A Comparative Study of the Catalytic Behaviour of Alkoxy-1,3,5-Triazapentadiene Copper(II) Complexes in Cyclohexane Oxidation


Abstract
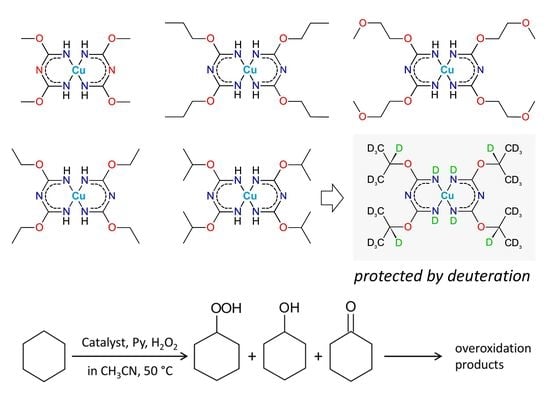
:
1. Introduction
2. Results
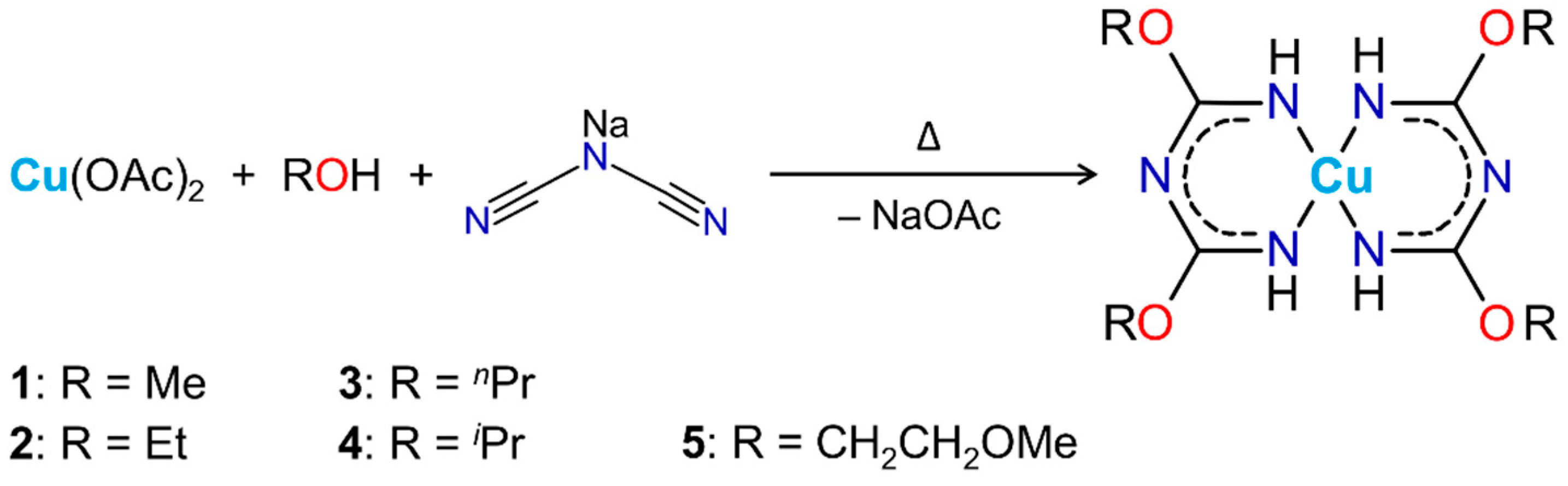
2.1. Synthesis of the Complexes
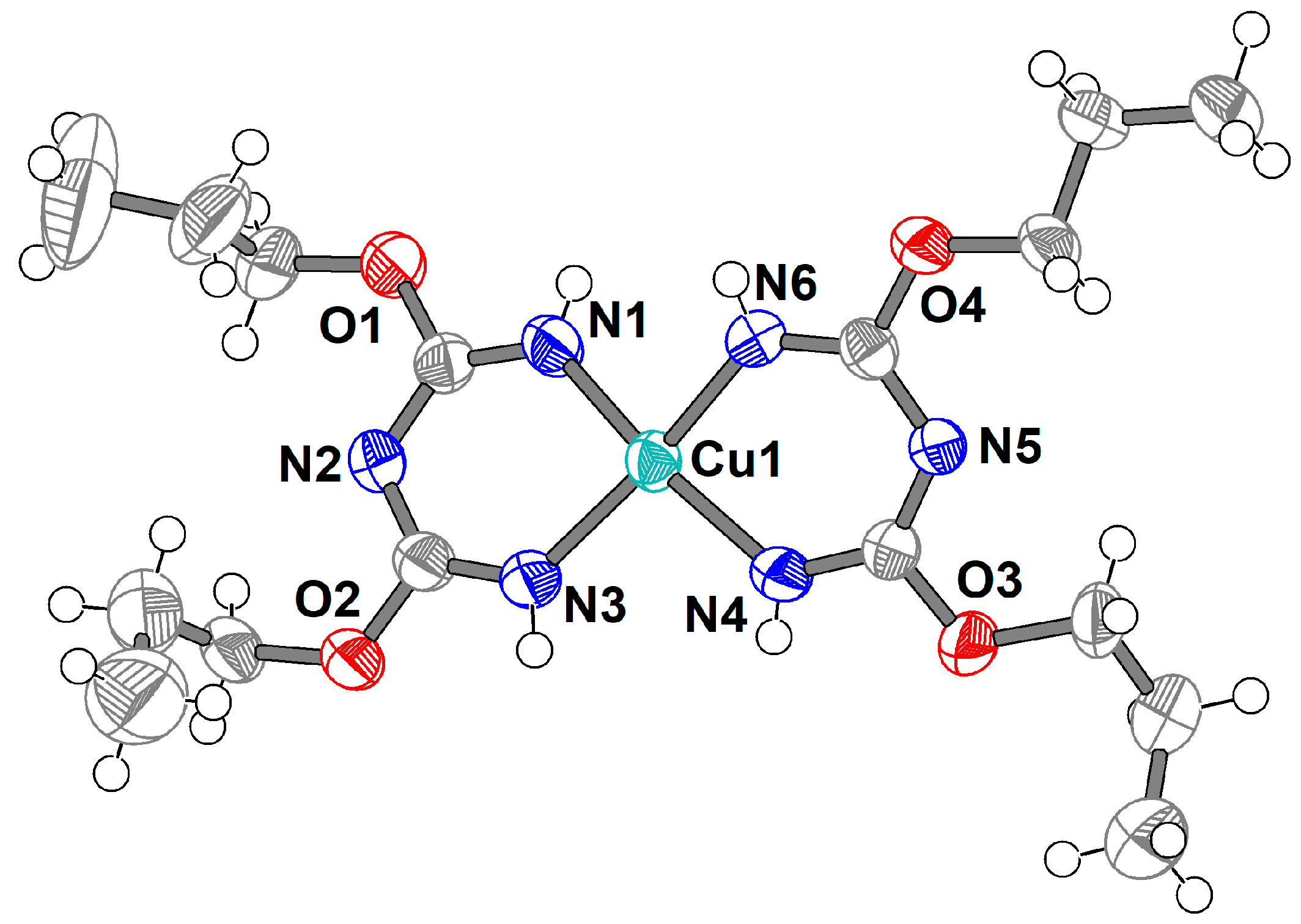
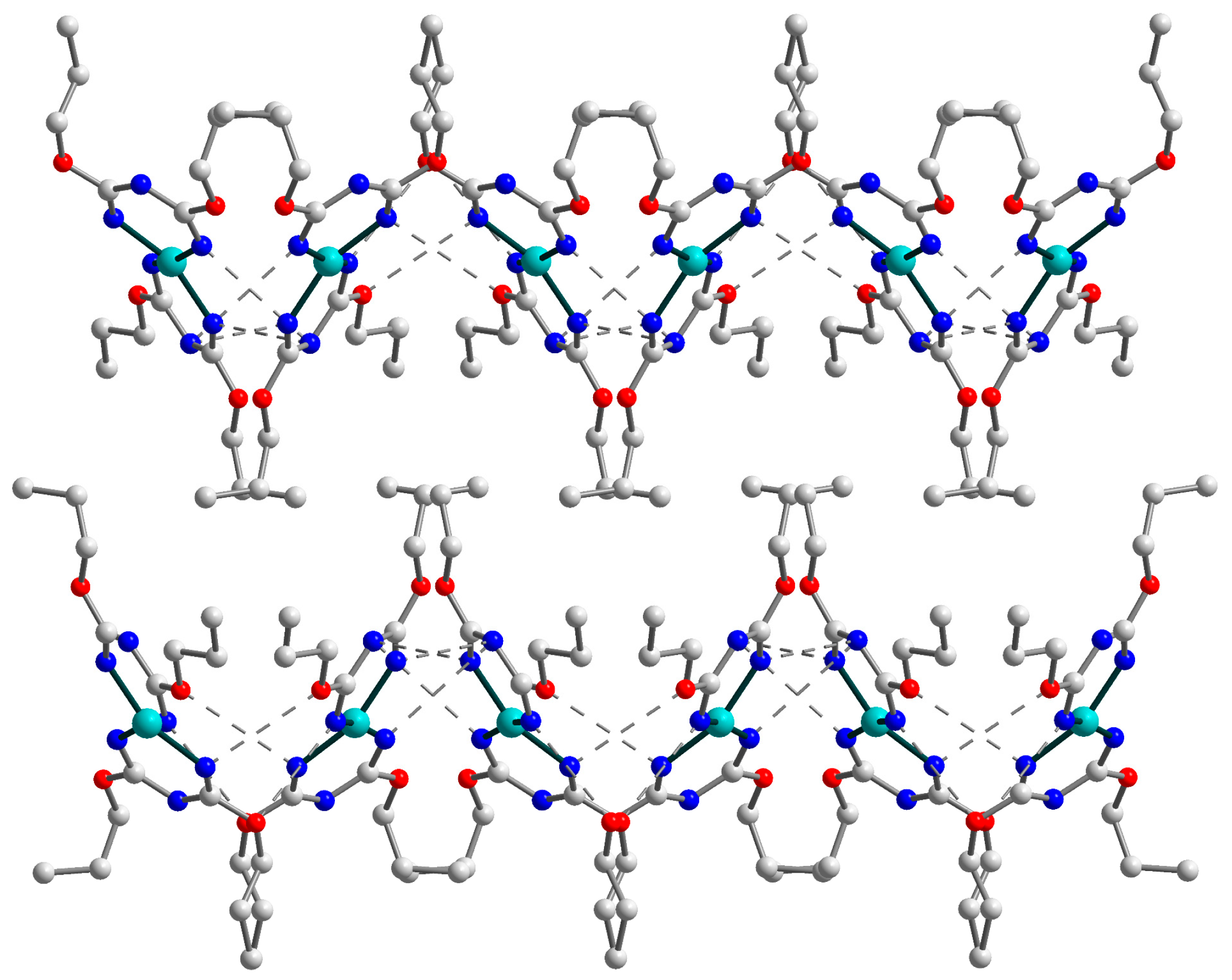
2.2. Crystal Structure of 3
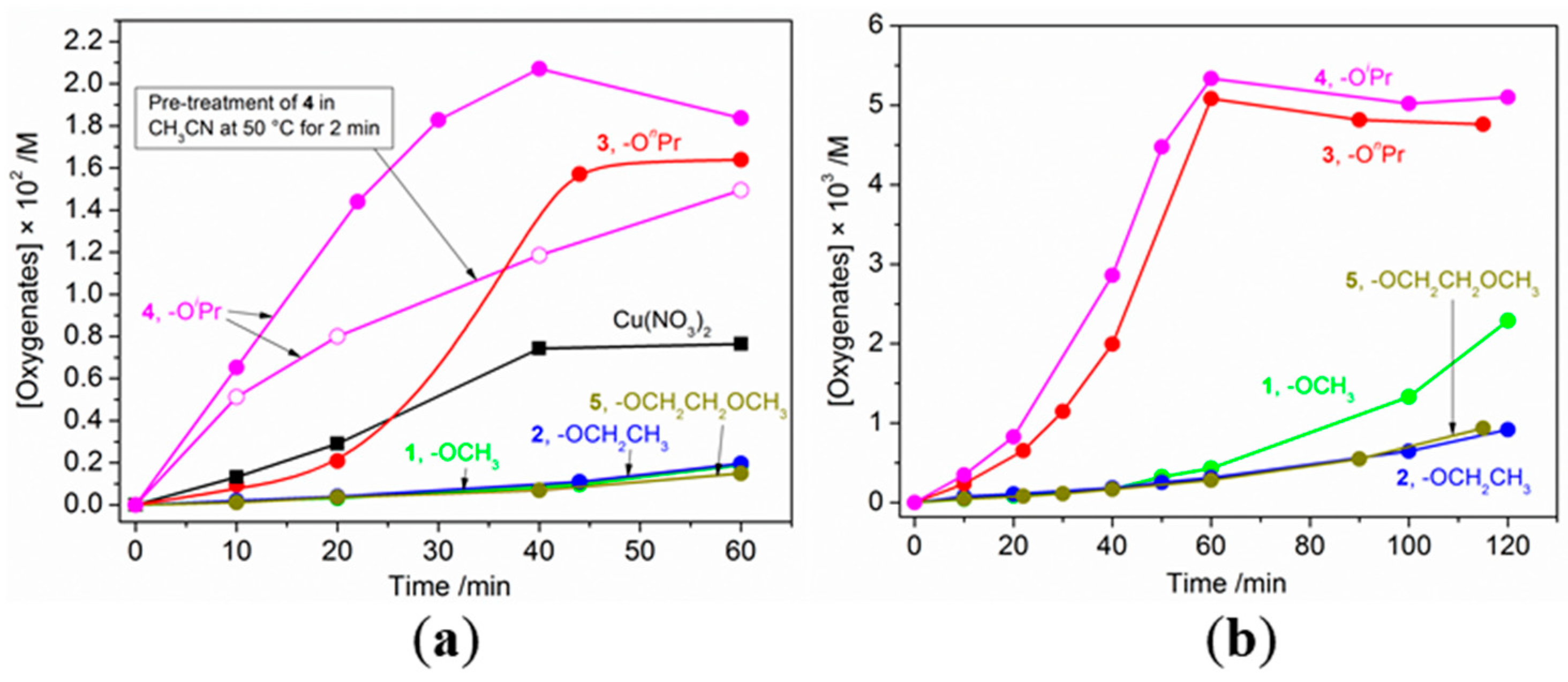
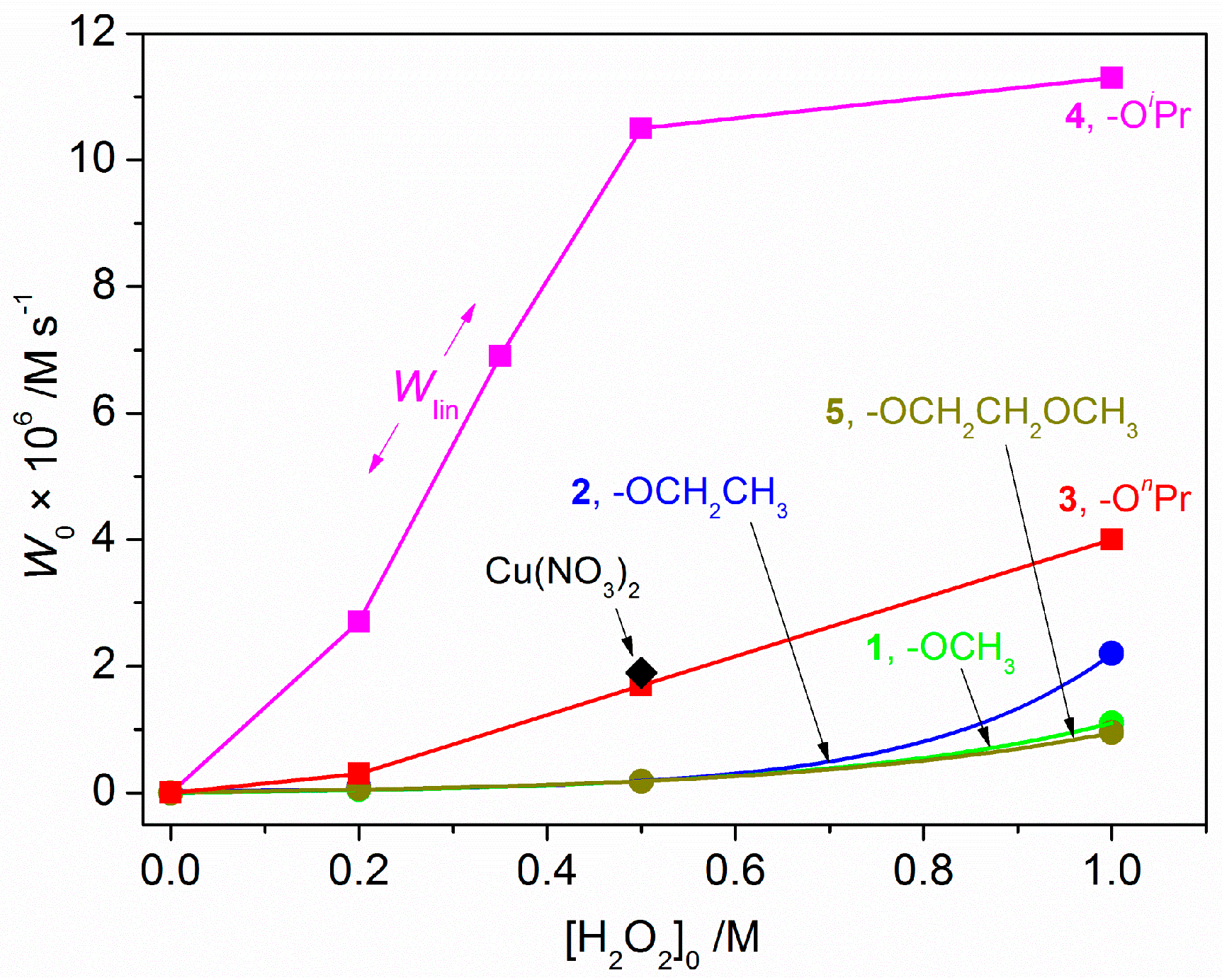
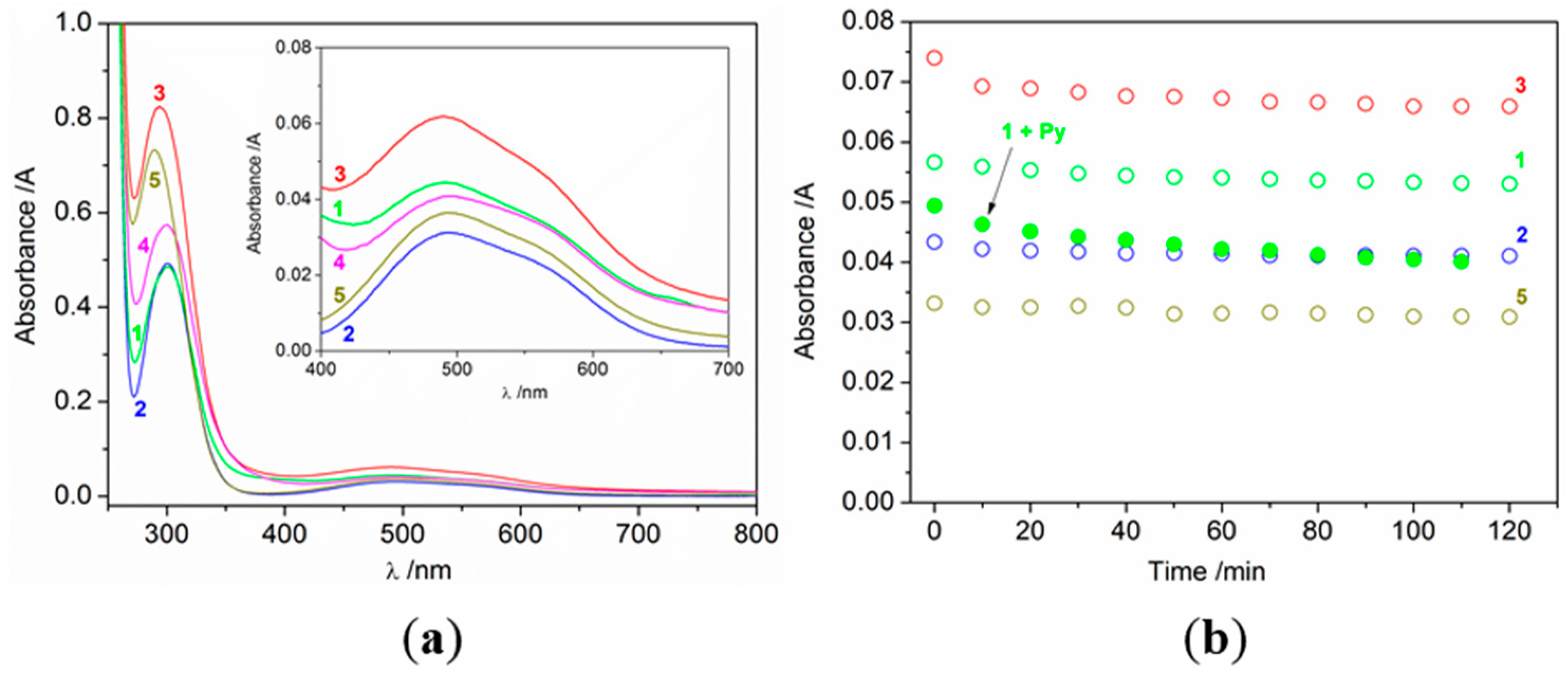
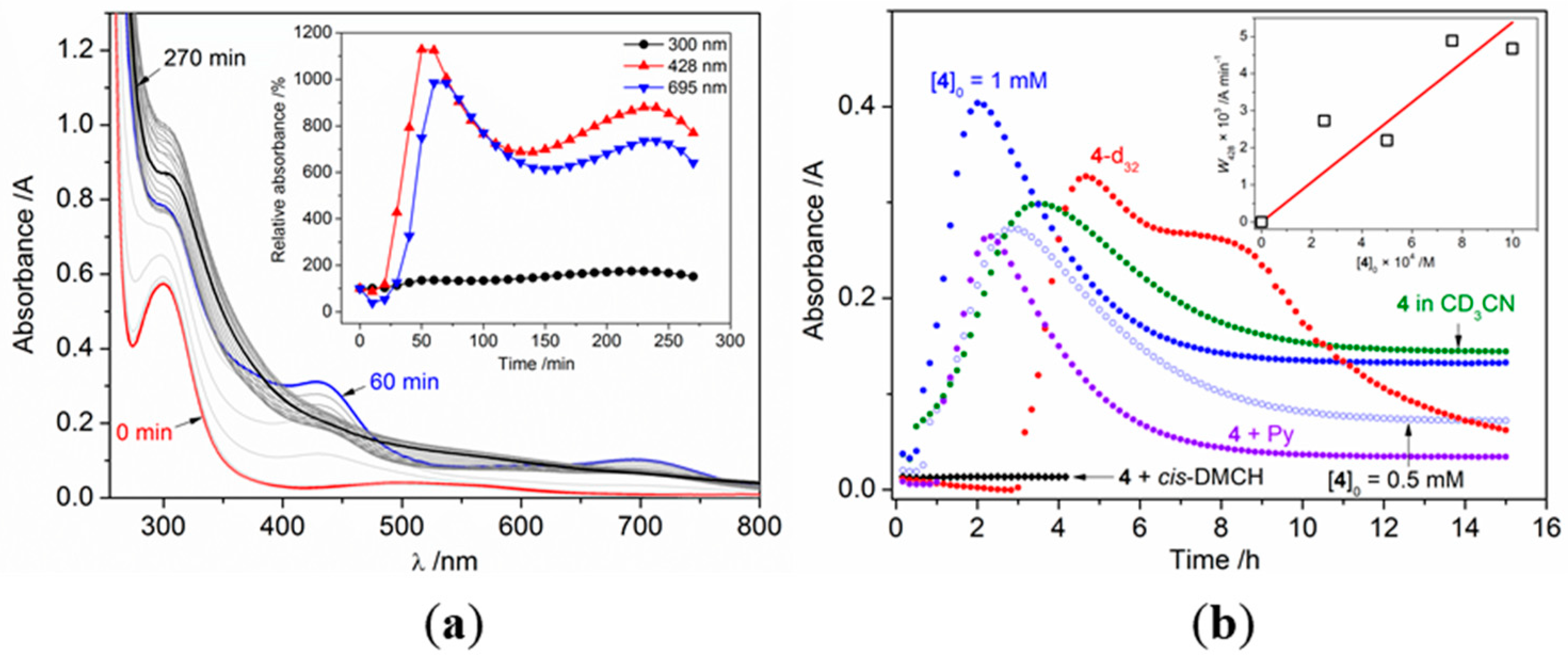
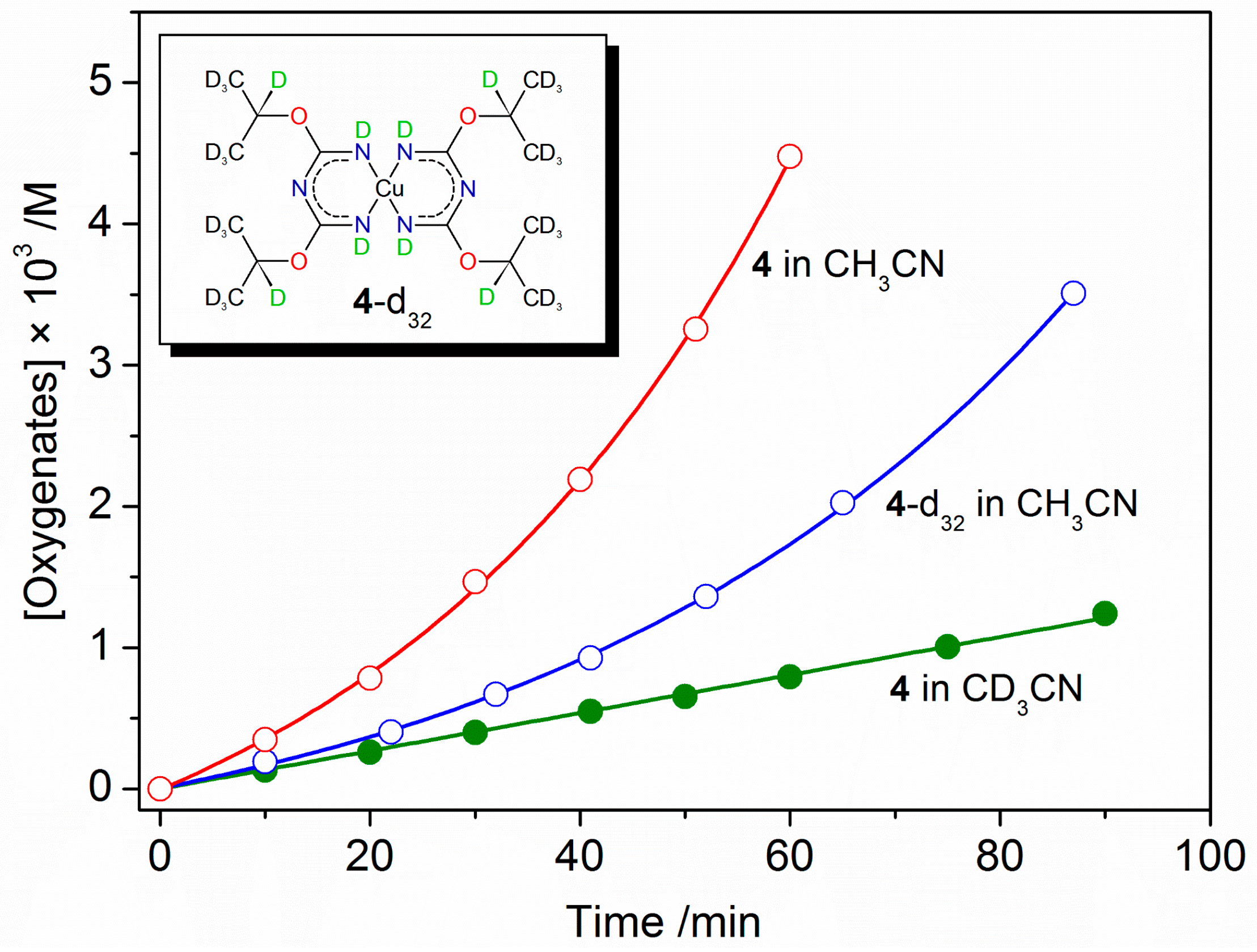
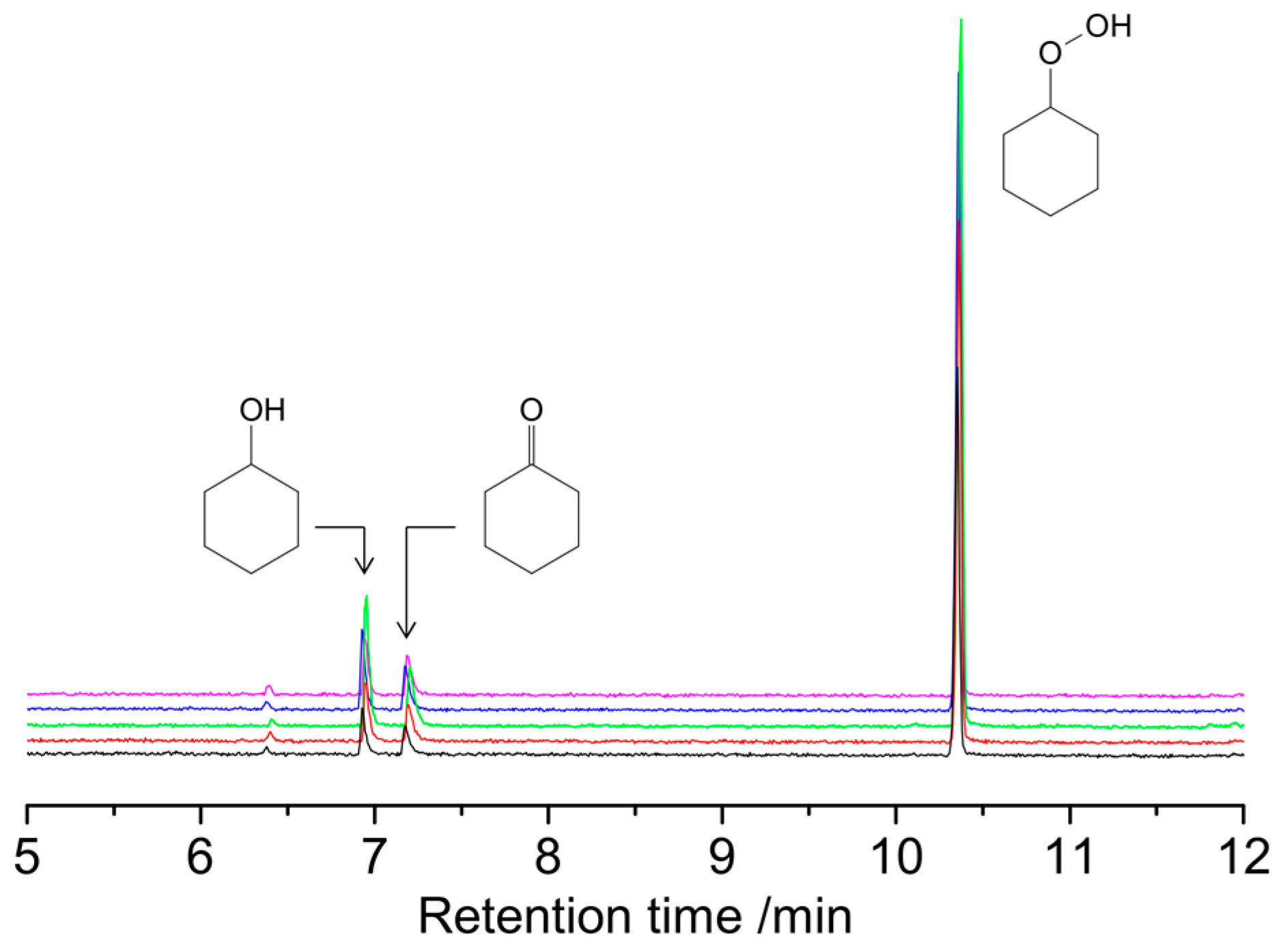
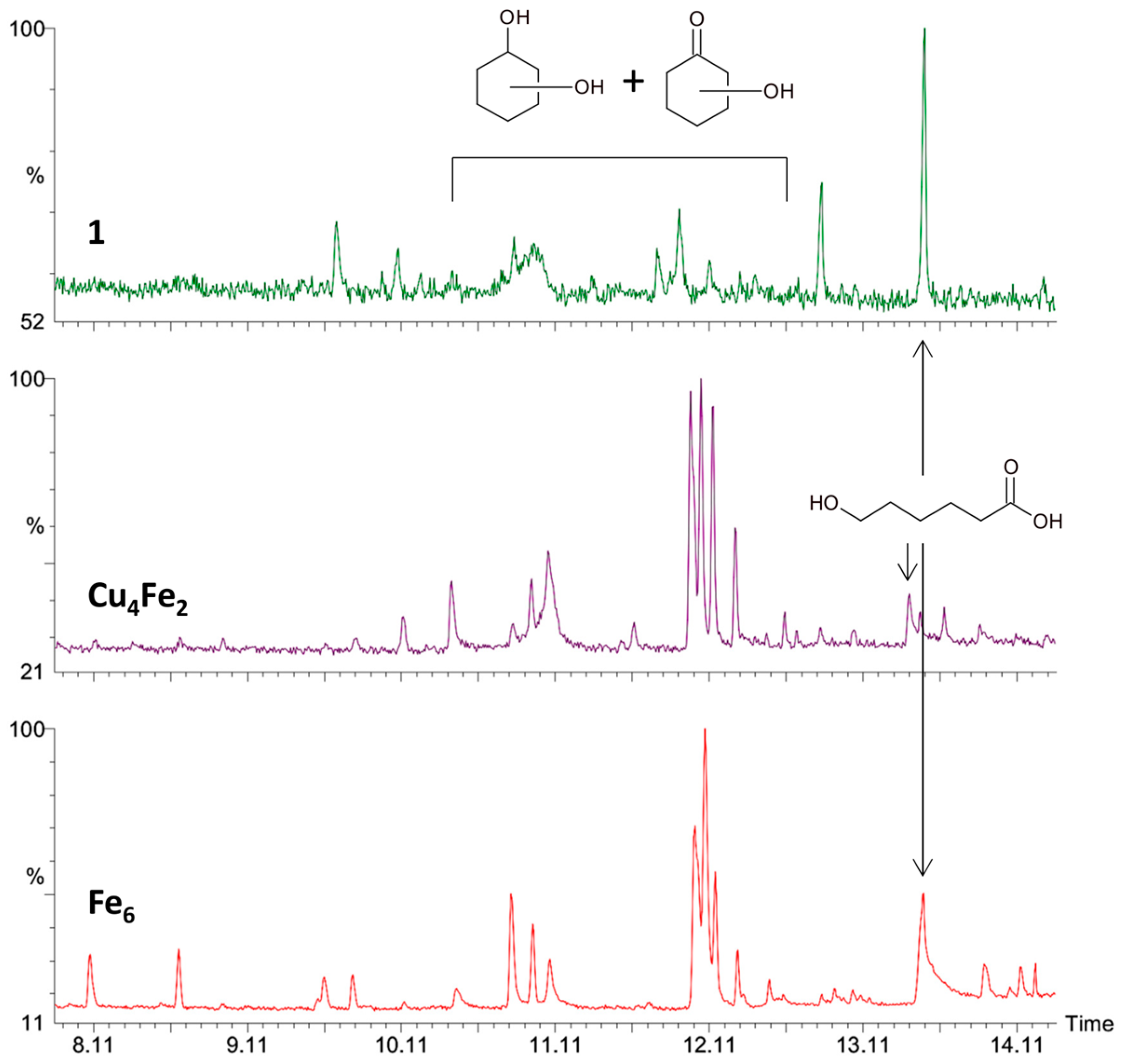
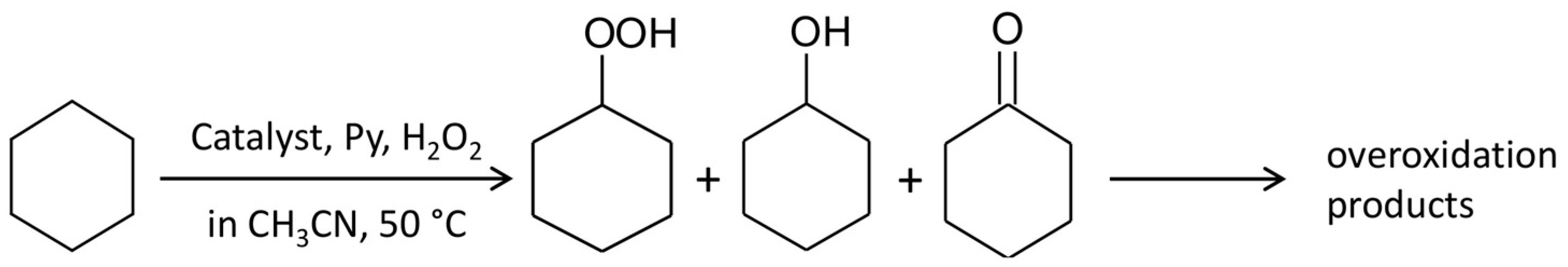
2.3. Catalytic Oxidation of Cyclohexane with H2O2
3. Materials and Methods
3.1. Reagents and Materials
3.2. Crystallography
3.3. Catalytic Oxidation of Alkanes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gandeepan, P.; Muller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Transition Metals for C–H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef] [PubMed]
- Karimov, R.R.; Hartwig, J.F. Transition-Metal-Catalyzed Selective Functionalization of C(sp3)–H Bonds in Natural Products. Angew. Chem. Int. Ed. 2018, 57, 4234–4241. [Google Scholar] [CrossRef] [PubMed]
- Labinger, J.A. Platinum-Catalyzed C–H Functionalization. Chem. Rev. 2017, 117, 8483–8496. [Google Scholar] [CrossRef] [PubMed]
- Cernak, T.; Dykstra, K.D.; Tyagarajan, S.; Vachal, P.; Krska, S.W. The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev. 2016, 45, 546–576. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, J.F.; Larsen, M.A. Undirected, Homogeneous C–H Bond Functionalization: Challenges and Opportunities. ACS Central Sci. 2016, 2, 281–292. [Google Scholar] [CrossRef]
- Nesterov, D.S.; Nesterova, O.V.; Pombeiro, A.J.L. Homo- and heterometallic polynuclear transition metal catalysts for alkane C–H bonds oxidative functionalization: Recent advances. Coord. Chem. Rev. 2018, 355, 199–222. [Google Scholar] [CrossRef]
- Crabtree, R.H. The stability of organometallic ligands in oxidation catalysis. J. Organomet. Chem. 2014, 751, 174–180. [Google Scholar] [CrossRef]
- Weiner, H.; Finke, R.G. An all-inorganic, polyoxometalate-based catechol dioxygenase that exhibits > 100,000 catalytic turnovers. J. Am. Chem. Soc. 1999, 121, 9831–9842. [Google Scholar] [CrossRef]
- Kim, W.; Yuan, G.; McClure, B.A.; Frei, H. Light Induced Carbon Dioxide Reduction by Water at Binuclear ZrOCoII Unit Coupled to Ir Oxide Nanocluster Catalyst. J. Am. Chem. Soc. 2014, 136, 11034–11042. [Google Scholar] [CrossRef]
- Geletii, Y.V.; Botar, B.; Koegerler, P.; Hillesheim, D.A.; Musaev, D.G.; Hill, C.L. An all-inorganic, stable, and highly active tetraruthenium homogeneous catalyst for water oxidation. Angew. Chem. Int. Ed. 2008, 47, 3896–3899. [Google Scholar] [CrossRef]
- Lv, H.; Song, J.; Geletii, Y.V.; Vickers, J.W.; Sumliner, J.M.; Musaev, D.G.; Koegerler, P.; Zhuk, P.F.; Bacsa, J.; Zhu, G.; et al. An Exceptionally Fast Homogeneous Carbon-Free Cobalt-Based Water Oxidation Catalyst. J. Am. Chem. Soc. 2014, 136, 9268–9271. [Google Scholar] [CrossRef] [PubMed]
- Oloo, W.N.; Que, L. Bioinspired Nonheme Iron Catalysts for C–H and C=C Bond Oxidation: Insights into the Nature of the Metal-Based Oxidants. Acc. Chem. Res. 2015, 48, 2612–2621. [Google Scholar] [CrossRef] [PubMed]
- Bryliakov, K.P.; Talsi, E.P. Active sites and mechanisms of bioinspired oxidation with H2O2, catalyzed by non-heme Fe and related Mn complexes. Coord. Chem. Rev. 2014, 276, 73–96. [Google Scholar] [CrossRef]
- Olivo, G.; Cusso, O.; Borrell, M.; Costas, M. Oxidation of alkane and alkene moieties with biologically inspired nonheme iron catalysts and hydrogen peroxide: From free radicals to stereoselective transformations. J. Biol. Inorg. Chem. 2017, 22, 425–452. [Google Scholar] [CrossRef] [PubMed]
- Nesterov, D.S.; Chygorin, E.N.; Kokozay, V.N.; Bon, V.V.; Boca, R.; Kozlov, Y.N.; Shul’pina, L.S.; Jezierska, J.; Ozarowski, A.; Pombeiro, A.J.L.; et al. Heterometallic CoIII4FeIII2 Schiff Base Complex: Structure, Electron Paramagnetic Resonance, and Alkane Oxidation Catalytic Activity. Inorg. Chem. 2012, 51, 9110–9122. [Google Scholar] [CrossRef] [PubMed]
- Kopylovich, M.N.; Pombeiro, A.J.L. Coordination chemistry of 1,3,5-triazapentadienes. Coord. Chem. Rev. 2011, 255, 339–355. [Google Scholar] [CrossRef]
- Bhunia, S.; Pawar, G.G.; Kumar, S.V.; Jiang, Y.W.; Ma, D.W. Selected Copper-Based Reactions for C–N, C–O, C–S, and C–C Bond Formation. Angew. Chem. Int. Ed. 2017, 56, 16136–16179. [Google Scholar] [CrossRef]
- Marchetti, F.; Pettinari, R.; Pettinari, C. Recent advances in acylpyrazolone metal complexes and their potential applications. Coord. Chem. Rev. 2015, 303, 1–31. [Google Scholar] [CrossRef]
- Shixallyev, N.Q.; Gurbanov, A.V.; Maharramov, A.M.; Mahmudov, K.T.; Kopylovich, M.N.; Martins, L.; Muzalevskiy, V.M.; Nenajdenko, V.G.; Pombeiro, A.J.L. Halogen-bonded tris(2,4-bis(trichloromethyl)-1,3,5-triazapentadienato)-M(III) M = Mn, Fe, Co complexes and their catalytic activity in the peroxidative oxidation of 1-phenylethanol to acetophenone. New J. Chem. 2014, 38, 4807–4815. [Google Scholar] [CrossRef]
- Shixaliyev, N.Q.; Maharramov, A.M.; Gurbanov, A.V.; Nenajdenko, V.G.; Muzalevskiy, V.M.; Mahmudov, K.T.; Kopylovich, M.N. Zinc(II)-1,3,5-triazapentadienate complex as effective catalyst in Henry reaction. Catal. Today 2013, 217, 76–79. [Google Scholar] [CrossRef]
- Figiel, P.J.; Kopylovich, M.N.; Lasri, J.; da Silva, M.F.C.G.; da Silva, J.J.R.F.; Pombeiro, A.J.L. Solvent-free microwave-assisted peroxidative oxidation of secondary alcohols to the corresponding ketones catalyzed by copper(II) 2,4-alkoxy-1,3,5-triazapentadienato complexes. Chem. Commun. 2010, 46, 2766–2768. [Google Scholar] [CrossRef] [PubMed]
- Kopylovich, M.N.; Lasri, J.; da Silva, M.; Pombeiro, A.J.L. Single-pot template transformations of cyanopyridines on a Pd-II centre: Syntheses of ketoimine and 2,4-dipyridyl-1,3,5-triazapentadiene palladium(II) complexes and their catalytic activity for microwave-assisted Suzuki-Miyaura and Heck reactions. Dalton Trans. 2009, 3074–3084. [Google Scholar] [CrossRef] [PubMed]
- Kopylovich, M.N.; Karabach, Y.Y.; da Silva, M.F.C.G.; Figiel, P.J.; Lasri, J.; Pombeiro, A.J.L. Alkoxy-1,3,5-triazapentadien(e/ato) Copper(II) Complexes: Template Formation and Applications for the Preparation of Pyrimidines and as Catalysts for Oxidation of Alcohols to Carbonyl Products. Chem. Eur. J. 2012, 18, 899–914. [Google Scholar] [CrossRef] [PubMed]
- Shul’pin, G.B. New Trends in Oxidative Functionalization of Carbon-Hydrogen Bonds: A Review. Catalysts 2016, 6, 50. [Google Scholar] [CrossRef]
- Wang, F.; Chen, P.H.; Liu, G.S. Copper-Catalyzed Radical Relay for Asymmetric Radical Transformations. Acc. Chem. Res. 2018, 51, 2036–2046. [Google Scholar] [CrossRef] [PubMed]
- Trammell, R.; Rajabimoghadam, K.; Garcia-Bosch, I. Copper-Promoted Functionalization of Organic Molecules: From Biologically Relevant Cu/O2 Model Systems to Organometallic Transformations. Chem. Rev. 2019, 119, 2954–3031. [Google Scholar] [CrossRef] [PubMed]
- Bilyachenko, A.N.; Kulakova, A.N.; Levitsky, M.M.; Korlyukov, A.A.; Khrustalev, V.N.; Vologzhanina, A.V.; Titov, A.A.; Dorovatovskii, P.V.; Shul’pina, L.S.; Lamaty, F.; et al. Ionic Complexes of Tetra- and Nonanuclear Cage Copper(II) Phenylsilsesquioxanes: Synthesis and High Activity in Oxidative Catalysis. ChemCatChem 2017, 9, 4437–4447. [Google Scholar] [CrossRef]
- Kulakova, A.N.; Korlyukov, A.A.; Zubavichus, Y.V.; Khrustalev, V.N.; Bantreil, X.; Shul’pina, L.S.; Levitsky, M.M.; Ikonnikov, N.S.; Shubina, E.S.; Lamaty, F.; et al. Hexacoppergermsesquioxanes as complexes with N-ligands: Synthesis, structure and catalytic properties. J. Organomet. Chem. 2019, 884, 17–28. [Google Scholar] [CrossRef]
- Tran, B.L.; Li, B.J.; Driess, M.; Hartwig, J.F. Copper-Catalyzed Intermolecular Amidation and Imidation of Unactivated Alkanes. J. Am. Chem. Soc. 2014, 136, 2555–2563. [Google Scholar] [CrossRef]
- Bakhoda, A.; Jiang, Q.; Badiei, Y.M.; Bertke, J.A.; Cundari, T.R.; Warren, T.H. Copper-Catalyzed C(sp3)–H Amidation: Sterically Driven Primary and Secondary C–H Site-Selectivity. Angew. Chem. Int. Ed. 2019, 58, 3421–3425. [Google Scholar] [CrossRef]
- Teng, F.; Sun, S.; Jiang, Y.; Yu, J.T.; Cheng, J. Copper-catalyzed oxidative C(sp3)–H/N–H coupling of sulfoximines and amides with simple alkanes via a radical process. Chem. Commun. 2015, 51, 5902–5905. [Google Scholar] [CrossRef] [PubMed]
- Chikkade, P.K.; Kuninobu, Y.; Kanai, M. Copper-catalyzed intermolecular C(sp3)–H bond functionalization towards the synthesis of tertiary carbamates. Chem. Sci. 2015, 6, 3195–3200. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, F.; McCann, S.D.; Wang, D.; Chen, P.; Stahl, S.S.; Liu, G. Enantioselective cyanation of benzylic C–H bonds via copper-catalyzed radical relay. Science 2016, 353, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Hutchings, G.J. Recent Advances in the Direct Synthesis of H2O2. ChemCatChem 2019, 11, 298–308. [Google Scholar] [CrossRef]
- Ranganathan, S.; Sieber, V. Recent Advances in the Direct Synthesis of Hydrogen Peroxide Using Chemical Catalysis—A Review. Catalysts 2018, 8, 22. [Google Scholar] [CrossRef]
- Bryliakov, K.P. Catalytic Asymmetric Oxygenations with the Environmentally Benign Oxidants H2O2 and O2. Chem. Rev. 2017, 117, 11406–11459. [Google Scholar] [CrossRef]
- Gamba, I.; Codola, Z.; Lloret-Fillol, J.; Costas, M. Making and breaking of the O–O bond at iron complexes. Coord. Chem. Rev. 2017, 334, 2–24. [Google Scholar] [CrossRef]
- Nesterova, O.V.; Nesterov, D.S.; Krogul-Sobczak, A.; da Silva, M.F.C.G.; Pombeiro, A.J.L. Synthesis, crystal structures and catalytic activity of Cu(II) and Mn(III) Schiff base complexes: Influence of additives on the oxidation catalysis of cyclohexane and 1-phenylehanol. J. Mol. Catal. A 2017, 426, 506–515. [Google Scholar] [CrossRef]
- Buvaylo, E.A.; Kokozay, V.N.; Vassilyeva, O.Y.; Skelton, B.W.; Nesterova, O.V.; Pombeiro, A.J.L. Copper(II) complex of the 2-pyridinecarbaldehyde aminoguanidine Schiff base: Crystal structure and catalytic behaviour in mild oxidation of alkanes. Inorg. Chem. Commun. 2017, 78, 85–90. [Google Scholar] [CrossRef]
- Low, S.; Becker, J.; Wurtele, C.; Miska, A.; Kleeberg, C.; Behrens, U.; Walter, O.; Schindler, S. Reactions of Copper(II) Chloride in Solution: Facile Formation of Tetranuclear Copper Clusters and Other Complexes That Are Relevant in Catalytic Redox Processes. Chem. Eur. J. 2013, 19, 5342–5351. [Google Scholar] [CrossRef]
- Nesterov, D.S.; Nesterova, O.V.; da Silva, M.F.C.G.; Pombeiro, A.J.L. Catalytic behaviour of a novel Fe(III) Schiff base complex in the mild oxidation of cyclohexane. Catal. Sci. Technol. 2015, 5, 1801–1812. [Google Scholar] [CrossRef]
- Rocha, B.G.M.; Kuznetsov, M.L.; Kozlov, Y.N.; Pombeiro, A.J.L.; Shul’pin, G.B. Simple soluble Bi(III) salts as efficient catalysts for the oxidation of alkanes with H2O2. Catal. Sci. Technol. 2015, 5, 2174–2187. [Google Scholar] [CrossRef]
- Novikov, A.S.; Kuznetsov, M.L.; Pombeiro, A.J.L.; Bokach, N.A.; Shul’pin, G.B. Generation of HO· Radical from Hydrogen Peroxide Catalyzed by Aqua Complexes of the Group III Metals [M(H2O)n]3+ (M = Ga, In, Sc, Y, or La): A Theoretical Study. ACS Catal. 2013, 3, 1195–1208. [Google Scholar] [CrossRef]
- Kirillova, M.V.; Kuznetsov, M.L.; Kozlov, Y.N.; Shu’pina, L.S.; Kitaygorodskiy, A.; Pombeiro, A.J.L.; Shul’pin, G.B. Participation of Oligovanadates in Alkane Oxidation with H2O2 Catalyzed by Vanadate Anion in Acidified Acetonitrile: Kinetic and DFT Studies. ACS Catal. 2011, 1, 1511–1520. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Kirillova, M.V.; Kozlov, Y.N.; Shul’pina, L.S.; Kudinov, A.R.; Pombeiro, A.J.L. Decamethylosmocene-catalyzed efficient oxidation of saturated and aromatic hydrocarbons and alcohols with hydrogen peroxide in the presence of pyridine. J. Catal. 2011, 277, 164–172. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Kozlov, Y.N.; Shul’pina, L.S.; Kudinov, A.R.; Mandelli, D. Extremely Efficient Alkane Oxidation by a New Catalytic Reagent H2O2/Os3(CO)12/Pyridine. Inorg. Chem. 2009, 48, 10480–10482. [Google Scholar] [CrossRef] [PubMed]
- Nam, W.; Lim, M.H.; Moon, S.K.; Kim, C. Participation of two distinct hydroxylating intermediates in iron(III) porphyrin complex-catalyzed hydroxylation of alkanes. J. Am. Chem. Soc. 2000, 122, 10805–10809. [Google Scholar] [CrossRef]
- Codola, Z.; Gamba, I.; Acuna-Pares, F.; Casadevall, C.; Clemancey, M.; Latour, J.-M.; Luis, J.M.; Lloret-Fillol, J.; Costas, M. Design of Iron Coordination Complexes as Highly Active Homogenous Water Oxidation Catalysts by Deuteration of Oxidation-Sensitive Sites. J. Am. Chem. Soc. 2019, 141, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Olivo, G.; Lanzalunga, O.; Di Stefano, S. Non-Heme Imine-Based Iron Complexes as Catalysts for Oxidative Processes. Adv. Synth. Catal. 2016, 358, 843–863. [Google Scholar] [CrossRef]
- Shul’pin, G.B. Metal-catalyzed hydrocarbon oxygenations in solutions: The dramatic role of additives: A review. J. Mol. Catal. A 2002, 189, 39–66. [Google Scholar] [CrossRef]
- Nesterova, O.V.; Kopylovich, M.N.; Nesterov, D.S. Stereoselective oxidation of alkanes with m-CPBA as an oxidant and cobalt complex with isoindole-based ligands as catalysts. RCS Adv. 2016, 6, 93756–93767. [Google Scholar] [CrossRef]
- Gunay, A.; Theopold, K.H. C–H Bond Activations by Metal Oxo Compounds. Chem. Rev. 2010, 110, 1060–1081. [Google Scholar] [CrossRef] [PubMed]
- Bailey, W.D.; Dhar, D.; Cramblitt, A.C.; Tolman, W.B. Mechanistic Dichotomy in Proton-Coupled Electron-Transfer Reactions of Phenols with a Copper Superoxide Complex. J. Am. Chem. Soc. 2019, 141, 5470–5480. [Google Scholar] [CrossRef] [PubMed]
- Dhar, D.; Yee, G.M.; Tolman, W.B. Effects of Charged Ligand Substituents on the Properties of the Formally Copper(III)–Hydroxide ([CuOH]2+) Unit. Inorg Chem 2018, 57, 9794–9806. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, N.; Tolman, W.B. [CuO]+ and [CuOH]2+ Complexes: Intermediates in Oxidation Catalysis? Acc. Chem. Res. 2015, 48, 2126–2131. [Google Scholar] [CrossRef] [PubMed]
- King, A.E.; Huffman, L.M.; Casitas, A.; Costas, M.; Ribas, X.; Stahl, S.S. Copper-Catalyzed Aerobic Oxidative Functionalization of an Arene C–H Bond: Evidence for an Aryl–Copper(III) Intermediate. J. Am. Chem. Soc. 2010, 132, 12068–12073. [Google Scholar] [CrossRef]
- Elwell, C.E.; Gagnon, N.L.; Neisen, B.D.; Dhar, D.; Spaeth, A.D.; Yee, G.M.; Tolman, W.B. Copper-Oxygen Complexes Revisited: Structures, Spectroscopy, and Reactivity. Chem. Rev. 2017, 117, 2059–2107. [Google Scholar] [CrossRef]
- McCann, S.D.; Stahl, S.S. Copper-Catalyzed Aerobic Oxidations of Organic Molecules: Pathways for Two-Electron Oxidation with a Four-Electron Oxidant and a One-Electron Redox-Active Catalyst. Acc. Chem. Res. 2015, 48, 1756–1766. [Google Scholar] [CrossRef] [Green Version]
- Kozlov, Y.N.; Nizova, G.V.; Shul’pin, G.B. The mechanism of hydrogen peroxide-induced aerobic oxidation of alkanes in catalysis by a vanadium complex and pyrazine-2-carboxylic acid. Russ. J. Phys. Chem. 2001, 75, 770–774. [Google Scholar]
- Shul’pin, G.B.; Kozlov, Y.N.; Nizova, G.V.; Suss-Frank, G.; Stanislas, S.; Kitaygorodskiy, A.; Kulikova, V.S. Oxidations by the reagent “O2-H2O2-vanadium derivative-pyrazine-2-carboxylic acid”. Part 12. Main features, kinetics and mechanism of alkane hydroperoxidation. J. Chem. Soc. Perkin Trans. 2 2001, 1351–1371. [Google Scholar] [CrossRef]
- Kozlov, Y.N.; Nizova, G.V.; Shul’pin, G.B. Oxidations by the reagent “O2-H2O2-vanadium derivative-pyrazine-2-carboxylic acid”—Part 14. Competitive oxidation of alkanes and acetonitrile (solvent). J. Mol. Catal. A 2005, 227, 247–253. [Google Scholar] [CrossRef]
- Hynes, A.J.; Wine, P.H. Kinetics and Mechanism of the Reaction of Hydroxyl Radicals with Acetonitrile Under Atmospheric Conditions. J. Phys. Chem. 1991, 95, 1232–1240. [Google Scholar] [CrossRef]
- Semenov, S.N.; Belding, L.; Cafferty, B.J.; Mousavi, M.P.S.; Finogenova, A.M.; Cruz, R.S.; Skorb, E.V.; Whitesides, G.M. Autocatalytic Cycles in a Copper-Catalyzed Azide-Alkyne Cycloaddition Reaction. J. Am. Chem. Soc. 2018, 140, 10221–10232. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, O.V.; Nesterov, D.S.; Vranovicova, B.; Boca, R.; Pombeiro, A.J.L. Heterometallic CuIIFeIII and CuIIMnIII alkoxobridged complexes revealing a rare hexanuclear M6(µ-X)7(µ3-X)2 molecular core. Dalton Trans. 2018, 47, 10941–10952. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Nesterov, D.S.; Shul’pina, L.S.; Pombeiro, A.J.L. A hydroperoxo-rebound mechanism of alkane oxidation with hydrogen peroxide catalyzed by binuclear manganese(IV) complex in the presence of an acid with involvement of atmospheric dioxygen. Inorg. Chim. Acta 2017, 455, 666–676. [Google Scholar] [CrossRef]
- Bilyachenko, A.N.; Levitsky, M.M.; Yalymov, A.I.; Korlyukov, A.A.; Vologzhanina, A.V.; Kozlov, Y.N.; Shul’pina, L.S.; Nesterov, D.S.; Pombeiro, A.J.L.; Lamaty, F.; et al. A heterometallic (Fe6Na8) cage-like silsesquioxane: Synthesis, structure, spin glass behavior and high catalytic activity. RSC Adv. 2016, 6, 48165–48180. [Google Scholar] [CrossRef]
- Vinogradov, M.M.; Kozlov, Y.N.; Bilyachenko, A.N.; Nesterov, D.S.; Shul’pina, L.S.; Zubavichus, Y.V.; Pombeiro, A.J.L.; Levitsky, M.M.; Yalymov, A.I.; Shul’pin, G.B. Alkane oxidation with peroxides catalyzed by cage-like copper(II) silsesquioxanes. New J. Chem. 2015, 39, 187–199. [Google Scholar] [CrossRef]
- Vinogradov, M.M.; Kozlov, Y.N.; Nesterov, D.S.; Shul’pina, L.S.; Pombeiro, A.J.L.; Shul’pin, G.B. Oxidation of hydrocarbons with H2O2/O2 catalyzed by osmium complexes containing p-cymene ligands in acetonitrile. Catal. Sci. Technol. 2014, 4, 3214–3226. [Google Scholar] [CrossRef]
- Gryca, I.; Czerwinska, K.; Machura, B.; Chrobok, A.; Shul’pina, L.S.; Kuznetsov, M.L.; Nesterov, D.S.; Kozlov, Y.N.; Pombeiro, A.J.L.; Varyan, I.A.; et al. High Catalytic Activity of Vanadium Complexes in Alkane Oxidations with Hydrogen Peroxide: An Effect of 8-Hydroxyquinoline Derivatives as Noninnocent Ligands. Inorg. Chem. 2018, 57, 1824–1839. [Google Scholar] [CrossRef]
- Wierzchowski, P.T.; Zatorski, L.W. Determination of cycle C6 and C7 peroxides and hydroperoxides by gas chromatography. Chromatographia 2000, 51, 83–86. [Google Scholar] [CrossRef]
- Bilyachenko, A.N.; Khrustalev, V.N.; Zubavichus, Y.V.; Shul’pina, L.S.; Kulakova, A.N.; Bantreil, X.; Lamaty, F.; Levitsky, M.M.; Gutsul, E.I.; Shubina, E.S.; et al. Heptanuclear Fe5Cu2-Phenylgermsesquioxane containing 2,2’-Bipyridine: Synthesis, Structure, and Catalytic Activity in Oxidation of C-H Compounds. Inorg. Chem. 2018, 57, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Guillois, K.; Mangematin, S.; Tuel, A.; Caps, V. Gold-catalyzed aerobic epoxidation of trans-stilbene in methylcyclohexane. Part II: Identification and quantification of a key reaction intermediate. Catal. Today 2013, 203, 111–115. [Google Scholar] [CrossRef]
- Astakhov, G.S.; Bilyachenko, A.N.; Korlyukov, A.A.; Levitsky, M.M.; Shul’pina, L.S.; Bantreil, X.; Lamaty, F.; Vologzhanina, A.V.; Shubina, E.S.; Dorovatovskii, P.V.; et al. High-Cluster (Cu9) Cage Silsesquioxanes: Synthesis, Structure, and Catalytic Activity. Inorg. Chem. 2018, 57, 11524–11529. [Google Scholar] [CrossRef] [PubMed]
- Kirillova, M.V.; Kuznetsov, M.L.; Romakh, V.B.; Shul’pina, L.S.; Frausto da Silva, J.J.R.; Pombeiro, A.J.L.; Shul’pin, G.B. Mechanism of oxidations with H2O2 catalyzed by vanadate anion or oxovanadium(V) triethanolaminate (vanadatrane) in combination with pyrazine-2-carboxylic acid (PCA): Kinetic and DFT studies. J. Catal. 2009, 267, 140–157. [Google Scholar] [CrossRef]
- Kirillova, M.V.; Kirillov, A.M.; Mandelli, D.; Carvalho, W.A.; Pombeiro, A.J.L.; Shul’pin, G.B. Mild homogeneous oxidation of alkanes and alcohols including glycerol with tert-butyl hydroperoxide catalyzed by a tetracopper(II) complex. J. Catal. 2010, 272, 9–17. [Google Scholar] [CrossRef]
- Ghosh, M.; Pattanayak, S.; Dhar, B.B.; Singh, K.K.; Panda, C.; Sen Gupta, S. Selective C–H Bond Oxidation Catalyzed by the Fe-bTAML Complex: Mechanistic Implications. Inorg. Chem. 2017, 56, 10852–10860. [Google Scholar] [CrossRef] [PubMed]
- Nesterov, D.S.; Nesterova, O.V.; Kopylovich, M.N.; Pombeiro, A.J.L. Pronounced retention of stereoconfiguration upon sp3 C–H bonds hydroxylation of dimethylcyclohexanes and decahydronaphthalenes with m-CPBA oxidant and a Co-phthalocyanine catalyst. Mol. Catal. 2018, 459, 8–15. [Google Scholar] [CrossRef]
- Nesterova, O.V.; Kasyanova, K.V.; Makhankova, V.G.; Kokozay, V.N.; Vassilyeva, O.Y.; Skelton, B.W.; Nesterov, D.S.; Pombeiro, A.J.L. Stereospecific sp3 C–H oxidation with m-CPBA: A CoIII Schiff base complex as pre-catalyst vs. its CoIIICdII heterometallic derivative. Appl. Catal. A 2018, 560, 171–184. [Google Scholar] [CrossRef]
- Nesterova, O.V.; Kasyanova, K.V.; Buvaylo, E.A.; Vassilyeva, O.Yu.; Skelton, B.W.; Nesterov, D.S.; Pombeiro, A.J.L. Heterometallic CoIIIZnII Schiff Base Catalyst for Mild Hydroxylation of C(sp3)–H Bonds of Unactivated Alkanes: Evidence for Dual Mechanism Controlled by the Promoter. Catalysts 2019, 9, 209. [Google Scholar] [CrossRef]
- Balamurugan, M.; Mayilmurugan, R.; Suresh, E.; Palaniandavar, M. Nickel(II) complexes of tripodal 4N ligands as catalysts for alkane oxidation using m-CPBA as oxidant: Ligand stereoelectronic effects on catalysis. Dalton Trans. 2011, 40, 9413–9424. [Google Scholar] [CrossRef]
- Hikichi, S.; Hanaue, K.; Fujimura, T.; Okuda, H.; Nakazawa, J.; Ohzu, Y.; Kobayashi, C.; Akita, M. Characterization of nickel(II)-acylperoxo species relevant to catalytic alkane hydroxylation by nickel complex with mCPBA. Dalton Trans. 2013, 42, 3346–3356. [Google Scholar] [CrossRef]
- Company, A.; Gomez, L.; Guell, M.; Ribas, X.; Luis, J.M.; Que, L.; Costas, M. Alkane hydroxylation by a nonheme iron catalyst that challenges the heme paradigm for oxygenase action. J. Am. Chem. Soc. 2007, 129, 15766–15777. [Google Scholar] [CrossRef] [PubMed]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cu1–N1 | 1.929(5) |
| Cu1–N3 | 1.933(4) |
| Cu1–N4 | 1.931(5) |
| Cu1–N6 | 1.935(5) |
| N1–Cu1–N3 | 87.8(2) |
| N1–Cu1–N4 | 160.6(2) |
| N1–Cu1–N6 | 93.1(2) |
| N3–Cu1–N4 | 96.0(2) |
| N3–Cu1–N6 | 165.5(2) |
| N4–Cu1–N6 | 87.9(2) |
| Catalytic System | Cyclohexane | Methylcyclohexane, 1°:2°:3° | cis-1,2-Dimethylcyclohexane | Adamantane 3°:2° | Proposed C–H Attacking Species | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| Yield, % b | TON c | cis/trans ratio | 1°:2°:3° | |||||
| 1/Py d/H2O2 | 13 | 27 | 1:7:17 | 1.2 | 1:7:34 | 3:1 | HO· | this work |
| 4/Py/H2O2 | 19 | 37 | 1:8:18 | 1.2 | 1:10:44 | 3:1 | HO· | this work |
| [CuII(HL1)(NO3)(DMF)](NO3)·H2O/Py/H2O2 | 22 | 43 | 1:2:16 | – | – | – | HO· | [38] |
| [CuII(L2)Cl2]·DMF/Py/H2O2 | 21 | 42 | – | 1.3 | 1:7:43 | 3:1 | HO· | [39] |
| [FeIII(HL1)Cl2(DMF)]Cl·DMF/HNO3/H2O2 | 37 | 900 | 1:6:16 | ~1.0 | – | – | HO· | [41] |
| [CoIII4FeIII2O(L3)8]·4DMF·H2O/HNO3/H2O2 | 26 | 3570 | 1:7:20 | – | – | – | HO· | [15] |
| (n-Bu4N)VVO3/H2SO4/H2O2 | – | – | 1:7:26 | ~1.0 | – | – | HO· | [74] |
| [VVO(OCH3)(L4)2]/PCA e/H2O2 | 39 | 897 | 1:5:17 | 1.3 | – | – | HO· | [69] |
| [OCuII4(L5)4(BOH)4][BF4]2/CF3COOH/H2O2 | 3 | 15 | 1:5:14 | 1.3 | – | – | HO· | [75] |
| [OCuII4(L5)4(BOH)4][BF4]2/TBHP f | 5 | 22 | 1:16:128 | 1.3 | – | – | tBuO· | [75] |
| [FeIII(L6)(Cl)](Et4N)2/TBHP | – | – | – | – | – | 3:1 | tBuO· | [76] |
| [FeIII(L6)(Cl)](Et4N)2/m-CPBA g | 2 | 5 | – | 30 | 69:1 | FeV=O | [76] | |
| [MnIV2(L7)2O3][PF6]2/HOAc/H2O2 | 46 | 1:26:200 | 2.9 | – | – | MnV=O | [65] | |
| [CoII(L8)](NO3)2/m-CPBA | – | – | – | 56 | 0:1:35 h | 21:1 | CoIV=O | [51] |
| [CoII(L9)]/HNO3/m-CPBA | 8 | 80 | – | 57 | 0:1:36 | – | CoIV=O or CoIII–OOC(O)Ar | [77] |
| [CoII(L9)]/HNO3/H2O2 | – | – | – | 1.1 | 0:1:35 | – | HO· | [77] |
| [CoIII(L10)3]·DMF/HNO3/m-CPBA | – | – | – | 59 | 0:1:32 | 22:1 | CoIII–OOC(O)Ar | [78] |
| [CoIIIZnII(L10)3Cl2]·CH3OH/HNO3/m-CPBA | – | – | – | 90 | 0:1:38 | 39:1 | CoIII–OOC(O)Ar | [79] |
| [CoIIICdII(L10)3Cl2]·0.5H2O/HNO3/m-CPBA | – | – | – | 78 | 0:1:40 | 23:1 | CoIII–OOC(O)Ar | [78] |
| [NiII(L11)(CH3CN)2](BPh4)2/m-CPBA | 9 | 622 | – | – | – | 13:1 | NiII–O· | [80] |
| [{NiII(L12)}(OH)2]/m-CPBA | 2 | 46 | 1:47:250 | – | – | – | NiII–OO(O)Ar or NiII–O· | [81] |
| [FeIII(L13)Cl]/m-CPBA | 0.3 | 0.9 | – | >72 | 0:1:58 | – | FeIII–OOC(O)Ar and FeIV=O | [47] |
| [FeII(L14)(CF3SO3)2]/H2O2 | 0.7 | 6.5 | – | 28 | – | 30:1 | FeV=O | [82] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nesterova, O.V.; Kopylovich, M.N.; Nesterov, D.S. A Comparative Study of the Catalytic Behaviour of Alkoxy-1,3,5-Triazapentadiene Copper(II) Complexes in Cyclohexane Oxidation. Inorganics 2019, 7, 82. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7070082
Nesterova OV, Kopylovich MN, Nesterov DS. A Comparative Study of the Catalytic Behaviour of Alkoxy-1,3,5-Triazapentadiene Copper(II) Complexes in Cyclohexane Oxidation. Inorganics. 2019; 7(7):82. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7070082
Chicago/Turabian StyleNesterova, Oksana V., Maximilian N. Kopylovich, and Dmytro S. Nesterov. 2019. "A Comparative Study of the Catalytic Behaviour of Alkoxy-1,3,5-Triazapentadiene Copper(II) Complexes in Cyclohexane Oxidation" Inorganics 7, no. 7: 82. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7070082