Redox-Active Manganese Pincers for Electrocatalytic CO2 Reduction


Department of Chemistry, University of Colorado Boulder, 215 UCB Boulder, CO 80309-0215, USA
*
Author to whom correspondence should be addressed.
Inorganics 2020, 8(11), 62; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8110062
Submission received: 17 October 2020
/
Revised: 5 November 2020
/
Accepted: 7 November 2020
/
Published: 11 November 2020
(This article belongs to the Special Issue Organomanganese Chemistry)
Abstract
:The decrease of total amount of atmospheric CO2 is an important societal challenge in which CO2 reduction has an important role to play. Electrocatalytic CO2 reduction with homogeneous catalysts is based on highly tunable catalyst design and exploits an abundant C1 source to make valuable products such as fuels and fuel precursors. These methods can also take advantage of renewable electricity as a green reductant. Mn-based catalysts offer these benefits while incorporating a relatively cheap and abundant first-row transition metal. Historically, interest in this field started with Mn(bpy-R)(CO)3X, whose performance matched that of its Re counterparts while achieving substantially lower overpotentials. This review examines an emerging class of homogeneous Mn-based electrocatalysts for CO2 reduction, Mn complexes with meridional tridentate coordination also known as Mn pincers, most of which contain redox-active ligands that enable multi-electron catalysis. Although there are relatively few examples in the literature thus far, these catalysts bring forth new catalytic mechanisms not observed for the well-established Mn(bpy-R)(CO)3X catalysts, and show promising reactivity for future studies.

1. Introduction
CO2 as a global challenge. Increased atmospheric levels of CO2 pose a significant threat to our environment, while continued societal dependence upon fossil fuels leads to ever-increasing concentrations of this greenhouse gas [1,2,3,4]. Addressing these problems is critical to avoid increased ocean acidification and the potentially disastrous consequences of average planetary temperature increases, which include rising sea levels, increased desert formation, and widespread species extinction [5]. Current projections indicate that CO2 emissions must reach net zero by 2070 to prevent exceeding the Paris Agreement benchmark of a 2 °C global temperature increase [4]. Furthermore, recent estimates based upon current emission trajectories place the likely average temperature increase for a doubling of atmospheric CO2 somewhere between 2.3 °C and 4.5 °C, with decreasing likelihood of remaining below 2 °C [6].
Several CO2-based methods of addressing this pressing problem exist, including the reduction of CO2 emissions (decarbonization), the removal of CO2 emissions through carbon capture and sequestration (CCS), and the recycling of emitted CO2 into new products [5]. Decarbonization via the replacement of CO2-emitting technologies with more environmentally benign alternatives will undoubtedly serve an important role in addressing climate change as key technologies such as wind and solar energy gradually replace their traditional fossil fuel-burning predecessors. However, these clean electricity technologies suffer from drawbacks such as intermittent production, meaning that their electrical outputs must be used immediately or lost in the absence of adequate storage technology [7]. Moreover, these technologies fail to address the persistent need for portable, combustible fuels in sectors such as transportation. In contrast, CCS aims to pull emitted CO2 either directly from emitting sources or from the atmosphere, trapping CO2 emissions rather than preventing them. While this strategy could theoretically result in net negative CO2 emissions, economic barriers often hinder large-scale implementation of carbon capture or necessitate its coupling to the further extraction of fossil fuels by using the captured CO2 in enhanced oil recovery (EOR) [8]. This presents a clear obstacle in the face of long-term problems such as continued societal dependence upon fossil fuels.
The recycling of emitted CO2 waste into value-added fuels or fuel precursors has the potential to address many of the above problems, creating a closed cycle from CO2 emissions back to organic fuels while exploiting a potentially valuable and abundant C1 source. Closing this cycle using CO2 as a chemical feedstock theoretically results in net-zero emissions and has the potential to financially incentivize the reduction of emissions, acting as a complement to carbon capture or decarbonization technologies. CO2 reduction (CO2R) can be coupled with carbon capture. Carbon capture and utilization (CCU) is a growing field predicted to use up to 700 million tons of CO2 per year by 2050 [8,9], serving as a complement to CCS.
Electrocatalysis as an answer to the challenge. Although many CO2R catalysts have been investigated, electrocatalytic CO2 reduction in particular presents further benefits. Many chemical CO2 reduction strategies have been explored but often require harsh conditions such as high temperature and pressure, high-energy reductants that are themselves energy-intensive to synthesize, or expensive precious metal catalysts, with relatively few reports of successful base metal-catalyzed reactions [10,11,12,13]. Electrocatalysis can operate under milder conditions while offering increased ability to interface with other emission mitigation strategies. Crucially, electrochemical methods are readily coupled to clean energy sources such as solar, enabling the production of solar fuels that effectively store intermittently generated energy for later use. This serves an important function in service of other renewable energy technologies while ensuring that the desired CO2 reduction is carried out with electrons as a green reductant. While research on the production of other solar fuels such as hydrogen gas also shows promising developments [14], the reduction of CO2 to liquid fuels, or precursors for such fuels, can exploit the existing infrastructure for liquid fossil fuels.
Electrocatalytic figures of merit. There are several figures of merit when comparing electrocatalysts, including overpotential, Faradaic efficiency (FE), selectivity, current density, and stability over time. FE is % measure of how much of the charge delivered to a system was used productively in the synthesis of product. Overpotential is an additional, distinct figure of merit that allows us to establish the necessary operating potential for a given catalyst to achieve a current density, usually defined at 10 mA cm−2. Current density is a metric intrinsically related to overpotential. It is most often the maximal current obtained from a known surface area under a certain set of experimental conditions (i.e., a set potential) and is expressed in units of mA cm−2, relying on estimations of electroactive surface area. Selectivity in the production of solar fuels is a key metric since protons and electrons, two of the ubiquitous reagents in such transformations, can combine to form hydrogen, rather than store hydrogen equivalents within the molecular framework of a storage molecule in the synthesis of a fuel. Faradaic efficiencies and selectivity are highlighted in this review, as they are the most common.
Molecular electrocatalysis for CO2 reduction has been explored extensively due to high tunability through molecular design, often leading to improved selectivity over its heterogeneous counterpart [15,16]. Appropriately designed homogeneous catalysts also avoid the problems of electrode poisoning and the high overpotentials associated with obtaining sufficient current density in heterogeneous electrochemical CO2 reduction [15,16,17]. Molecular catalysts based upon a variety of metals have been assessed for CO2 reduction, particularly from group VII through group X [17]. Although several 4d and 5d metals such as Re [18,19,20], Ru [21,22,23,24,25,26,27], and Ir [28,29,30,31] have been investigated for CO2 reduction with success, first-row transition metals are often preferred to their second- or third-row counterparts due to their higher earth abundance and lower cost. For this reason, established catalysts based upon the third-row group VII metal Re are frequently substituted with analogues using Mn, the third most abundant transition metal in the Earth’s crust [32]. Unlike group-based catalyst families using later transition metals, group VII catalysts frequently exhibit CO2R activity regardless of whether the metal center is Mn or Re [17]. This offers valuable opportunities for comparison and insight.
Tridentate ligands that coordinate in a mer fashion, or pincers, have also resulted in a variety of complexes with demonstrated activity for CO2 reduction [33]. Because of the potential for high selectivity and activity while maintaining lower costs, the present review focuses specifically on the current state of research on homogeneous electrocatalytic Mn pincer complexes for CO2 reduction. This specific class of catalysts is of interest because they incorporate tridentate redox non-innocent ligands that can act as electron reservoirs, improve catalyst stability, and often result in mechanisms that diverge from more commonly reported bidentate bpy-based Mn systems.
2. Mn-Based Electrocatalysts in CO2 Reduction
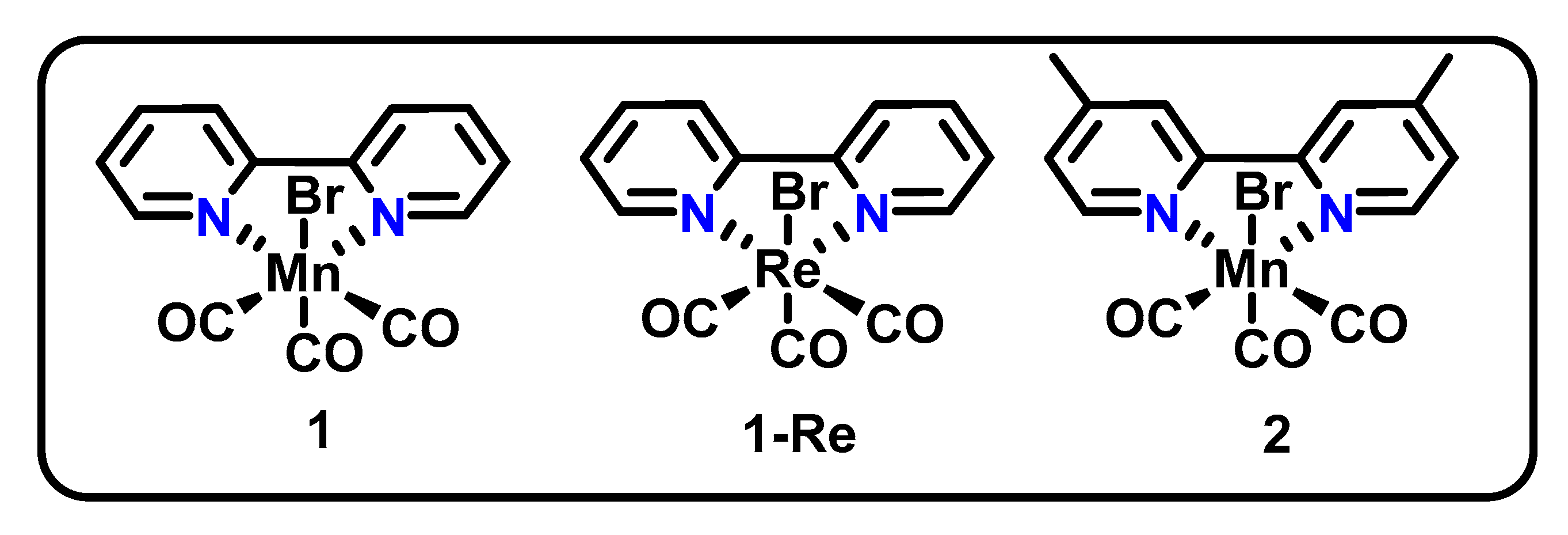
The first reported Mn-based CO2R catalysts were fac-Mn(bpy-R)(CO)3Br (bpy-R = 2,2′-bipyridine (bpy) (1) or 4,4′-dimethyl-2,2′-bipyridine (dmbpy (2), Figure 1) [34], structural analogues of a Re-based catalyst originally reported in 1984 [35]. Synthesis of 1 was initially reported in 1979 [36], but the complex was not studied for CO2R catalysis until 2011 [34]. The fac-M(bpy-R)(CO3)X catalysts therefore established promising structural motifs for CO2R catalysis, inspiring many further reports that decorated the parent bpy ligand to modulate electronic or steric effects of the bidentate ligand, or substituted other X-type ligands for bromide [19,20,37,38,39]. A key element of these catalysts is the use of redox-active bpy ligands, enabling the storage of an additional e- equivalent within the ligand framework. This surpasses the typical one-electron capacity common to abundant first-row transition metals in the +1 state. This enables multielectron catalysis by the complex [40], a necessary feature for CO2R reactions that require a minimum of two electrons. This catalytic design is a strategy commonly employed in reaction development, conferring “nobility” to the first-row metals [40,41]. Catalysts of this class most often produce CO with high FE for the reaction shown in Equation (1),
while other catalysts may exhibit products more consistent with reactions shown in Equations (2)–(4),
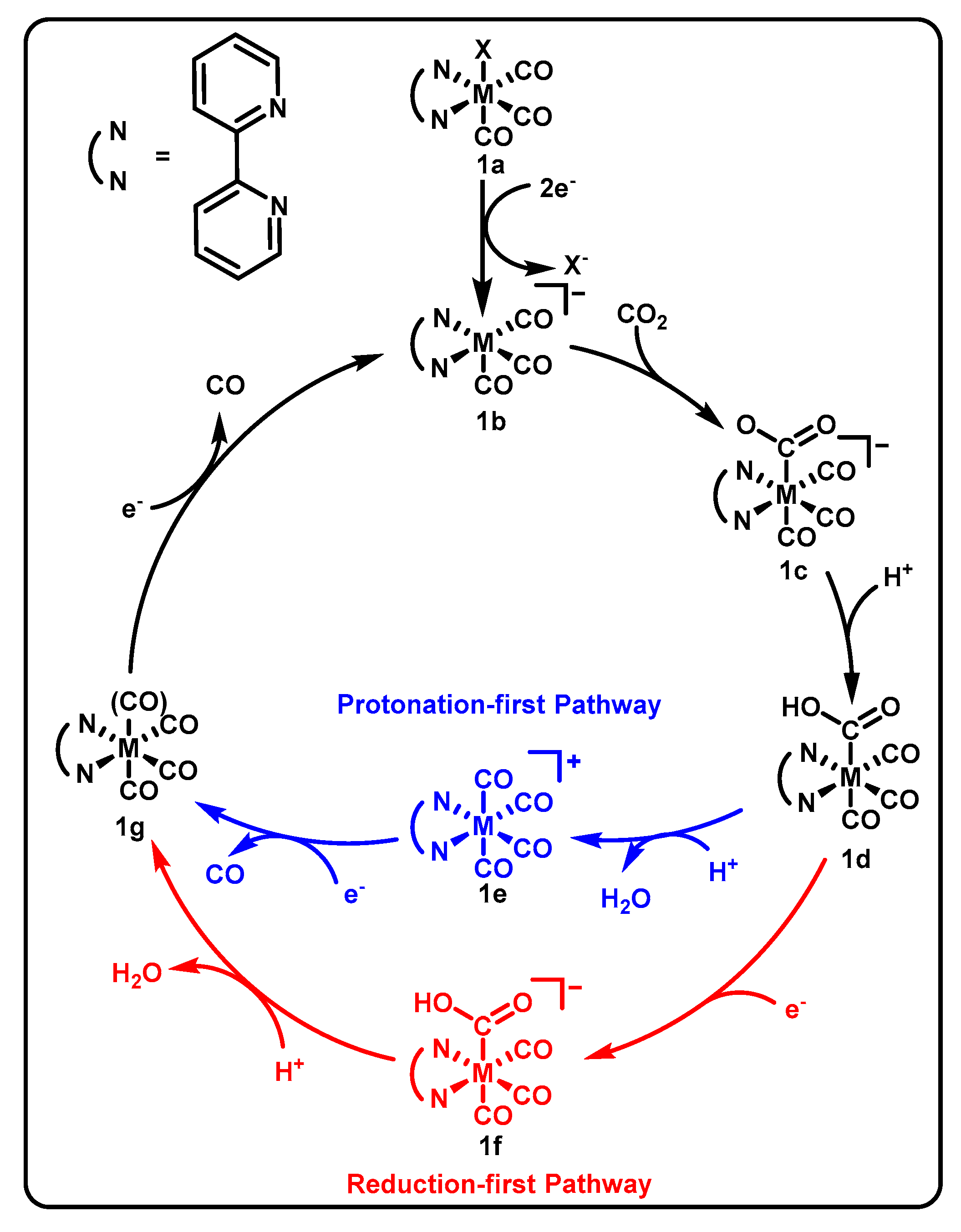
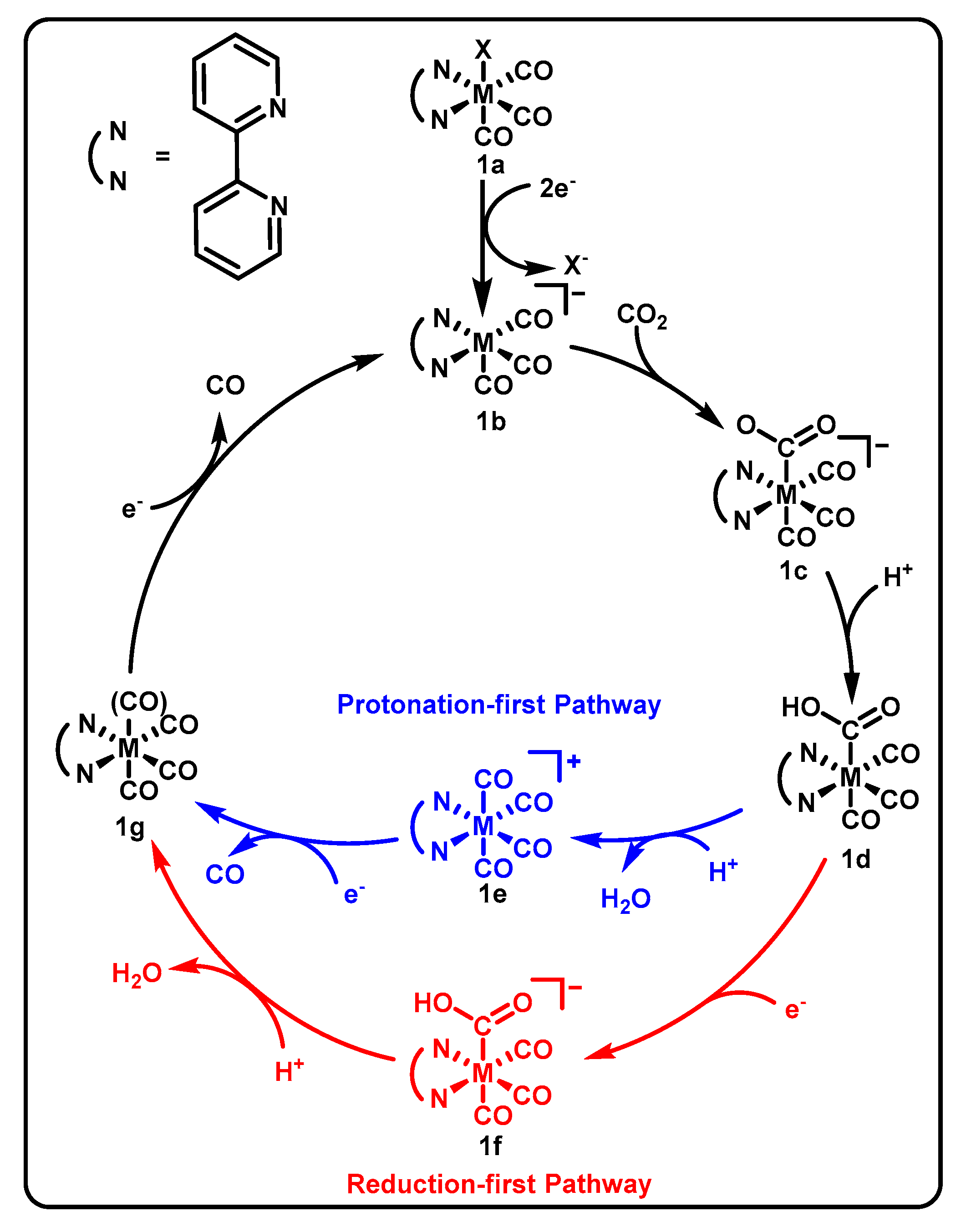
This established class of Mn-based electrocatalysts has been studied in depth, yielding mechanistic insights as depicted in Scheme 1. Entrance into the catalytic cycle is proposed to involve reduction by two electrons and X-ligand loss, after which CO2 substrate may bind to the metal center and become protonated [42]. After this stage, the mechanism diverges into two possible paths, i.e., the reduction-first pathway (in red, proceeding through structure f in Scheme 1) or the protonation-first pathway (in blue, proceeding through structure e in Scheme 1), the latter of which may occur at more positive potentials due to increased favorability of reducing a cationic species [17].
Catalyst 2 (Figure 1) was found to operate at potentials 400 mV positive of its Re analogue in electrochemical experiments in 95/5 MeCN/H2O and 0.1 M tetrabutylammonium perchlorate (TBAP) electrolyte, with comparable FE and selectivity. This is thought to be due to a sequence of steps upon entrance into the catalytic cycle (the conversion from 1a to 1b in Scheme 1) [42]. The Re catalyst 1-Re requires the addition of a second equivalent of electrons prior to the dissociation of X− due to the increased stability of the singly reduced [Re(bpy)(CO)3X]•− species, whereas the Mn version of the catalyst 1 more readily gives up X− after only a single reduction to form either [Mn(bpy)(CO)3]0 or a dimerized form of the catalyst. This facilitates a second reduction at a more positive potential to yield 1b. However, the Mn complex exhibits lower current densities and requires the use of a sacrificial proton source, unlike its Re counterpart 1-Re [34,35], which was shown to most likely operate via the reduction-first pathway (Scheme 1, red). The reduction-first mechanistic pathway diverges from the protonation-first one by the acceptance of an additional reducing equivalent by 1d, leading to the formation of 1f instead of protonation leading to 1e. The reduction step of the reduction-first pathway 1d→1f is followed by a protonation/dehydration step to lead to the formation of 1g, which in the case of Re retains all four carbonyl ligands. The Mn derivative is likely to lose one CO ligand to reach a 5-coordiante 1g after the 1e→1g step. The active catalyst 1b is regenerated after an additional reduction, which in the case of Re occurs with loss of a CO ligand.
Nonetheless, installation of various substituents on the bpy ligand of the Mn-based catalyst have been shown to minimize overpotential by sterically preventing dimerization [38] or promoting the protonation-first pathway through use of hydrogen-bonding groups [39] (Scheme 1 blue), or to improve catalytic rate by increasing electron donation into the metal center [37].
With a promising background for Mn-based electrocatalysis established, the present review discusses budding research developments in Mn pincer complexes, an emerging class of Mn-based CO2R electrocatalysts that illustrate novel reactivity motifs in the synthesis of solar fuels. Similarly to the M(bpy)(CO)3X catalysts, this class of complexes employs polydentate redox-active ligands for use as electron reservoirs that can enable multi-electron catalyses at metal centers that would normally be limited to one-electron-steps. Metals in the 1+ oxidation state of group VII are examples of such species as they would normally be able to accept only one electron equivalent before reaching their M0 state. While a simple 2,2′ bipyridine can act as an electron reservoir, the expansion of the ligand from a simple bidentate bpy derivative to a larger, conjugated tridentate pincer can additionally leverage the chelate effect to improve chemical and thermal stability of the complex and strength of the ligand-metal binding, provide additional opportunities to tune electron donation into the metal center, and enable novel mechanistic avenues for product formation [43].
3. Pincers in Mn CO2 Reduction Electrocatalysts
Mn pincer complexes based upon a variety of ligand frameworks are discussed herein and organized by coordination type. Table 1 presents a summary of the key examples discussed below to allow for ease of comparison between them as well as the previously discussed benchmark Mn(bpy-R)(CO)3X catalysts. For each catalyst type, a brief discussion of analogous complexes based upon other metals centers is offered to examine the relationship between metal center identity and activity. The abbreviation of the pincer type is based on the elements in contact with the metal center- for example, 3b is an ONN pincer with O, N and N being the elements of the pincer ligand that are attached to the metal.
3.1. ONN Pincer Binding in Mn(bpy-R)(CO)3X Catalysts
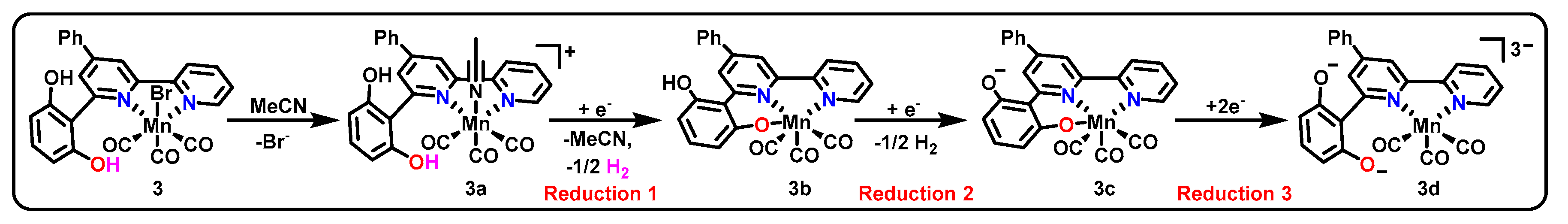
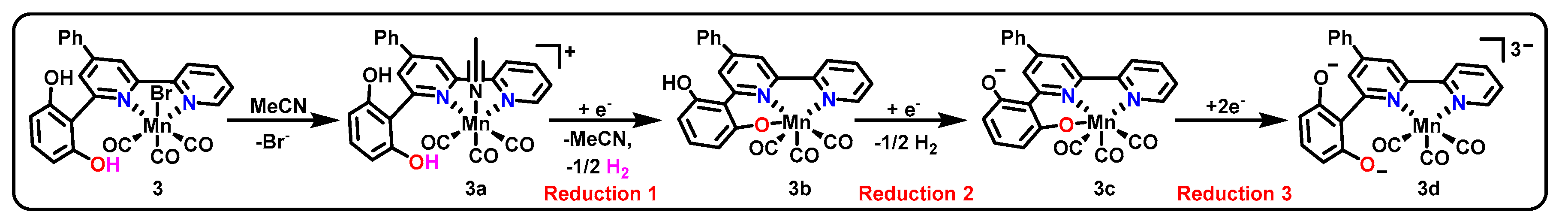
Among the many examples of Mn(bpy-R)(CO)3X catalysts reported, one example from 2014 (3, Scheme 2 and Table 1) that was initially believed to behave in a similar fashion to other catalysts of the class was later found to incorporate pincer intermediates [44,45]. Catalyst 3 was found to produce CO with 70% FE and formate/formic acid with 22% FE with the ligand as the sole proton source under an applied bias of −1.8 V vs. Fc/Fc+. This performance, yielding a turnover frequency (TOF) of 1.4 s−1 and turnover number (TON) of 19 for CO production, compared favorably with the original Mn(bpy)(CO)3Br catalyst. Cyclic voltammetry (CV) is an electrochemical technique that allows us to diagnose potentials necessary for the introduction of reducing equivalents into the molecular frameworks of catalysts. Such data showed that complex 3 underwent three reductions at −1.2 V vs. saturated calomel electrode (SCE), −1.51 V vs. SCE, and −1.66 V vs. SCE. Both the second and third reductive features showed gradual attenuation of current at higher scan rates, indicating the existence of a chemical step preceding each of the two reductions [44]. Although the chemical step preceding the second reduction was initially presumed to be dimerization (as expected for Mn(bpy-R)(CO)3X catalysts), later studies using IR and UV/VIS spectroelectrochemistry (SEC) illustrate that this was not the case [45].
Scheme 2 presents the proposed pathway for catalyst activation for entrance into the catalytic cycle [45]. Although this example is based upon a bidentate pre-catalyst and bidentate active catalytic state, the ability of the phenoxide group to coordinate into the metal center is proposed to play a significant role in two intermediates, namely 3b and 3c, and may be responsible for the notable mechanistic differences between this catalyst and the canonical Mn(bpy-R)(CO)3X catalysts. The ability of the ligand to become tridentate and donate an additional two electrons to the metal may provide an alternative route toward a stable 18-electron count for the complex, thus no longer favoring formation of a Mn–Mn bond.
Additionally, the ligand itself can act as a proton source for catalysis, explaining the observation of formate production in the absence of added proton source [44]. In the presence of an external proton source, the products of catalysis for the complex were also found to be variable depending upon the proton source acidity [45]. While added water resulted in highly selective CO formation (90%), increasing acidity of the added proton source was found to increase the ratio of formate to CO. By changing the proton source from water to phenol, the CO and formate FE were changed from 90% and 4% to 15% and 39%, respectively, with the intermediate-acidity 3,3,3-trifluoroethanol (TFE) giving intermediate values of 48% and 36%, respectively. Efficiency for hydrogen production increases concomitantly with increasing efficiency for formate production, suggesting water as the best proton source for obtaining single-product selectivity as well as the highest overall efficiency.
Despite intriguing mechanistic insights and good FE for CO production under optimized conditions, 3 still lacks optimal catalyst stability. Bulk electrolysis of 3 shows an appreciable decrease in current, likely due to catalyst deactivation, within an hour of initiating electrolysis at −1.8 V vs. SCE in dry MeCN/tetrabutylammonium hexafluorophosphate (TBA PF6) solution [44]. Furthermore, data for turnovers in excess of 28 for any given CO2R product have not been reported for the catalyst [45]. While this work presents an interesting contrast to other Mn(bpy-R)-based systems, there is still further work to be done in examining routes to stable complexes that employ this framework.
3.2. NNN and tpy-Based Pincers
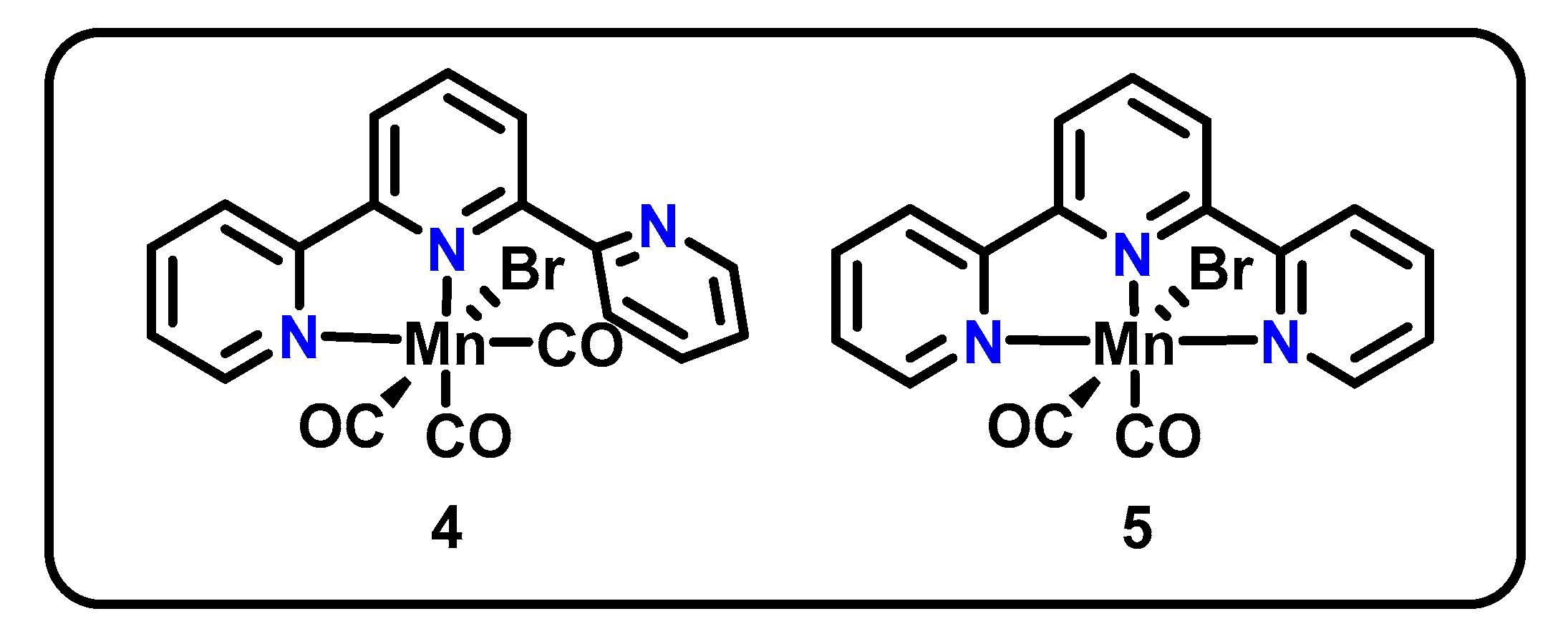
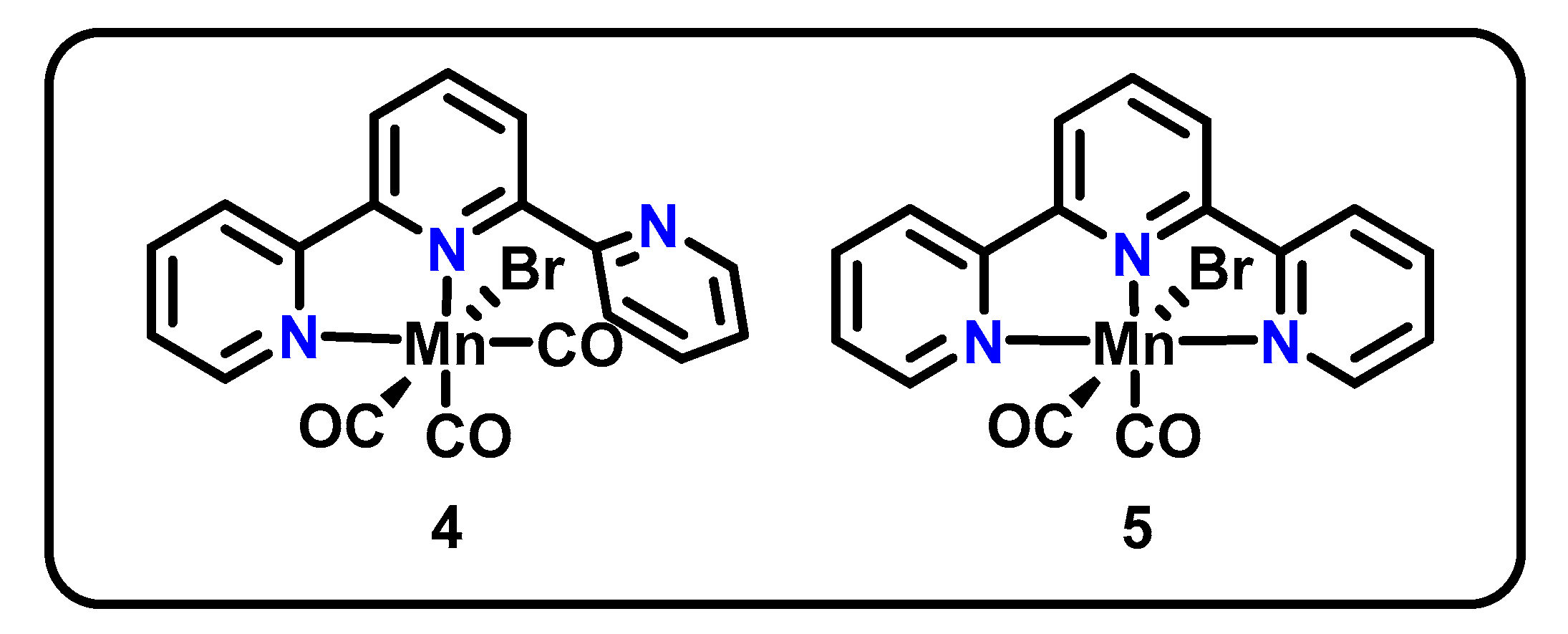
Another direct descendant of the Mn(bpy-R)(CO)3X family is the Mn(tpy) pincer. Extending the classic bpy structure to contain a third binding site yields 2,2′:6′,2′′-terpyridine (tpy), which has been shown to be a good ligand for CO2R catalysts of both Mn and Re. Building on an original report by Chardon-Noblat in 2014 [46], Machan and Kubiak studied CO2R activity of two such catalysts in 2016, namely Mn(κ2-tpy)(CO)3Br (4) and Mn(κ3-tpy)(CO)2Br (5) (Figure 2) [47].
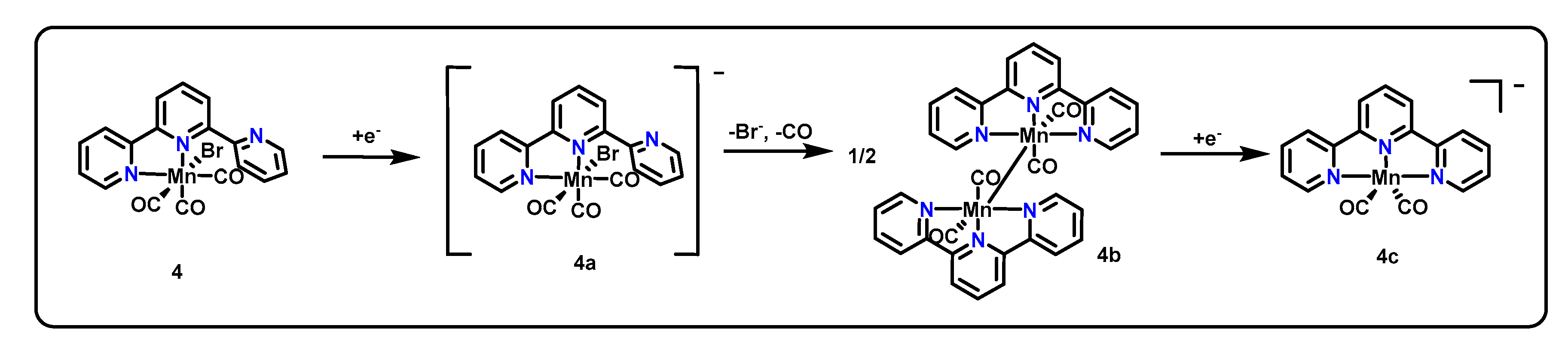
Using CV, complex 4 shows reductive features at −1.56 V and −1.77 V vs. Fc/Fc+ and irreversible oxidation features on the return sweep at −1.57 V and −0.99 V vs. Fc/Fc+. Faster scan rates revealed an additional feature at −1.63 V vs. Fc/Fc+. The CV response of 5 was remarkably similar, with irreversible reduction features at −1.56 V and −1.75 V and irreversible oxidation features at −1.57 V and −0.95 V vs. Fc/Fc+. Fast scan rates again revealed an additional irreversible feature at −1.69 V vs. Fc/Fc+. For each complex, the second irreversible reductions (−1.77 V for 4 and −1.75 V for 5) and second irreversible oxidations (1–0.99 V; 2–0.95 V vs. Fc/Fc+) were attributed to the reduction and oxidation (respectively) of a dimerized species (4b) presumably via 4a, as shown in Scheme 3, using evidence from chemical reduction experiments and IR spectroelectrochemistry (SEC). From a scan rate analysis, the formation of this dimer was concluded to occur on a similar timescale to that of the CV experiment, a much slower timescale than that observed for catalysts with simple bidentate bpy ligands. Therefore, both 4 and 5 were found to access the same dimerized intermediate and active catalyst state 4c as shown for 4 in Scheme 3. The authors conclude that the bidentate pre-catalyst 4 therefore can perform CO2R using a κ3 pincer-type coordination mode.
The dimerization, along with an observed first order in CO2, suggest a large number of similarities between the mechanism of CO2R for these complexes and that of the original Mn(bpy-R)(CO)3X catalysts [47]. However, the notably slower dimerization of this system allows for competing pathways between dimer formation and CO2 binding, enabling access to lower overpotentials. The success of the catalysts was further evidenced through 129% FE for CO for 4 (value exceeds 100% due to loss of a CO equivalent from pre-catalyst) and 93% FE for CO for 5. However, these values were reflective of bulk electrolyses conducted through only the first 4.1 turnovers of the catalyst. Thereafter, catalyst degradation was observed, lowering overall FE and again pointing to stability issues with the catalyst.
In addition to group VII metals, terpy pincers of Ru have also been reported for CO2R catalysis and have even been shown to produce C2 products when cooled to −20 °C [21,51]. Narayanan et al. also reported an additional NNN-type pincer complex of Ni for CO2 reduction with aldimine binding rather than additional pyridyl side wings [52].
3.3. PNP Pincers
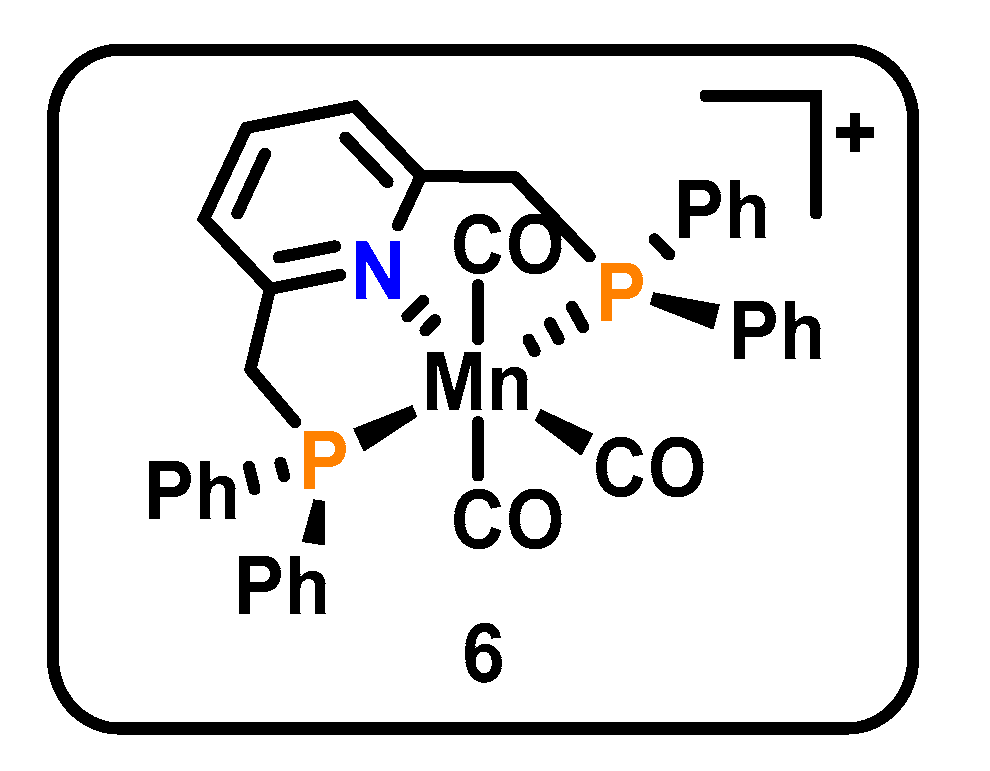
In a larger departure from the classical bpy-based catalysts, Rao et al. reported a CO2R catalyst with an Mn center with PNP coordination to a pincer ligand (6, Figure 3) [48]. This catalyst produces both CO and carbonate, and interestingly is inhibited by H2O addition, a clear contrast between this catalyst and its Mn(bpy)(CO)3Br predecessor. While few mechanistic insights were offered, the catalyst CVs reveal similar overpotential requirements to Mn(bpy)(CO)3Br, with a catalytic onset of approximately −2.0 V vs. Fc/Fc+. Similarly, the catalyst yielded excellent FE for CO in the absence of water (96% over 125 min). Despite unknowns, this system operates via a clearly different mechanism other than previous Mn-based CO2R examples. Catalyst 6 highlights the potentially significant contributions of pincer frameworks to the field in discovering alternate routes to CO2 reduction.
3.4. CNC Pincers
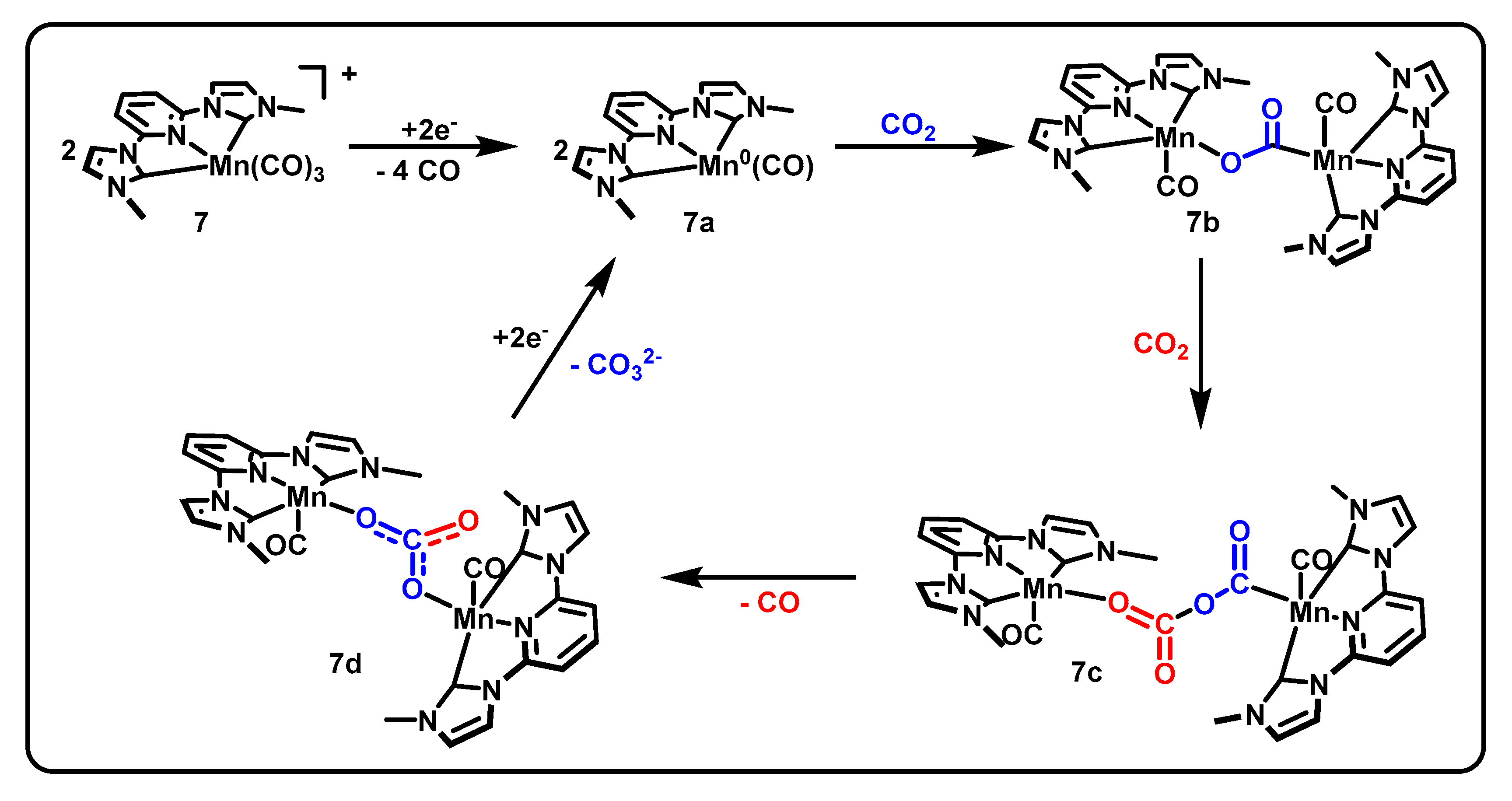
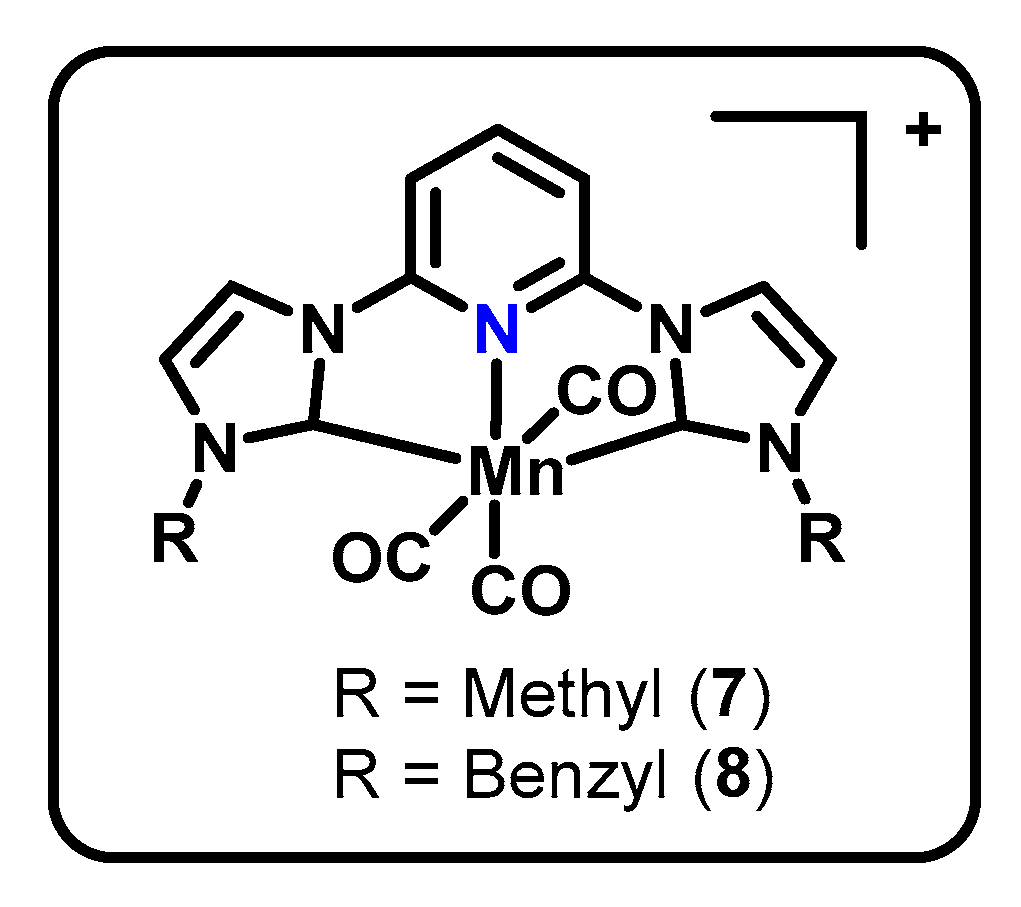
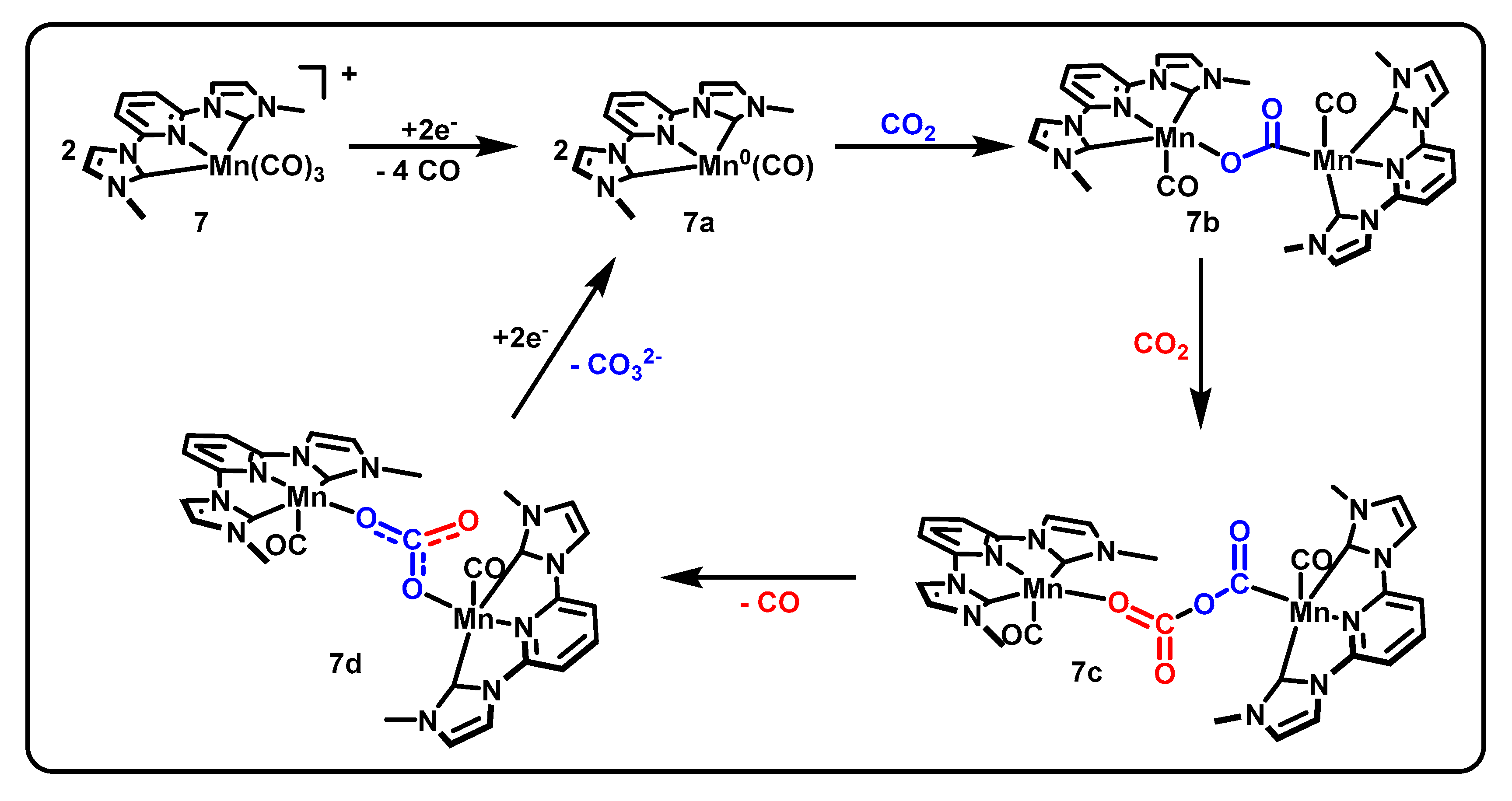
Pincers incorporating metal–carbon bonds from N-heterocyclic carbenes (NHCs) have generated interest due to the strong sigma donation available from NHCs, opening the possibility for increased electron density on the metal center and thus increased reactivity for CO2 reduction. In 2018, the following two CNC manganese catalysts using NHC frameworks to promote CO2R were reported: 7 [MnCNCMe(CO)3]Br and 8 [MnCNCBn(CO)3]Br (Figure 4) [49]. The activity of these catalysts diverges substantially from the proposed mechanisms of the benchmark Mn(bpy-R)(CO)3X, resulting in protonless operation and production of carbonate, CO32− [50], as shown in Scheme 4. The molecular structure of 7 from X-ray crystallography indicated the expected octahedral coordination and outer-sphere bromide. A geometric distortion of the pyridine ring was also observed, suggesting a delocalized molecular orbital and the ligand acting as an electron reservoir.
CV experiments demonstrated activity under an atmosphere of CO2 both with and without a proton source present. Current densities up to 16 mA/cm2 are obtained in the absence of protons. Compared to activity under inert atmosphere, 8 [MnCNCMe(CO)3]Br and 7 [MnCNCBn(CO)3]Br displayed 18- and 21-fold current increases under CO2, respectively. Turnover was shown to occur with 1e- per manganese center, based on results from the combined techniques of diffusion ordered spectroscopy (DOSY) and normal pulse voltammetry (NPV) as described by Donadt et al. [54].
Initial controlled potential electrolysis experiments at −2.3 V vs. Fc/Fc+ determined an FE of 87% ± 3% and 86% ± 4%, respectively, for the methyl- and benzyl-substituted complexes and further product analysis by derivatization and GC–MS–EI found carbonate to be the second product of the protonless reaction with the above 95% current efficiencies. Further CV experiments determined a second order in CO2 by using the resulting currents from varying CO2 concentrations as relative rates and a half-order in catalyst by varying catalyst concentration. The latter determination was the first example of the Burés normalized timescale method [50] being used to determine the order in catalyst from voltammetry data. The proposed mechanism (Scheme 4) begins with the reduction of two equivalents of the metal complex 7 followed by the loss of two CO each to form a square planar Mn0 species 7a. One equivalent of CO2 is then bound between two manganese centers to form 7b before a second CO2 inserts into the complex to arrive at 7c. A final rearrangement produces a bimetallic carbonate complex 7d, which then releases the carbonate and regenerates the square planar Mn0 species 7a.
Other CNC ligand frameworks have been investigated for CO2 reduction. A Re analogue, [ReCNCBn(CO)3]Cl, was synthesized and investigated but, unlike other Re analogues, showed little catalytic response to CO2 [55]. This behavior is unlike previous studied group VII catalysts, as discussed above. Density Functional Theory (DFT) calculations showed that between the reduced forms (7a for Mn) of the two complexes, the Mn complex demonstrates increased metalloradical character (66%) while the Re has a much lower radical character on the metal (38%), suggesting that the reduced Re complex may not be sufficiently nucleophilic for CO2 reduction [50]. A second explanation for this observation would be that the CO ligands are too strongly bound to allow coordination of a new CO2 ligand. Calculations also showed that the Mn complex has unique access to a square planar intermediate with a single bound CO that is not accessible to the Re complex, possibly due to differences in orbital populations upon reduction.
Group X metals, however, have shown some precedence for successful CO2R by CNC pincer complexes having square planar or pseudo-square planar geometries [33,56,57,58]. While many early examples based on Pd exhibit low efficiencies for CO2 reduction, instead favoring hydrogen evolution [56,57,58], progress has been made with current PdCNC pincer complexes exceeding FE of 50% for CO [59].
4. Conclusions
The use of pincer-type ligands for Mn-based CO2R electrocatalysts presents a promising future direction for CO2R catalysis. Current vignettes of CO2R have been reported with CNC, NNN, and PNP ligand frameworks, along with an interesting report of ONN intermediate pincer binding. These complexes incorporate the relatively abundant and low-cost Mn in place of expensive and rare noble metals while frequently exhibiting high catalytic rates and good selectivity for CO2R under optimized conditions. The use of pincer-type ligands enables novel mechanistic pathways that diverge from those of the oft-discussed Mn(bpy)(CO)3X catalysts whose performance serves as a benchmark for other Mn-based catalysts.
Despite these advances and promising preliminary results, there is still much work to be performed to achieve improved overpotentials and catalyst stability. Many of the catalysts discussed herein are sensitive to water or oxygen or do not have extensive data reported regarding their long-term stability. While FE for the reported bulk electrolysis experiments shows excellent CO2 conversion, these values are indicative of short timescales on the order of single hours and catalyst turnovers lying only in the double digits. Future work will likely focus on identifying the limits of catalyst stability and improve this metric to make the catalysts more industrially relevant.
Funding
The authors would like to thank the University of Colorado for the generous funding of this research.
Acknowledgments
T.M. would like to thank the Department of Chemistry at CU Boulder for the Sewall Fellowship.
Conflicts of Interest
The authors declare no conflict of interest.
References
- IPCC. Climate Change 2014: Synthesis Report; Intergovernmental Panel on Climate Change: Geneva, Switzerland, 2014.
- Cox, P.M.; Betts, R.A.; Jones, C.D.; Spall, S.A.; Totterdell, I.J. Acceleration of global warming due to carbon-cycle feedbacks in a coupled climate model. Nature 2000, 408, 184–187. [Google Scholar] [CrossRef]
- Solomon, S.; Plattner, G.-K.; Knutti, R.; Friedlingstein, P. Irreversible climate change due to carbon dioxide emissions. Proc. Natl. Acad. Sci. USA 2009, 106, 1704–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IPCC. Global Warming of 1.5 °C; WHO: Geneva, Switzerland, 2018.
- Mikkelsen, M.; Jørgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Sherwood, S.; Webb, M.J.; Annan, J.D.; Armour, K.C.; Forster, P.M.; Hargreaves, J.C.; Hegerl, G.; Klein, S.A.; Marvel, K.D.; Rohling, E.J.; et al. An assessment of Earth’s climate sensitivity using multiple lines of evidence. Rev. Geophys. 2020, 58, e2019RG000678. [Google Scholar] [CrossRef] [PubMed]
- Whittingham, M.S. History, Evolution, and Future Status of Energy Storage. Proc. IEEE 2012, 100, 1518–1534. [Google Scholar] [CrossRef]
- Global CCS Institute. The Global Status of CCS: 2019; Global Carbon Capture and Storage Institute Ltd: Melbourne, Australia, 2019. [Google Scholar]
- Mac Dowell, N.; Fennell, P.S.; Shah, N.; Maitland, G.C. The role of CO2 capture and utilization in mitigating climate change. Nat. Clim. Chang. 2017, 7, 243–249. [Google Scholar] [CrossRef] [Green Version]
- Kar, S.; Goeppert, A.; Kothandaraman, J.; Prakash, G.K.S. Manganese-Catalyzed Sequential Hydrogenation of CO2 to Methanol via Formamide. ACS Catal. 2017, 7, 6347–6351. [Google Scholar] [CrossRef]
- Schneidewind, J.; Adam, R.; Baumann, W.; Jackstell, R.; Beller, M. Low-Temperature Hydrogenation of Carbon Dioxide to Methanol with a Homogeneous Cobalt Catalyst. Angew. Chem. Int. Ed. 2017, 56, 1890–1893. [Google Scholar] [CrossRef]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kuhl, S.; Havecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.L.; et al. The Active Site of Methanol Synthesis over Cu/ZnO/Al2O3 Industrial Catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef]
- Yang, X.; Chen, X. Chapter 5—Mechanistic Insights and Computational Prediction of Base Metal Pincer Complexes for Catalytic Hydrogenation and Dehydrogenation Reactions. In Pincer Compounds; Morales-Morales, D., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 101–110. [Google Scholar]
- Gao, S.; Fan, W.; Liu, Y.; Jiang, D.; Duan, Q. Artificial water-soluble systems inspired by [FeFe]-hydrogenases for electro- and photocatalytic hydrogen production. Int. J. Hydrog. Energy 2020, 45, 4305–4327. [Google Scholar] [CrossRef]
- Peterson, A.A.; Nørskov, J.K. Activity Descriptors for CO2 Electroreduction to Methane on Transition-Metal Catalysts. J. Phys. Chem. Lett. 2012, 3, 251–258. [Google Scholar] [CrossRef]
- Nam, D.-H.; De Luna, P.; Rosas-Hernández, A.; Thevenon, A.; Li, F.; Agapie, T.; Peters, J.C.; Shekhah, O.; Eddaoudi, M.; Sargent, E.H. Molecular enhancement of heterogeneous CO2 reduction. Nat. Mater. 2020, 19, 266–276. [Google Scholar] [PubMed]
- Jiang, C.; Nichols, A.W.; Machan, C.W. A look at periodic trends in d-block molecular electrocatalysts for CO2 reduction. Dalton Trans. 2019, 48, 9454–9468. [Google Scholar] [CrossRef] [PubMed]
- Smieja, J.M.; Kubiak, C.P. Re(bipy-tBu) (CO)3Cl−improved Catalytic Activity for Reduction of Carbon Dioxide: IR-Spectroelectrochemical and Mechanistic Studies. Inorg. Chem. 2010, 49, 9283–9289. [Google Scholar] [CrossRef]
- Sung, S.; Kumar, D.; Gil-Sepulcre, M.; Nippe, M. Electrocatalytic CO2 Reduction by Imidazolium-Functionalized Molecular Catalysts. J. Am. Chem. Soc. 2017, 139, 13993–13996. [Google Scholar] [CrossRef]
- Clark, M.L.; Cheung, P.L.; Lessio, M.; Carter, E.A.; Kubiak, C.P. Kinetic and Mechanistic Effects of Bipyridine (bpy) Substituent, Labile Ligand, and Brønsted Acid on Electrocatalytic CO2 Reduction by Re(bpy) Complexes. ACS Catal. 2018, 8, 2021–2029. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, C.; Weinberg, D.R.; Kang, P.; Concepcion, J.J.; Harrison, D.P.; Brookhart, M.S.; Meyer, T.J. Electrocatalytic reduction of CO2 to CO by polypyridyl ruthenium complexes. Chem. Commun. 2011, 47, 12607–12609. [Google Scholar] [CrossRef]
- Machan, C.W.; Sampson, M.D.; Kubiak, C.P. A Molecular Ruthenium Electrocatalyst for the Reduction of Carbon Dioxide to CO and Formate. J. Am. Chem. Soc. 2015, 137, 8564–8571. [Google Scholar] [CrossRef]
- Johnson, B.A.; Maji, S.; Agarwala, H.; White, T.A.; Mijangos, E.; Ott, S. Activating a Low Overpotential CO2 Reduction Mechanism by a Strategic Ligand Modification on a Ruthenium Polypyridyl Catalyst. Angew. Chem. Int. Ed. Engl. 2016, 55, 1825–1829. [Google Scholar] [CrossRef]
- Chen, Z.; Kang, P.; Zhang, M.-T.; Meyer, T.J. Making syngas electrocatalytically using a polypyridyl ruthenium catalyst. Chem. Commun. 2014, 50, 335–337. [Google Scholar] [CrossRef]
- Chen, Z.; Concepcion, J.J.; Brennaman, M.K.; Kang, P.; Norris, M.R.; Hoertz, P.G.; Meyer, T.J. Splitting CO2 into CO and O2 by a single catalyst. Proc. Natl. Acad. Sci. USA 2012, 109, 15606–15611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daryanavard, M.; Hadadzadeh, H.; Weil, M.; Farrokhpour, H. Electrocatalytic reduction of CO2 to CO in the presence of a mononuclear polypyridyl ruthenium(II) complex. J. CO2 Util. 2017, 17, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Lilio, A.M. Bi-Functional Molecular Catalysts for CO2 Reduction; UC San Diego: San Diego, CA, USA, 2015. [Google Scholar]
- Hu, G.; Jiang, J.J.; Kelly, H.R.; Matula, A.J.; Wu, Y.; Romano, N.; Mercado, B.Q.; Wang, H.; Batista, V.S.; Crabtree, R.H.; et al. Surprisingly big linker-dependence of activity and selectivity in CO2 reduction by an iridium(I) pincer complex. Chem. Commun. 2020, 56, 9126–9129. [Google Scholar] [CrossRef] [PubMed]
- Kang, P.; Cheng, C.; Chen, Z.; Schauer, C.K.; Meyer, T.J.; Brookhart, M. Selective Electrocatalytic Reduction of CO2 to Formate by Water-Stable Iridium Dihydride Pincer Complexes. J. Am. Chem. Soc. 2012, 134, 5500–5503. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Sun, C.; Sun, N.; Meng, L.; Chen, D. Theoretical mechanism studies on the electrocatalytic reduction of CO2 to formate by water-stable iridium dihydride pincer complex. Dalton Trans. 2013, 42, 5755. [Google Scholar] [CrossRef] [PubMed]
- Feller, M.; Gellrich, U.; Anaby, A.; Diskin-Posner, Y.; Milstein, D. Reductive Cleavage of CO2 by Metal–Ligand-Cooperation Mediated by an Iridium Pincer Complex. J. Am. Chem. Soc. 2016, 138, 6445–6454. [Google Scholar] [CrossRef] [PubMed]
- Weast, R.C. CRC Handbook of Chemistry and Physics; CRC Press: Boca Raton, FL, USA, 1988. [Google Scholar]
- Eberhardt, N.A.; Guan, H. Reduction of CO2 Mediated or Catalyzed by Pincer Complexes; Elsevier: Amsterdam, The Netherlands, 2018; pp. 67–99. [Google Scholar]
- Bourrez, M.; Molton, F.; Chardon-Noblat, S.; Deronzier, A. [Mn(bipyridyl)(CO)3Br]: An Abundant Metal Carbonyl Complex as Efficient Electrocatalyst for CO2 Reduction. Angew. Chem. Int. Ed. 2011, 50, 9903–9906. [Google Scholar] [CrossRef]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Electrocatalytic reduction of carbon dioxide mediated by Re(bipy)(CO)3Cl (bipy = 2,2′-bipyridine). J. Chem. Soc. Chem. Commun. 1984, 328–330. [Google Scholar] [CrossRef]
- Staal, L.H.; Oskam, A.; Vrieze, K. The syntheses and coordination properties of M(CO)3X(DAB) (M = Mn, Re; X = Cl, Br, I.; DAB = 1,4-diazabutadiene). J. Organomet. Chem. 1979, 170, 235–245. [Google Scholar] [CrossRef]
- Smieja, J.M.; Sampson, M.D.; Grice, K.A.; Benson, E.E.; Froehlich, J.D.; Kubiak, C.P. Manganese as a Substitute for Rhenium in CO2 Reduction Catalysts: The Importance of Acids. Inorg. Chem. 2013, 52, 2484–2491. [Google Scholar] [CrossRef]
- Sampson, M.D.; Kubiak, C.P. Manganese Electrocatalysts with Bulky Bipyridine Ligands: Utilizing Lewis Acids to Promote Carbon Dioxide Reduction at Low Overpotentials. J. Am. Chem. Soc. 2016, 138, 1386–1393. [Google Scholar] [CrossRef]
- Ngo, K.T.; McKinnon, M.; Mahanti, B.; Narayanan, R.; Grills, D.C.; Ertem, M.Z.; Rochford, J. Turning on the Protonation-First Pathway for Electrocatalytic CO2 Reduction by Manganese Bipyridyl Tricarbonyl Complexes. J. Am. Chem. Soc. 2017, 139, 2604–2618. [Google Scholar] [CrossRef]
- Luca, O.R.; Crabtree, R.H. Redox-active ligands in catalysis. Chem. Soc. Rev. 2013, 42, 1440–1459. [Google Scholar] [CrossRef]
- Chirik, P.J.; Wieghardt, K. Radical Ligands Confer Nobility on Base-Metal Catalysts. Science 2010, 327, 794–795. [Google Scholar] [CrossRef] [PubMed]
- Riplinger, C.; Sampson, M.D.; Ritzmann, A.M.; Kubiak, C.P.; Carter, E.A. Mechanistic Contrasts between Manganese and Rhenium Bipyridine Electrocatalysts for the Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2014, 136, 16285–16298. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.A.W.; Green, K.-A.; Nelson, P.N.; Lorraine, S.C. Review: Pincer ligands—Tunable, versatile and applicable. Polyhedron 2018, 143, 11–27. [Google Scholar] [CrossRef]
- Franco, F.; Cometto, C.; Ferrero Vallana, F.; Sordello, F.; Priola, E.; Minero, C.; Nervi, C.; Gobetto, R. A local proton source in a [Mn(bpy-R)(CO)3Br]-type redox catalyst enables CO2 reduction even in the absence of Brønsted acids. Chem. Commun. 2014, 50, 14670–14673. [Google Scholar] [CrossRef] [PubMed]
- Franco, F.; Cometto, C.; Nencini, L.; Barolo, C.; Sordello, F.; Minero, C.; Fiedler, J.; Robert, M.; Gobetto, R.; Nervi, C. Local Proton Source in Electrocatalytic CO2 Reduction with [Mn(bpy-R)(CO)3Br] Complexes. Chem. A Eur. J. 2017, 23, 4782–4793. [Google Scholar] [CrossRef] [Green Version]
- Compain, J.-D.; Bourrez, M.; Haukka, M.; Deronzier, A.; Chardon-Noblat, S. Manganese carbonyl terpyridyl complexes: Their synthesis, characterization and potential application as CO-release molecules. Chem. Commun. 2014, 50, 2539–2542. [Google Scholar] [CrossRef]
- Machan, C.W.; Kubiak, C.P. Electrocatalytic reduction of carbon dioxide with Mn(terpyridine) carbonyl complexes. Dalton Trans. 2016, 45, 17179–17186. [Google Scholar] [CrossRef]
- Rao, G.K.; Pell, W.; Korobkov, I.; Richeson, D. Electrocatalytic reduction of CO2 using Mn complexes with unconventional coordination environments. Chem. Commun. 2016, 52, 8010–8013. [Google Scholar] [CrossRef] [PubMed]
- Myren, T.H.T.; Lilio, A.M.; Huntzinger, C.G.; Horstman, J.W.; Stinson, T.A.; Donadt, T.B.; Moore, C.; Lama, B.; Funke, H.H.; Luca, O.R. Manganese N-Heterocyclic Carbene Pincers for the Electrocatalytic Reduction of Carbon Dioxide. Organometallics 2019, 38, 1248–1253. [Google Scholar] [CrossRef]
- Myren, T.H.T.; Alherz, A.; Thurston, J.R.; Stinson, T.A.; Huntzinger, C.G.; Musgrave, C.B.; Luca, O.R. Mn-Based Molecular Catalysts for the Electrocatalytic Disproportionation of CO2 into CO and CO32–. ACS Catal. 2020, 10, 1961–1968. [Google Scholar] [CrossRef]
- Nagao, H.; Mizukawa, T.; Tanaka, K. Carbon-Carbon Bond Formation in the Electrochemical Reduction of Carbon Dioxide Catalyzed by a Ruthenium Complex. Inorg. Chem. 1994, 33, 3415–3420. [Google Scholar] [CrossRef]
- Narayanan, R.; McKinnon, M.; Reed, B.R.; Ngo, K.T.; Groysman, S.; Rochford, J. Ambiguous electrocatalytic CO2 reduction behaviour of a nickel bis(aldimino)pyridine pincer complex. Dalton Trans. 2016, 45, 15285–15289. [Google Scholar] [CrossRef]
- Vogt, M.; Nerush, A.; Diskin-Posner, Y.; Ben-David, Y.; Milstein, D. Reversible CO2 binding triggered by metal–ligand cooperation in a rhenium(I) PNP pincer-type complex and the reaction with dihydrogen. Chem. Sci. 2014, 5, 2043–2051. [Google Scholar] [CrossRef]
- Donadt, T.B.; Lilio, A.M.; Stinson, T.A.; Lama, B.; Luca, O.R. DOSY NMR and Normal Pulse Voltammetry for the Expeditious Determination of Number of Electrons Exchanged in Redox Events. Chem. Sel. 2018, 3, 7410–7415. [Google Scholar] [CrossRef]
- Myren, T.H.T.; Alherz, A.; Stinson, T.A.; Huntzinger, C.G.; Lama, B.; Musgrave, C.; Luca, O.R. Metalloradical intermediates in electrocatalytic reduction of CO2 to CO: Mn versus Re bis-N-heterocyclic carbene pincers. Dalton Trans. 2020, 49, 2053–2057. [Google Scholar] [CrossRef]
- Therrien, J.A.; Wolf, M.O. The Influence of para Substituents in Bis(N-Heterocyclic Carbene) Palladium Pincer Complexes for Electrocatalytic CO2 Reduction. Inorg. Chem. 2017, 56, 1161–1172. [Google Scholar] [CrossRef]
- Therrien, J.A.; Wolf, M.O.; Patrick, B.O. Polyannulated Bis(N-heterocyclic carbene)palladium Pincer Complexes for Electrocatalytic CO2 Reduction. Inorg. Chem. 2015, 54, 11721–11732. [Google Scholar] [CrossRef]
- Therrien, J.A.; Wolf, M.O.; Patrick, B.O. Electrocatalytic Reduction of CO2 with Palladium Bis-N-heterocyclic Carbene Pincer Complexes. Inorg. Chem. 2014, 53, 12962–12972. [Google Scholar] [CrossRef] [PubMed]
- Therrien, J.A.; Wolf, M.O.; Patrick, B.O. Synthesis and comparison of nickel, palladium, and platinum bis(N-heterocyclic carbene) pincer complexes for electrocatalytic CO2 reduction. Dalton Trans. 2018, 47, 1827–1840. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structures of fac-Mn(bpy)(CO)3Br (1) and fac-Mn(dmbpy)(CO)3Br (2).

Scheme 1.
Proposed mechanistic pathways of M(bpy)(CO)3X structural Group 7 analogues of catalyst 1, where M = Re (1-Re) or Mn (1) [42]. The protonation-first pathway is shown in blue, whereas the reduction-first pathway is shown in red.
Scheme 1.
Proposed mechanistic pathways of M(bpy)(CO)3X structural Group 7 analogues of catalyst 1, where M = Re (1-Re) or Mn (1) [42]. The protonation-first pathway is shown in blue, whereas the reduction-first pathway is shown in red.
Scheme 2.
Proposed mechanism of activation of 3 for entrance into a catalytic cycle for the production of CO and formate.
Scheme 2.
Proposed mechanism of activation of 3 for entrance into a catalytic cycle for the production of CO and formate.
Scheme 3.
Proposed mechanism for the reductive activation of 4.

Figure 3.
Structure of the PNP Mn pincer catalyst 6 [48].
Figure 3.
Structure of the PNP Mn pincer catalyst 6 [48].

Figure 4.
Structures of complexes 7 ([MnCNCMe(CO)3]Br) and 8 ([MnCNCBn(CO)3]Br).

Scheme 4.
Proposed mechanism of [MnCNCMe(CO)3]Br, adapted from [50] with permission.
Scheme 4.
Proposed mechanism of [MnCNCMe(CO)3]Br, adapted from [50] with permission.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of pincer catalysts discussed.
| (Pre-)Catalyst | Pincer Type | Oxidation States | Products (FE, %) | Bulk Electrolysis Conditions | Proton Source | Ref. |
|---|---|---|---|---|---|---|
| 3a (intermediate, Scheme 2) | ONN | Mn(I), Mn(0) | CO (70%), HCOO− (22%), H2O | −1.8 V vs. SCE, 4 h, MeCN | none | [44] |
| CO (90%), HCOO− (4%) | −1.5 V vs. SCE, 2 h, MeCN, 0.1M TBA PF6 | 2.7 M H2O | [45] | |||
| CO (48%), HCOO− (36%) | −1.5 V vs. SCE, 2 h, MeCN, 0.1 M TBA PF6 | 2.7 M TFE | ||||
| CO (15%), HCOO− (39%) | −1.5 V vs. SCE, 2 h, MeCN, 0.1 M TBA PF6 | 2.7 M phenol | ||||
| Mn(κ2-tpy)(CO)3Br (4, Figure 2) | NNN | Mn(I), Mn(0) | CO (129%) 1, H2O | −2.2 V vs. Fc/Fc+, MeCN, 0.1 M TBA PF6, 4.1 turnovers | 0.5 M phenol | [46,47] |
| Mn(κ3-tpy)(CO)2Br (5, Figure 2) | NNN | Mn(I), Mn(0) | CO (93%), H2O | −2.2 V vs. Fc/Fc+, MeCN, 0.1 M TBA PF6, 4.1 turnovers | 0.5 M phenol | |
| MnPNP(CO)3 (6, Figure 3) | PNP | Mn(I), Mn(0) | CO (96%), CO32− | −2.3 V vs. Fc/Fc+, 125 min, MeCN, 0.1 M TBA PF6 | none | [48] |
| MnCNCMe (7, Figure 4) | CNC | Mn(I), Mn(0) | CO (87%), CO32− | −2.3 V vs. Fc/Fc+, MeCN, 0.1 M TBA PF6 | none | [49] |
| MnCNCBn (8, Figure 4) | CNC | Mn(I), Mn(0) | CO (86%), CO32− (93%) | −2.3 V vs. Fc/Fc+, MeCN, 0.1 M TBA PF6 | none | [49,50] |
| Mn(dmbpy)(CO)3X (2, Figure 1) | - | Mn(I), Mn(0) | CO (100%), H2O | 138 C, −1.70 V vs. Ag/AgNO3, 1mM cat in 95/5 MeCN/H2O, 0.1 M TBAP | 5% H2O | [34] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Petersen, H.A.; Myren, T.H.T.; Luca, O.R. Redox-Active Manganese Pincers for Electrocatalytic CO2 Reduction. Inorganics 2020, 8, 62. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8110062
AMA Style
Petersen HA, Myren THT, Luca OR. Redox-Active Manganese Pincers for Electrocatalytic CO2 Reduction. Inorganics. 2020; 8(11):62. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8110062
Chicago/Turabian StylePetersen, Haley A., Tessa H. T. Myren, and Oana R. Luca. 2020. "Redox-Active Manganese Pincers for Electrocatalytic CO2 Reduction" Inorganics 8, no. 11: 62. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8110062
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.