Hydrosilylation of Aldehydes by a Manganese α-Diimine Complex
by
 ,
,
Veeranna Yempally
1 ,
,
Azal Shahbaz
1,
Wai Yip Fan
2,
Sherzod T. Madrahimov
1,* and
Ashfaq A. Bengali
1,* 1
Science Department, Texas A&M University at Qatar, PO Box 23874 Doha, Qatar
2
Department of Chemistry, 3 Science Drive, National University of Singapore, Singapore 117543, Singapore
*
Authors to whom correspondence should be addressed.
Inorganics 2020, 8(11), 61; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8110061
Submission received: 21 October 2020
/
Revised: 5 November 2020
/
Accepted: 6 November 2020
/
Published: 10 November 2020
(This article belongs to the Special Issue Organomanganese Chemistry)
Abstract
:This paper describes the catalytic activity of air stable and easy to handle manganese complexes towards the hydrosilylation of aldehydes. These catalysts incorporate a bulky diazabutadiene ligand and exhibit good functional group tolerance and chemoselectivity in the hydrosilylation of aldehydes, utilizing primary silanes as the reducing agent. The reactions proceed with turnover frequencies approaching 150 h−1 in some instances, similar to those observed for other manganese-based catalysts. The conversion of aromatic aldehydes to the corresponding alcohols was found to be more efficient than that for the analogous aliphatic systems.
1. Introduction
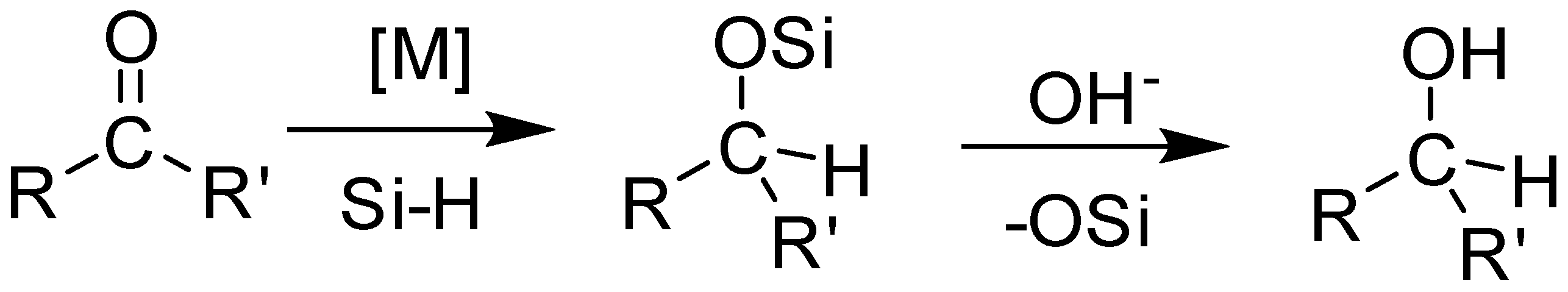
Hydrosilylation, the largest volume industrial reaction using homogenous catalysts, is an important step in the generation of valuable building blocks for the synthesis of organic chemicals and functional materials (Scheme 1) [1,2,3,4]. The formation of silyl ether protected alcohols in a single step with a high atom economy using easy to handle liquid hydrosilanes, makes this reaction an attractive alternative to hydrogenation [5,6,7]. The past decade has seen a dramatic rise in the base metal-catalyzed hydrosilylation of alkenes and carbonyl compounds, with efficiencies rivaling those observed for precious metal catalysts [8,9,10,11,12]. Recently, manganese-based catalysts have been employed in the hydrosilylation of carbonyl compounds [13,14,15]. Manganese, the fourth most earth abundant element by mass, is inexpensive and bio-compatible, thus making the resulting organometallic complexes attractive catalysts for organic transformations, including hydrosilylation reactions.
Several manganese complexes incorporating nitrogen-based chelating and other ligands have been developed as catalysts for the conversion of aldehydes and ketones into alcohols [14]. For example, Lugan et al. demonstrated that manganese complexes bearing NHC ligands are efficient photocatalysts for the reduction of ketones with diphenylsilanes under UV irradiation at room temperature [16,17]. Similarly, Cutler and coworkers utilized manganese carbonyl complexes containing acyl ligands for the efficient reduction of carbonyl compounds [18], while Chung et al. reported the generation of a pre-catalyst from a manganese tricarbonyl complex with an indenyl ligand for the hydrosilylation of ketones [19,20]. Currently, the highest reported turnover number (up to 76,800 h−1) for a first-row metal-based hydrosilylation catalyst is for a bis(imino)pyridine ligated manganese carbonyl complex [21].
Manganese-based carbonyl complexes with diazabutadiene (DAB) ligands are effective catalysts for the electrochemical reduction of CO2, and have been extensively studied for their promise as visible light-assisted CO release agents (photo-CORM) [22,23,24,25,26]. However, these complexes have not yet been investigated as hydrosilylation catalysts for the reduction of aldehydes and ketones. Our interest in the application of these complexes towards the hydrosilylation of carbonyl compounds stemmed from a recent report by Royo et al. that highlighted the use of the cationic [Mn(CO)3(bis-NHCMe)(NCMe)]BF4 complex for this reaction [27]. The authors demonstrated that the NHC ligand enhanced catalytic performance, for the reduction of ketones and aldehydes by phenylsilane. An important role for the acetonitrile ligand was also proposed, since [Mn(CO)3(bis-NHCMe)(NCMe)]BF4 exhibited improved catalytic activity compared to Mn(CO)3(bis-NHCMe)Br in the reduction of acetophenone.
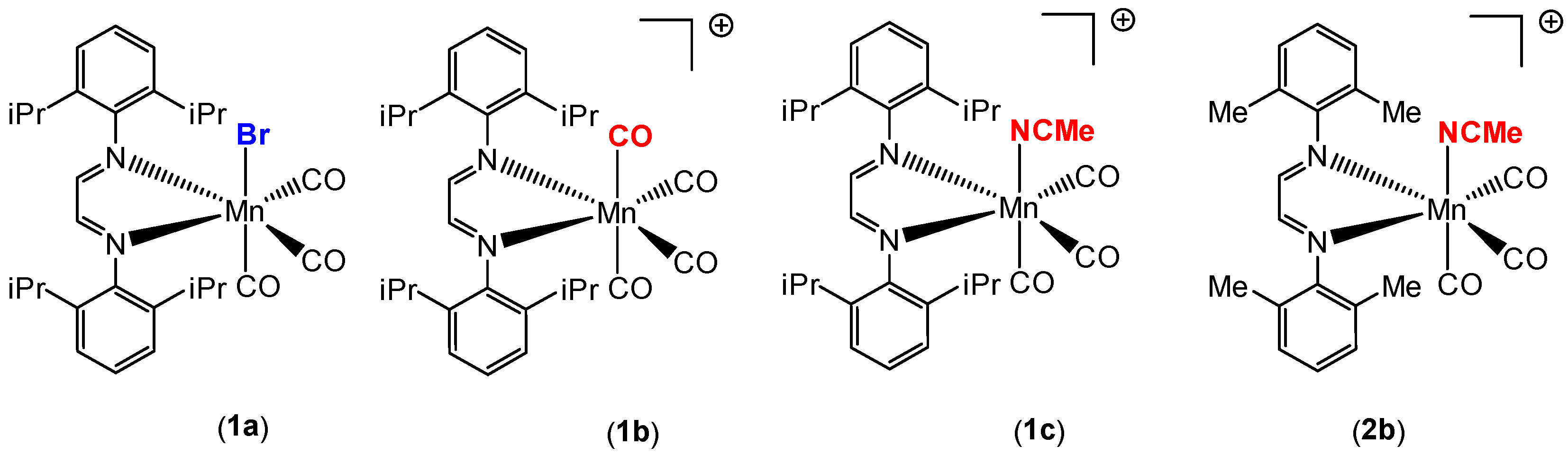
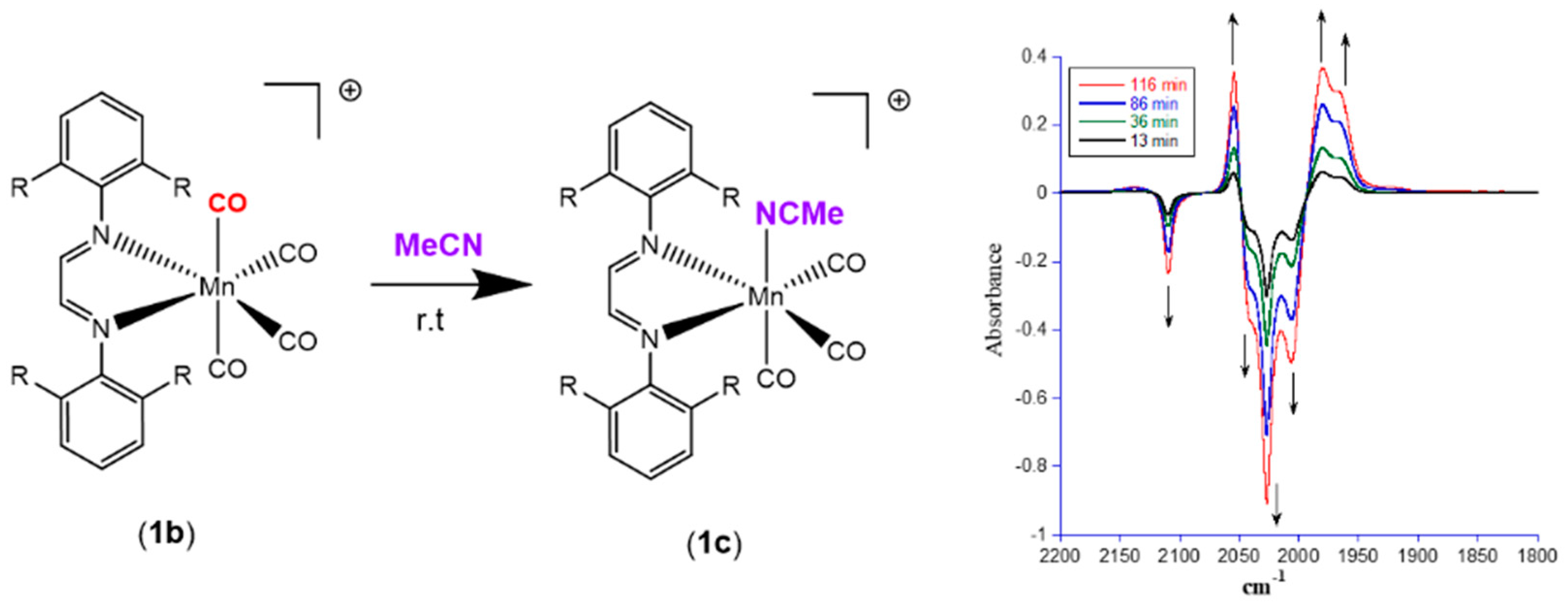
Our group recently synthesized several manganese carbonyl complexes containing a bulky DAB ligand [R-DAB = (N,N-bis(2,6-R)-1,4-diaza-1,3-butadiene), R = iPr2Ph, MePh], fac-Mn(Br)(CO)3(iPr2Ph-DAB) (1a), [Mn(CO)4(iPr2Ph-DAB)][PF6] (1b), [Mn(CO)3(NCMe)(iPr2Ph-DAB)][PF6] (1c), and [Mn(CO)3(NCMe)(Me2Ph-DAB)][PF6] (2b) for potential application as CO2 reduction catalysts and photo-CORMs (Figure 1) [28]. Compound 1a showed remarkable photoreactivity, and efficient CO release was observed upon visible light irradiation. The cationic complex 1b with a sterically encumbered DAB ligand was found to be air stable, easy to handle, and have an exceptionally labile CO group such that room temperature dissolution in acetonitrile solvent resulted in the generation of 1c (Figure 2) [28].
The facile generation of a vacant coordination site (implied by the lability of the CO ligand), together with the similarity between 1c and Royo’s catalyst prompted us to investigate the applicability of these complexes in the catalytic hydrosilylation of aldehydes and ketones. It was envisioned that a rapid substitution of the CO or MeCN ligand in these complexes by phenylsilane or substrate would generate an easily accessible pre-catalyst, which could subsequently reduce aldehyde or ketone groups to yield silyl ethers. Additionally, the unique redox properties of the DAB ligand were expected to support changes in the oxidation state of the metal center in this process. In the current study, the ability of these complexes to promote the hydrosilylation of aldehydes and ketones is reported as a function of the ancillary ligands in the primary (1a–c) and secondary (2b) coordination sphere of the metal center. Complexes 1c and 2b were found to have activities similar to those reported for Royo’s catalyst, and provide another example for the application of manganese-based catalysts in the reduction of aldehydes to alcohols.
2. Results and Discussion
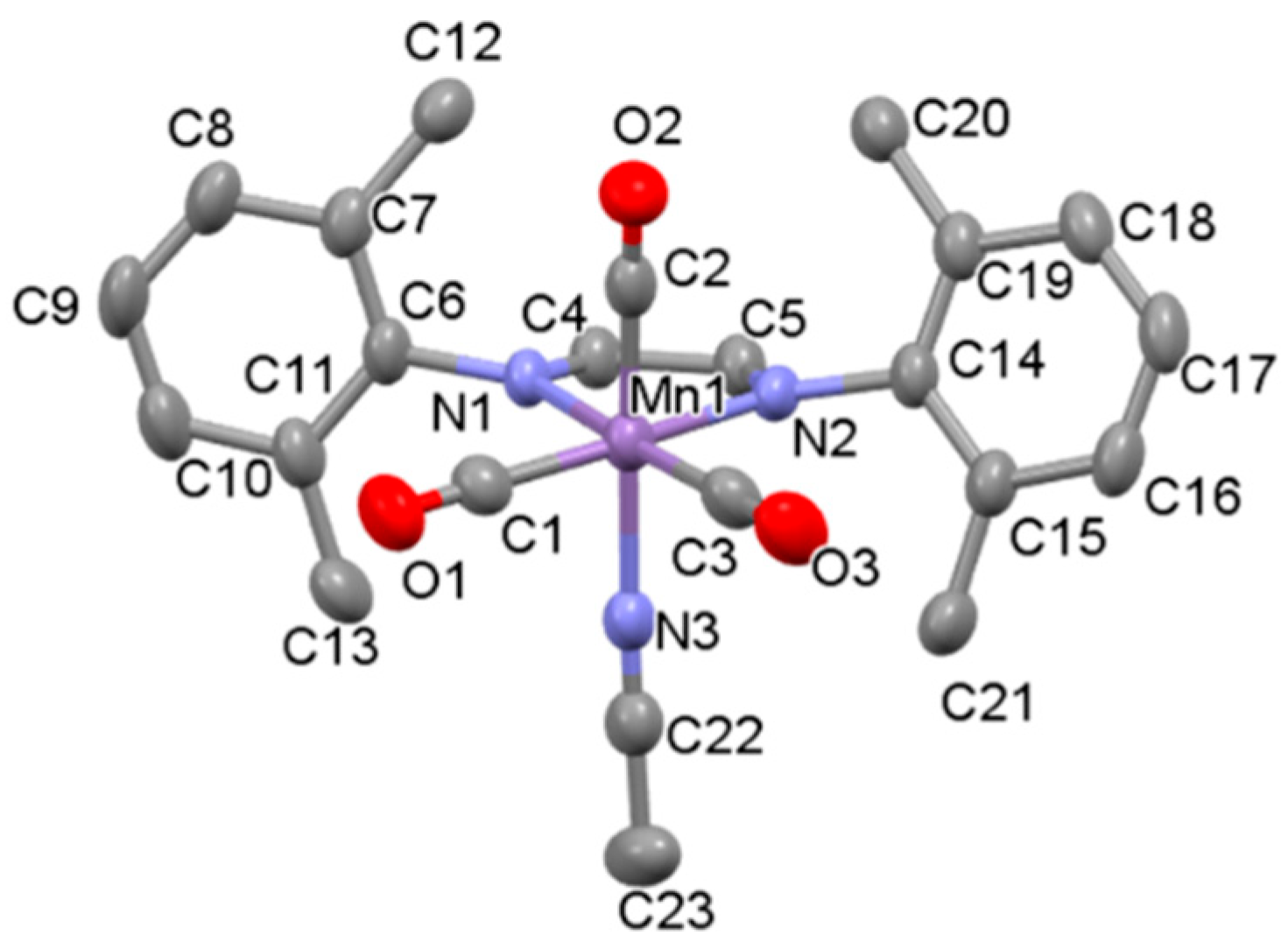
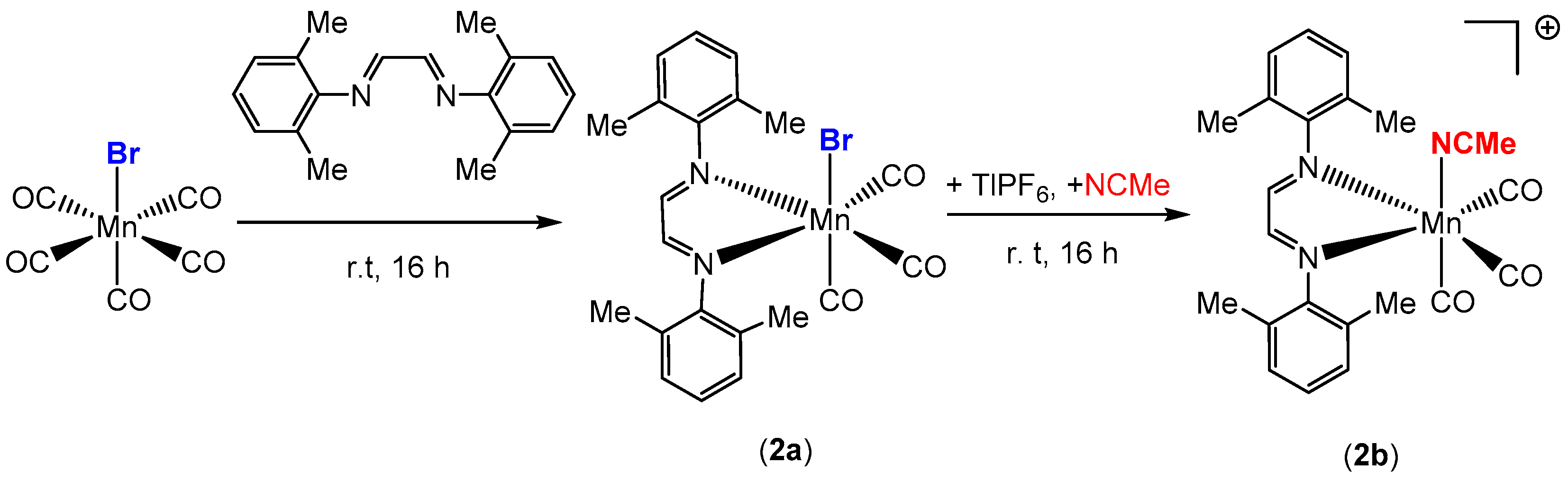
The synthesis, characterization, and photochemical reactivity of compounds 1a–c has been reported previously by our group [28]. In this paper, these studies were extended to explore the catalytic activity of complexes 1a–c towards the hydrosilylation of aldehydes. Additionally, compound 2b, with the sterically less crowded Me2Ph-DAB ligand, was synthesized from the precursor compound Mn(Me2Ph-DAB)(CO)3Br, 2a, as shown in Scheme 2, to investigate the role of steric influence in catalytic activity. Both compounds, 2a and 2b, were isolated and characterized by single crystal X-ray diffraction, FT-IR and 1H NMR spectroscopy. The X-ray structures of 2a and 2b are shown in Figure 3 and Figure S1, respectively.
The FTIR spectrum of complex 2b in dichloromethane solvent displays characteristic facial carbonyl stretching frequencies of 2053 cm−1, 1978 cm−1 and 1963 cm−1, almost identical to those of compound 1c [28]. Compound 2b is stable in air, and unlike 1c, can be manipulated under visible light conditions without noticeable decomposition. Similar to other reported cationic manganese complexes with an acetonitrile ligand [24], 2b has an octahedral structure, and its overall geometry is similar to that of 1c. However, reduced steric crowding results in the greater C(2)–Mn(1)–N(3) bond angle of 175.99 in 2b compared to 170.59 in 1c. Consistent with a closer approach of the acetonitrile ligand towards the less sterically encumbered metal center, the Mn1-N3 bond length in 2b (2.009 Å) is slightly shorter than in 1c (2.030 Å). These findings are consistent with the expectation that, relative to iPr2Ph-DAB, the reduced steric profile of the Me2Ph-DAB ligand should result in a more accessible Mn center in 2b compared to 1c. As discussed later, the observed structural differences in 2b and 1c can be correlated with differences in the catalytic activities of the two complexes.
2.1. Catalyst Optimization
Due to the previously determined lability of the CO ligand in 1b, the initial screening and optimization was performed with this catalyst by monitoring the reaction of trans-cinnamaldehyde with phenylsilane. The reactions were conducted at 80 °C, with progress monitored by GC-MS at regular time intervals. Under these conditions, the hydrosilylation reaction proceeded with an optimal catalyst loading of 2.5 mol % and with high chemoselectivity, as side products arising from the reduction of the alkene group were not observed. The aldehyde group was selectively reduced to the corresponding alcohol (following hydrolysis with 2M aqueous NaOH), as indicated by 1H NMR and GC-MS analysis, with 98% conversion after 22 h with a moderate TON = 40 in acetonitrile solvent (Table 1, entry 2). Surprisingly, the alcohol was the major product, even without hydrolysis, with minor amounts of the silylether observed under these conditions, along with some unidentified species. A control experiment performed without the metal catalyst showed no conversion, thereby confirming the involvement of 1b in the catalytic process (Table 1, entry 7). The use of tertiary and secondary silanes also resulted in the reduction of trans-cinnamaldehyde, but as observed previously for other manganese-based systems [13], complete conversion required significantly longer reaction times (e.g., 92 h for Ph2SiH2).
Several solvents, including 1,2-dichloroethane, toluene and THF, were used, but a significant improvement in catalytic activity was not observed in comparison to performing the reaction in acetonitrile. However, thereactions conducted under neat (solvent free) conditions proceeded at a significantly faster rate (2 h, 98% for 1b, Table 1, entry 2) under similar catalytic conditions. These results suggest that acetonitrile inhibits the reaction, most likely by competing with silane and substrate for binding to the Mn center, so that in the absence of solvent the reactions proceed at a faster rate.
2.2. Catalyst Comparison
In acetonitrile solvent and under identical reaction conditions, 2b showed superior catalytic activity compared to 1b and 1c, whereas negligible product formation was observed with catalyst 1a (Table 1, entries 1–4). For example, the reaction turnover frequency (TOF) in the case of 2b was significantly higher than for 1b and 1c (13 h−1 vs 1.8 h−1). Furthermore, unlike the other complexes, catalytic activity was also observed at room temperature. The higher reactivity of 2b is most likely related to the reduced steric bulk of the Me2Ph-DAB ligand compared to iPr2-DAB, which results in a less sterically congested site at the metal center facilitating the binding of silane and aldehyde at different stages in the reduction reaction. However, the observed difference in reactivity between 1b, 1c and 2b disappeared when the reactions were conducted under neat conditions, resulting in an almost complete reduction within two hours in all three cases (Table 1, entries 2–4). For 1c and 2b, catalytic activity with almost complete conversion to the alcohol under neat conditions was maintained with catalyst loadings as low as 1 mol % (Table 1, entries 5 and 6), resulting in TOF values of 45 h−1 and 48 h−1, respectively.
In acetonitrile solvent, both 1b and 1c selectively reduced the aldehyde group, with 98% conversion into the alcohol after 22 h. However, a higher conversion to the alcohol was observed during the first two hours of the reaction in the case of 1c. As mentioned earlier, previous studies have shown that the CO group in 1b is exceptionally labile, and is replaced by acetonitrile at room temperature within two hours. Thus, it is reasonable to expect that in the early stages of the reaction, 1b is converted to 1c prior to silane activation, resulting in a lower amount of alcohol production during the initial phase of the reaction in this case.
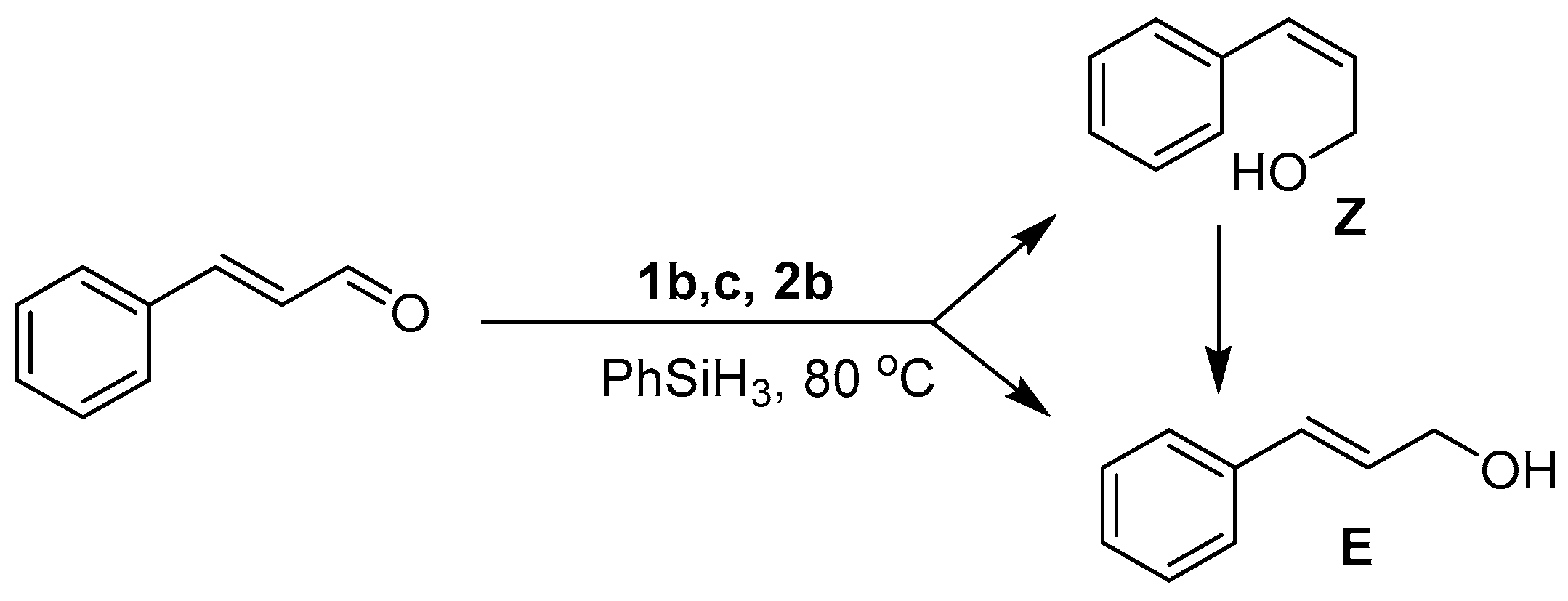
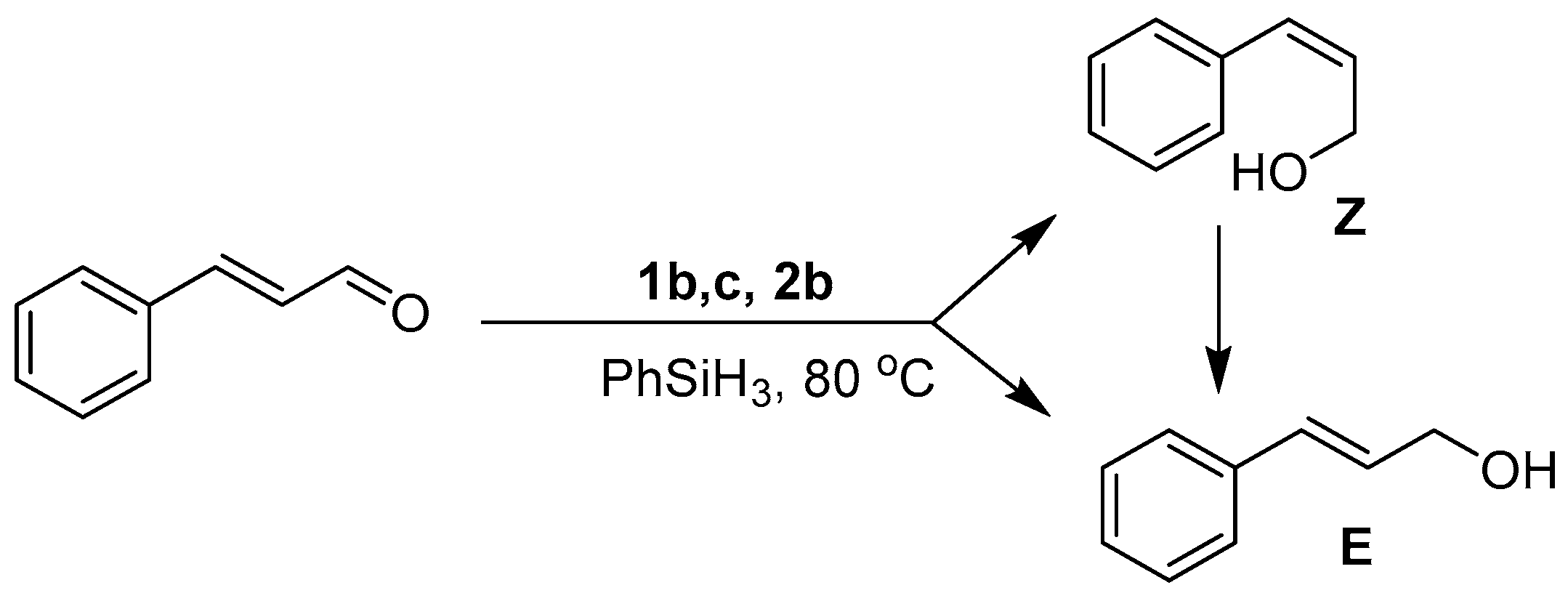
In all instances (except 1a), the GC-MS spectra obtained after ten minutes of reaction showed the presence of both the E and Z isomers of the alcohol, with only the thermodynamically favored E form isolated upon reaction completion (Figure 4). Intriguingly, the distribution ratio of isomers at this time depends upon the catalyst’s identity with E:Z ratios of 38:62 (1b), 67:33 (1c), and 70:30 (2b) (Figures S2–S4). Observation of the Z form of the alcohol during the early stages of the reaction raises the interesting possibility that catalysts 1b, 1c and 2b initially promote the trans → cis isomerization of trans-cinnamaldehyde, following which the cis isomer is hydrosilylated to yield the Z form of cinnamyl alcohol. Because of restricted rotation about the C=C bond in trans-cinnamaldehyde, such an isomerization is unlikely without disruption of the conjugated π system, which may occur upon alkene binding to the Mn center. The observed variation in the initial distribution of the E:Z ratios points to the existence of subtle kinetic effects arising from differences in the ligand structure around the Mn center. While it is difficult to further rationalize this trend with the preliminary data at hand, these observations are the subject of further on-going experimental and theoretical investigations into the mechanism of the reactions.
2.3. Substrate Scope
Since 2b was found to be the most effective catalyst (in acetonitrile solvent), substrate scope was assessed with this catalyst and the results are shown in Table 2. Under solvent-free conditions, 2b exhibited a high activity towards the reduction of benzaldehyde (Table 2, entry 1). Even at a relatively low catalyst loading of 0.1 mol %, 90% conversion was observed within a few hours, resulting in a respectable TOF value of 150 h−1. Similarly, the hydrosilylation of the carbonyl group of p-nitro-, p-bromo-, and p-methoxybenzaldehyde, to yield the corresponding alcohol, proceeded with high yield and selectivity, demonstrating good functional group tolerance (entries 2–4). Under similar conditions, p-benzaldehyde with an electron-donating group (–OCH3) was reduced more readily than with an electron withdrawing group (–Br, –NO2). Given that ortho and para substituents exhibit similar directing effects, the comparable TOF values for the reduction of p- and o-nitrobenzaldehyde (entries 4 and 5) to the respective alcohols further indicates that the electronic properties of the substituents on the aromatic ring, and not their steric profile, influence the efficiency of the reaction. Heteroatom substituted aromatic aldehydes, such as 2-pyridinecarboxaldehyde, 2-thiophenecarboxaldehyde, 2-pyrrolecarboxyaldehyde and chlorofurfural (entries 10–13), were also reduced to the corresponding alcohols by employing catalyst 2b, but with lower yields. These sub-optimal catalytic results can be rationalized by the expected deactivation of the catalyst following substrate coordination to the Mn center through the heteroatom in competition with silane or aldehyde binding.
To examine the influence of the aromatic ring upon the hydrosilylation reaction, the reduction of aliphatic aldehydes, such as 1-heptanal (entry 14) and 2-methylpentanal, was attempted. While the GC-MS spectrum after 3 h in both cases indicated the formation of multiple unidentified side products, evidence for the formation of either the silyl ether or alcohol (upon hydrolysis) was not obtained even after prolonged reaction times (24 h). This difference in reactivity between the aliphatic and aromatic systems is likely due to conjugation in the latter case, which serves to weaken the aldehyde C=O bond, thereby facilitating hydrosilylation.
The results also demonstrate that catalyst 2b is significantly more efficient at reducing aldehydes in comparison to ketones. For example, under similar conditions and in dramatic contrast to the reduction of benzaldehyde, the conversion of acetophenone to the corresponding alcohol proceeded with only a 55% yield after 41 h (entry 7). Similarly reduced reactivity was also observed for benzophenone reduction (entry 6). Furthermore, 2b was unable to catalyze the reduction of 2-methoxyacetophenone even after 41 h.
2.4. Proposed Mechanism
For many systems, the mechanism of hydrosilylation has been thoroughly investigated, and the initial steps include the oxidative addition of an Si–H bond across the metal center. To confirm that Si–H bond activation is also a key step in the current system, experiments were conducted to detect the presence of a Mn–H bond upon the reaction of catalyst with silane. The stoichiometric (1:1) reaction of catalyst 1b with PhSiH3 in the absence of aldehyde was followed by 1H NMR in acetonitrile-d3. At room temperature, no change in the spectrum was observed after the addition of phenylsilane. However, upon heating the mixture to 80 °C, the color of the solution changed from dark red to dark yellow, and a peak at δ = −5.68 ppm, assigned to the hydride resonance of the putative Mn(H)(SiH2Ph) intermediate, was observed (Figure S6). The position of the hydride resonance is in a region similar to that observed for the related Mn–silane complexes [17,27]. Unfortunately, attempts to isolate this intermediate were unsuccessful. While an Ojima like hydrosilylation mechanism is likely in the present case, further mechanistic investigations with supporting DFT modeling are ongoing and will be reported in due course.
3. Materials and Methods
3.1. General
Unless otherwise stated, all experiments were performed in the absence of light. Anhydrous solvents (MeCN, CH2Cl2, Et2O) and Schlenk techniques were used for the synthesis of all the complexes. Manganese pentacarbonyl bromide (Mn(CO)5Br) and thallium hexafluorophosphate (TlPF6) were purchased from Strem chemicals, and the ligands iPr2Ph-DAB and Me2Ph-DAB were purchased from Sigma-Aldrich (St. Louis, MI, USA). All reagents were used as received without further purification. NMR spectra were recorded on a Bruker Avance II 400 spectrometer (Billerica, MA, USA) while GC-MS data were obtained using an Agilent GC-MS spectrometer (Santa Clara, CA, USA). 1H, 19F, and 13C NMR were referenced to residual solvent resonances. Infrared spectra were obtained on a Bruker vertex 80 FTIR spectrometer utilizing a 0.75 mm path length temperature-controlled cell (Harrick Scientific) with CaF2 windows. Compounds 1a, 1b, and 1c were synthesized according to the reported procedure [28].
3.2. fac-Mn(Br)(CO)3(Me2Ph-DAB) (2a)
A round-bottom flask was charged with 0.315 g of Mn(CO)5Br (1.15 mmol), 0.300 g of Me2Ph-DAB (1.14 mmol), and 20 mL of anhydrous diethyl ether under a nitrogen atmosphere. The solution was left overnight with constant stirring at room temperature. The solvent was evaporated to yield a solid dark blue-colored residue which was washed with cold hexane (3 × 2 mL) and dried under vacuum to afford 0.496 g of a dark blue powder (90% yield). Dark blue-colored crystals of suitable quality for single crystal X-ray diffraction were grown from slow evaporation of the solvent (1:1 dichloromethane/hexane) at 25 °C. IR data in dichloromethane (νCO in cm−1): 2035 (s), 1971 (s), 1930 (m). NMR data in CDCl3: 1H δ 8.32 (s, 2 H), 7.13 (m, 6 H), 2.55 (s, 6 H of CH3), 2.26 (s, 6 H of CH3).
3.3. fac-[Mn(CO)3(Me2Ph-DAB)(CH3CN)]PF6 (2b)
A round-bottom flask was charged with 0.500 g of 2a (1.08 mmol), 0.725 g TlPF6 (2.08 mmol), and 50 mL of anhydrous dichloromethane under a nitrogen atmosphere. To the above reaction mixture, 1 mL of anhydrous acetonitrile was added via a syringe. The solution was left overnight with constant stirring at room temperature. The reaction mixture was filtered through a Celite pad to remove thallium bromide and evaporated to yield an orange residue which was washed several times with diethyl ether and dried in vacuo to afford 0.450 g (76% yield) of an orange solid, 2b. Orange crystals of suitable quality for X-ray diffraction were grown from the slow evaporation of a solvent mixture (hexane/dichloromethane (1:1) at 25 °C in an nitrogen-filled KIYON Glovebox (Kiyon Glovebox System, Seoul, South Korea). IR data in dichloromethane (νCO in cm−1): 2053 (s), 1978 (m), 1963 (br). NMR data in CD2Cl2: 1H δ 8.44 (s, 2 H), 7.17 (m, 6 H), 2.35 (s, 3 H of MeCN), 2.30 (s, 6 H of DAB-Me), 2.24 (s, 6 H of DAB-Me).
3.4. X-ray Crystallography
Diffraction data for 2a and 2b were obtained by placing the crystals under streaming nitrogen (100 K) in a Bruker-AXS Smart Apex CCD single-crystal diffractometer. Data were collected at 100(2) K using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The space groups were determined based on systematic absences and intensity statistics. The structures were solved by direct methods and refined by fullmatrix least-squares on F2. Anisotropic displacement parameters were determined for all non-hydrogen atoms. Hydrogen atoms were placed at idealized positions and refined with fixed isotropic displacement parameters. The following is a list of programs used: data collection and cell refinement, APEX2 [29]; data reductions, SAINTPLUS version 6.63 [30]; absorption correction, SADABS [31]; structural solutions, SHELXS-97 [32]; structural refinement, SHELXL-97 [33]; graphics and publication materials, Mercury version 3.0 [34]. The CIF files, tables, and CHECKCIF outputs are available in the Supporting Information (Tables S1–S6).
3.5. General Procedure for Hydrosilylation of Carbonyl Compounds Employing Catalyst 2b
In an inert air glove box, an 8 mL scintillation vial was charged with an appropriate amount of catalyst 2b (1.0 mol % or 2.5 mol %). The vial was charged with 1.0 mmol of PhSiH3 and 0.78 mmol of the carbonyl compound (aldehyde or ketone). The progress of the reaction was monitored by GC-MS. After completion of the reaction, the reaction mixture was treated with 5.0 mL of dichloromethane solvent. The resulting reaction mixture was then hydrolyzed with 2.0 M aqueous NaOH solution at room temperature for 2 h. The product was extracted from the dichloromethane layer and dried over anhydrous magnesium sulphate. The solvent was removed by rotary evaporation (37 °C) under reduced pressure to isolate the corresponding alcohol. The conversion yield was determined by NMR spectroscopy following the addition of 0.51 M MeOH in a capillary tube as an internal standard. A similar procedure was followed for hydrosilylation with catalysts 1a–1c. The product alcohol 1H NMR spectra for all the substrates used in this study were in agreement with the literature-reported data [21].
4. Conclusions
Air stable and easy to handle manganese complexes incorporating a bulky DAB ligand were found to be attractive hydrosilylation catalysts. In the presence of these catalysts, the efficient hydrosilylation of aldehydes with good functional group tolerance and chemoselectivity was observed with primary silanes as the reducing agent. The reactions proceeded without the need for an external activating agent. In some cases, direct alcohol product formation was observed by GC-MS even without hydrolysis. Aldehydes such as benzaldehyde and cinnamaldehyde were converted into corresponding alcohols at the moderate catalytic loading of 0.1 mol %, with turn-over frequencies approaching 150 h−1, comparable to other Mn-based catalysts. The conversion of aromatic aldehydes was more efficient than for the aliphatic systems. Mechanistic studies are on-going but there has been an initial detection of a Mn–H intermediate, suggesting a classical Ojima-like mechanism for the catalytic reaction.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2304-6740/8/11/61/s1, Figure S1: X-ray crystal structure for compound fac-Mn(Br)(CO)3(MePh-DAB) (2a), Figure S2: GC-MS of the reaction mixture (Cinnamaldehyde + Silane) at 80 °C in the presence of catalyst 1b after 10 min, Figure S3: GC-MS of the reaction mixture (Cinnamaldehyde + Silane) at 80 °C in the absence of catalyst 1c after 10 min, Figure S4: GC-MS of the reaction mixture (Cinnamaldehyde + Silane) at 80 °C in the absence of catalyst 1d after 10 min, Figure S5: GC-MS of the reaction mixture (Cinnamaldehyde + Silane) at 80 °C in the absence of catalyst, Figure S6: 1H-NMR spectrum of catalyst 1b with a stoichiometric amount of PhSiH3 at RT in acetonitrile-d3, Tables S1–S6: X-Ray crystallographic data, bond angles, and bond lengths for Compounds 2a and 2b.
Author Contributions
Conceptualization, V.Y.; Formal analysis, V.Y.; Funding acquisition, W.Y.F.; Methodology, V.Y. and W.Y.F.; Supervision, A.A.B. and S.T.M.; Validation, A.S.; Writing—original draft, V.Y.; Writing—review & editing, A.A.B., S.T.M. and W.Y.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a grant from the Qatar National Research Fund (QNRF) under the National Priorities Research Program, grant number 8-094-1-018.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Noles, A.F.; Hubert, A.J. Industrial Applications of Homogenous Catalysis; Mortreaux, A., Petit, F., Eds.; Springer: Amsterdam, The Netherlands, 1985; pp. 80–91. [Google Scholar]
- Roy, A.K. A Review of recent progress in homogenous catalyzed hydrosilylation. Adv. Organomet. Chem. 2007, 55, 1–59. [Google Scholar]
- Plueddemann, E.P. Silane Coupling Agents, 2nd ed.; Plenum Press: New York, NY, USA, 1991. [Google Scholar]
- Uvarov, V.M.; de Vekki, D.A. Recent progress in the development of catalytic systems for homogenous and asymmetric hydrosilyylation of ketones. J. Organomet. Chem. 2020, 923, 121415. [Google Scholar] [CrossRef]
- Marciniec, B. Hydrosilylation of Alkenes and Their Derivatives. In Hydrosilylation: A Comprehensive Review on Recent Advances; Springer: Berlin/Heidelberg, Germany, 2010; pp. 3–51. [Google Scholar]
- Ojima, I.; Li, Z.; Zhu, J. Recent advances in the hydrosilylation and related reactions. In The Chemistry of Organic Silicon Compounds; Rappoport, Z., Apeloig, Y., Eds.; Wiley: New York, NY, USA, 1998; Volume 2, pp. 1687–1792. [Google Scholar]
- De Vries, J.G.; Elsevier, C.J. The Handbook of Homogeneous Hydrogenation; Wiley-VCH: Weinheim, Germany, 2007; pp. 1–3. [Google Scholar]
- Tendrreau, A.M.; Atienza, C.C.H.; Weller, K.J.; Nye, S.A.; Lewis, K.M.; Delis, J.G.P.; Chirik, P.J. Iron catalysts for selective Anti-Markovnikov alkene hydrosilylation using tertiary silanes. Science 2012, 335, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, Y.; Shimada, S. Hydrosilylation reaction of olefins: Recent advances and perspectives. RSC Adv. 2015, 5, 20603–20616. [Google Scholar] [CrossRef]
- Chirik, P.J. Modern Alchemy: Replacing precious metals with Iron in catalytic alkene and carbonyl hydrogenation reactions. In Catalysis without Precious Metals; Bullock, R.M., Ed.; Wiley-VCH: Weinheim, Germany, 2010; pp. 83–142. [Google Scholar]
- Sun, J.; Deng, L. Cobalt Complex-Catalyzed Hydrosilylation of Alkenes and Alkynes. ACS Catal. 2016, 6, 290–300. [Google Scholar] [CrossRef]
- Bauer, I.; Knolker, H.J. Iron Catalysis in Organic Synthesis. Chem. Rev. 2015, 115, 3170–3387. [Google Scholar] [CrossRef] [PubMed]
- Valyaev, D.A.; Lavigne, G.; Lugan, N. Manganese organometallic compounds in homogeneous catalysis: Past, present, and prospects. Coord. Chem. Rev. 2016, 308, 191–235. [Google Scholar] [CrossRef]
- Trovitch, R.J. The Emergence of Manganese-Based Carbonyl Hydrosilylation Catalysts. Acc. Chem. Res. 2017, 50, 2842–2852. [Google Scholar] [CrossRef]
- Yang, X.; Wang, C. Manganese-Catalyzed Hydrosilylation Reactions. Chem. Asian J. 2018, 13, 2307–2315. [Google Scholar] [CrossRef]
- Zheng, J.; Elangovan, S.; Valyaev, D.A.; Brousses, R.; Cesar, V.; Sortias, J.B.; Darcel, C.; Lugan, N.; Lavigne, G. Hydrosilylation of Aldehydes and Ketones Catalyzed by Half-Sandwich Manganese(I) N-Heterocyclic Carbene Complexes. Adv. Synth. Catal. 2014, 356, 1093–1097. [Google Scholar] [CrossRef]
- Valyaev, D.A.; Wei, D.; Elangovan, S.; Cavailles, M.; Dorcet, V.; Sortais, J.B.; Darcel, C.; Lugan, N. Half-Sandwich Manganese Complexes Bearing Cp Tethered N-Heterocyclic Carbene Ligands: Synthesis and Mechanistic Insights into the Catalytic Ketone Hydrosilylation. Organometallics 2016, 35, 4090–4098. [Google Scholar] [CrossRef]
- Hanna, P.K.; Gregg, B.T.; Cutler, A.R. Manganese carbonyl compounds as hydrosilation catalysts for organoiron acyl complexes. Organometallics 1991, 10, 31–33. [Google Scholar] [CrossRef]
- Son, S.U.; Paik, S.J.; Lee, I.S.; Chung, Y.K.; Seok, W.K.; Lee, H.N. Chemistry of [(1H-hydronaphthalene)Mn(CO)3]: The Role of Ring-Slippage in Substitution, Catalytic Hydrosilylation, and Molecular Crystal Structure of [(η3-C10H9)Mn(CO)3P(OMe)3]. Organometallics 1999, 18, 4114–4118. [Google Scholar] [CrossRef]
- Son, S.U.; Paik, S.J.; Chung, Y.K. Hydrosilylation of ketones catalyzed by tricarbonyl(naphthalene)manganese cation. J. Mol. Catal. A 2000, 151, 87–90. [Google Scholar] [CrossRef]
- Mukhopadhyay, T.K.; Flores, M.; Groy, T.L.; Trovitch, R.J. A Highly Active Manganese Precatalyst for the Hydrosilylation of Ketones and Esters. J. Am. Chem. Soc. 2014, 136, 882–885. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Tory, J.; Hartl, F. Electrocatalytic Reduction of Carbon Dioxide with a Manganese(I) Tricarbonyl Complex Containing a Nonaromatic α-Diimine Ligand. Organometallics 2014, 33, 5002–5008. [Google Scholar] [CrossRef]
- Sampson, M.D.; Kubaik, C.P. Manganese electrocatalysts with bulky bipyridine ligands: Utilizing Lewis acids to promote carbon dioxide reduction at low overpotentials. J. Am. Chem. Soc. 2016, 138, 1386–1393. [Google Scholar] [CrossRef]
- Smieja, J.M.; Sampson, M.D.; Grice, K.A.; Benson, E.E.; Froehlich, J.D.; Kubaik, C.P. Manganese as a Substitute for Rhenium in CO2 Reduction Catalysts: The Importance of Acids. Inorg. Chem. 2013, 52, 2484. [Google Scholar] [CrossRef]
- Gonzalez, M.A.; Carrington, S.J.; Fry, N.L.; Martinez, J.L.; Mascharak, P.K. Syntheses, structures, and properties of new manganese carbonyls as photoactive CO-releasing molecules: Design strategies that lead to CO photolability in the visible region. Inorg. Chem. 2012, 51, 11930–11940. [Google Scholar] [CrossRef]
- Sakla, R.; Singh, A.; Kaushik, R.; Kumar, P.D.; Jose, A. Allosteric Regulation in Carbon Monoxide (CO) Release: Anion Responsive CO-Releasing Molecule (CORM) Derived from (Terpyridine) phenol Manganese Tricarbonyl Complex with Colorimetric and Fluorescence Monitoring. Inorg. Chem. 2019, 58, 10761–10768. [Google Scholar] [CrossRef]
- Pinto, M.; Friães, S.; Franco, F.; Lioret-Fillol, J.; Royo, B. Manganese N-Heterocyclic Carbene Complexes for Catalytic Reduction of Ketones with Silanes. ChemCatChem 2018, 10, 2734–2740. [Google Scholar] [CrossRef]
- Yempally, V.; Kyran, S.J.; Raju, R.K.; Fan, W.Y.; Brothers, E.N.; Darensbourg, D.J.; Bengali, A.A. Thermal and Photochemical Reactivity of Manganese Tricarbonyl and Tetracarbonyl Complexes with a Bulky Diazabutadiene Ligand. Inorg Chem. 2014, 53, 4081–4088. [Google Scholar] [CrossRef]
- APEX2, v 2009.7−0; Bruker AXS, Inc.: Madison, WI, USA, 2007.
- SAINTPLUS: Program for Reduction of Area Detector Data, 1034, v 6.63; Bruker AXS Inc.: Madison, WI, USA, 2007.
- Sheldrick, G.M. SADABS: Program for Absorption Correction of 1036 Area Detector Frames; Bruker AXS Inc.: Madison, WI, USA, 2001. [Google Scholar]
- Sheldrick, G.M. SHELXS-97: Program for Crystal Structure 1038 Solution; Universitat Gottingen: Gottingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXL-97: Program for Crystal Structure 1040 Refinement; Universitat Gottingen: Gottingen, Germany, 1997. [Google Scholar]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
Scheme 1.
Metal catalyzed hydrosilylation of organic carbonyls.
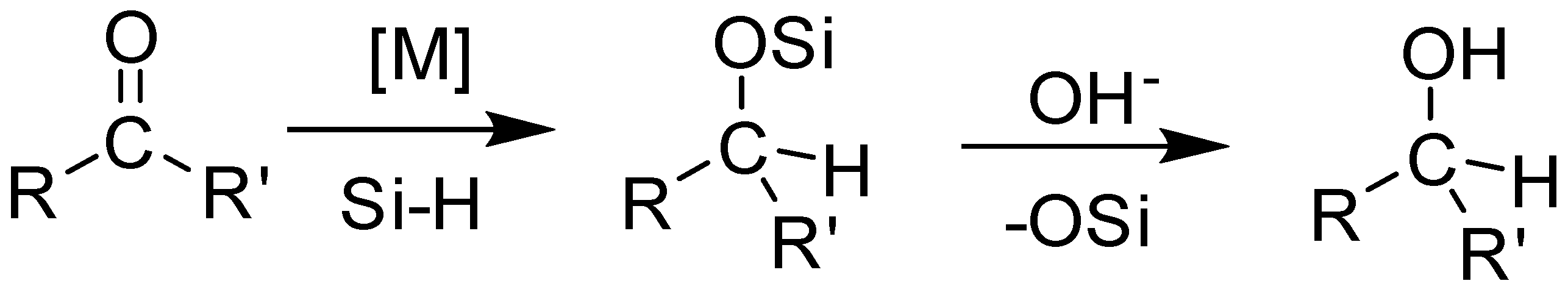
Figure 1.
Manganese carbonyl catalysts employed in the hydrosilylation of aldehydes in this work.
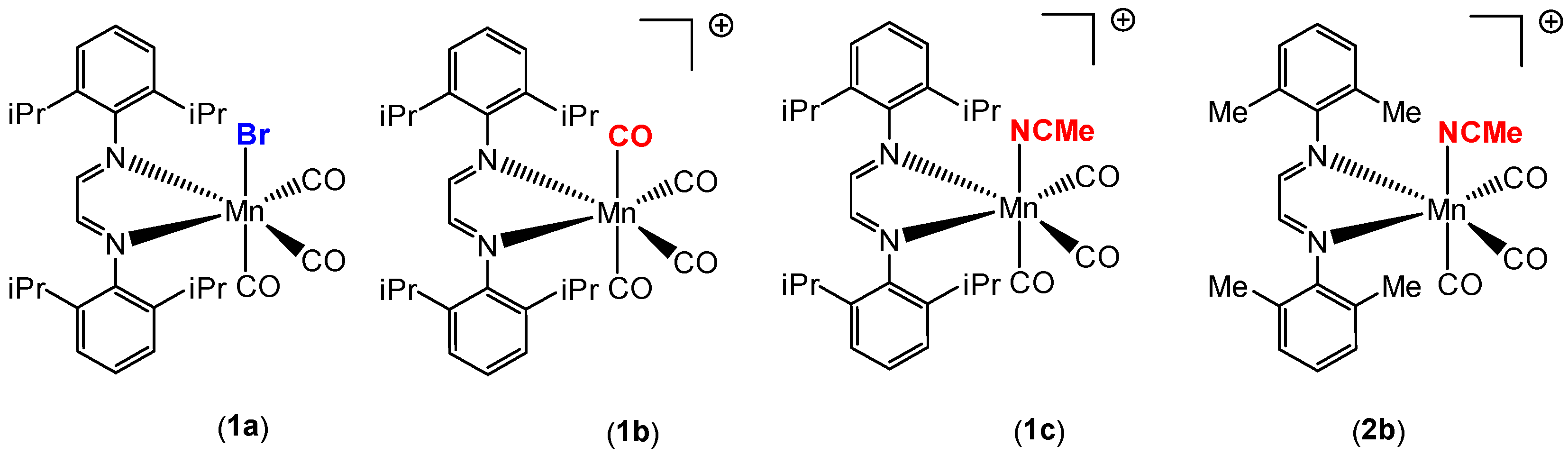
Figure 2.
Facile substitution of a carbonyl ligand in 1b by acetonitrile at 298 K.

Scheme 2.
Synthesis of manganese carbonyl complex 2b.
Figure 3.
Thermal ellipsoid (50% probability level) plot of the solid-state structure of 2b with select atom labeling. Hydrogen atoms and the PF6 ion are omitted for clarity. Selected bond lengths (Å): Mn1–C1 1.824, Mn1–C2 1.832, Mn1–C3 1.822, Mn1–N1 2.046, Mn1–N2 2.045. Selected bond angles (deg): C2–Mn1–N3 175.99, C1–Mn1–N1 94.45, C3–Mn1–N1 174.99, C1–Mn1–N2 172.12.
Figure 3.
Thermal ellipsoid (50% probability level) plot of the solid-state structure of 2b with select atom labeling. Hydrogen atoms and the PF6 ion are omitted for clarity. Selected bond lengths (Å): Mn1–C1 1.824, Mn1–C2 1.832, Mn1–C3 1.822, Mn1–N1 2.046, Mn1–N2 2.045. Selected bond angles (deg): C2–Mn1–N3 175.99, C1–Mn1–N1 94.45, C3–Mn1–N1 174.99, C1–Mn1–N2 172.12.

Figure 4.
Formation of E and Z isomers of cinnamyl alcohol after 10 min of reaction followed by full conversion to the E isomer at reaction completion.
Figure 4.
Formation of E and Z isomers of cinnamyl alcohol after 10 min of reaction followed by full conversion to the E isomer at reaction completion.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Hydrosilylation of trans-cinnamaldehyde catalyzed by 1a–c, 2b a.

| Entry | Catalyst | Solvent | Time (h) | Conversion(%) b |
|---|---|---|---|---|
| 1 | 1a | CH3CN | 18 (2) | 2 (5) |
| 2 | 1b | CH3CN | 22 (2) | 98 (98) |
| 3 | 1c | CH3CN | 22 (2) | 98 (98) |
| 4 | 2b | CH3CN | 3 (2) | 98 (98) |
| 5 | 1c c | Neat | 2.0 | 90 |
| 6 7 | 2b c no catalyst | neat neat | 2.0 24 | 95 n.r. |
Values in parenthesis refer to neat conditions. a Reaction conditions: silane (1.2 mmol), substrate (1.0 mmol). b Determined by 1H NMR on the basis of cinnamaldehyde consumption (MeOH as internal standard) after hydrolysis of the reaction mixture with 2 M NaOH. c Reaction performed with 1.0 mol % of catalyst.
Table 2.
Hydrosilylation of aldehydes and ketones catalyzed by 2b with PhSiH3a.
| Entry | Substrate | mol % | Time (h) | Conversion (%) b |
|---|---|---|---|---|
| 1 |  | 2.5 1.0 0.1 | 3.0 3.0 6.0 | 99 94 90 * |
| 2 |  | 1.0 | 3.0 | 97 |
| 3 |  | 1.0 | 5.0 | 99 |
| 4 |  | 1.0 2.5 | 19 7.0 | 77 95 * |
| 5 |  | 2.5 | 7.0 | 87 * |
| 6 |  | 2.5 | 41 | 50 |
| 7 |  | 2.5 | 41 | 55 |
| 8 |  | 2.5 | 41 | n. r. |
| 9 |  | 2.5 1.0 | 3.0 3.0 | 99 98 |
| 10 |  | 2.5 | 6.5 | 71 |
| 11 |  | 2.5 | 6.0 | 87 |
| 12 |  | 2.5 | 6.5 | 15 |
| 13 |  | 2.5 | 6.5 | 53 |
| 14 |  | 2.5 | 24 | n.r. |
a Reaction conditions: silane (1.2 mmol), substrate (1.0 mmol). b Determined by 1H NMR on the basis of consumption of substrate (MeOH as internal standard) after hydrolysis of the reaction mixture with 2 M NaOH. All reactions were performed under a nitrogen atmosphere and neat conditions.* isolated yield after purification.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yempally, V.; Shahbaz, A.; Fan, W.Y.; Madrahimov, S.T.; Bengali, A.A. Hydrosilylation of Aldehydes by a Manganese α-Diimine Complex. Inorganics 2020, 8, 61. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8110061
AMA Style
Yempally V, Shahbaz A, Fan WY, Madrahimov ST, Bengali AA. Hydrosilylation of Aldehydes by a Manganese α-Diimine Complex. Inorganics. 2020; 8(11):61. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8110061
Chicago/Turabian StyleYempally, Veeranna, Azal Shahbaz, Wai Yip Fan, Sherzod T. Madrahimov, and Ashfaq A. Bengali. 2020. "Hydrosilylation of Aldehydes by a Manganese α-Diimine Complex" Inorganics 8, no. 11: 61. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8110061
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.