a (SCH2)2P=O Moiety. Organometallics 2013, 32, 4523–4530. [Google Scholar] [CrossRef]
- Abul-Futouh, H.; El-khateeb, M.; Görls, H.; Asali, K.J.; Weigand, W. Selenium Makes the Difference: Protonation of [FeFe]-Hydrogenase Mimics with Diselenolato Ligands. Dalton Trans. 2017, 46, 2937–2947. [Google Scholar] [CrossRef]
- Abul-Futouh, H.; Zagranyarski, Y.; Müller, C.; Schulz, M.; Kupfer, S.; Görls, H.; Elkhateeb, M.; Gräfe, S.; Dietzek, B.; Peneva, K.; et al. [FeFe]-Hydrogenase H-cluster Mimics Mediated by Naphthalene Monoimide Derivatives of Peri-Substituted Dichalcogenides. Dalton Trans. 2017, 46, 11180–11191. [Google Scholar] [CrossRef]
- Abul-Futouh, H.; Almazahreh, L.R.; Harb, M.K.; Görls, H.; El-khateeb, M.; Weigand, W. [FeFe]-Hydrogenase H-Cluster Mimics with Various –S(CH2)nS– Linker Lengths (n = 2–8): A Systematic Study. Inorg. Chem. 2017, 56, 10437–10451. [Google Scholar] [CrossRef]
- Trautwein, R.; Almazahreh, L.R.; Görls, H.; Weigand, W. The Influence of OH Groups in [Fe(CO)3]2[(µ-ECH2)2C(CH2OH)2] (E = S, Se) Complexes toward the Cathodic Process. Z. Anorg. Allg. Chem. 2013, 639, 1512–1519. [Google Scholar] [CrossRef]
- Almazahreh, L.; Imhof, W.; Talarmin, J.; Schollhammer, P.; Görls, H.; El-Khateeb, M.; Weigand, W. Ligand Effects on the Electrochemical Behavior of [Fe2(CO)5(L){μ-(SCH2)2(Ph)P=O}] (L = PPh3, P(OEt)3) Hydrogenase Model Complexes. Dalton Trans. 2015, 44, 7177–7189. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian09; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. 1988, B37, 785–789. [Google Scholar] [CrossRef]
- Dolg, M.; Stoll, H.; Preuss, H. A combination of quasirelativistic pseudopotential and ligand field calculations for lanthanoid compounds. Theor. Chim. Acta 1993, 85, 441–450. [Google Scholar] [CrossRef]
- Bergner, A.; Dolg, M.; Küchle, W.; Stoll, H.; Preuss, H. Ab initio energy-adjusted pseudopotentials for elements of groups 13–17. Mol. Phys. 1993, 80, 1431–1441. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comp. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Kütt, A.; Rodima, T.; Saame, J.; Raamat, E.; Mäemets, V.; Kaljurand, I.; Koppel, I.A.; Garlyauskayte, R.Y.; Yagupolskii, Y.L.; Yagupolskii, L.M.; et al. Equilibrium acidities of superacids. J. Org. Chem. 2011, 76, 391–395. [Google Scholar] [CrossRef]
- Almazahreh, L.; Trautwein, R.; Görls, H.; Weigand, W. Steric effect of the dithiolato linker on the reduction mechanism of [Fe2(CO)6{μ-(XCH2)2CRR′}] hydrogenase models (X = S, Se). Dalton Trans. 2015, 44, 18780–18794. [Google Scholar]
- Peng, B.; Li, Q.S.; Xie, Y.; King, R.B.; Schaefer, H.F., III. Unsaturated trinuclear ruthenium carbonyls: Large structural differences between analogous carbonyl derivatives of the first, second, and third row transition metals. Dalton Trans. 2008, 6977–6986. [Google Scholar] [CrossRef]
- Feng, X.; Gu, J.; Xie, Y.; King, R.B.; Schaefer, H.F., III. Homoleptic Carbonyls of the Second-Row Transition Metals: Evaluation of Hartree–Fock and Density Functional Theory Methods. J. Chem. Theory Comput. 2007, 3, 1580–1587. [Google Scholar] [CrossRef] [PubMed]
- Imhof, W.; Anders, E.; Göbel, A.; Görls, H. A Theoretical Study on the Complete Catalytic Cycle of the Hetero-Pauson-Khand-Type [2+2+1] Cycloaddition Reaction of Ketimines, Carbon Monoxide and Ethylene Catalyzed by Iron Carbonyl Complexes. Chem. Eur. J. 2003, 9, 1166–1181. [Google Scholar] [CrossRef] [PubMed]
- Imhof, W.; Anders, E. Regioselectivity in Iron-Catalyzed [2+2+1] Cycloadditions: A DFT Investigation of Substituent Effects in 1,4-Diazabutadienes. Chem. Eur. J. 2004, 10, 5717–5729. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 1− | 12− | 12−(Isomer) | P1 | |
|---|---|---|---|---|---|
| Fe1–Fe2 | 235.0 | 287.7 | 350.2 | 271.6 | 306.0 |
| Fe1–S1 | 231.3 | 236.4 | 241.8 | 233.2 | - |
| Fe1–S2 | 231.3 | 235.9 | 239.9 | - | - |
| Fe2–S1 | 231.8 | 235.8 | 240.0 | 234.9 | 245.1 |
| Fe2–S2 | 231.8 | 236.9 | 242.1 | 243.6 | 241.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abul-Futouh, H.; Imhof, W.; Weigand, W.; Almazahreh, L.R. Electrochemical and Computational Insights into the Reduction of [Fe2(CO)6{µ-(SCH2)2GeMe2}] Hydrogenase H-Cluster Mimic. Inorganics 2019, 7, 50. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7040050
Abul-Futouh H, Imhof W, Weigand W, Almazahreh LR. Electrochemical and Computational Insights into the Reduction of [Fe2(CO)6{µ-(SCH2)2GeMe2}] Hydrogenase H-Cluster Mimic. Inorganics. 2019; 7(4):50. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7040050
Chicago/Turabian StyleAbul-Futouh, Hassan, Wolfgang Imhof, Wolfgang Weigand, and Laith R. Almazahreh. 2019. "Electrochemical and Computational Insights into the Reduction of [Fe2(CO)6{µ-(SCH2)2GeMe2}] Hydrogenase H-Cluster Mimic" Inorganics 7, no. 4: 50. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7040050