“Intelligent” Pt Catalysts Based on Thin LaCoO3 Films Prepared by Atomic Layer Deposition


1
Chemical and Biomolecular Engineering, University of Pennsylvania, Philadelphia, PA 19104, USA
2
Materials Science and Engineering, University of Pennsylvania, Philadelphia, PA 19104, USA
*
Author to whom correspondence should be addressed.
Inorganics 2019, 7(9), 113; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7090113
Submission received: 13 August 2019
/
Revised: 9 September 2019
/
Accepted: 10 September 2019
/
Published: 12 September 2019
(This article belongs to the Section Inorganic Solid-State Chemistry)
Abstract
:LaCoO3 films were deposited onto MgAl2O4 powders by atomic layer deposition (ALD) and then used as catalyst supports for Pt. X-ray diffraction (XRD) showed that the 0.5 nm films exhibited a perovskite structure after redox cycling at 1073 K, and scanning transmission electron microscopy and elemental mapping via energy-dispersive X-ray spectroscopy (STEM/EDS) data demonstrated that the films covered the substrate uniformly. Catalysts prepared with 3 wt % Pt showed that the Pt remained well dispersed on the perovskite film, even after repeated oxidations and reductions at 1073 K. Despite the high Pt dispersion, CO adsorption at room temperature was negligible. Compared with conventional Pt on MgAl2O4, the reduced forms of the LaCoO3-containing catalyst were highly active for the CO oxidation and water gas shift (WGS) reactions, while the oxidized catalysts showed much lower activities. Surprisingly, the reduced catalysts were much less active than the oxidized catalysts for toluene hydrogen. Catalysts prepared from thin films of Co3O4 or La2O3 exhibited properties more similar to Pt/MgAl2O4. Possible reasons for how LaCoO3 affects properties are discussed.
1. Introduction
Sintering is a serious problem with supported metal catalysts in high-temperature applications, such as in automotive emissions control, because it leads to a loss of catalytically active surface area [1]. The concept of “intelligent” catalysts, in which the catalytic metals are supported on mixed oxides which have a perovskite structure, was developed as a possible solution to this problem [2]. The idea behind intelligent catalysts is that catalytic metals can become part of the perovskite lattice for some perovskites under oxidizing conditions, then return to the surface under reducing conditions. This “ingress” of large metal particles into the perovskite and “egress” of smaller metal particles out of the lattice could allow redispersion of the catalyst and restoration of activity. However, there are two serious limitations that prevent the practical application of the concept. First, the perovskites that are of interest for this application typically have low surface areas, at least after high-temperature cycling. Second, it has been shown that nucleation and subsequent formation of metal particles frequently occurs in the bulk of the perovskite, not at the surface, so that the metal particles that are formed are not accessible to reactants [3,4,5].
To avoid the problems associated with bulk perovskites, our group has been preparing thin-film perovskites, between 0.5 and 1.0 nm in thickness, on high-surface-area substrates using atomic layer deposition (ALD). The deposited films retain a high surface area, and because the typical particle size for metal catalysts is greater than 1 nm, nucleation of particles in the bulk of the oxide can be avoided. In past published works, films of CaTiO3 and LaFeO3 were grown on MgAl2O4 and then used as catalyst supports for Pd [6], Ni [7], and Pt [8]. The CaTiO3 and LaFeO3 films were found to uniformly cover the MgAl2O4 support and to be stable to cycling between oxidizing and reducing conditions (redox cycling) at temperatures as high as at least 1073 K.
Previous work with the thin-film versions of intelligent catalysts exhibited a number of interesting catalytic properties that are similar to what is observed in their bulk counterparts. For example, following an initial calcination at 1073 K, both CaTiO3- and LaFeO3-supported catalysts reduced at 773 K showed poor catalytic activity, but the same catalysts reduced at 1073 K were very active. This kind of behavior would be expected for bulk perovskite supports, since high temperatures are required for egress of the metal cations following high-temperature oxidation. Similar to Ni catalysts formed by ex-solution, Ni supported on CaTiO3 films showed extreme tolerance against coking in the presence of dry methane [7]. Finally, similar to what is observed with bulk perovskites [9], Pt, but not Pd, was found to interact strongly with CaTiO3 [8].
One of the more interesting perovskites to consider as a catalyst support is LaCoO3. LaCoO3 is much more easily reduced than either CaTiO3 or LaFeO3 [10], implying that there should be a much higher concentration of oxygen vacancies under even mild reducing conditions. This could impact the ease with which metals enter and leave the perovskite lattice. Co oxides are also known to be catalytically active for reactions such as CO and methane oxidation [11,12] and can also promote the activity of Pt for other reactions [13]. CO oxidation is important for pollution control applications [14,15]. Therefore, we set out to examine the properties of Pt on LaCoO3 films and to compare those properties to Pt on LaFeO3. The LaCoO3 films were again prepared by ALD on MgAl2O4 and characterized by catalyst testing and various physical characterization methods.
2. Results
2.1. Catalyst Characterization
The growth rate for La2O3 on MgAl2O4 in our system was reported to be 3.4 × 1017 La atoms/cycle·m2 (0.017 nm La2O3/cycle) in a previous work [6]. The weight changes associated with deposition of Co3O4 films on MgAl2O4 are shown in Figure S1a, which demonstrates that the amount Co3O4 that was added per cycle was independent of the number of cycles and equal to 5.0 × 1017 Co atoms/cycle·m2 (0.013 nm Co3O4/cycle, assuming uniform film growth and a bulk density for the film). This growth rate was the same as that measured previously for Co3O4 on γ-Al2O3 [16]. Therefore, in order to grow LaCoO3 films with the correct stoichiometry, we alternated between three ALD cycles of La2O3 and two ALD cycles of Co3O4. Figure S1b shows the sample weight as a function of this alternating set of ALD cycles and demonstrates that the weight changes for the mixed oxide were consistent with that predicted from the sum of the individual oxides.
Samples were prepared with either 30 wt % LaCoO3 (0.3 g of La2O3 and 0.15 g of Co3O4 on 1 g of MgAl2O4), 20 wt % La2O3, or 20 wt % Co3O4. After this, one ALD cycle of Pt was added to each of these materials to produce materials that were roughly 3 wt % Pt. A base-case sample was also prepared with one ALD cycle of Pt on MgAl2O4. Each of the samples was then exposed to five oxidation–reduction cycles, in which it was alternately oxidized in air at 1073 K for 2 h and then reduced in dry H2 at 1073 K for 1 h. Each of these samples is listed in Table 1, along with its BET surface area and calculated film thickness.
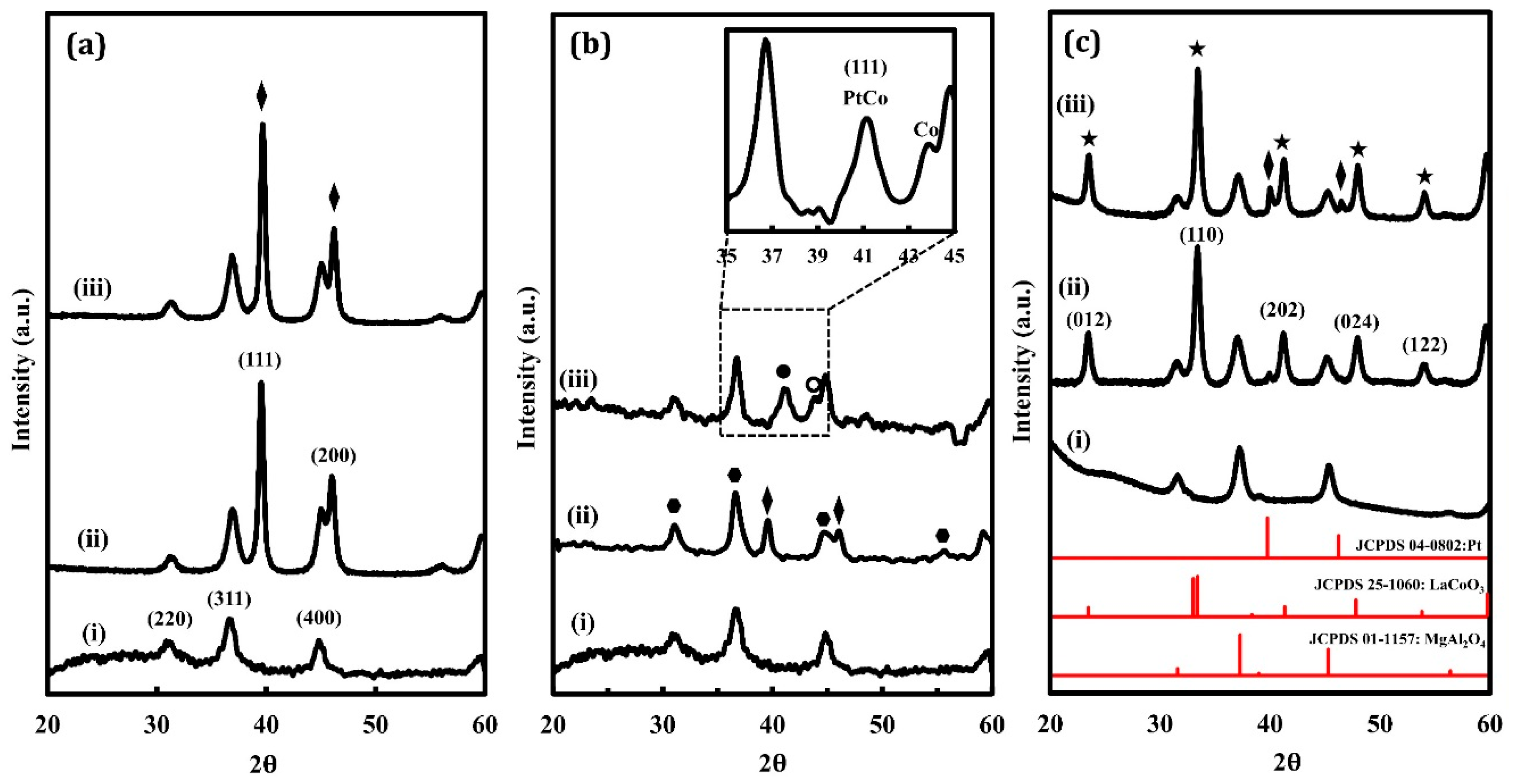
XRD patterns of the oxidized and reduced Pt/LCO/MAO, Pt/Co/MAO, and Pt/MAO samples are shown in Figure 1, with the pattern for MgAl2O4 shown at the bottom of each set of patterns for comparison. In addition to the features associated with MgAl2O4, the Pt/MAO sample (Figure 1a) exhibited intense Pt peaks for both the oxidized and reduced samples. This is consistent with the fact that large Pt particles are formed by high-temperature redox cycling [9]. Because bulk Pt is difficult to oxidize, XRD patterns for the oxidized and reduced samples remained unchanged. The XRD patterns for Pt/Co/MAO are shown in Figure 1b. The oxidized sample exhibited Pt features at 39° and 46° 2θ, but it was difficult to compare the intensities of these peaks with that of the substrate due to the overlap in peaks for Co3O4 and MgAl2O4. However, when this sample was reduced, the features associated with Pt shifted to 41.4° 2θ due to the formation of a Pt–Co alloy. There was also a small shoulder in the XRD pattern at 44.2° 2θ, which was likely associated with metallic Co.
XRD patterns for the Pt/LCO/MAO are shown in Figure 1c. The most intense features were those associated with the perovskite phase. An estimate of the crystallite size of the LaCoO3 using the Scherrer equation and the peak at 33° 2θ gave a value of 11 nm for both the oxidized and reduced samples. The other big difference between this and the other two samples was that the Pt peak at 39° 2θ was very small on the oxidized sample and was only slightly larger on the reduced sample, despite the fact that all three samples had a similar Pt loading. Also, the reduced sample showed no evidence of Pt–Co alloy formation.
High-angle annular dark field (HAADF)/STEM images of the oxidized and reduced Pt/LCO/MAO sample are shown in Figure 2, along with the corresponding EDS mappings. Again, both of these samples had been oxidized and reduced five times at 1073 K before the final treatment. The image of the oxidized sample, shown Figure 2a, is not easily distinguishable from that of the unmodified MgAl2O4. There was no evidence for larger particles. The EDS maps show that Pt, La, and Co were uniformly present over most of the surface. With relatively few exceptions, the EDS intensities for these three elements follow those of the Mg and Al. The STEM image of the reduced sample in Figure 2b is largely unchanged, but the EDS mappings show differences in the elemental positions, with Co appearing to have agglomerated into 10 nm islands. Pt and La remained relatively well distributed over the surface. Although some regions of the sample also showed large Pt particles (Figure S2), the concentration of these particles was insufficient to account for the Pt content of the sample but may account for the Pt XRD peak in Figure 1c. That LaCoO3 could at least partially decompose under harsh reducing conditions is expected [17], but it is surprising this was not also observed in the XRD measurements. The XRD showed no change in the perovskite peak intensities and no evidence of new phases.
2.2. CO Oxidation Rates
Past work with CO oxidation over perovskite-supported, ex-solution catalysts exhibited a high sensitivity to the pretreatment conditions [6,7,8]. Following high-temperature (1073 K) oxidation and relatively mild (773 K) reduction, the catalysts were inactive, but activity was restored upon high-temperature reduction. This behavior is unusual for supported metals and appears to be due to the high-temperature requirements for driving the metals into and out of the perovskite lattice. It is notable that this cycling between active and inactive states only occurs for those metal–perovskite pairs in which the metal can become part of the perovskite lattice [8]. Therefore, we employed similar CO oxidation measurements on the catalysts of this study to see whether these would also show evidence for ex-solution.
Figure 3 and Figure S3 show CO oxidation rates over Pt/LCO/MAO, Pt/La/MAO, Pt/Co/MAO, and Pt/MAO samples after various redox treatments. These samples were initially oxidized at 873 K to remove precursor ligands but were not otherwise redox cycled so that we could observe changes in the catalysts upon these various pretreatments. The rates were obtained under differential conditions with a constant gas hourly space velocity (GHSV) of 60,000 mL·g−1·h−1 (100 mL/min total flow rate, 100 mg catalyst). In order to maintain differential conditions, rates were measured at lower temperatures on the more active catalysts.
The effect of redox treatments for Pt/MAO, shown in both the Arrhenius plots and light-off profiles, has been discussed in a previous publication [8]. The sample exhibited similar activities as the conventional Pt catalysts, with results from the measurements reproduced in Figure S3. Initially, the rates were reasonably high due to the fact that the Pt dispersion was greater than 20%. However, high-temperature oxidation caused a precipitous drop in the activity and rates were not restored by high-temperature reduction. Additional redox cycling caused further drops in the activity, and after five redox cycles, the rates had dropped by a factor of 50. Chemisorption measurements indicated that the loss in activity could be completely explained by a loss of Pt dispersion.
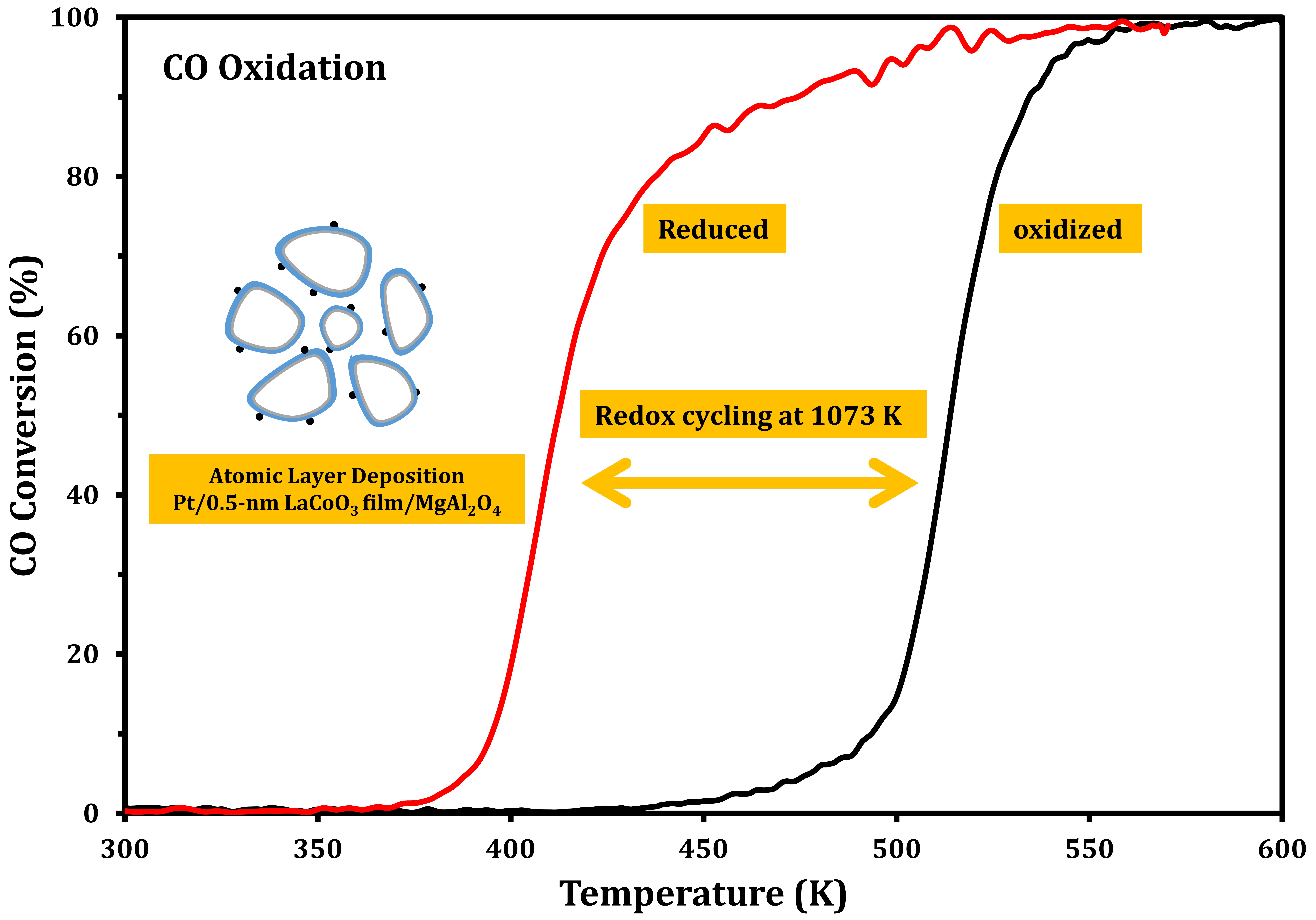
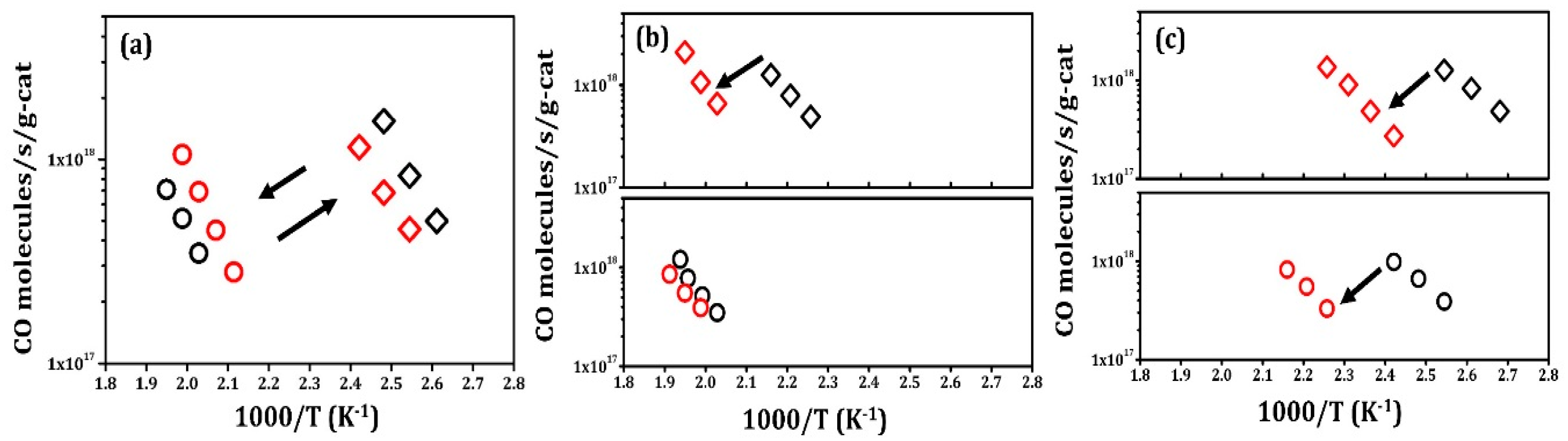
Rate data for Pt/LCO/MAO are shown in Figure 3a and reveal completely different characteristics. To begin these experiments, the catalyst was first reduced at 1073 K in 10% H2–He. Similar to what has been observed in other ex-solution catalysts, this reduced catalyst showed very high activity, approximately 5 times higher than that observed in fresh Pt/MAO. When this catalyst was oxidized at 1073 K in 10% O2–He, the activity dropped by almost a factor of 100. The light-off curves for CO oxidation over oxidized and reduced Pt/LCO/MAO (sixth redox cycle) are shown in Figure S4, confirming that there was a large difference in activity between the oxidized and reduced samples. Also, as shown in Figure S5, reduction at 773 K did not significantly restore activity, but reduction at 1073 K brought the rates back to their initial value. The activity of this catalyst could be cycled between active and inactive states, with minimal changes, even after five redox cycles.
CO oxidation rates on Pt/La/MAO and Pt/Co/MAO are shown in Figure 3b,c. Initially, the Pt/La/MAO exhibited properties similar to that of fresh Pt/MAO and the rates simply decreased after high-temperature treatments. After five redox cycles, Pt/La/MAO remained somewhat more active than Pt/MAO, suggesting that La2O3 suppresses Pt sintering to some extent. The Pt/Co/MAO sample was more interesting. The initial activity after 1073 K reduction was a factor of 2 times higher than even the reduced Pt/LCO/MAO. This agrees with previous work that has shown Co promotes Pt catalysts for CO oxidation [18]. However, repeated oxidation–reduction cycles again caused the activity of this catalyst to drop. Interestingly, the reduced catalysts were always somewhat more active than the oxidized ones.
In the absence of promoters (e.g., the effects of ceria are discussed in reference [19]), CO oxidation rates on Pt catalysts have been shown to be strictly proportional to Pt surface area [20]. Therefore, we performed CO chemisorption on the oxidized and reduced samples that had undergone five redox cycles in order to better understand the changes observed in Figure 3. Pt dispersions were also estimated from the normalized reaction rates reported in the literature [21]. These values are shown in Table 2. Similar to what was reported previously for Pt on CaTiO3 films [8], there was no CO chemisorption on either the oxidized or reduced Pt/LCO/MAO sample. Based on reaction rates, the Pt dispersion on the reduced Pt/LCO/MAO should have been 40%. In contrast to this, the agreement between Pt dispersions measured by CO chemisorption and estimated from the normalized reaction rates was very good for each of the other catalysts.
The failure to observe CO chemisorption on the Pt/LCO/MAO sample suggests that the reaction rate expression should be very different. With noninteracting supports, CO oxidation on Pt is inversely proportional to the CO partial pressure due to the fact that the surface is saturated with CO. The rate limiting step is O2 adsorption [18], which in turn is limited by the availability of empty sites on the Pt. Based on the lack of CO chemisorption, the reaction order for CO on the reduced Pt/LCO/MAO sample is expected to be different. In Figure 4, differential rates are plotted at 393 and 423 K as a function of the CO partial pressure for a fixed O2 pressure of 7.6 Torr. At both temperatures, there was a transition from a slightly negative order to the zeroth order. The dependence on CO partial pressure was clearly much weaker on Pt/LCO/MAO than on conventional Pt catalysts, suggesting that the reaction mechanism is different.
2.3. WGS and Toluene Hydrogenation
Because changes in the adsorption properties could affect other reactions, we measured rates for toluene hydrogenation and WGS on the Pt/LCO/MAO catalyst as a function of the treatment conditions. These two reactions test the ability of the catalyst to activate H2 and H2O and are therefore complementary to CO oxidation, which is limited by O2 dissociative adsorption. Pt-based catalysts are generally very active for hydrogenation of unsaturated organic molecules; however, a recent study of Pt on CaTiO3 films showed surprisingly low rates for toluene hydrogenation [8]. While Pt by itself is not a good WGS catalyst due to the inability of Pt to dissociate water, support interactions can lead to interfacial sites that are highly active. For example, ceria-supported Pt can be highly active for WGS and has received considerable attention for this application [22,23].
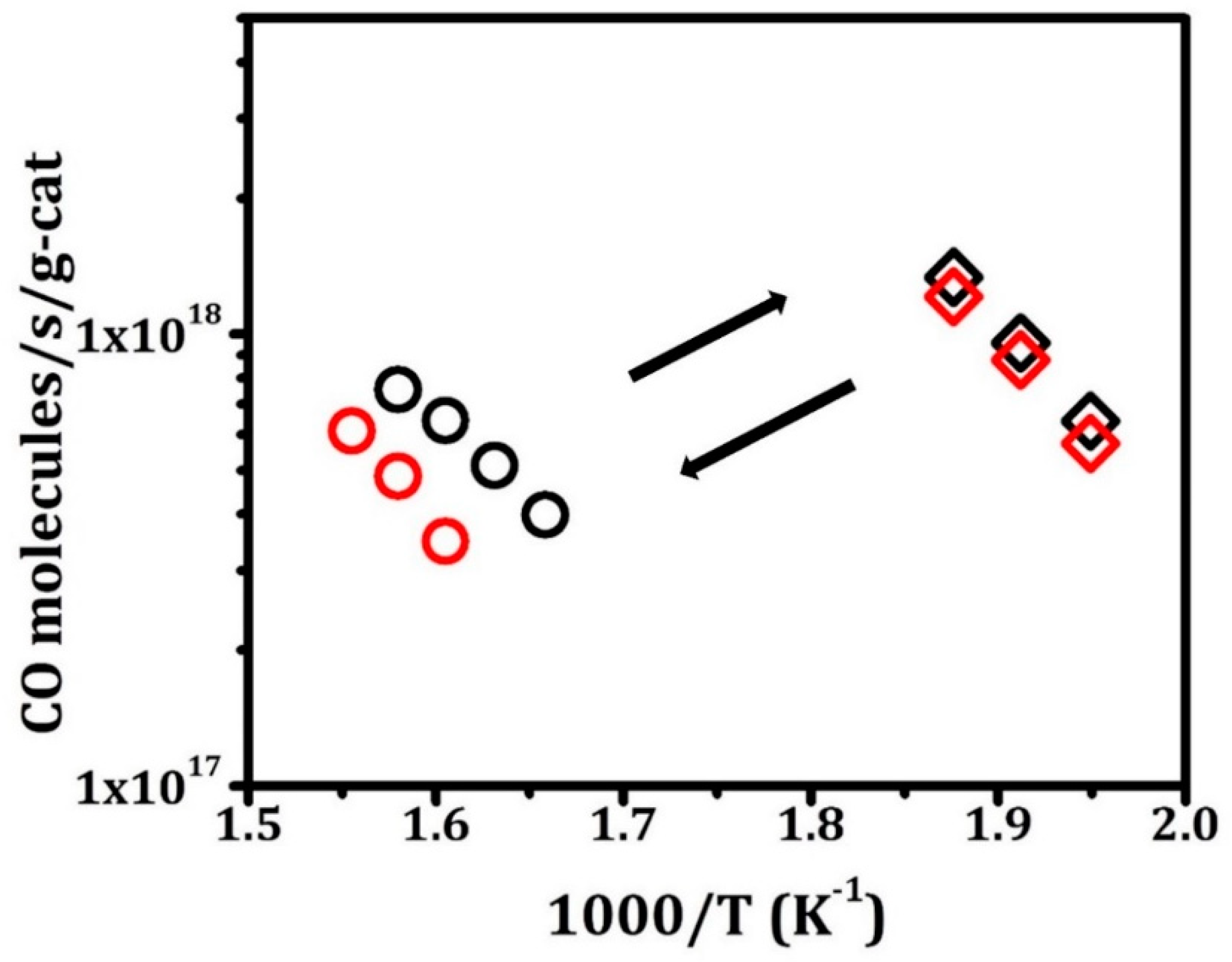
Differential WGS rates are reported for Pt/LCO/MAO in Figure 5 on samples that were again reduced or oxidized at 1073 K, after one or five redox cycles. Similar to what was observed for CO oxidation, the reduced catalysts were roughly 50 times more active than the oxidized catalyst, and redox cycling did not significantly affect the rates. The difference in activities was confirmed by the light-off profiles, shown in Figure S6. It is also noteworthy that rates on the reduced catalysts approached those observed with ceria-supported Pt and were much more active than Pt on noninteracting supports. This is demonstrated in Figure S7, which compares rates on Pt/γ-Al2O3, Pt/CeO2/γ-Al2O3, and the reduced Pt/LCO/MAO, all of which had similar Pt loadings and were calcined at 1073 K. The fact that Pt/CeO2/γ-Al2O3 and reduced Pt/LCO/MAO were comparable implies that the LaCoO3 phase plays a role in the reaction.
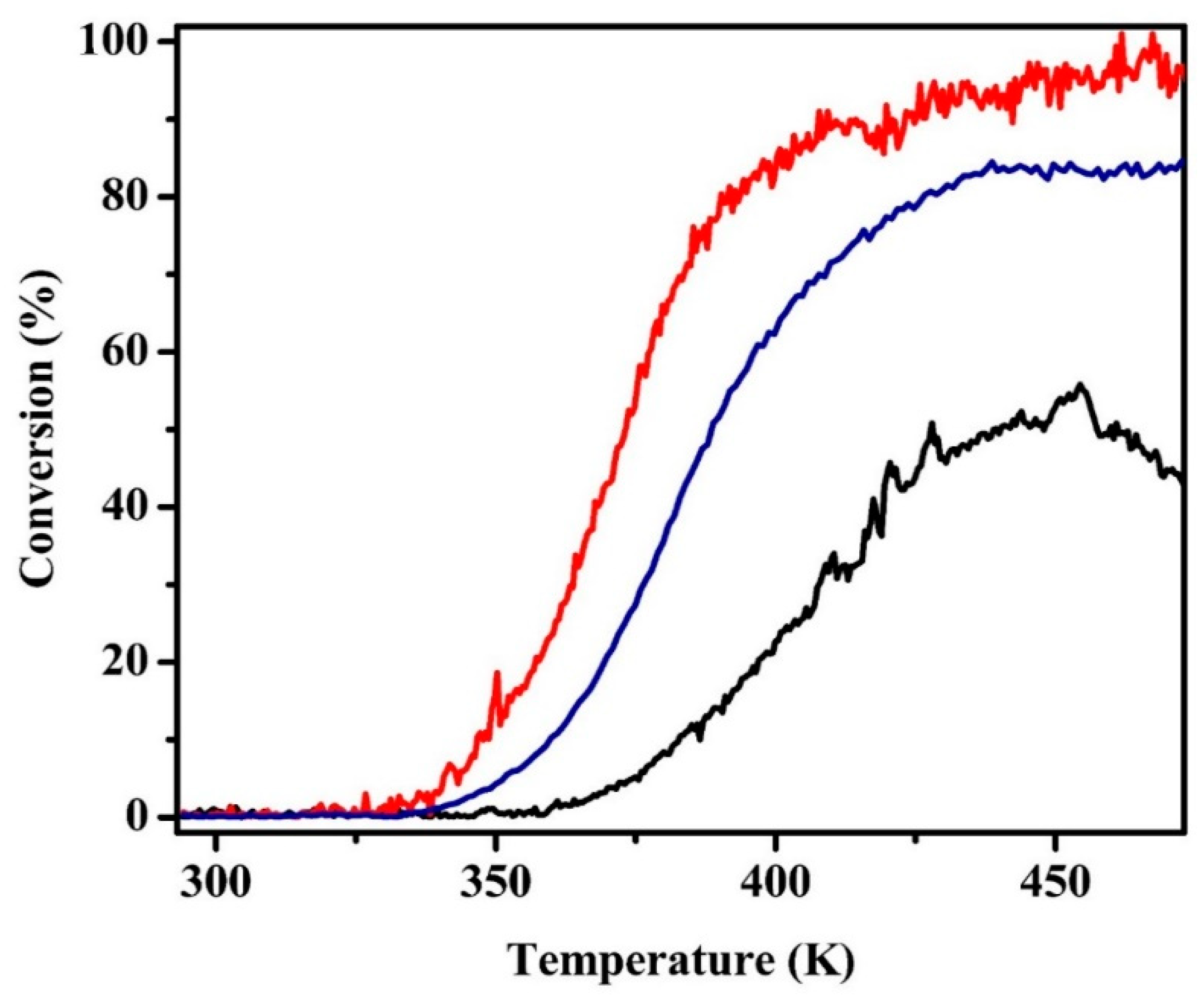
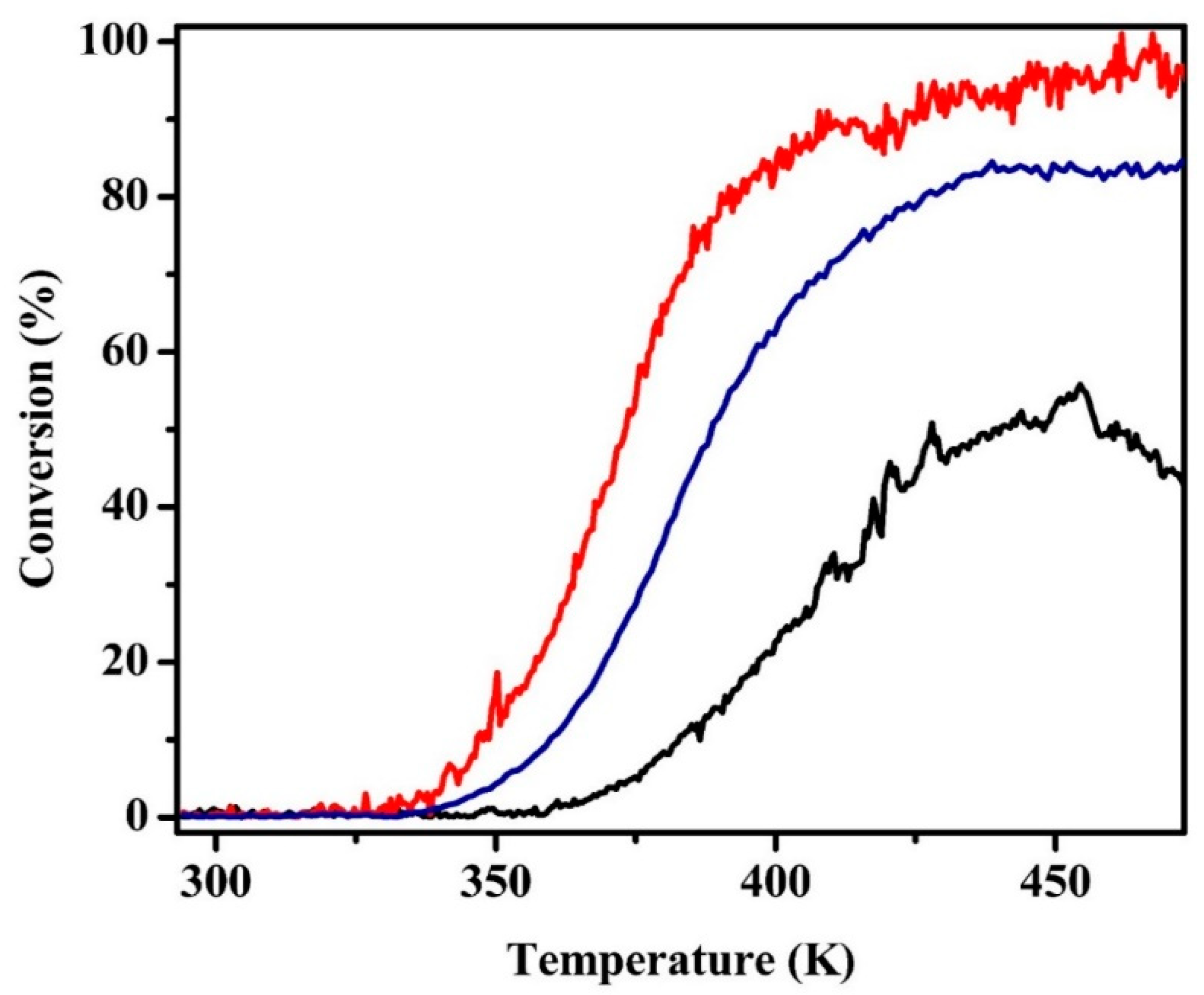
Light-off curves for toluene hydrogenation are shown in Figure 6 for the Pt/MAO and the reduced and oxidized Pt/LCO/MAO catalysts. The Pt/MAO catalyst used here was calcined and reduced at 1073 K prior to measuring the rates. This data are surprising for multiple reasons. First, rates on Pt/MAO were shifted to lower temperatures by nearly 50 degrees compared with rates on reduced Pt/LCO/MAO. For a typical reaction activation energy of 100 kJ/mol, a 50-degree temperature shift corresponds to a factor of 70 difference in rates. Second, the oxidized catalyst was more active for this reaction than the reduced catalyst. Because the STEM/EDS results suggested that the LaCoO3 phase can at least partially decompose upon high-temperature reduction, which in turn could lead to Pt–Co alloy formation, we also measured rates on the reduced Pt/Co/MAO sample (Figure S8). Rates on Pt/Co/MAO were comparable to those of Pt/MAO, showing that alloy formation cannot explain the low rates observed with Pt/LCO/MAO.
3. Discussion
The results of this study demonstrate that thin films of LaCoO3 can be formed by ALD and that these films can exhibit interesting properties as supports for Pt catalysts. Catalysts prepared from these thin films retain a high Pt dispersion (as evidenced from the STEM/EDS data) following high-temperature redox cycling, and the reduced form of the catalyst is highly active for CO oxidation and WGS. However, many questions remain about the nature of the support interactions in these materials and they are clearly not a drop-in replacement for conventional Pt catalysts, given their low activity for hydrogenation reactions.
There are many similarities between the results shown here and those reported previously for Pt and Pd on CaTiO3 and LaFeO3 films [6,8]. In each of these systems, high reduction and oxidation temperatures were required to transition the catalysts between their active and inactive states. For example, in each case, a catalyst placed in its inactive state by oxidation at 1073 K could not be restored by reduction at 773 K but could be restored by reduction at 1073 K. Furthermore, despite the fact that each of the perovskite-based catalysts was highly active for CO oxidation, CO adsorption was suppressed in each case. Unlike conventional Pt and Pd catalysts, each of the perovskite-based catalysts maintained their activity following many oxidation–reduction cycles. Similar to what was observed for Pt on CaTiO3, hydrogenation of toluene was strongly suppressed. Finally, the properties of Pt catalysts supported on the individual oxides (e.g., Pt/Co/MAO and Pt/La/MAO) were completely different from that observed for the mixed oxide support. The similarity between results for catalysts prepared from the thin-film perovskites and the catalyst prepared by ex-solution of bulk perovskites strongly suggests Pt interacts with the thin films in a similar manner to that with the bulk perovskites.
It is interesting to ask why the perovskite phase is important in these systems. With a 0.5 nm film, bulk incorporation of the Pt seems unlikely. However, it is possible that the Pt could incorporate into lattice positions at the surface. These atoms would then be bonded strongly to the oxide cations making up the perovskite, so that they would still be present at the surface to catalyze reactions. The bonding to the perovskite would clearly affect their adsorption and reaction properties. It is somewhat surprising that perovskites as different as CaTiO3 and LaCoO3 would show such similar properties.
One question about the LaCoO3 system is that the STEM/EDS results seemed to indicate that the LaCoO3 decomposed and segregated into Co-rich islands. However, this is not consistent with either the XRD data or the reaction results. Regarding XRD data, not only did the perovskite peaks remain intense following high-temperature reduction, but we saw no evidence for the Pt–Co alloy formation that occurred with Pt on the Co3O4 films. The catalytic properties of the Pt/LCO/MAO catalyst were also distinctly different from those of Pt/La/MAO or Pt/Co/MAO. One possibility is that the Co-rich islands observed in the STEM/EDS results retained their perovskite structure, probably with a La deficiency, since the La did not show similar segregation.
While the concept of an “intelligent” catalyst is not new, the ability to prepare perovskite-supported catalysts with high surface areas and no remaining catalyst in the bulk is new. While we do not fully understand what gives rise to their properties, these materials are clearly novel and interesting.
4. Experimental Methods
4.1. Sample Preparation
The MgAl2O4 support used in this study was prepared in our laboratory using a precipitation method that has been described in greater detail in previous publications [6]. Prior to use, it was calcined in air at 1173 K for 12 h to ensure stability. The final MgAl2O4 powder had a Brunauer–Emmett–Teller (BET) surface area of 120 m2/g and exhibited only peaks that are characteristic of MgAl2O4 in X-ray diffraction (XRD).
La2O3, Co3O4, and LaCoO3 films were deposited onto the MgAl2O4 support by ALD using a home-built, static system that has been described in more detail elsewhere [24]. The ALD precursors used in this study were La(TMHD)3 (Strem Chemicals, Inc., Newburyport, MA, USA) and Co(TMHD)3 (Strem Chemicals, Inc). To generate sufficient vapor pressures, the evacuated La(TMHD)3 and Co(TMHD)3 precursor powders were heated to 523 K. An ALD cycle began by exposing the evacuated MgAl2O4 substrate to vapor from one of the precursors for ~5 min to ensure a complete surface reaction. After evacuation to remove excess precursor, the substrate was placed in a muffle furnace to oxidize the precursor ligands. Oxidation was performed at 873 K for 3 min for La and at 773 K for 5 min for Co. This procedure was repeated the desired number of times. Growth rates for each oxide film were determined by measuring the sample weights every five cycles. The film thicknesses were estimated from the mass of the film, assuming it had the bulk density and formed a uniform coating over then entire surface.
For water gas shift (WGS) studies, we prepared a Pt/CeO2/γ-Al2O3 sample for comparison. CeO2 was again added by ALD, using Ce(TMHD)4 (Strem Chemicals, Inc.) as the precursor. The CeO2/Al2O3 sample was identical to that used in a previous work [24]. The CeO2 loading was 20 wt %, corresponding to a film thickness of roughly 0.5 nm, and the BET surface area of the sample was 89 m2/g after calcination to 773 K.
Pt was added to the supports using ALD to ensure good initial contact between the Pt and the surface. One ALD cycle using Platinum(II) acetylacetonate (Strem Chemicals, Inc.) was sufficient to achieve the desired loading. In all cases, the Pt was deposited onto the as-prepared oxide films. In other words, the CeO2/γ-Al2O3 and Co3O4/MgAl2O4 films had been calcined to only 773 K and the La2O3/MgAl2O4 and LaCoO3/MgAl2O4 to only 873 K prior to adding the Pt. After addition of the Pt precursor, the samples were oxidized at 573 K to remove the ligands.
4.2. Characterization Methods
XRD was performed on a Rigaku Smartlab diffractometer (Rigaku, Tokyo, Japan) with a Cu Kα X-Ray source (λ = 0.15416 nm). Specific surface areas of samples were measured using BET isotherms with N2 at 77 K (Home-built). Scanning transmission electron microscopy (STEM) and elemental mapping via energy-dispersive X-ray spectroscopy (EDS) were performed with a JEOL JEM-F200 STEM (JEOL Ltd., Tokyo, Japan) operated at 200 kV. The powder specimens were diluted in isopropanol and put on lacey carbon films on copper grids (Electron Microscopy Sciences). The Pt dispersions were determined by CO chemisorption at room temperature, assuming adsorption of one CO molecule per surface Pt. Prior to adsorption, catalysts were reduced in 200 Torr H2 at 473 K. The loading of Pt on each sample was measured using inductively coupled plasma optical emission spectrometry (ICP-OES), equipped with a Mod Lichte nebulizer (SPECTRO Analytical Instruments Inc, Mahwah, NJ, USA).
Steady-state, differential reaction rates for CO oxidation and WGS were measured over 0.1 g samples in a 1/4 in., quartz, tubular flow reactor. Products were detected with an on-line gas chromatograph (SRI8610C) equipped with a Hayesep Q column and a thermal conductivity detector (TCD), using He as the carrier gas. Prior to reaction measurements, the catalysts were pretreated in either oxidizing (10% O2 in He) or reducing (10% H2 in He) conditions for 1 h at 1073 K. For CO oxidation, the total flow rate was maintained at 100 mL/min using a He carrier, with partial pressures of CO and O2 adjusted to achieve the desired partial pressures. The dependence of the rates on temperature was determined using fixed partial pressures of 25 Torr CO and 12.5 Torr O2, and reaction orders for CO were measured by varying the CO flow rate for fixed temperatures and O2 partial pressures. For WGS, the partial pressures of CO and H2O were both maintained at 25 Torr, using He as a carrier gas flowing through a water bubbler at 307 K.
Light-off reaction profiles for CO oxidation were measured over 0.1 g samples in a separate tubular flow reactor with an on-line quadrupole mass spectrometer (SRS-RGA-100) to continuously measure product compositions. The partial pressures of CO and O2 were maintained at 25 and 12.5 Torr, respectively, in a total flow of 100 mL/min, with He as the carrier gas. The temperature ramp rate was 10 K/min. Light-off curves for WGS reactions were measured with the same setup, with the partial pressures of CO and H2O both kept at 25 Torr. The temperature ramp rate was again 10 K/min. For light-off curves of toluene hydrogenation, the toluene partial pressure was kept at ~7 Torr, achieved by passing the H2 component through a toluene bubbler that was kept at 273 K. The temperature ramp rate was maintained at 10 K/min from room temperature to 473 K.
5. Conclusions
Thin films of LaCoO3 were successfully deposited onto MgAl2O4 using ALD. When reduced at high temperatures, Pt catalysts prepared from thin-film supports were highly active for CO oxidation and WGS and remained highly stable to redox cycling at 1073 K. The properties of the LaCoO3-supported Pt catalysts showed important similarities to Pt on CaTiO3 and LaFeO3 films but were not similar to Pt on Co3O4 or La2O3 films, implying that the perovskite structure of the thin-film supports is important for determining their properties.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2304-6740/7/9/113/s1, Figure S1: The mass gain of (a) Co3O4 and (b) LaCoO3 on MgAl2O4 as a function of ALD cycles. Figure S2: High-angle annular dark field STEM image and EDS maps of Mg, Al, La, Co, and Pt on reduced Pt/LCO/MAO after five redox cycles, taken from the region indicated by the green box. The scale bars represent 20 nm. Figure S3: Steady-state, differential reaction rates for CO oxidation with 25 Torr of CO and 12.5 Torr O2 for 0.1 g sample of Pt/MAO. Figure S4: Light-off curves for CO oxidation over 0.1 g samples of oxidized (black) and reduced (red) Pt/LCO/MAO after five redox cycles. Figure S5: Steady-state, differential reaction rates for CO oxidation with 25 Torr of CO and 12.5 Torr O2 for 0.1 g sample of Pt/LCO/MAO. Figure S6: Light-off curves for WGS over 0.1 g samples of oxidized (black) and reduced (red) Pt/LCO/MAO after five redox cycles. Figure S7: Steady-state, differential reaction rates over 0.1 g samples of (orange circles) 1073 K calcined Pt/CeO2/γ-Al2O3, (black circles) 1073 K calcined Pt/γ-Al2O3, and (diamond) reduced Pt/LCO/MAO after five redox cycles for water gas shift with partial pressures of CO and H2O both kept at 25 Torr. Figure S8: Light-off profiles for toluene hydrogenation for 0.1 g samples of (blue) reduced Pt/Co/MAO and (black) reduced Pt/LCO/MAO. Samples have been pretreated with five redox cycles prior to measurements.
Author Contributions
Conceptualization, R.J.G. and X.M.; methodology, R.J.G and X.M.; software, A.C.F.; formal analysis, X.M.; investigation, X.M.; data curation, X.M and A.C.F.; writing—original draft preparation, X.M.; writing—review and editing, R.J.G., X.M., A.C.F. and E.A.S; supervision, R.J.G.; funding acquisition, R.J.G and E.A.S.
Funding
This work was funded by the Department of Energy, Office of Basic Energy Sciences, Chemical Sciences, Geosciences and Biosciences Division, Grant No. DE-FG02-13ER16380. The STEM work was carried out in part at the Singh Center for Nanotechnology, part of the National Nanotechnology Coordinated Infrastructure Program, which is supported by the National Science Foundation grant NNCI-1542153. A.F. and E.A.S. acknowledge support from Integrated Mesoscale Architectures for Sustainable Catalysis (IMASC), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under Award # DE-SC0012573.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Farrauto, R.J.; Deeba, M.; Alerasool, S. Gasoline automobile catalysis and its historical journey to cleaner air. Nat. Catal. 2019, 2, 603–613. [Google Scholar] [CrossRef]
- Nishihata, Y.; Mizuki, J.; Akao, T.; Tanaka, H.; Uenishi, M.; Kimura, M.; Okamoto, T.; Hamada, N. Self-regeneration of a Pd-perovskite catalyst for automotive emissions control. Nature 2002, 418, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Katz, M.B.; Dai, S.; Zhang, K.; Du, X.; Graham, G.W.; Pan, X.Q. New Atomic-Scale Insight into Self-Regeneration of Pt-CaTiO3 Catalysts: Incipient Redox-Induced Structures Revealed by a Small-Angle Tilting STEM Technique. J. Phys. Chem. C 2017, 121, 17348–17353. [Google Scholar] [CrossRef]
- Dai, S.; Zhang, S.; Katz, M.B.; Graham, G.W.; Pan, X.Q. In Situ Observation of Rh-CaTiO3 Catalysts during Reduction and Oxidation Treatments by Transmission Electron Microscopy. ACS Catal. 2017, 7, 1579–1582. [Google Scholar] [CrossRef]
- Neagu, D.; Oh, T.-S.; Miller, D.N.; Ménard, H.; Bukhari, S.M.; Gamble, S.R.; Gorte, R.J.; Vohs, J.M.; Irvine, J.T. Nano-socketed nickel particles with enhanced coking resistance grown in situ by redox exsolution. Nat. Commun. 2015, 6, 8120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onn, T.M.; Monai, M.; Dai, S.; Fonda, E.; Montini, T.; Pan, X.Q.; Graham, G.W.; Fornasiero, P.; Gorte, R.J. Smart Pd Catalyst with Improved Thermal Stability Supported on High-Surface-Area LaFeO3 Prepared by Atomic Layer Deposition. J. Am. Chem. Soc. 2018, 140, 4841–4848. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Jang, J.B.; Zhang, L.; Stach, E.A.; Gorte, R.J. Improved Coking Resistance of “Intelligent” Ni Catalysts Prepared by Atomic Layer Deposition. ACS Catal. 2018, 8, 7679–7687. [Google Scholar] [CrossRef]
- Lin, C.; Foucher, A.C.; Ji, Y.; Curran, C.D.; Stach, E.A.; McIntosh, S.; Gorte, R.J. “Intelligent” Pt Catalysts Studied on High-Surface-Area CaTiO3 Films. ACS Catal. 2019, 9, 7318–7327. [Google Scholar] [CrossRef]
- Tanaka, H.; Taniguchi, M.; Uenishi, M.; Kajita, N.; Tan, I.; Nishihata, Y.; Mizuki, J.I.; Narita, K.; Kimura, M.; Kaneko, K. Self-regenerating Rh- and Pt-based perovskite catalysts for automotive-emissions control. Angew. Chem. Int. Ed. 2006, 45, 5998–6002. [Google Scholar] [CrossRef]
- Bidrawn, F.; Kungas, R.; Vohs, J.M.; Gorte, R.J. Modeling Impedance Response of SOFC Cathodes Prepared by Infiltration. J. Electrochem. Soc. 2011, 158, B514–B525. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.W.; Li, Y.; Liu, Z.Q.; Haruta, M.; Shen, W.J. Low-temperature oxidation of CO catalysed by Co3O4 nanorods. Nature 2009, 458, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Bahlawane, N. Kinetics of methane combustion over CVD-made cobalt oxide catalysts. Appl. Catal. B-Environ. 2006, 67, 168–176. [Google Scholar] [CrossRef]
- Mergler, Y.J.; van Aalst, A.; van Delft, J.; Nieuwenhuys, B.E. Promoted Pt catalysts for automotive pollution control: Characterization of Pt/SiO2, Pt/CoOx/SiO2, and Pt/MnOx/SiO2 catalysts. J. Catal. 1996, 161, 310–318. [Google Scholar] [CrossRef]
- Arena, F.; Chio, R.D.; Espro, C.; Fazio, B.; Palella, A.; Spadaro, L. A New Class of MnCeOx Materials for the Catalytic Gas Exhausts Emission Control: A Study of the CO Model Compound Oxidation. Top. Catal. 2019, 62, 259–265. [Google Scholar] [CrossRef]
- Arena, F.; Chio, R.D.; Espro, C.; Fazio, B.; Palella, A.; Spadaro, L. A definitive assessment of the CO oxidation pattern of a nanocomposite MnCeOx catalyst. React. Chem. Eng. 2018, 3, 293–300. [Google Scholar] [CrossRef]
- Mao, X.; Foucher, A.; Stach, E.A.; Gorte, R.J. A Study of Support Effects for CH4 and CO Oxidation over Pd Catalysts on ALD-Modified Al2O3. Catal. Lett. 2019, 149, 905–915. [Google Scholar] [CrossRef]
- Steiger, P.; Alxneit, I.; Ferri, D. Nickel incorporation in perovskite-type metal oxides-Implications on reducibility. Acta Mater. 2019, 164, 568–576. [Google Scholar] [CrossRef]
- Mergler, Y.; Hoebink, J.; Nieuwenhuys, B. CO Oxidation over a Pt/CoOx/SiO2 Catalyst: A Study Using Temporal Analysis of Products. J. Catal. 1997, 167, 305–313. [Google Scholar] [CrossRef]
- Cargnello, M.; Doan-Nguyen, V.V.T.; Gordon, T.R.; Diaz, R.E.; Stach, E.A.; Gorte, R.J.; Fornasiero, P.; Murray, C.B. Control of Metal Nanocrystal Size Reveals Metal-Support Interface Role for Ceria Catalysts. Science 2013, 341, 771–773. [Google Scholar] [CrossRef] [Green Version]
- Gorte, R.J. Ceria in Catalysis: From Automotive Applications to the Water Gas Shift Reaction. AIChE J. 2010, 56, 1126–1135. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Goodman, D.W. High-Pressure Catalytic Reactions over Single-Crystal Metal-Surfaces. Surf. Sci. Rep. 1991, 14, 1–107. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Willigan, R.R.; Dardas, Z.; Vanderspurt, T.H. Water gas shift activity of noble metals supported on ceria-zirconia oxides. AIChE J. 2006, 52, 1888–1894. [Google Scholar] [CrossRef]
- Bunluesin, T.; Gorte, R.J.; Graham, G.W. Studies of the water-gas-shift reaction on ceria-supported Pt, Pd, and Rh: Implications for oxygen-storage properties. Appl. Catal. B-Environ. 1998, 15, 107–114. [Google Scholar] [CrossRef]
- Onn, T.M.; Kungas, R.; Fornasiero, P.; Huang, K.; Gorte, R.J. Atomic Layer Deposition on Porous Materials: Problems with Conventional Approaches to Catalyst and Fuel Cell Electrode Preparation. Inorganics 2018, 6, 34. [Google Scholar] [CrossRef]
Figure 1.
X-ray diffraction (XRD) patterns of (a) Pt/MAO, (b) Pt/Co/MAO, and (c) Pt/LCO/MAO: (i) unmodified MgAl2O4, (ii) oxidized Pt-containing sample after five redox cycles at 1073 K, and (iii) reduced Pt-containing sample after five redox cycles at 1073 K. Metallic Pt features are marked with diamonds, Co3O4 with pentagons, Pt–Co alloy with solid circles, metallic Co with hollow circles, and the perovskite with stars.
Figure 1.
X-ray diffraction (XRD) patterns of (a) Pt/MAO, (b) Pt/Co/MAO, and (c) Pt/LCO/MAO: (i) unmodified MgAl2O4, (ii) oxidized Pt-containing sample after five redox cycles at 1073 K, and (iii) reduced Pt-containing sample after five redox cycles at 1073 K. Metallic Pt features are marked with diamonds, Co3O4 with pentagons, Pt–Co alloy with solid circles, metallic Co with hollow circles, and the perovskite with stars.
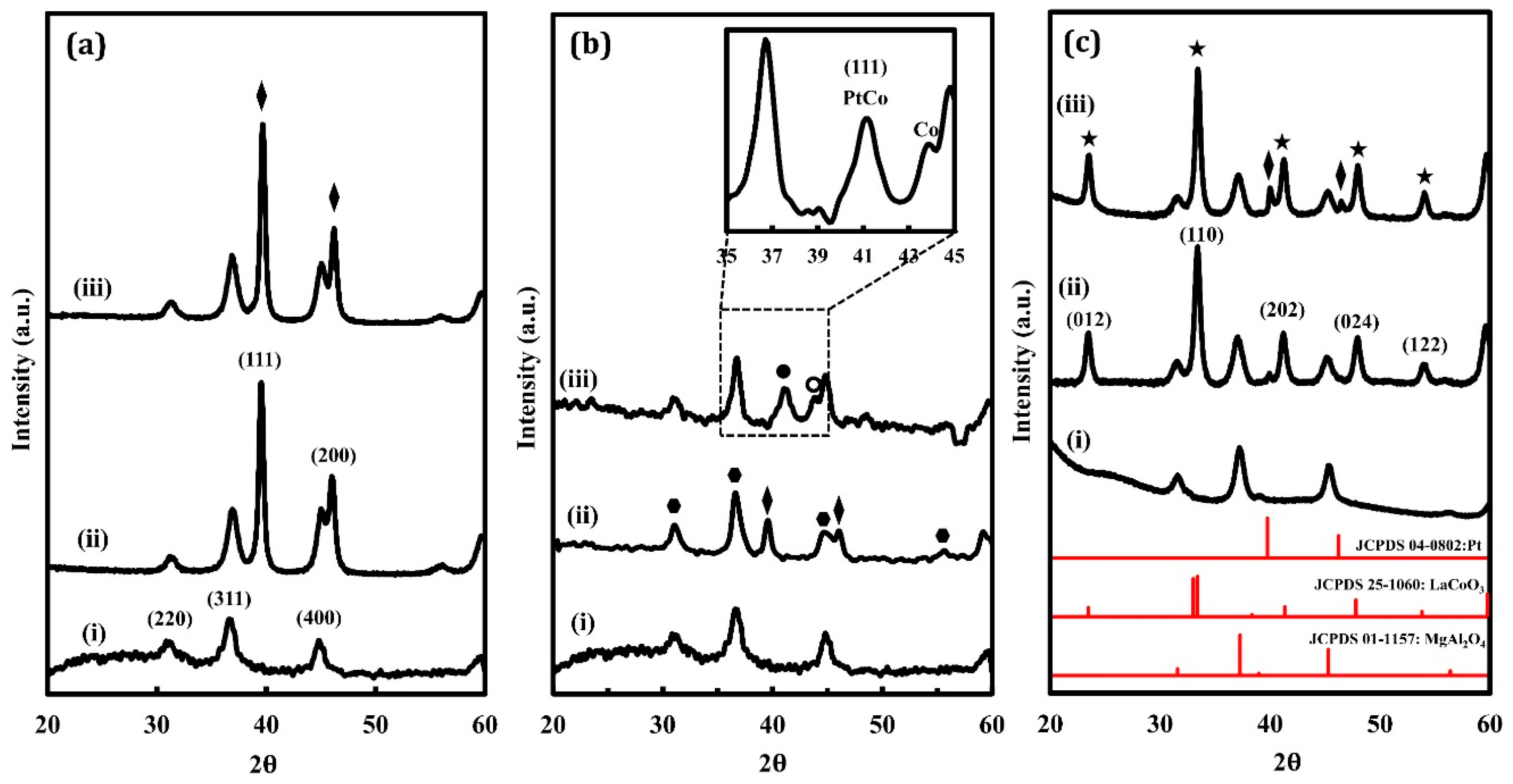
Figure 2.
(a) High-angle annular dark field (HAADF)/scanning transmission electron microscopy (STEM) image and energy-dispersive X-ray spectroscopy (EDS) maps of Mg, Al, La, Co, and Pt on oxidized Pt/LCO/MAO after five redox cycles, taken from the region indicated by the green box. (b) HAADF/STEM image and EDS maps of Mg, Al, La, Co, and Pt on reduced Pt/LCO/MAO after five redox cycles, taken from the region indicated by the green box. The scale bar in the HAADF/STEM image represents 50 nm, while the scale bars in EDS mappings represent 25 nm.
Figure 2.
(a) High-angle annular dark field (HAADF)/scanning transmission electron microscopy (STEM) image and energy-dispersive X-ray spectroscopy (EDS) maps of Mg, Al, La, Co, and Pt on oxidized Pt/LCO/MAO after five redox cycles, taken from the region indicated by the green box. (b) HAADF/STEM image and EDS maps of Mg, Al, La, Co, and Pt on reduced Pt/LCO/MAO after five redox cycles, taken from the region indicated by the green box. The scale bar in the HAADF/STEM image represents 50 nm, while the scale bars in EDS mappings represent 25 nm.

Figure 3.
Steady-state, differential reaction rates for CO oxidation with 25 Torr of CO and 12.5 Torr O2 for 0.1 g samples of (a) Pt/LCO/MAO, (b) Pt/La/MAO, and (c) Pt/Co/MAO. Rates measured on samples after oxidation in 10% O2–He at 1073 K for 1 h are marked in circles; rates measured on samples after reduction in 10% H2–He at 1073 K for 1 h are marked in diamonds. Black symbols denote the first redox cycle, while red symbols denote the fifth cycle.
Figure 3.
Steady-state, differential reaction rates for CO oxidation with 25 Torr of CO and 12.5 Torr O2 for 0.1 g samples of (a) Pt/LCO/MAO, (b) Pt/La/MAO, and (c) Pt/Co/MAO. Rates measured on samples after oxidation in 10% O2–He at 1073 K for 1 h are marked in circles; rates measured on samples after reduction in 10% H2–He at 1073 K for 1 h are marked in diamonds. Black symbols denote the first redox cycle, while red symbols denote the fifth cycle.
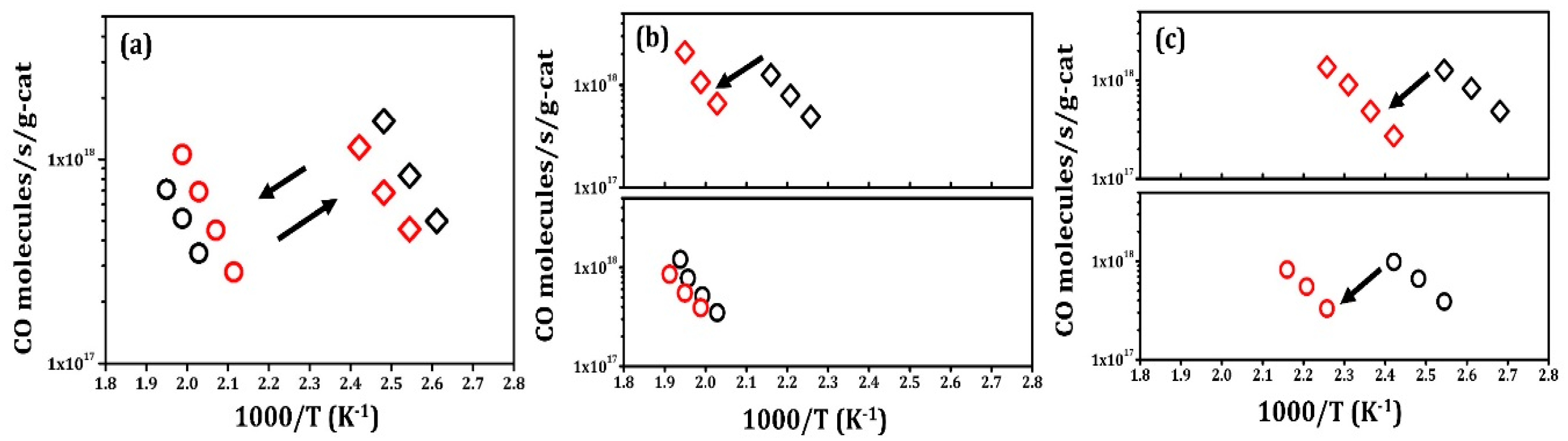
Figure 4.
Dependence of CO oxidation rates at different temperatures, at constant P(O2) = 7.6 Torr, over 0.1 g sample of reduced Pt/LCO/MAO after five redox cycles at 1073 K.
Figure 4.
Dependence of CO oxidation rates at different temperatures, at constant P(O2) = 7.6 Torr, over 0.1 g sample of reduced Pt/LCO/MAO after five redox cycles at 1073 K.

Figure 5.
Steady-state, differential reaction rates over 0.1 g sample of Pt/LCO/MAO for water gas shift with partial pressures of CO and H2O both kept at 25 Torr. Rates measured on sample after oxidation in 10% O2–He at 1073 K for 1 h are marked in circles; rates measured on samples after reduction in 10% H2–He at 1073 K for 1 h are marked in diamonds. Black symbols denote the first redox cycle, while red symbols denote the fifth cycle.
Figure 5.
Steady-state, differential reaction rates over 0.1 g sample of Pt/LCO/MAO for water gas shift with partial pressures of CO and H2O both kept at 25 Torr. Rates measured on sample after oxidation in 10% O2–He at 1073 K for 1 h are marked in circles; rates measured on samples after reduction in 10% H2–He at 1073 K for 1 h are marked in diamonds. Black symbols denote the first redox cycle, while red symbols denote the fifth cycle.

Figure 6.
Light-off profiles for toluene hydrogenation for 0.1 g samples of (red) oxidized Pt/MAO, (blue) oxidized Pt/LCO/MAO, and (black) reduced Pt/LCO/MAO. Samples had been pretreated with five redox cycles prior to measurements.
Figure 6.
Light-off profiles for toluene hydrogenation for 0.1 g samples of (red) oxidized Pt/MAO, (blue) oxidized Pt/LCO/MAO, and (black) reduced Pt/LCO/MAO. Samples had been pretreated with five redox cycles prior to measurements.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Properties of samples used in this study.
| BET S.A. (±2 m2/g) | Metal Oxide Loading (±1.5 wt %) | Pt Loading (±0.2 wt %) | Film Thickness (nm) | |
|---|---|---|---|---|
| Pt/LCO/MAO | 58 | 30 | 2.9 | 0.5 |
| Pt/La/MAO | 83 | 20 | 2.7 | 0.3 |
| Pt/Co/MAO | 81 | 20 | 2.8 | 0.3 |
| Pt/MAO | 118 | - | 3.1 | - |
Abbreviation: BET S.A.—Brunauer–Emmett–Teller surface area.
Table 2.
Summary of Pt dispersion (%) measured by CO chemisorption or estimated based on dispersion and activity of Pt (111) in the literature for catalysts after the fifth reduction and oxidation pretreatments at 1073 K.
Table 2.
Summary of Pt dispersion (%) measured by CO chemisorption or estimated based on dispersion and activity of Pt (111) in the literature for catalysts after the fifth reduction and oxidation pretreatments at 1073 K.
| 1073 K Oxidized | 1073 K Reduced | |||
|---|---|---|---|---|
| Measured | Estimated | Measured | Estimated | |
| Pt/LCO/MAO | - | 2 | - | 40 |
| Pt/MAO | 0.1 | 0.1 | 0.1 | 0.4 |
| Pt/Co/MAO | 7 | 6.5 | 13 | 13 |
| Pt/La/MAO | 1.3 | 1.4 | 1.4 | 2.8 |
-: Not able to be measured.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mao, X.; Foucher, A.C.; Stach, E.A.; Gorte, R.J. “Intelligent” Pt Catalysts Based on Thin LaCoO3 Films Prepared by Atomic Layer Deposition. Inorganics 2019, 7, 113. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7090113
AMA Style
Mao X, Foucher AC, Stach EA, Gorte RJ. “Intelligent” Pt Catalysts Based on Thin LaCoO3 Films Prepared by Atomic Layer Deposition. Inorganics. 2019; 7(9):113. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7090113
Chicago/Turabian StyleMao, Xinyu, Alexandre C. Foucher, Eric A. Stach, and Raymond J. Gorte. 2019. "“Intelligent” Pt Catalysts Based on Thin LaCoO3 Films Prepared by Atomic Layer Deposition" Inorganics 7, no. 9: 113. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics7090113
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.