Electronic Structure of Cubane-Like Vanadium–Nitrogen Cationic Clusters [V4N4]+ and [V6N6]+


1
College of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China
2
State Key Laboratory of Explosion Science and Technology, Beijing Institute of Technology, Beijing 100081, China
3
Department of Chemistry, University of Oxford, South Parks Road, Oxford OX1 3QZ, UK
*
Author to whom correspondence should be addressed.
Inorganics 2019, 7(4), 52; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7040052
Submission received: 20 March 2019
/
Revised: 2 April 2019
/
Accepted: 4 April 2019
/
Published: 12 April 2019
(This article belongs to the Special Issue Applications of Density Functional Theory in Inorganic Chemistry)
Abstract
:Density Functional Theory and Complete Active Space Self-Consistent Field (CASSCF) methodologies are used to explore the electronic structure of the cationic V–N clusters, [V4N4]+ and [V6N6]+, that have been identified in recent mass spectrometric experiments. Our calculations indicate that both clusters are based on cubane-like fragments of the rock-salt lattice. In the smaller [V4N4]+ cluster, the V–V bonding is delocalized over the tetrahedron, with net bond orders of 1/3 per V–V bond. In [V6N6]+, in contrast, the V–V bonding is strongly localized in the central V2N2 unit, which has a short V=V double bond. CASSCF calculations reveal that both localized and delocalized V–V bonds are highly multi-configurational.
1. Introduction
Small clusters of transition metal nitrides, MxNy, represent the simplest models for the active sites in the bulk nitrides that have an important role in heterogeneous catalysis [1,2,3]. Amongst these, vanadium nitride is a particularly important example, and catalytic applications of both the bulk material and thin films have been reported [4,5,6,7]. Metal–metal bonding remains very strong in bulk VN, to the extent that it is an extremely good conductor [8], and the solid also features a high concentration of nitrogen vacancies, giving a range of stoichiometries VNx, where x ≤ 1 [9,10]. Indeed, low-valent binary compounds of the early transition metal elements are, in general, stabilized by extensive metal–metal bonding; examples include TiN [11] and VO [12], as well as the low-temperature form of VO2 [13]. The nature of the surface sites on non-stoichiometric VN remains a matter of debate, but it is clear that V–V bonding has the potential to drive extensive surface reconstruction that may impact the catalytic performance [1]. In light of this extreme non-stoichiometry in the solid state, small molecular clusters with V:N ratios close to 1 (i.e., VxNy, x ≈ y) provide an important opportunity to explore the interplay of metal–metal bonding, structure, magnetism and catalytic competence of the vanadium nitride phase. In recent work by Hirabayashi and Ichihashi, particular members of the [VxNy]+ family were shown to be highly active catalysts for ammonia decomposition [14]. Clusters with V:N ratios close to 1.0 were formed by sputtering metal targets with accelerated Xe+ beams in the presence of N2. Prominent peaks in the mass spectrum were assigned to [V3N3]+, [V4N4]+, [V5N5]+ and [V6N6]+, as well as the vanadium-rich species [V3N2]+, [V4N3]+, [V5N4]+ and [V6N5]+. The subsequent reactivity of these ions with NH3 depends critically on their composition, and the equiatomic clusters [V4N4]+ and [V6N6]+ appear to be particularly inert, binding one or two molecules of NH3 without showing any N–H bond cleavage. All other ions, in contrast, lose an equivalent of H2 from the bound NH3 to form [VxNy(NH)]+. The relative inertness of [V4N4]+ and [V6N6]+ led Hirabayashi and Ichihashi to propose structures based on cubane-like architectures, which represent fragments of the rock-salt lattice of the parent nitride—VN. Cubane-like motifs are, in fact, relatively common in gas-phase clusters of binary compounds of the alkali and alkaline earth metals and can be viewed as intermediate stages in the crystal growth process [15,16,17]. (NaCl)x clusters, for example, have been shown to adopt rock-salt motifs [18], as have a range of alkaline earth metal-oxide clusters, such as [MxOx]+, [Mx+1Ox]+ and [MxOx]2+ (M = Ca, Mg). Amongst the transition metal elements, the nitrides [TixNx]0/+ (x = 4, 6 and 9) show similar structural features [11,19,20].
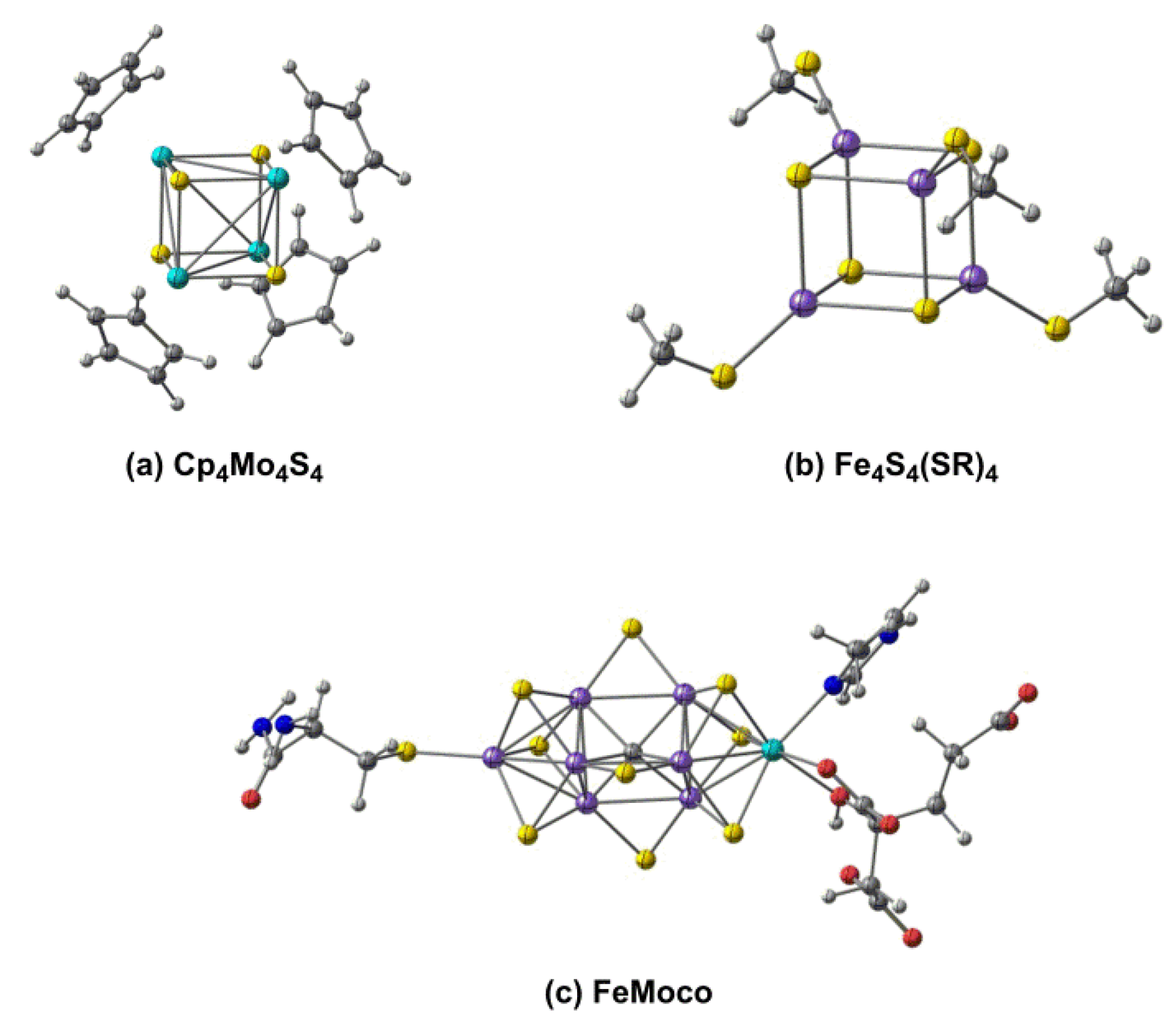
Our interest in the electronic structure of these cubane clusters lies in the extent to which they are stabilized by metal–metal bonding. Stable, coordinatively saturated cubane clusters are known across much of the transition series (see Figure 1 for selected examples), and many have important roles in biology. Fe4S4 clusters, for example, are ubiquitous in biological electron transfer [21] and were amongst the first to be studied using the broken-symmetry approach pioneered by Noodleman [22]. More recently, the remarkable structure of the Fe7Mo double cubane in the FeMo cofactor in nitrogenase has been revealed [23,24], as has the importance of Mn3Ca cubane motifs in water oxidation [25]. The presence of multiple bridging ligands between the metals in all these clusters means that whilst metal–metal bonding is possible, it is not absolutely essential for stability, and as a result, the bonding in cubanes spans the entire spectrum from strong covalent to weak exchange coupling. The cyclopentadienyl-capped clusters Cp4M4E4 (M = Cr, Mo; E = O, S) [26] are a case in point wherein delocalized covalent bonding in the molybdenum systems gives way to weak exchange coupling in the chromium analogues.
In this paper, we explore the electronic structures of two of the clusters identified in the mass-spectrometric experiments of Hirabayashi and Ichihashi, [V4N4]+ and [V6N6]+, both of which have been proposed to adopt cubane-like architectures on the basis of their reactivity (or lack thereof) towards ammonia dehydrogenation. The coordinatively unsaturated nature of the metal centres leads to a relatively small splitting within the d-orbital manifold, and hence to a very rich electronic structure with many accessible electronic states. From a computational perspective, the description of metal–metal bonding therefore represents a very considerable challenge, and the extent to which single-determinant approaches, such as density functional theory, are appropriate remains to be established. Gorelsky [27] has explored this issue in the context of a vanadium dimer, V2(C5H5)2(pentalene), where both the geometry and the singlet–triplet splitting were found to be well described by the gradient-corrected (GGA) BP86 functional and also by the meta-GGA TPSS functional. The inclusion of exact Hartree–Fock exchange in the B3LYP functional, for example, causes a very substantial weakening of the V–V bond, which appears inconsistent with the available experimental data. In recent work, we identified a number of other cases where the inclusion of exact exchange in the functional provides a description of metal–metal bonding that is qualitatively different from the picture derived from GGA or meta-GGA functionals [28]. Here, we report results obtained using a range of different density functionals and compare these to the electronic structure picture that emerges from a parallel series of calculations performed with the CASSCF methodology.
2. Results
2.1. Electronic Structure of [V4N4]+
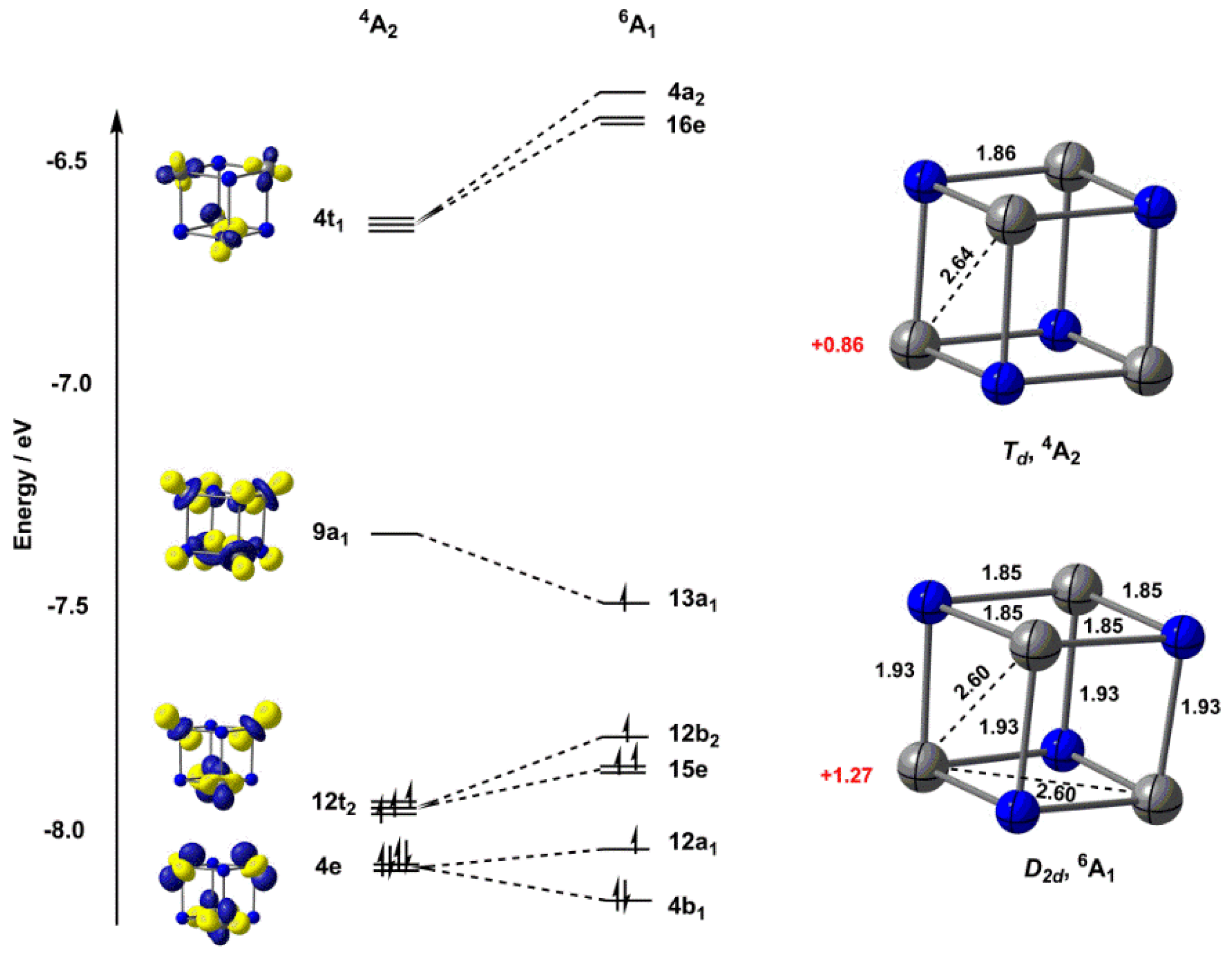
For [V4N4]+, we identified two closely-spaced states, both of which are based on a tetrahedral architecture (Figure 2). The first of these is a 4A2 state with rigorous Td symmetry: the six V–V bond lengths are identical at 2.65 Å (using the TPSS functional [29]). In the alternative sextet state, 6A1, the tetrahedron is slightly elongated along one of the two-fold axes, giving four long (1.93 Å) and eight short (1.85 Å) V–N bonds and overall D2d symmetry. The average V–V bond lengths in the 6A1 state are somewhat smaller than those in 4A2 (2.60 Å vs. 2.65 Å), while the average V–N bond lengths are marginally longer (1.88 Å vs. 1.86 Å). The origin of these structural patterns is apparent in the Kohn–Sham orbital diagram, also shown in Figure 2. In the perfect tetrahedron, the V orbitals form a basis for a1 and t2 representations (the z axis is defined locally here as the vector passing through each V atom and the centre of mass of the cluster). The a1 combination is bonding with respect to the V–V interactions but also strongly V–N antibonding, and as a result, it lies above the weakly V–V bonding t2 set. The orbitals, in contrast, are aligned tangentially around the surface of the tetrahedron, and form a basis for e + t1 + t2 representations, the doubly degenerate e orbital being strongly V–V bonding. In the 4A2 state, the two components of the 4e orbital are doubly occupied, while the three components of 12t2 each contain a single electron, giving rise to a perfectly symmetric structure with six equivalent V–V bond lengths of 2.65 Å. Promotion of a single electron from the 4e orbital into 9a1 results in an orbitally degenerate 6E state, and hence to a first-order Jahn–Teller instability that drives the distortion to D2d symmetry (and thus to the 6A1 state). The population of the 9a1 orbital also strengthens the V–V bonds while at the same time weakening the V–N bonds, leading to a global contraction of the V4 tetrahedron.
The structural trends identified in the previous paragraph are broadly independent of the choice of functional (Table 1), but the relative energies of the states are not: with the TPSS functional, the 6A1 state is the global minimum, 0.19 eV lower in energy than 4A2, while with the BP86 functional, the order is reversed, with 4A2 0.08 eV below 6A1. These behaviours appear to be generally characteristic of the broad classes of functionals (Table 1): the meta-GGAs, TPSS and M06L [30] favour the 6A1 state, while all three GGA functionals (BP86 [31,32], with and without dispersion corrections (Becke–Johnson D3 set [33]), and BLYP [34]) favour 4A2. The hybrid B3LYP functional [35] strongly stabilizes the state of higher multiplicity, a trend that has been observed many times in the literature [36]. Clearly, the balance between one-electron terms, favouring double occupation of the most bonding orbitals, and electron–electron repulsions, favouring higher multiplicities, is a delicate one that challenges even those functionals that have previously proven suitable for metal–metal bonded systems (TPSS and BP86) [27].
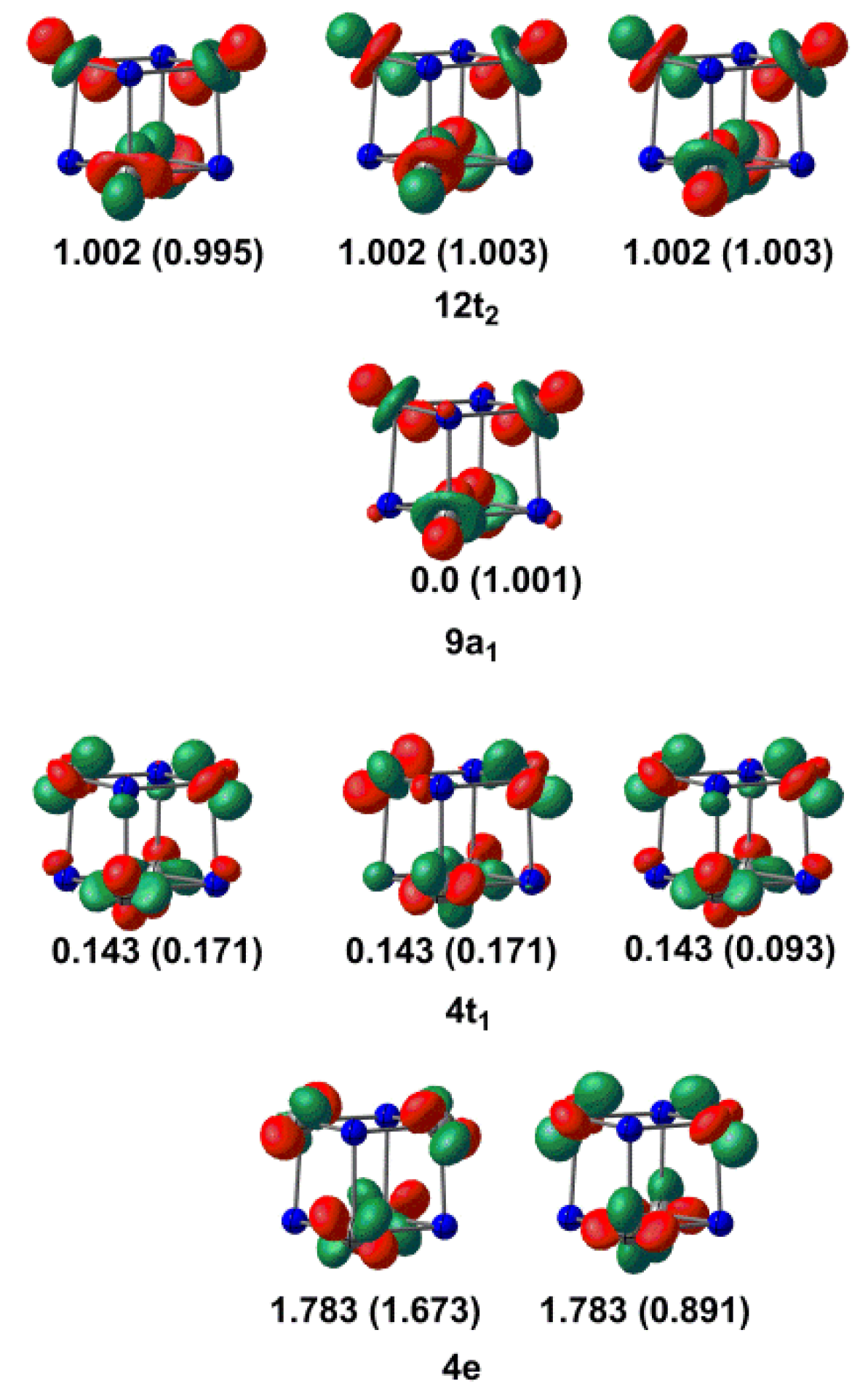
In light of the conflicting conclusions about the relative stabilities of 4A2 and 6A1 that emerged from the DFT treatment, we have turned to multi-configurational approaches (specifically the Complete Active Space Self-Consistent Field (CASSCF) methodology) to offer an alternative perspective on the V–V bonding. In both cases, the calculations were performed using the geometry of the corresponding state obtained from the calculations with the TPSS functional, although the precise geometry of a given state is rather independent of the exact choice (with the exception of the M06L functional, which tends to afford rather shorter V–V bonds). The 4A2 and 6A1 states were described using a CAS(7,9) active space, where the nine orbitals include the 4e, 9a1 and 12t2 orbitals in Figure 2, and also the three additional orbitals of the 4t1 set that are derived from the tangential set. The natural orbitals (the orbitals that diagonalise the one-particle density matrix [37,38]) and their occupation numbers are shown in Figure 3 (the orbitals shown are for the 4A2 state, but the iso-surface plots are almost indistinguishable from those for the 6A1 alternative). The three t2 orbitals had occupations close to 1.0, but the rather low occupations of the 4e orbitals (1.783) and the compensating high values for the 4t1 set (0.143) are indicative of strong multi-configurational character. Indeed, the single configuration shown in the Kohn–Sham orbital diagram makes up only 78% of the ground-state wavefunction, the remainder being largely contributions from double excitations from 4e to 4t1: these orbitals are bonding–antibonding combinations of the same set of basis functions, and so the strong correlations are consistent with Roos’s proposals for an appropriate choice of active space [39,40]. The natural orbital occupations for the 6A1 state (shown in parentheses in Figure 3) confirm a rather similar picture of the bonding: the three components of the 12t2 orbital were singly occupied, as were the 9a1 orbital and one of the two components of 4e. The one remaining pair in the 4e orbital was strongly correlated with the three members of the 4t1 orbital, but their overall occupations were reduced accordingly. In summary, the picture of bonding that emerged from the CASSCF calculations was qualitatively similar to the DFT perspective, with the caveat that the multi-configurational approach highlights the very strong static correlation within the doubly occupied V–V bonding orbitals. At the CASSCF level, the 4A2 state was less stable than the 6A1 alternative by 4.78 eV, but the incorporation of dynamic correlation using N-electron valence state perturbation (NEVPT2) theory reversed the order, such that the 4A2 state was more stable by 5.88 eV.
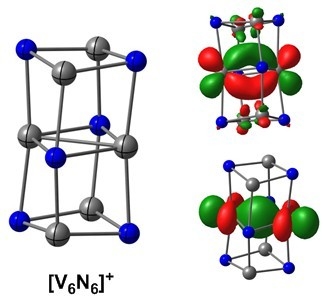
2.2. Electronic Structure of [V6N6]+
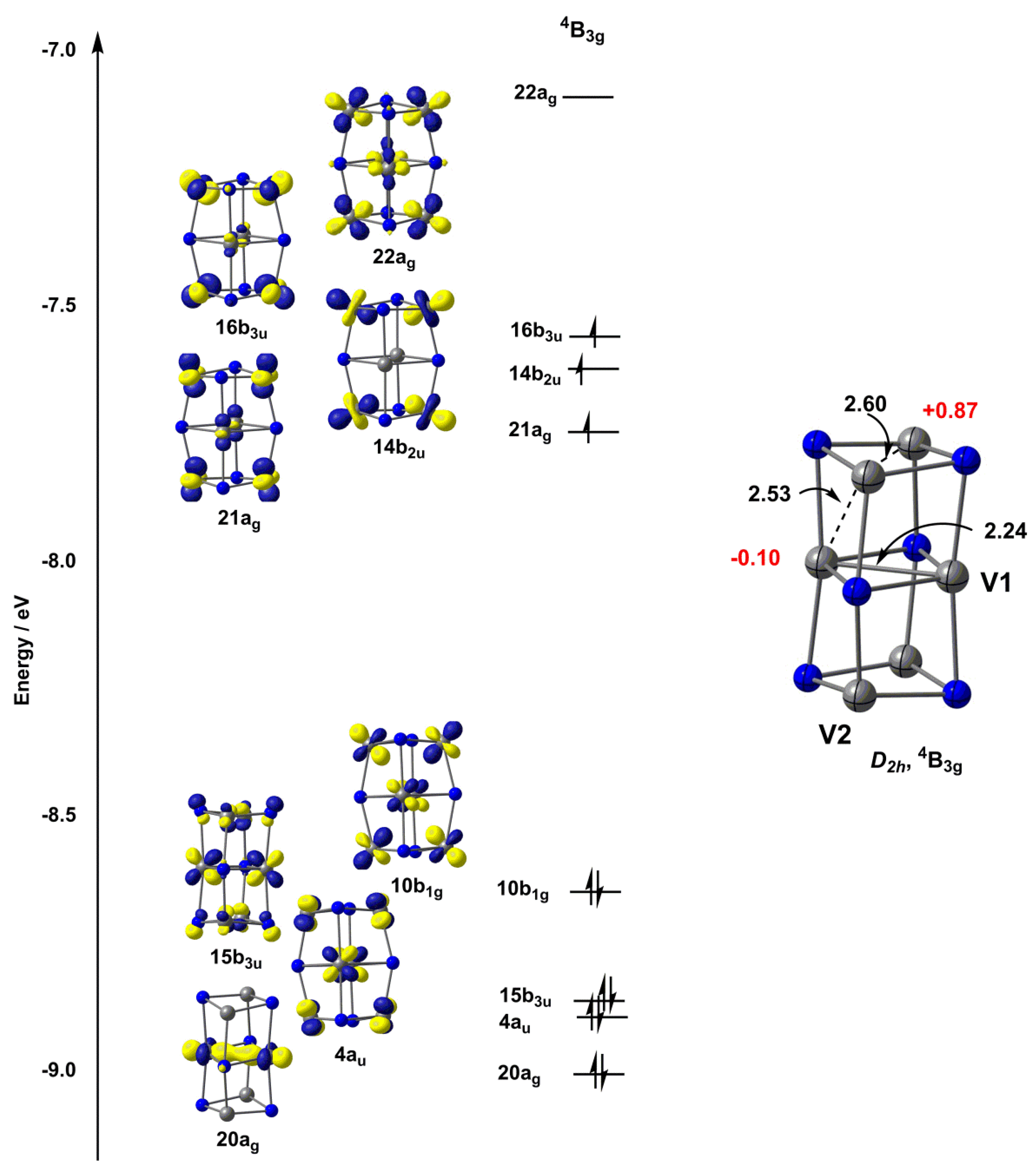
Turning now to the larger [V6N6]+ cluster, we considered a range of structures that have been proposed in the literature for related species, including the columnar fragment of the rock-salt structure proposed by Hirabayashi and Ichihashi (D2h symmetry) and also a hexagonal prismatic structure with two staggered V3N3 hexagons (D3d symmetry), which has been identified as the global minimum for the Al6N6 cluster [41]. In the case of [V6N6]+, this hexagonal structure proved to be over 3 eV higher in energy than the D2h alternative for all functionals, and was not considered further. As was the case for the [V4N4]+ cluster, we found a quartet ground state (4B3g) with overall D2h symmetry, with all alternative spin states substantially higher in energy. The V–V bond in the central layer of the cluster (V1–V1) is strikingly short at 2.23 Å (Table 2), a value that is indicative of strong multiple bonding. The bonds between the central and upper/lower layers (V1–V2) were somewhat longer, at 2.52 Å, whilst those within the top and bottom layers (V2–V2) are longer still, at 2.60 Å. All V–V contacts are, however, shorter than those found in either the 4A2 or 6A1 states of [V4N4]+. The Kohn–Sham orbitals for the 4B3g state, shown in Figure 4, confirm that, of the eleven available V 3d valence electrons, eight are paired in orbitals with significant V–V bonding character. Amongst the doubly occupied set, the 20ag and 15b3u orbitals are localized in the central V2N2 plane, and have strong V–V σ and V–V π character, respectively, consistent with the presence of a V=V double bond. The 4au and 10b1g orbitals, in contrast, are bonding with respect to all eight V1–V2 contacts between the central plane and the upper/lower layers of the column, accounting for the relatively short distances of 2.51 Å. The remaining three valence electrons occupy the 21ag, 14b2u and 16b3u orbitals, none of which are strongly V–V bonding or antibonding. There are, therefore, many qualitative parallels between the 4A2 state of [V4N4]+ and the 4B3g state of [V6N6]+: in both cases, some of the available electrons are paired up to form V–V covalent bonds, while the remainder are distributed singly over approximately non-bonding orbitals. The proportion of electrons in the two sets of orbitals changes as a function of the cluster size: for the 4A2 state of [V4N4]+, four of the seven available electrons are involved in V–V bonding, while in the 4B3g state of [V6N6]+ this fraction rises to 8/11, and the resulting increase in net bond order is behind the generally shorter bond lengths in the larger cluster.
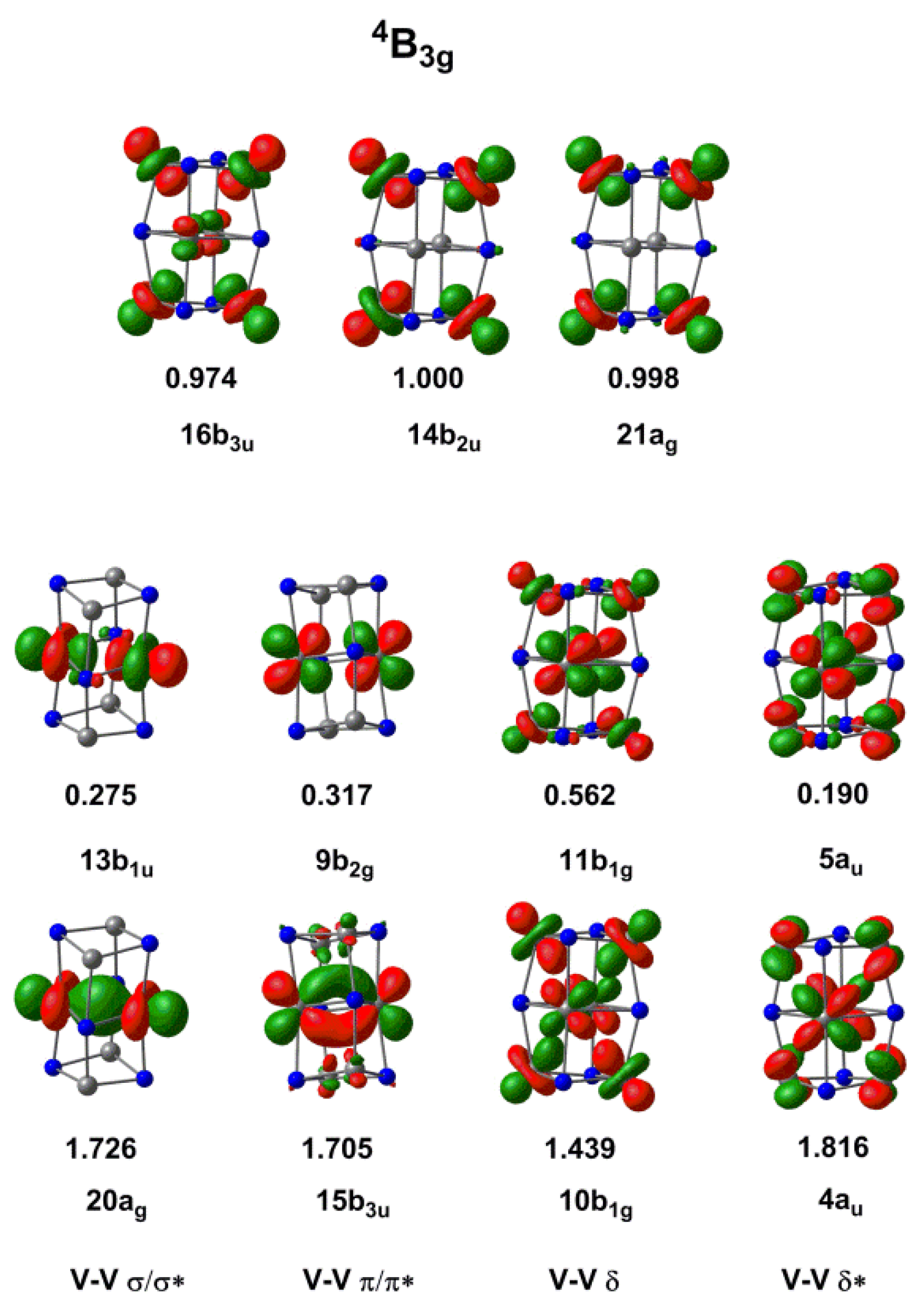
The CASSCF wavefunction for the 4B3g state of [V6N6]+ shown in Figure 5 revealed a qualitatively similar story about the V–V bonding, relative to the one established for [V4N4]+. We used a CAS(11,11) active space in which all four doubly occupied bonding orbitals in the Kohn–Sham scheme find antibonding counterparts in the virtual manifold that allow for strong correlations. These are most striking for the V–V σ and π bonds in the central V2N2 plane (occupations of the bonding–antibonding pairs are 1.726/0.275 (σ/σ*, 20ag/13b1u) and 1.705/0.317 (π/π*, 15b3u/9b2g)). Natural orbital occupations that differ dramatically from 2.0 and 0.0 are typical in clusters with multiple metal–metal bonds [42], and are highly indicative of weak bonding. The electrons in the 4au and 10b1g orbitals that mediate the bonding between the layers are also strongly correlated (1.439/0.562 and 1.816/0.190 for the 10b1g/11b1g and 4au/5au pairs, respectively). The three remaining singly occupied orbitals are then 21ag, 14b2u and 16b3u, all of which have occupations almost equal to 1.0. These three singly occupied orbitals bear a striking resemblance to the three singly occupied components of the t2 orbital in the 4A2 state of [V4N4]+.
3. Discussion
The two clusters considered here, [V4N4]+ and [V6N6]+, are both based on fragments of the rock-salt structure, as anticipated by Hirabayashi and Ichihashi in their original report of the mass-spectrometry experiments [14]. In the smaller cluster, quartet and sextet states are very close in energy, and meta-GGA functionals tend to favour the sextet, while GGA functionals predict the quartet to be the ground state. The B3LYP functional, as is typically the case, strongly stabilizes the higher spin state. Our analysis of the Kohn–Sham orbitals indicates that in the 4A2 state, the cluster is stabilized by two pairs of electrons in the V–V bonding 4e orbital, giving a net V–V bond order of 1/3 and a perfectly tetrahedral geometry. The sextet (6A1), in contrast, is D2d-symmetric, with the distortion away from a perfect tetrahedron being driven by a first-order Jahn–Teller instability. The V–V bonds are shorter and stronger in the 6A1 state as a result of the single occupation of the V–V bonding 9a1 orbital, but this is compensated for by a weakening of the V–N bonds. CASSCF calculations (CAS(9,7) active space) are consistent with the picture of bonding that emerges from the DFT calculations, but also reveal the strongly multi-configurational nature of the V–V bonding, which leads to low (<<2.0) occupancies of the 4e orbitals with compensating populations in the V–V antibonding 4t1 set. The ground state of the larger [V6N6]+ cluster appears to be less controversial: a 4B3g state is the most stable for all functionals considered here, and also at the NEVPT2 level. The cluster is stabilized by a strong V=V double bond between the metal atoms in the central V2N2 layer, a feature that emerges irrespective of the chosen theoretical model. In the DFT calculations, the 20ag (V–V σ) and 15b3u (V–V π) orbitals are doubly occupied, and the three unpaired electrons in the 4B3g ground state are localized on the upper and lower layers. To a first approximation, the cluster can therefore be viewed as a diamagnetic central V2N2 unit, with radical character delocalized over the upper and lower planes. CASSCF calculations support this basic model, but the multi-configurational character of the V=V double bond is again prominent, with occupations of the σ and π bonding and antibonding orbitals around 1.70 and 0.30. The balance between localized multiple V–V bonding in [V6N6]+ and the more delocalized situation in [V4N4]+ hints at a subtle relationship between structural and electronic properties, which may have important consequences for the reactivity of the nitrogen-deficient clusters, [Vn+1Nn]+. These will be the subject of subsequent studies.
4. Computational Methods
All DFT calculations reported in this paper were performed using Gaussian 16, revision A.03 [43] while the CASSCF calculations were done with ORCA, version 4.0 [44]. Density functional calculations were performed using Ahlrichs’s def2-TZVP basis set [45] on V and N either using the meta-GGA TPSS or M06L functionals, the GGA alternatives, BLYP or BP86, or the hybrid B3LYP. CASSCF calculations were performed using the same TZVP basis set and the RIJCOSX approximation to the exchange terms [46]. Dynamic correlation was accounted for using the N-electron valence state perturbation theory (NEVPT2) [47]. All optimised energies and geometries are summarised in Table S1, Supplementary Materials.
5. Conclusions
In this paper we used a combination of density functional theory and correlated ab initio theory (CASSCF) to explore the nature of the bonding in two clusters, [V4N4]+ and [V6N6]+, which have been observed in mass-spectrometry experiments. Unlike clusters with similar composition, both [V4N4]+ and [V6N6]+ are remarkably resistant to further reaction with NH3, suggesting that they share some unique structural and/or electronic features. Our calculations confirm that the two clusters are based on cubic architectures and can be viewed, to a first approximation, as fragments of the rock-salt lattice. However, it is clear that direct covalent V–V bonding plays a very substantial part in stabilizing these structures, just as it does in the low-valent solid phases with similar compositions, such as VN and VO. This bonding is particularly well-developed in the quartet ground state of the [V6N6]+ cluster, where a localized V=V double bond is present in the waist of the cluster. The DFT and CASSCF approaches offer broadly equivalent pictures of the metal–metal bonding, but the multi-configurational approach highlights the very strong static correlation that is typical of metal–metal bonds, particularly amongst the first transition series. The strong V–V bonds, along with the very symmetric structure, are consistent with the observed resistance of these two clusters to further reactions. Future work will focus on the interplay between V–V bonding and catalytic activity for the less symmetric nitrogen-deficient clusters, [V4N3]+ and [V6N5]+, both of which are substantially more reactive [14].
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2304-6740/7/4/52/s1, Table S1: Total energies and optimized Cartesian coordinates for all compounds described in the paper.
Author Contributions
Investigation, J.E.M. and P.H.; Project administration, J.E.M.; Supervision, J.E.M. and J.-G.Z.; Writing—original draft, P.H.; Writing—review & editing, J.E.M.
Funding
Piao He’s stay in Oxford was funded by the China Scholarship Council.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hargreaves, J.S.J. Heterogeneous catalysis with metal nitrides. Coord. Chem. Rev. 2013, 257, 2015–2031. [Google Scholar] [CrossRef]
- Guo, J.; Wang, P.; Wu, G.; Wu, A.; Hu, D.; Xiong, Z.; Wang, J.; Yu, P.; Chang, F.; Chen, Z.; et al. Lithium imide synergy with 3d transition-metal nitrides leading to unprecedented catalytic activities for ammonia decomposition. Angew. Chem. Int. Ed. 2015, 54, 2950–2954. [Google Scholar] [CrossRef]
- Lotz, C.R.; Sebba, F. Energies of activation for decomposition of ammonia catalysed by the nitrides of the 4th series transition elements. Trans. Faraday Soc. 1957, 53, 1246–1252. [Google Scholar] [CrossRef]
- Liao, M.Y.; Gotoh, Y.; Tsuji, H.; Ishikawa, J. Crystallographic structure and composition of vanadium nitride films deposited by direct sputtering of a compound target. J. Vac. Sci. Technol. A 2004, 22, 146–150. [Google Scholar] [CrossRef]
- Abghoui, Y.; Garden, A.L.; Howalt, J.G.; Vegge, T.; Skúlason, E. Electroreduction of N2 to ammonia at ambient conditions on mononitrides of Zr, Nb, Cr, and V: A DFT guide for experiments. ACS Catal. 2016, 6, 635–646. [Google Scholar] [CrossRef]
- Choi, D.; Blomgren, G.E.; Kumta, P.N. Fast and reversible surface redox reaction in nanocrystalline vanadium vitride supercapacitors. Adv. Mater. 2006, 18, 1178–1182. [Google Scholar] [CrossRef]
- McGill, W.J.; Sebba, F. The kinetics of ammonia decomposition over vanadium nitride. J. Catal. 1963, 2, 104–108. [Google Scholar] [CrossRef]
- Zhou, X.; Shang, C.; Gu, L.; Dong, S.; Chen, X.; Han, P.; Li, L.; Yao, J.; Liu, Z.; Xu, H.; et al. Mesoporous coaxial titanium nitride–vanadium nitride fibers of core–shell structures for high-performance supercapacitors. ACS Appl. Mater. Interfaces 2011, 3, 3058–3063. [Google Scholar] [CrossRef] [PubMed]
- Indlekofer, G.; Mariot, J.M.; Lengauer, W.; Beauprez, E.; Oelhafen, P.; Hague, C.F. Electronic structure of stoichiometric and substoichiometric vanadium nitrade from photoelectron spectroscopy. Solid State Commun. 1989, 72, 419–423. [Google Scholar] [CrossRef]
- Wu, G.; Yang, M.; Guo, X.; Wang, J. Comparative DFT study of N2 and NO adsorption on vanadium clusters Vn (n = 2–13). J. Comput. Chem. 2012, 33, 1854–1861. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.V.; Khanna, S.N. Structure and stability of TinNm clusters. Phys. Rev. B 1996, 54, 2240–2243. [Google Scholar] [CrossRef]
- Jakubikova, E.; Rappé, A.K.; Bernstein, E.R. Density functional theory study of small vanadium oxide clusters. J. Phys. Chem. A 2007, 111, 12938–12943. [Google Scholar] [CrossRef]
- Aetukuri, N.B.; Gray, A.X.; Drouard, M.; Cossale, M.; Gao, L.; Reid, A.H.; Kukreja, R.; Ohldag, H.; Jenkins, C.A.; Arenholz, E.; et al. Control of the metal–insulator transition in vanadium dioxide by modifying orbital occupancy. Nat. Phys. 2013, 9, 661–666. [Google Scholar] [CrossRef]
- Hirabayashi, S.; Ichihashi, M. Adsorption and dehydrogenation of ammonia on vanadium and niobium nitride cluster cations. Int. J. Mass Spectrom. 2016, 407, 86–91. [Google Scholar] [CrossRef]
- Pralong, V. Lithium intercalation into transition metal oxides: A route to generate new ordered rock salt type structure. Prog. Solid State Chem. 2009, 37, 262–277. [Google Scholar] [CrossRef]
- Burdett, J.K.; Mitchell, J.F. Nonstoichiometry in early transition metal compounds with the rock salt structure. Prog. Solid State Chem. 1995, 23, 131–170. [Google Scholar] [CrossRef]
- Liu, Q.Q.; Pan, H.L.; Tao, W.H.; Wu, H.T. New rock salt structure dielectric material Li2Ni3TiO6 at microwave frequency. J. Mater. Sci: Mater. Electron. 2017, 28, 9893–9899. [Google Scholar] [CrossRef]
- Nakamura, A.; Ukita, M.; Shimoda, N.; Furushima, Y.; Toyoura, K.; Matsunaga, K. First-principles calculations on slip system activation in the rock salt structure: Electronic origin of ductility in silver chloride. Philos. Mag. 2017, 97, 1281–1310. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Castleman, A.W., Jr. Growth of titanium nitride: From clusters to microcrystals. J. Chem. Phys. 1993, 98, 231–235. [Google Scholar] [CrossRef]
- Marksteiner, P.; Weinberger, P.; Neckel, A.; Zeller, R.; Dederichs, P.H. Electronic structure of substoichiometric carbides and nitrides of titanium and vanadium. Phys. Rev. B 1986, 33, 812–822. [Google Scholar] [CrossRef]
- Beinert, H.; Holm, R.H.; Münck, E. Iron–sulfur clusters: Nature’s modular, multipurpose structures. Science 1997, 277, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Noodleman, L. A model for the spin states of high-potential iron–sulfur [Fe4S4]3+ proteins. Inorg. Chem. 1988, 27, 3677–3679. [Google Scholar] [CrossRef]
- Lancaster, K.M.; Roemelt, M.; Ettenhuber, P.; Hu, Y.; Ribbe, M.W.; Neese, F.; Bergmann, U.; DeBeer, S. X-ray emission spectroscopy evidences a central carbon in the nitrogenase iron-molybdenum cofactor. Science 2011, 334, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Bjornsson, R.; Neese, F.; DeBeer, S. Revisiting the Mössbauer isomer shifts of the FeMoco cluster of nitrogenase and the cofactor charge. Inorg. Chem. 2017, 56, 1470–1477. [Google Scholar] [CrossRef]
- Krewald, V.; Neese, F.; Pantazis, D.A. On the magnetic and spectroscopic properties of high-valent Mn3CaO4 cubanes as structural units of natural and artificial water-oxidizing catalysts. J. Am. Chem. Soc. 2013, 135, 5726–5739. [Google Scholar] [CrossRef] [PubMed]
- McGrady, J.E. Periodic trends in metal–metal bonding in cubane clusters, (C5H5)4M4E4 [M = Cr, Mo, E = O, S]. J. Chem. Soc. Dalton Trans. 1999, 1393–1400. [Google Scholar] [CrossRef]
- Gorelsky, S.I. Complexes with a single metal–metal bond as a sensitive probe of quality of exchange-correlation functionals. J. Chem. Theor. Comput. 2012, 8, 908–914. [Google Scholar] [CrossRef]
- Spivak, M.; Arcisauskaite, V.; López, X.; McGrady, J.E.; de Graaf, C. A multiconfigurational approach to the electronic structure of trichromium extended metal atom chains. Dalton Trans. 2017, 46, 6202–6211. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.M.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401:1–146401:4. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101:1–194101:18. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Stefan, G.; Stephan, E.; Lars, G. Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Paulsen, H.; Trautwein, A.X. Density functional theory calculations for spin-crossover complexes. Top. Curr. Chem. 2012, 235, 197–219. [Google Scholar]
- Davidson, E.R. Natural orbitals. Adv. Quant. Chem. 1972, 6, 235–266. [Google Scholar]
- Davidson, E.R. Properties and uses of natural orbitals. Rev. Mod. Phys. 1972, 44, 451–464. [Google Scholar] [CrossRef]
- Roos, B.O. The complete active space self-consistent field method and its applications in electronic structure calculations. Adv. Chem. Phys. 1987, 69, 399–445. [Google Scholar]
- Veryazov, V.; Malmqvist, P.A.; Roos, B.O. How to select active space for multiconfigurational quantum chemistry? Int. J. Quantum Chem. 2011, 111, 3329–3338. [Google Scholar] [CrossRef]
- Kandalam, A.K.; Blanco, M.A.; Pandey, R. Theoretical study of AlnNn, GanNn, and InnNn (n = 4, 5, 6) clusters. J. Phys. Chem. B 2002, 106, 1945–1953. [Google Scholar] [CrossRef]
- Li, M.G.; Dzubak, A.L.; Mulla, A.; Brogden, D.W.; Berry, J.F.; Gagliardi, L. Assessing metal–metal multiple bonds in Cr–Cr, Mo–Mo, and W–W compounds and a hypothetical U–U compound: A quantum chemical study comparing DFT and multireference methods. Chem. Eur. J. 2012, 18, 1737–1749. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Neese, F. An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix. J. Comp. Chem. 2003, 24, 1740–1747. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Evangelisti, S.; Leininger, T.; Malrieu, J.-P. Introduction of N-electron valence states for multireference perturbation theory. J. Chem. Phys. 2001, 114, 10252–10264. [Google Scholar] [CrossRef]
Figure 1.
Clusters with cubane-like architectures: (a) the Cp4M4E4 architecture, (b) the Fe4S4 cubanes and (c) the FeMo cofactor of nitrogenase.
Figure 1.
Clusters with cubane-like architectures: (a) the Cp4M4E4 architecture, (b) the Fe4S4 cubanes and (c) the FeMo cofactor of nitrogenase.

Figure 2.
Optimised structures (TPSS/def2-TZVP) and Kohn–Sham molecular orbital diagrams for the quartet (4A2) and sextet (6A1) states of [V4N4]+. Bond lengths (in Å) are shown in black, Mulliken spin densities are shown in red. The levels of the 6A1 state have been shifted upwards by 0.59 eV to bring the barycentre of the 12a1–4b1 pair into coincidence with the 4e level of 4A2.
Figure 2.
Optimised structures (TPSS/def2-TZVP) and Kohn–Sham molecular orbital diagrams for the quartet (4A2) and sextet (6A1) states of [V4N4]+. Bond lengths (in Å) are shown in black, Mulliken spin densities are shown in red. The levels of the 6A1 state have been shifted upwards by 0.59 eV to bring the barycentre of the 12a1–4b1 pair into coincidence with the 4e level of 4A2.

Figure 3.
Natural orbital occupations for the 4A2 and 6A1 states (in parentheses) of [V4N4]+ with a (7,9) complete active space.
Figure 3.
Natural orbital occupations for the 4A2 and 6A1 states (in parentheses) of [V4N4]+ with a (7,9) complete active space.

Figure 4.
Optimised structure (TPSS/def2-TZVP) and Kohn–Sham molecular orbital diagram for the quartet (4B3g) state of [V6N6]+. Bond lengths (in Å) are shown in black, Mulliken spin densities are shown in red.
Figure 4.
Optimised structure (TPSS/def2-TZVP) and Kohn–Sham molecular orbital diagram for the quartet (4B3g) state of [V6N6]+. Bond lengths (in Å) are shown in black, Mulliken spin densities are shown in red.

Figure 5.
Natural orbital occupations for the 4B3g state of [V6N6]+ with a CAS(11,11) active space.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Relative energies, V–V bond lengths and Mulliken spin densities for the 4A2 and 6A1 states of [V4N4]+. The energetic reference point is taken as the more stable of the two states.
Table 1.
Relative energies, V–V bond lengths and Mulliken spin densities for the 4A2 and 6A1 states of [V4N4]+. The energetic reference point is taken as the more stable of the two states.
| Functional | 4A2 | 6A1 | ||||
|---|---|---|---|---|---|---|
| V–V/Å | ρ(V) | Erel/eV | V–V/Å | ρ(V) | Erel/eV | |
| TPSS | 2.64 | 0.86 | +0.19 | 2.60 | 1.27 | 0 |
| M06L | 2.62 | 0.90 | +0.38 | 2.56 | 1.29 | 0 |
| BP86 | 2.65 | 0.85 | 0 | 2.61 | 1.28 | +0.08 |
| BP86-D3 | 2.65 | 0.85 | 0 | 2.61 | 1.28 | +0.09 |
| BLYP | 2.68 | 0.85 | 0 | 2.63 | 1.28 | +0.05 |
| B3LYP | 2.62 | 0.70 | +0.65 | 2.62 | 1.36 | 0 |
Table 2.
V–V bond lengths and Mulliken spin densities for the 4B3g state of [V6N6]+.
| Functional | 4B3g | |
|---|---|---|
| V–V/Å 1 | ρ(V) 2 | |
| TPSS | 2.23, 2.52, 2.60 | −0.10, 0.87 |
| M06L | 2.23, 2.51, 2.58 | −0.12, 0.92 |
| BP86 | 2.24, 2.53, 2.62 | −0.03, 0.85 |
| BLYP | 2.27, 2.55, 2.64 | −0.05, 0.86 |
| B3LYP | 2.24, 2.53, 2.59 | −0.39, 1.07 |
1 the three bond lengths correspond to V1–V1, V1–V2 and V2–V2 in Figure 4; 2 spin densities are for V1 and V2.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
He, P.; Zhang, J.-G.; McGrady, J.E. Electronic Structure of Cubane-Like Vanadium–Nitrogen Cationic Clusters [V4N4]+ and [V6N6]+. Inorganics 2019, 7, 52. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7040052
AMA Style
He P, Zhang J-G, McGrady JE. Electronic Structure of Cubane-Like Vanadium–Nitrogen Cationic Clusters [V4N4]+ and [V6N6]+. Inorganics. 2019; 7(4):52. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7040052
Chicago/Turabian StyleHe, Piao, Jian-Guo Zhang, and John E. McGrady. 2019. "Electronic Structure of Cubane-Like Vanadium–Nitrogen Cationic Clusters [V4N4]+ and [V6N6]+" Inorganics 7, no. 4: 52. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7040052
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.