Ferrocenyl Migrations and Molecular Rearrangements: The Significance of Electronic Charge Delocalization

School of Chemistry, University College Dublin, D04 V1W8 Belfield, Dublin 4, Ireland
Inorganics 2020, 8(12), 68; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8120068
Submission received: 27 November 2020
/
Revised: 9 December 2020
/
Accepted: 9 December 2020
/
Published: 11 December 2020
(This article belongs to the Special Issue Ferrocene and Its Derivatives: Celebrating the 70th Anniversary of Its Discovery)
Abstract
:The enhanced stabilization of a carbocationic site adjacent to a ferrocenyl moiety was recognized within a few years of the discovery of sandwich compounds. While a detailed understanding of the phenomenon was the subject of some early debate, researchers soon took advantage of it to control the ease and direction of a wide range of molecular rearrangements. We, here, discuss the progress in this area from the pioneering studies of the 1960s, to more recent applications in chromatography and analytical detection techniques, and currently in the realm of bioactive organometallic complexes. Several classic reactions involving ferrocenyl migrations, such as the pinacol, Wolff, Beckmann, and Curtius, are discussed, as well as the influence of the ferrocenyl substituent on the mechanisms of the Nazarov, Meyer-Schuster, benzoin, and Stevens rearrangements. The preparation and isomerizations of ferrocenyl-stabilized vinyl cations and vinylcyclopropenes, together with the specific cyclization of acetylcyclopentadienyl-metal derivatives to form 1,3,5-substituted benzenes, demonstrate the versatility and generality of this approach.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
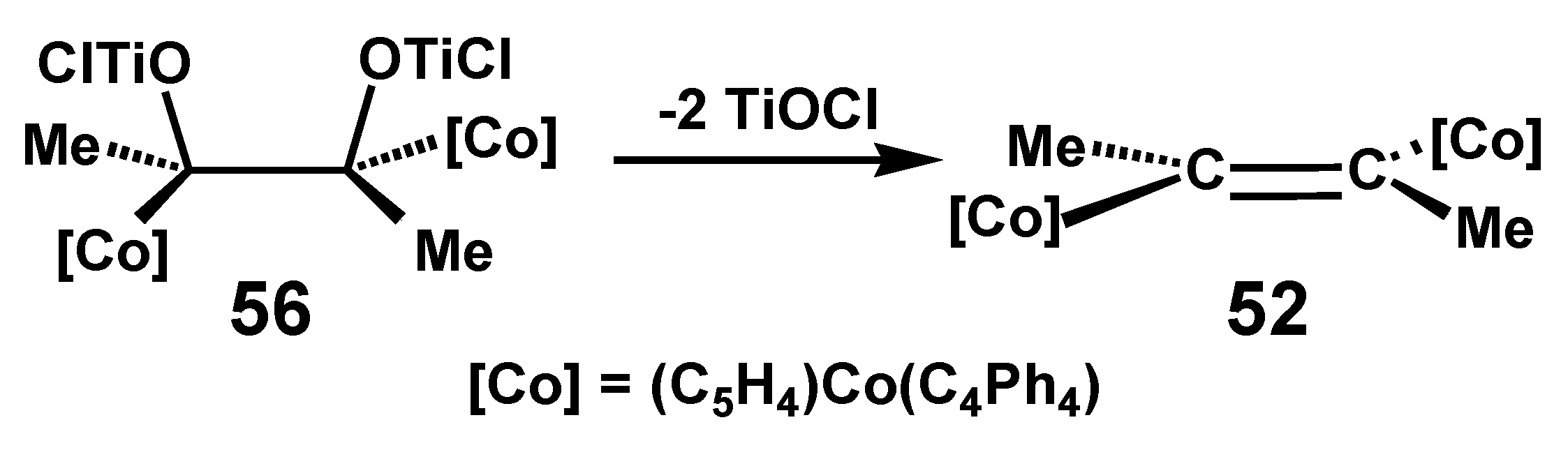{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
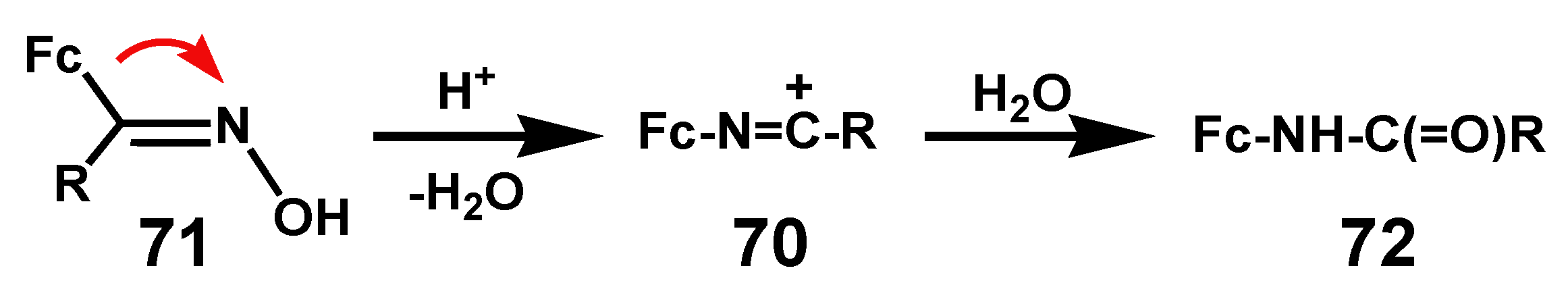{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The ability of the ferrocenyl moiety to stabilize an adjacent carbocationic center has long been attributed to delocalization of the electron deficiency onto the iron atom [1]. The X-ray crystallographically established [2,3] tendency of the cationic α-carbon atom to lean towards the metal as in 1, was originally interpreted in terms of an overlap of the vacant 2p orbital on carbon with a filled 3d orbital of suitable symmetry on iron [4]. Taking a more extreme view, one could regard the system as a neutral fulvene ligand coordinated to a [(C5H5)Fe]+ unit, as depicted in 2 (Figure 1), but this has been criticized based on the low barrier to rotation about the exocyclic bond in some cases [5,6].
Fortunately, a comprehensive overview of this, sometimes controversial, phenomenon has been presented by Gleiter, Bleiholder, and Rominger [7]. Nevertheless, it is this character of charge delocalization that facilitates a number of molecular rearrangements or ferrocenyl migrations, as we shall attempt to demonstrate.
2. Ferrocenyl-Mediated Carbocationic Rearrangements
2.1. Isomerizations of Secondary to Tertiary Carbocations
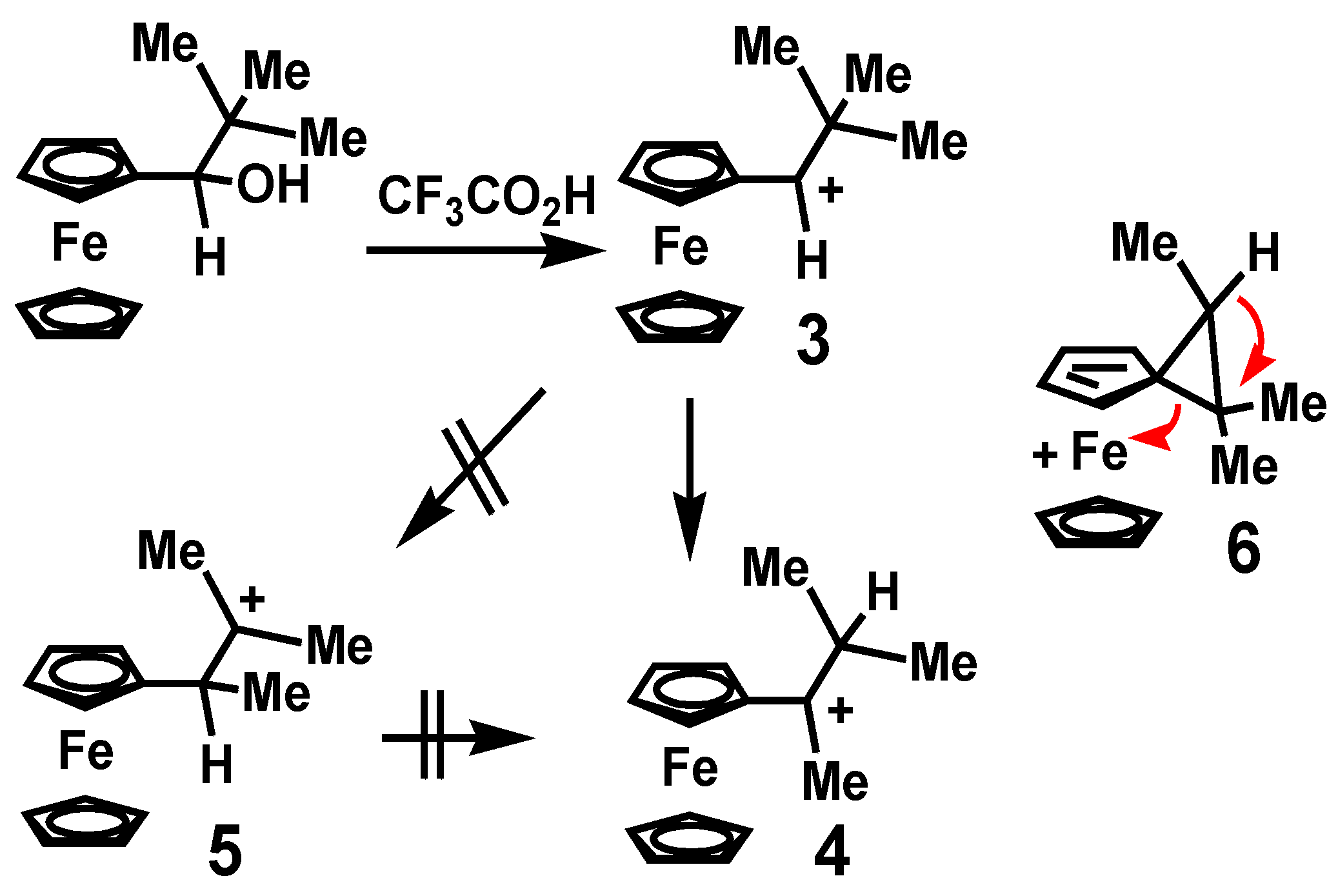
Following pioneering studies by Cais [8], dissolution of ferrocenyl-substituted alcohols in trifluoroacetic acid allowed the acquisition of well-resolved 1H NMR spectra by several other groups [9,10,11], and revealed the unusual stability of the carbocations so generated. Subsequently, in a series of ground-breaking papers, Watts and his group reported the rearrangement behavior of a number of alkylferrocenyl carbocations upon protonation of their precursor alcohols [12].
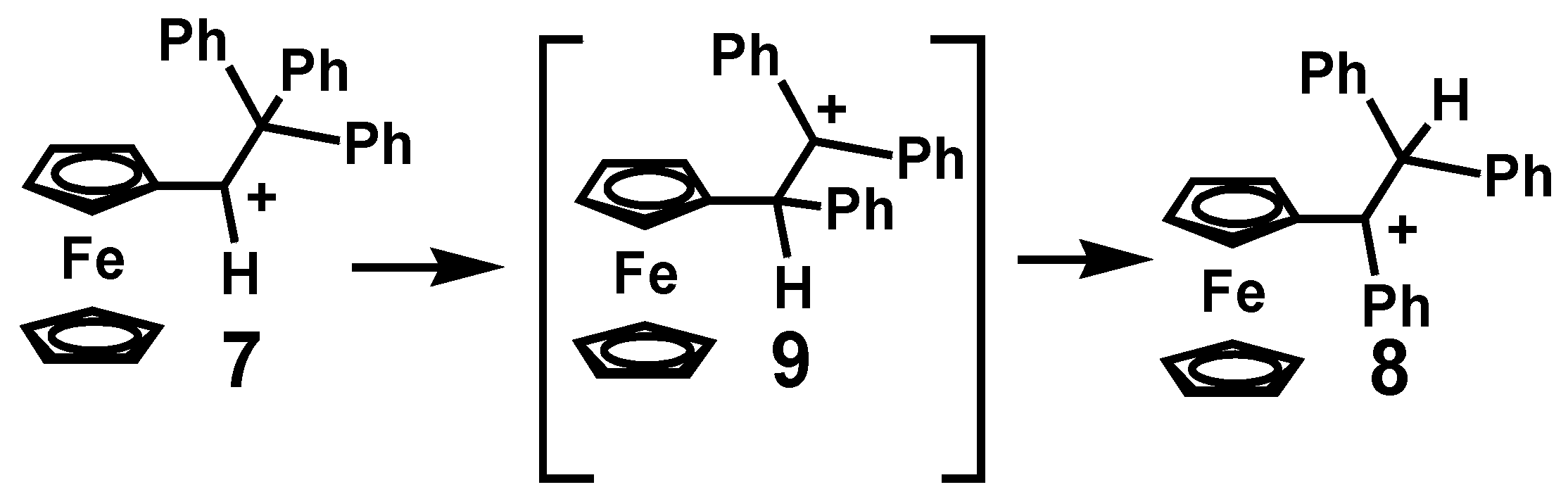
Typically, the quantitative conversion of the ferrocenyl-stabilized secondary carbocation 3 into its tertiary isomer 4, is in accord with the accepted hierarchy of stable carbocations. At first sight, this appears to be a double shift rearrangement whereby an alkyl migration to form the β-ferrocenyl cation 5 is followed by a [1,2]-hydrogen shift to yield the observed product. However, the authors did not prefer this mechanism since the intermediate rearrangement product, 5, would undoubtedly be thermodynamically disfavored relative to the ferrocenyl-stabilized starting cation 3. Instead, they proposed that the initial methyl migration might proceed synchronously with the formation of a short-lived spiro-diene species 6 (depicted in Scheme 1) which could reopen with concomitant hydrogen migration, thus generating the observed final product [13]. In such a scenario, the cationic charge would be delocalized onto the metal in the transition state. The analogous rearrangement of [Fc–CH–CPh3]+, 7, into [Fc–CPh–CHPh2]+, 8, was also observed (Scheme 2), but in this case the doubly-benzylic cation [Fc–CHPh–CPh2]+, 9, might be a viable intermediate [14].
2.2. Rearrangements of Vinyl and Allyl Cations
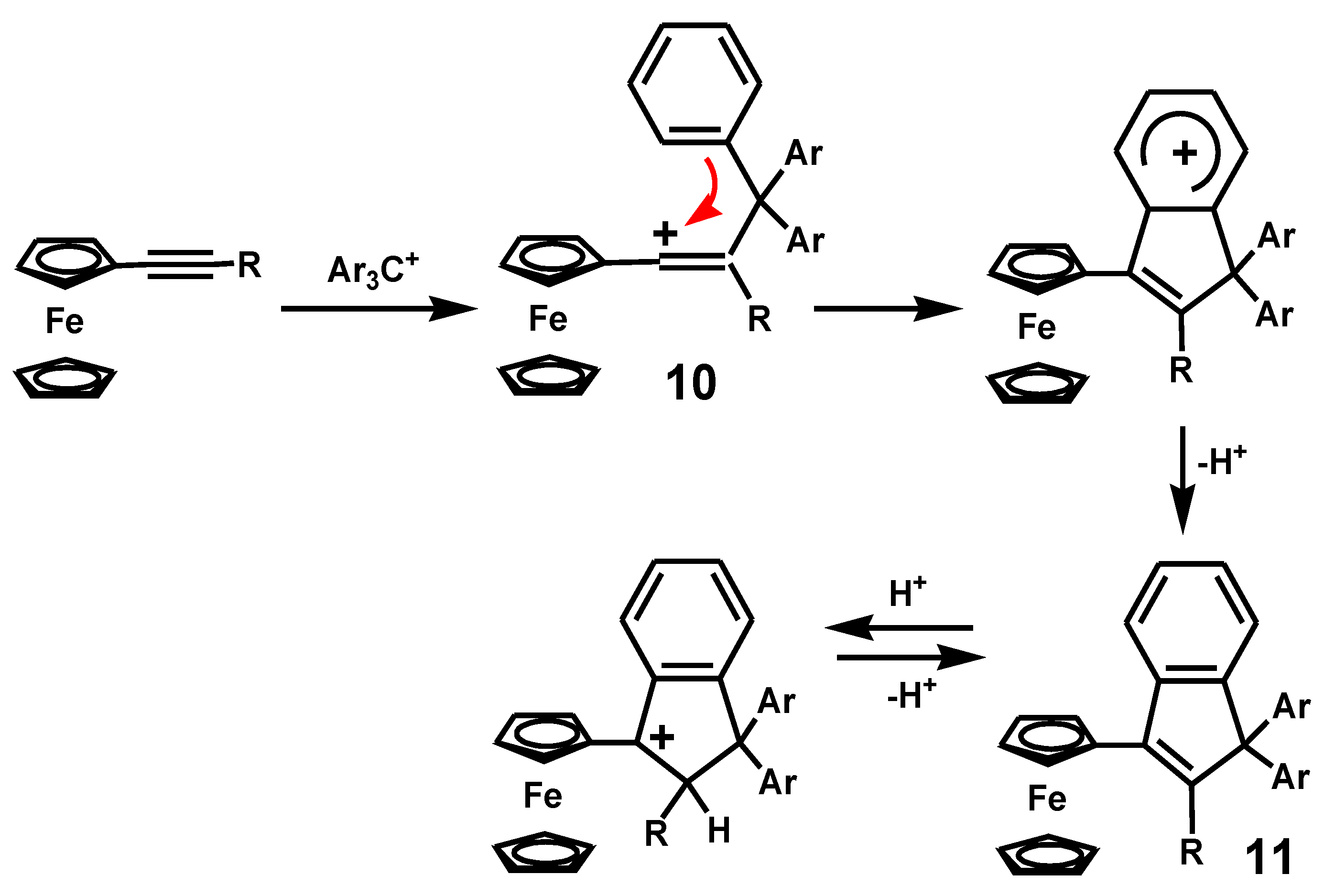
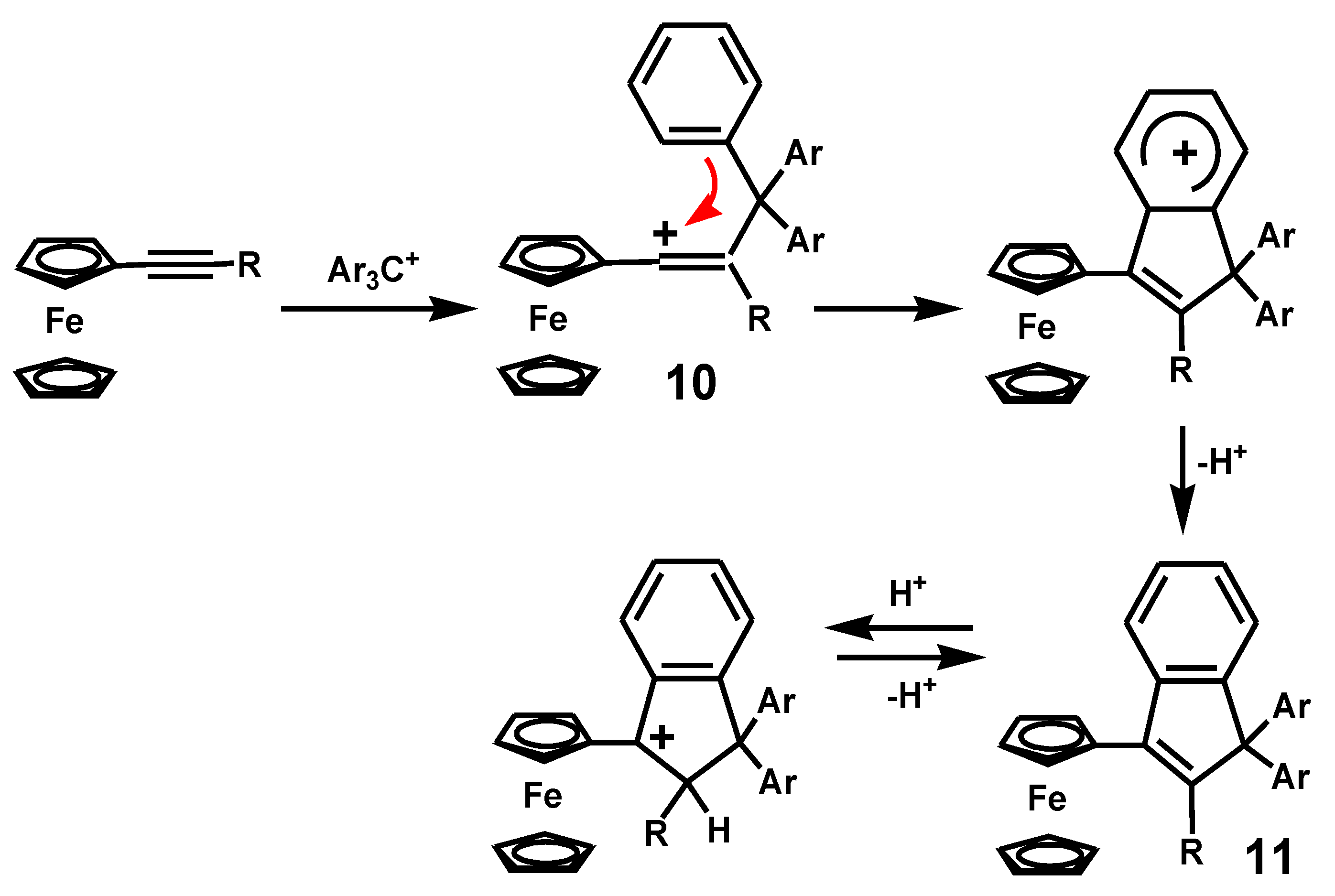
This work was elegantly extended to include the addition reactions of trityl cations to alkynylferrocenes to generate ferrocenyl-stabilized vinyl carbocations, 10, that underwent cyclization to form indenes, 11 [15]. The thermodynamic driving force in each case is the positioning of a carbocationic site adjacent to the ferrocenyl moiety, as shown in Scheme 3.
In a variant of this concept, the diferrocenyl-alkynes Fc–C≡C–CH2–Fc, 12a, and Fc–C≡C–C(=O)–Fc, 12b, were treated with trifluoroacetic acid to form the ferrocenyl-stabilized vinyl cations, 13a,b [15]. In these systems, cyclization occurred by electrophilic attack on the alkyl- or acyl-linked ferrocenyl substituent to form (η5-cyclopentadienyl)(3-ferrocenylpentalenyl)iron, 14a, or (η5-cyclopentadienyl)-(3-ferrocenyl-1-oxo-pentalenyl)iron, 14b, respectively, as depicted in Scheme 4.
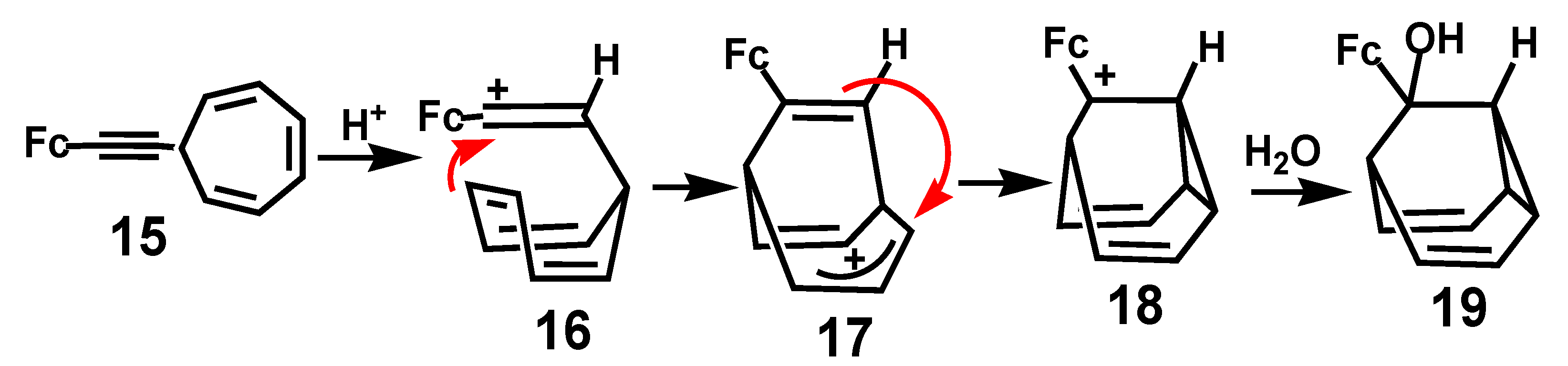
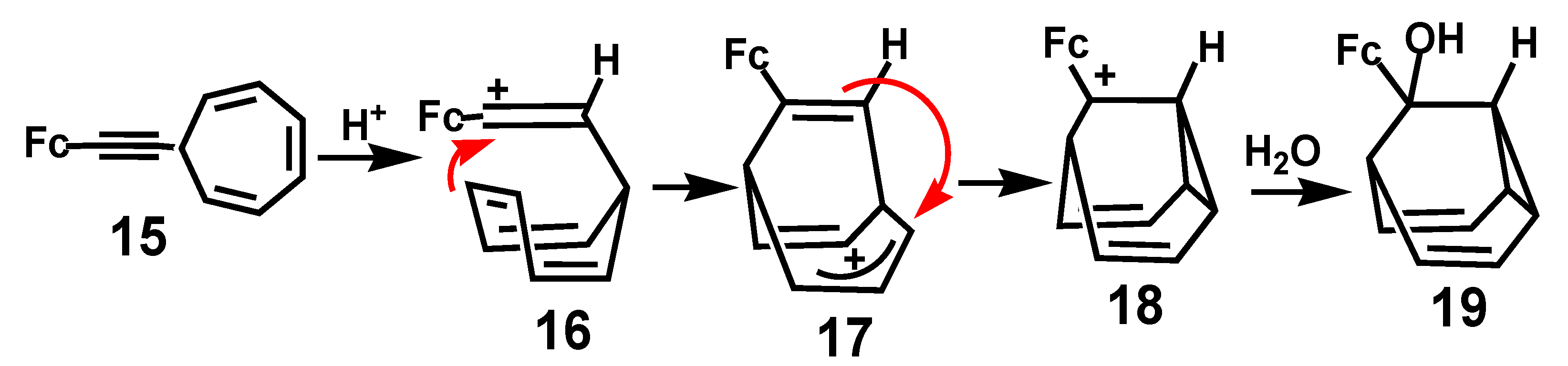
In another remarkable example, protonation of the alkynylcycloheptatriene derivative, 15, yielded a ferrocenyl-stabilized vinyl cation, 16, that eventually yielded a tricyclic alcohol. The proposed mechanism (Scheme 5) involves attack by a double bond of the trienyl unit at the vinylic cation to generate the 6-ferrocenylbicyclo [3.2.2]nona-3,6,8-trien-2-yl cation, 17; subsequent cyclopropyl ring closure yields the ferrocenyl-stabilized cation 18, and ultimately the alcohol 19 [15].
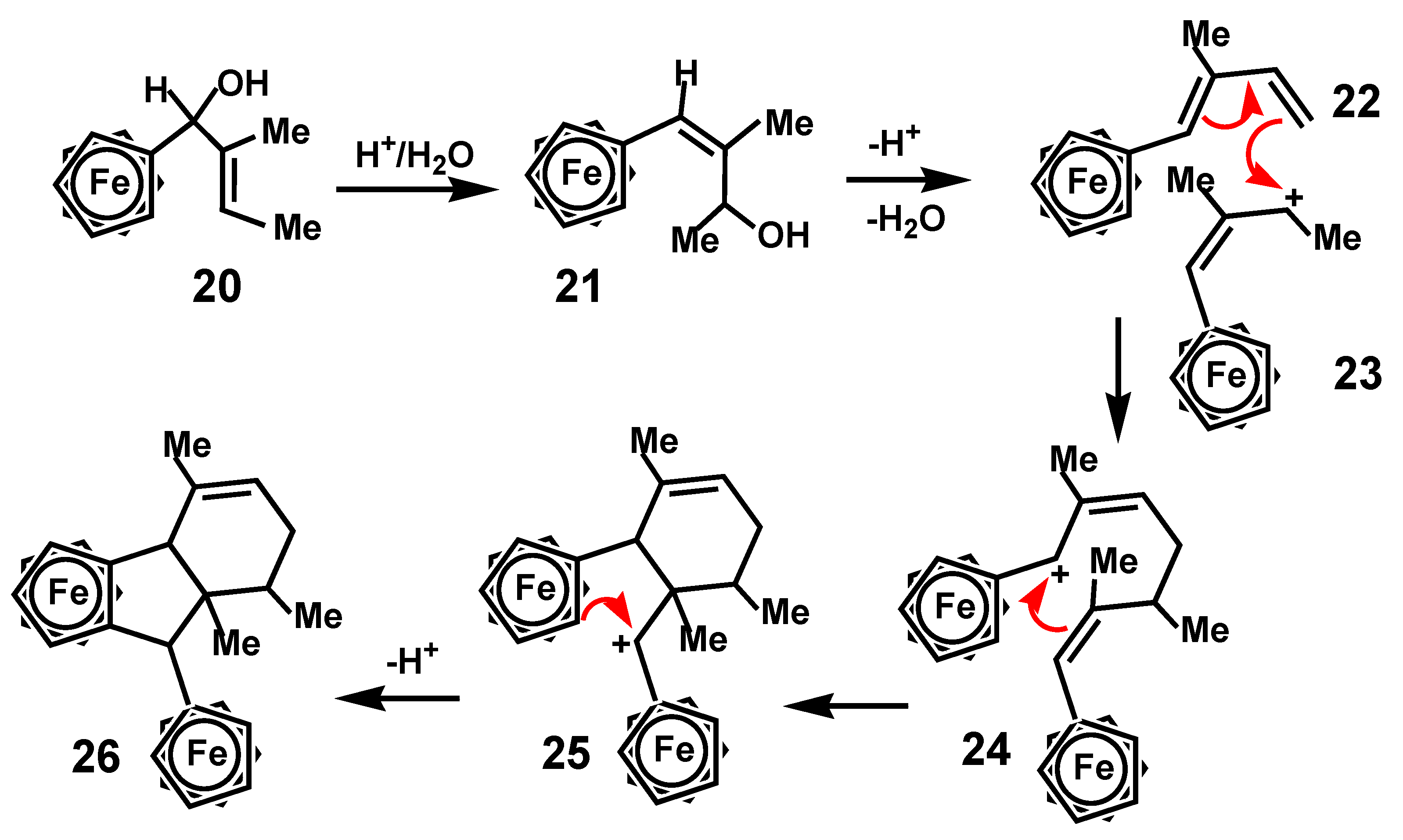
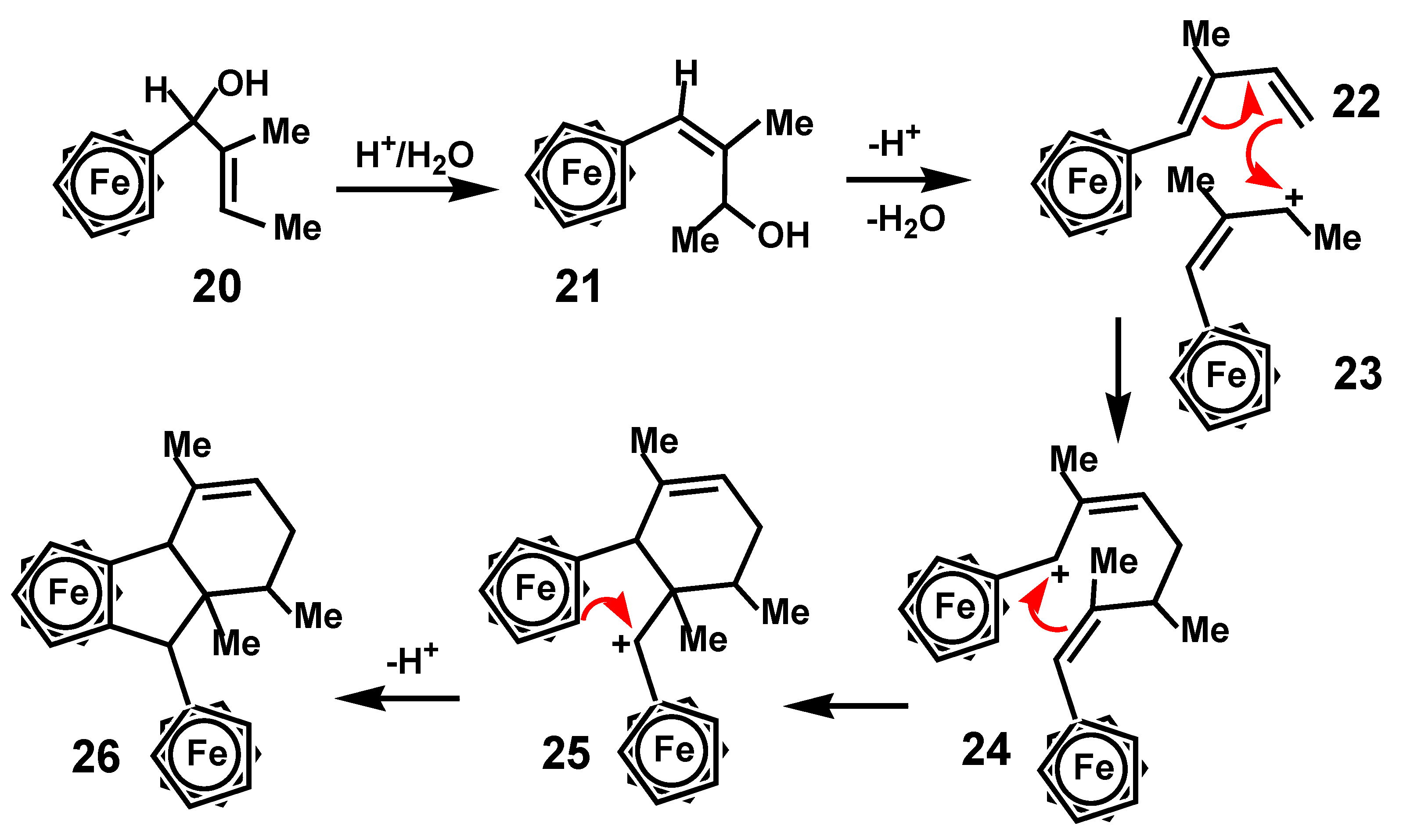
Another unexpected cyclization leading to formation of a polycyclic system was reported by the Schottenberger group who prepared the ferrocenyl allyl alcohol 20 that underwent an allyl rearrangement thus forming a vinylferrocene, 21, thereby positioning the hydroxy substituent near the chain terminus. Treatment with concentrated sulfuric acid yielded as one of the products a dimer whose structure, shown in Scheme 6, was established by X-ray crystallography [16]. The authors advanced a mechanism whereby elimination of water formed the diene 22, which then participated in a [4 + 2] Diels-Alder cycloaddition with the ferrocenyl allyl cation 23 prior to eventual indene formation. One might perhaps more realistically envisage a stepwise process involving electrophilic attack by the allyl cation on the diene to form the ferrocenyl-stabilized intermediate 24, closure of the six-membered ring to generate a different cationic site adjacent to the other ferrocenyl moiety, as in 25, and finally ring closure onto ferrocene, thus yielding the observed final product 26.
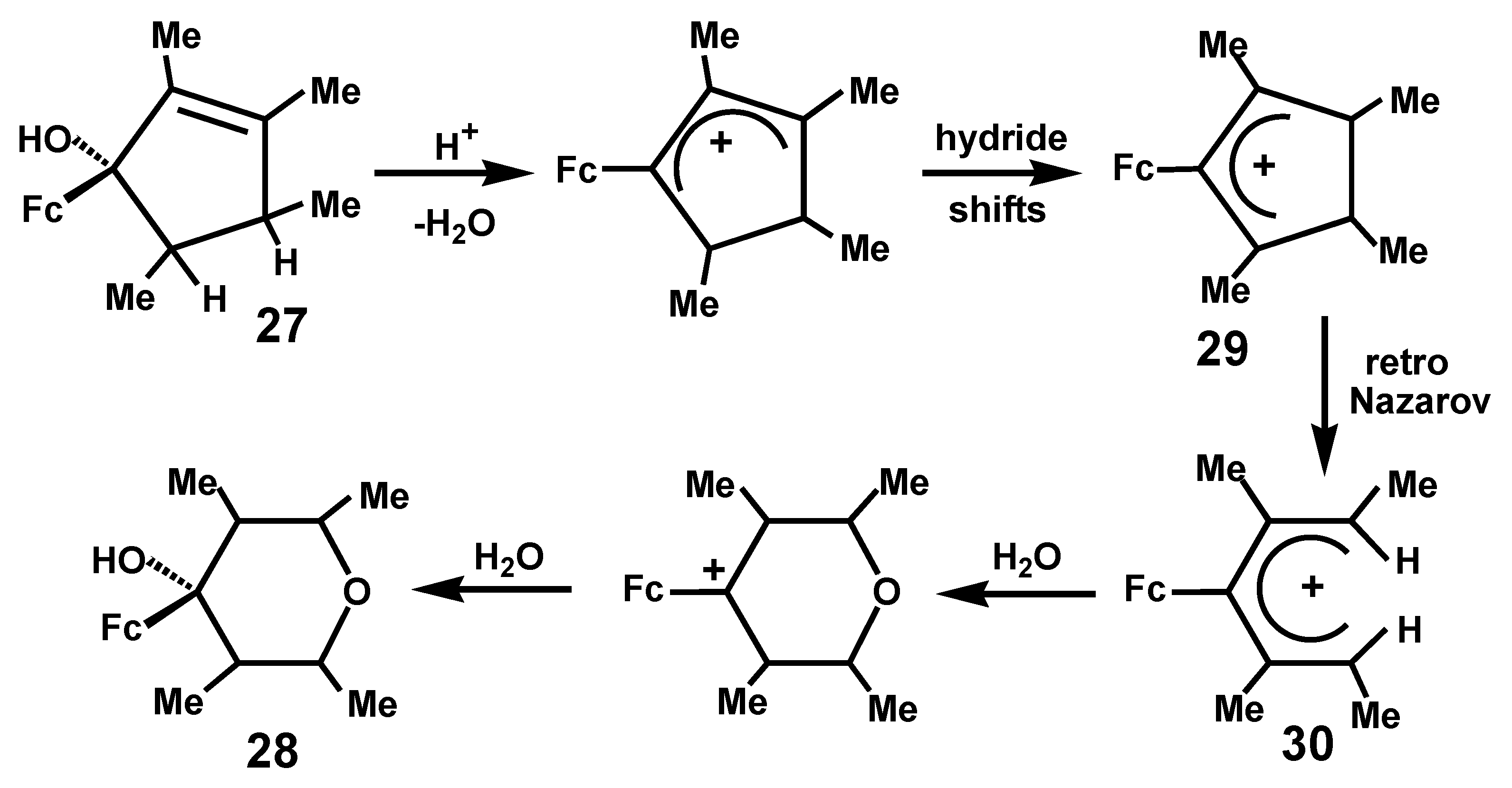
A second fascinating observation from the same laboratory resulted from the protonation of 1-ferrocenyl-2,3,4,5-tetramethylcyclopent-2-en-1-ol, 27, to form 4-ferrocenyl-4-hydroxy-2,3,5,6-tetra-methyltetrahydropyran, 28 [16]. The proposed mechanism involves initial elimination of water together with a series of hydride shifts to form the ferrocenyl-allyl cation 29 that can ring-open, in a reversal of the normal Nazarov cyclization, to generate the divinylferrocenyl cation 30. Now, aqueous hydrolysis to form the tetrahydropyran framework is followed by addition of a second molecule of water to furnish the final product, 28, as illustrated in Scheme 7.
2.3. Ferrocene-Mediated Cyclization to form Trisubstituted Aromatics, 1,3,5-C6H3(MLn)3
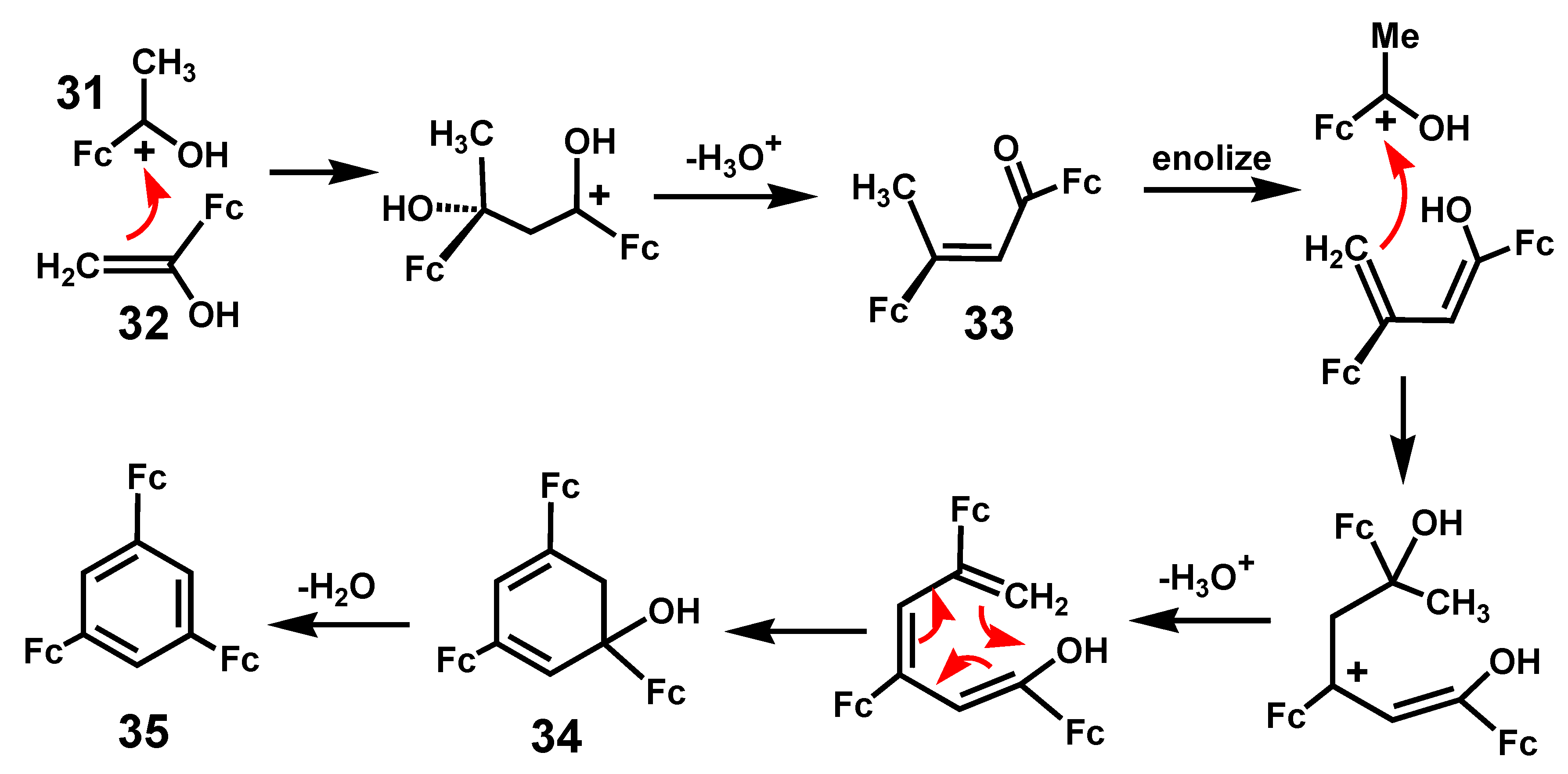
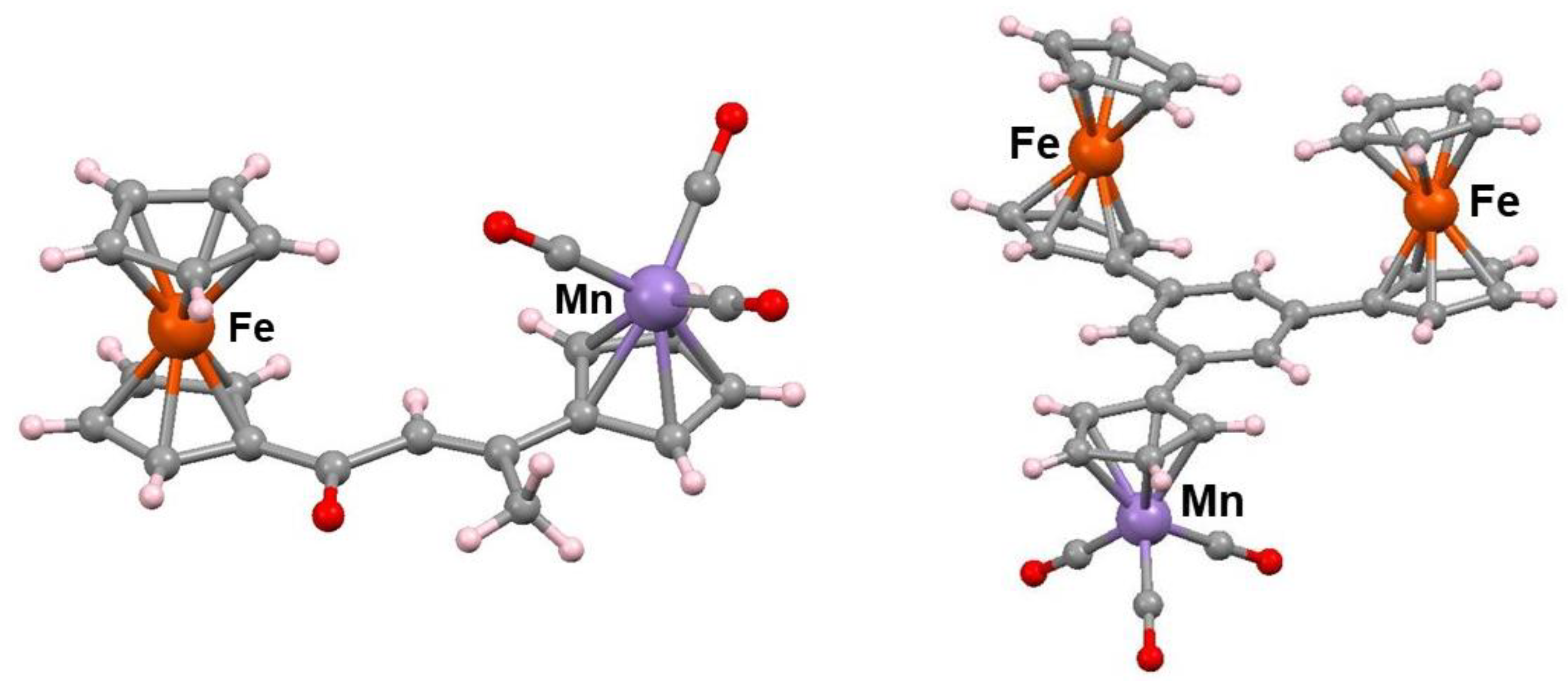
Treatment of acetylferrocene with SiCl4/ethanol provides a convenient route to the specific formation of 1,3,5-triferrocenylbenzene [17]. This contrasts with the transition metal-catalyzed trimerizations of ethynylferrocene which yield a mixture of 1,2,4 and 1,3,5 isomers [18,19]. The proposed mechanism shown in Scheme 8 involves initial reaction of the cation [FcC(OH)CH3]+, 31, with the enol form of the ketone, 32; this process continues in a stepwise manner as the chain gradually grows, via intermediates 33 and 34, prior to the final dehydration leading to 35. This is a general reaction and has also been exploited in the case of (acetylcyclopentadienyl)-manganesetricarbonyl which, likewise, yields the 1,3,5-trisubstituted aromatic [17]. Moreover, mixed species are also accessible, as typified by the Fe2Mn example, 36, shown in Figure 2; indeed, the bimetallic Fe–Mn precursor, 37, corresponding to 33 in Scheme 7 has also been characterized by X-ray crystallography [20]. This approach has also been used to prepare C3h-symmetric polycyclic systems [17].
In some ways, the ferrocenyl group behavior may be compared to that of apparently related molecular fragments, such as (η5-C5H4)Mn(CO)3 or (η6-C6H5)Cr(CO)3, which can also accommodate the build-up of positive charge on an adjacent atom [21] as, for example, in the above-mentioned preparation of 1,3,5-C6H3[(C5H4)Mn(CO)3]3 analogous to 1,3,5-triferrocenylbenzene [17]. However, this is not necessarily a true comparison since the metal tricarbonyl complexes can also stabilize an α-carbanion through delocalization of the negative charge onto the carbonyl ligands by enhanced back-donation of electron density into the π* manifold [22]. This is most evident in metal carbonyl infrared stretching frequencies, νCO, that decrease dramatically in the anions relative to their values in the corresponding cations.
3. Rearrangements of Vinylcyclopropenes
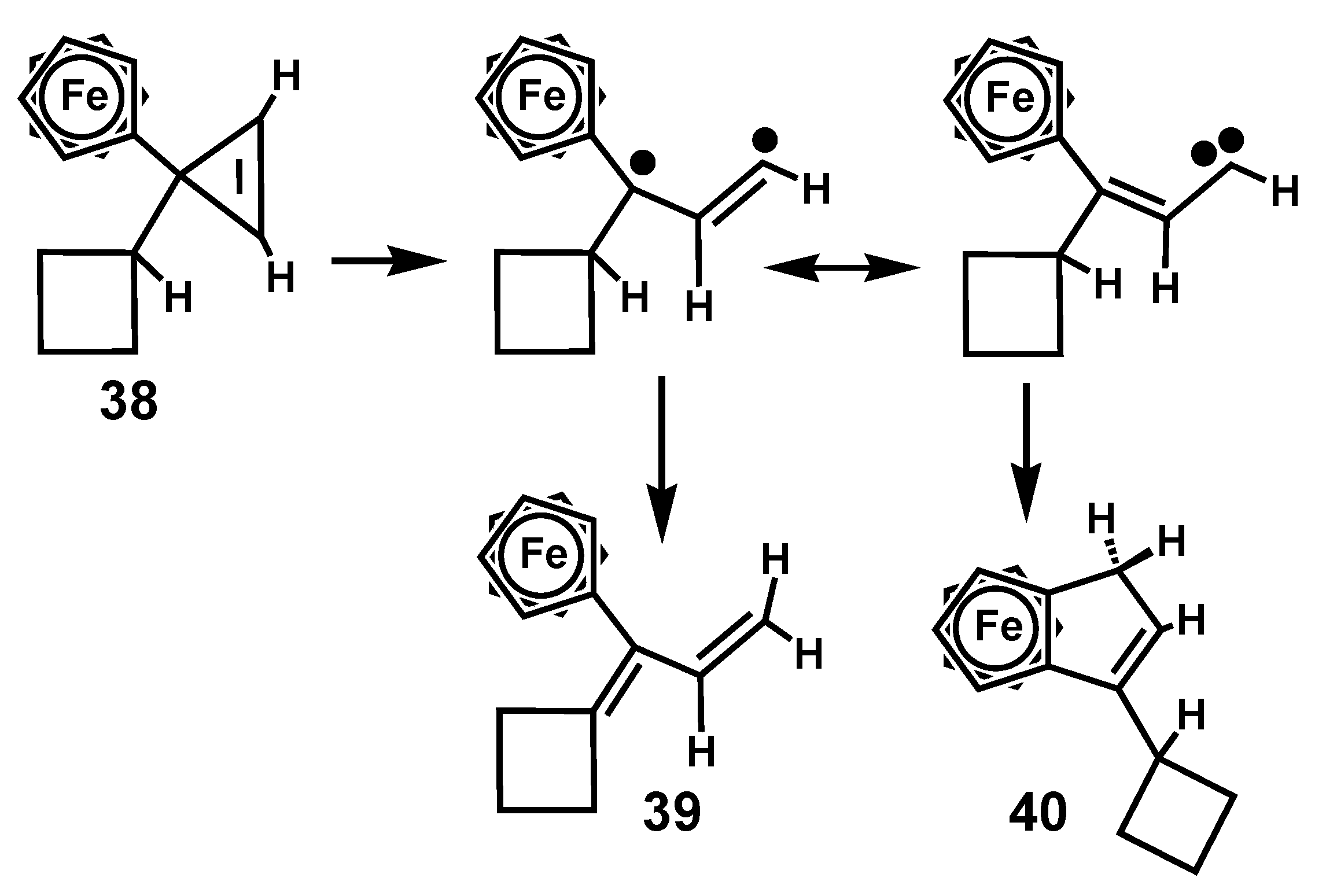
The thermal instability of vinylcyclopropenes is markedly enhanced when they also bear ferrocenyl substituents. Thus, decomposition of 3-cyclobutyl-3-ferrocenylcyclopropene, 38, brought about rearrangement even at 0 °C to give 3-cyclobutyliden-3-ferrocenylpropene, 39, and 3-cyclobutyl-1H-cyclopentaferrocene, 40 [23]. Mechanistically, one can invoke a ferrocenyl-stabilized diradical species or a vinyl carbene, as in Scheme 9. The products can therefore arise either by hydrogen migration, or by attack on the ferrocenyl unit to form 39 or 40, respectively.
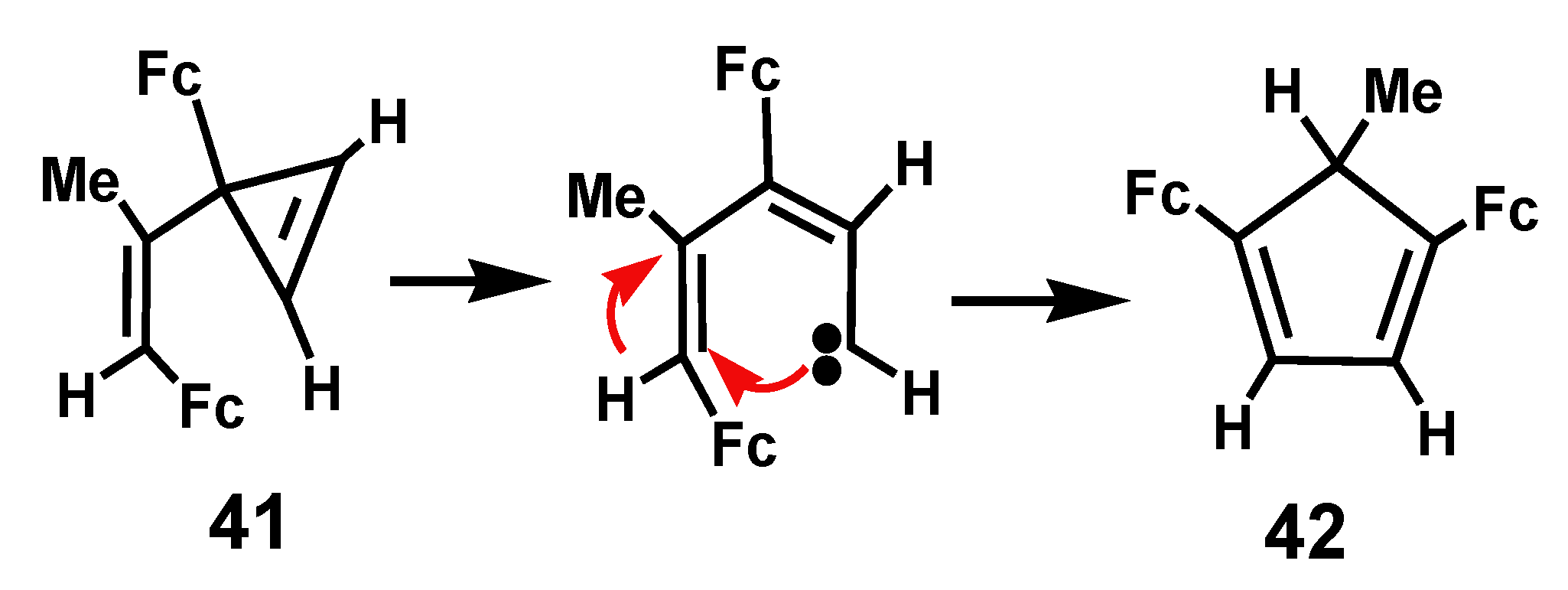
In the closely related system, 3-ferrocenyl-3-(2-ferrocenyl-1-methylvinylcyclopropene, 41, which bears two ferrocenyl substituents, rearrangement occurs at 20 °C in benzene solution to yield 1,4-diferrocenyl-5-methylcyclopentadiene, 42 (Scheme 10). Once again, the intermediacy of a vinyl carbene or a diradical species has been suggested [24].
4. Ferrocenyl Migration onto an Electron-Deficient Carbon Center
4.1. Pinacol Rearrangements
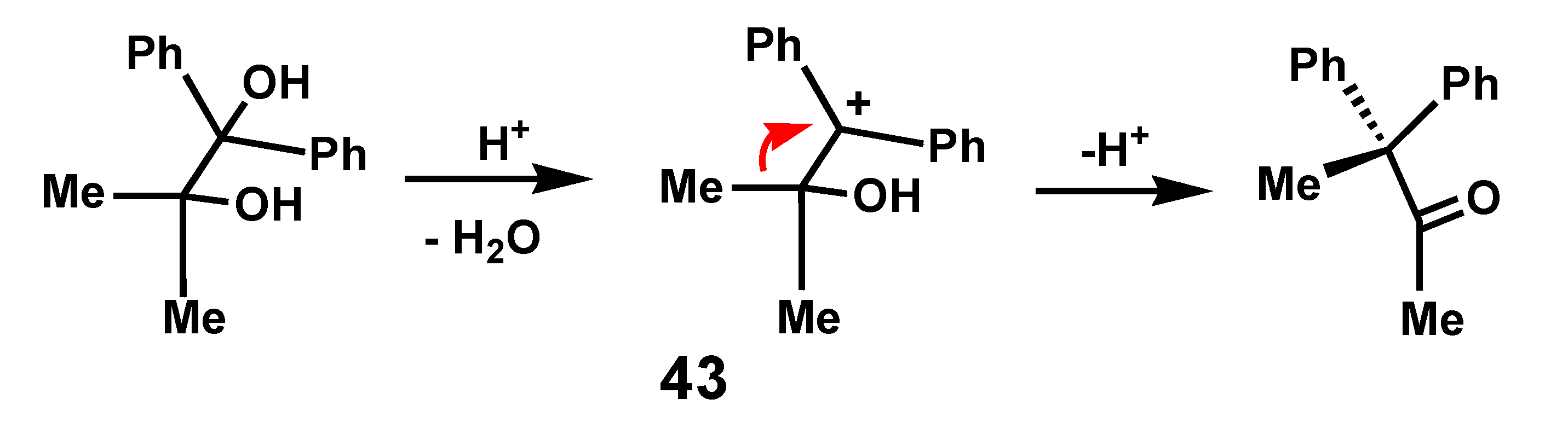
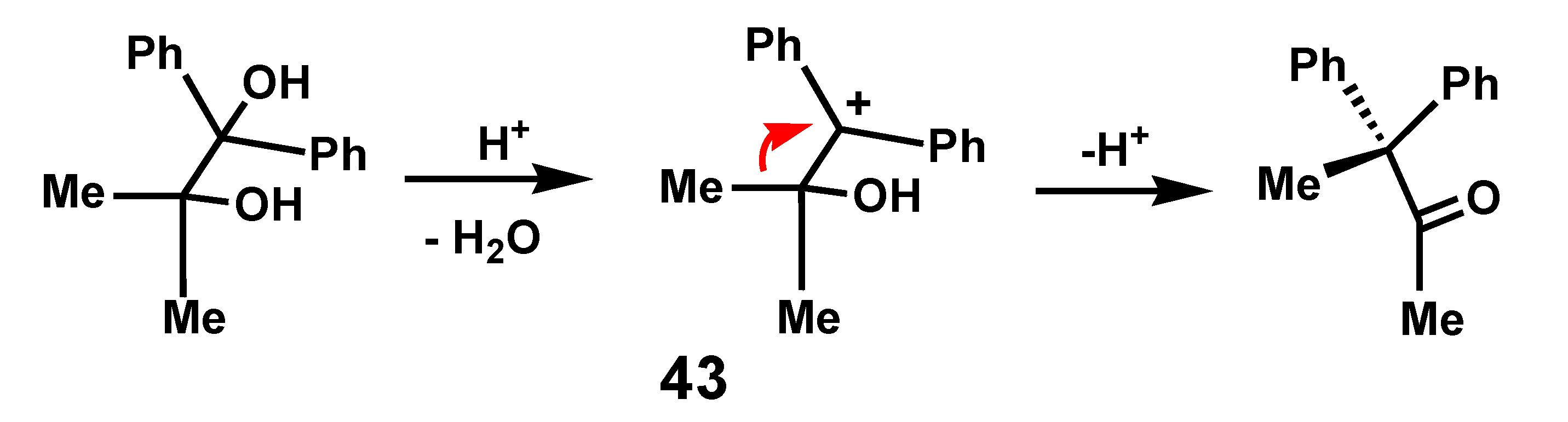
Migratory aptitude is a widely invoked parameter to account for the selectivity and ease of molecular rearrangements [25]. Typically, in cationic species, it is commonly assumed that a phenyl migration is favored over that of its methyl counterpart because of charge delocalization in the phenonium ion transition state. However, such generalizations must be treated with caution. For example, upon protonation of the pinacol Ph2C(OH)-CMe2OH, it is a methyl that preferentially migrates (Scheme 11), not because the alkyl group has a better migratory aptitude than the aryl substituent, but rather that the more favored carbocation, 43, is doubly benzylic [26].
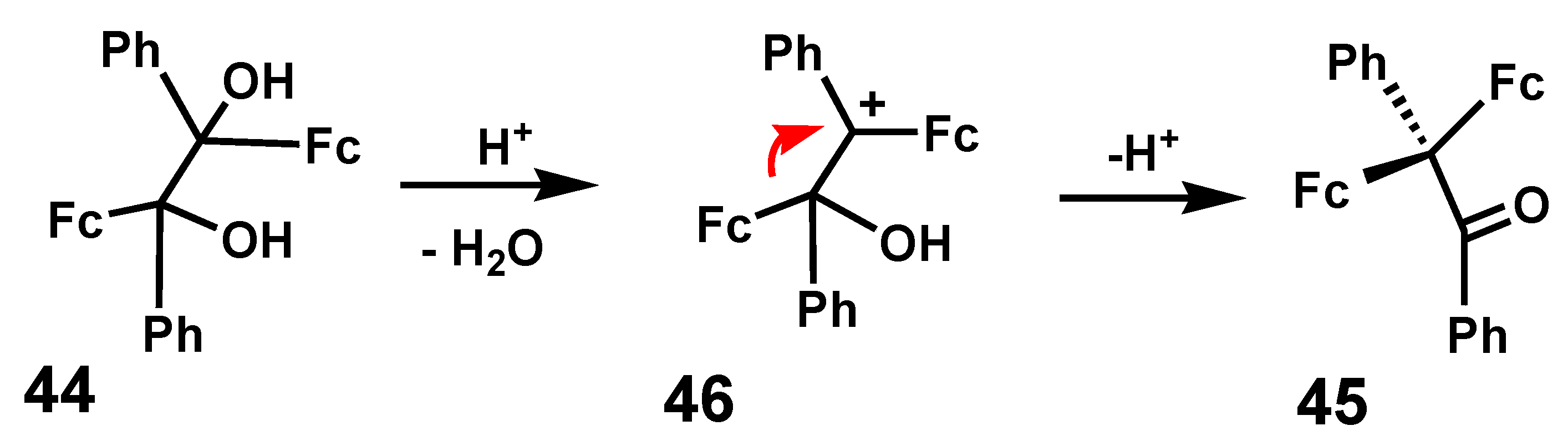
To make a fair comparison, for example between phenyl and ferrocenyl, one must eliminate such extraneous factors. This was first discussed by Weliky and Gould [27] in 1956 who prepared 1,2-diferrocenyl-1,2-diphenyl-ethanediol, 44, by treatment of benzoylferrocene with cobalt dichloride in the presence of a Grignard reagent. They noted that this pinacol “rearranged with remarkable ease (merely upon passing gaseous HCl over its solution in benzene) to the pinacolone 45.” Clearly, ferrocenyl was favored over phenyl migration, as in Scheme 12. Later work by several groups [28,29,30] revealed, not surprisingly, that ferrocenyl migration was also favored over an alkyl shift (R = Me, Et, nPr, nBu). Subsequently, however, it was found that phenyl migration in cation 46 is indeed observable, but that the ferrocenyl shift was preferred by a factor of ~8. Interestingly, it was noted that the rearrangement behavior of the threo and erythro isomers of 44 was identical [31].
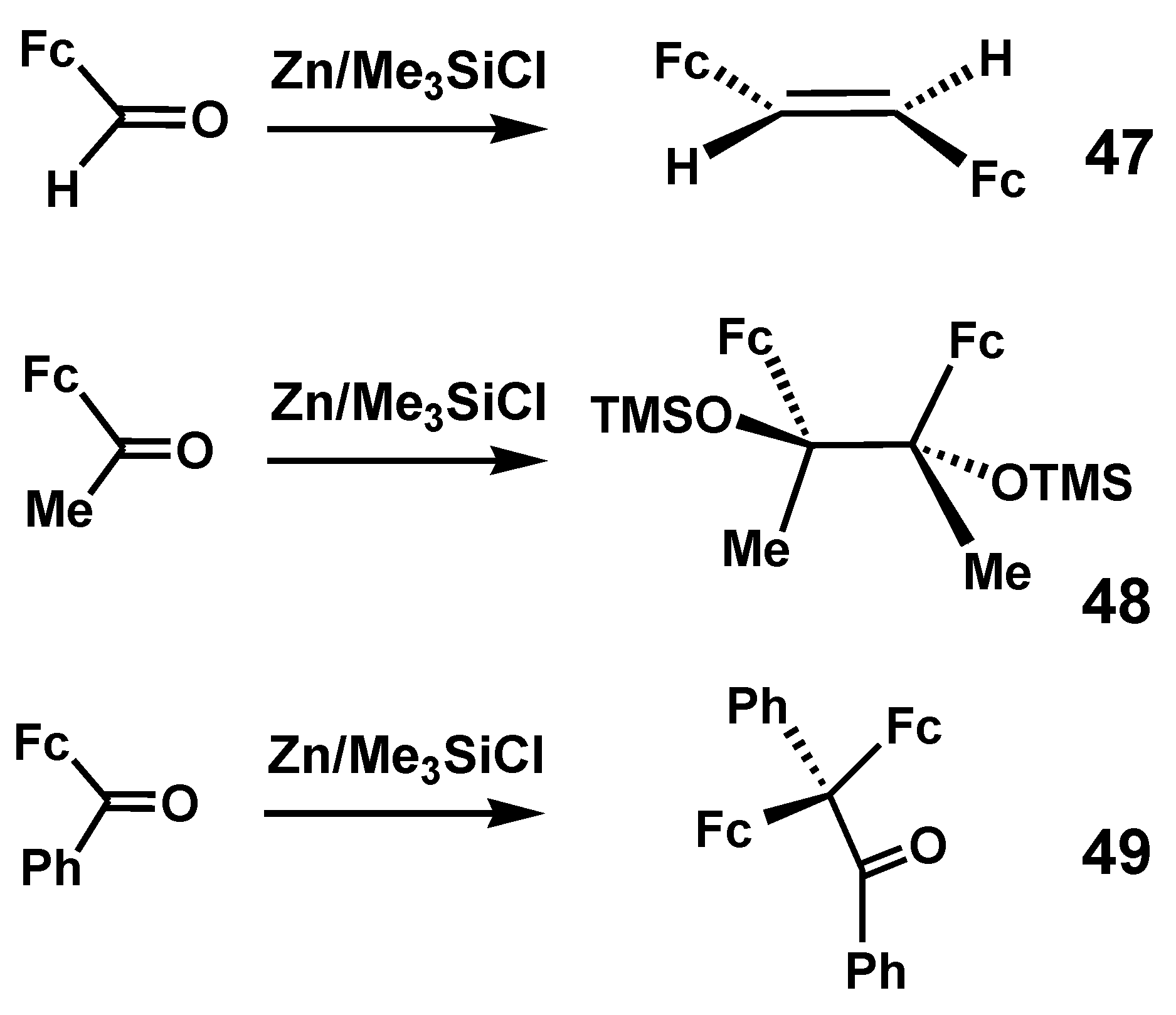
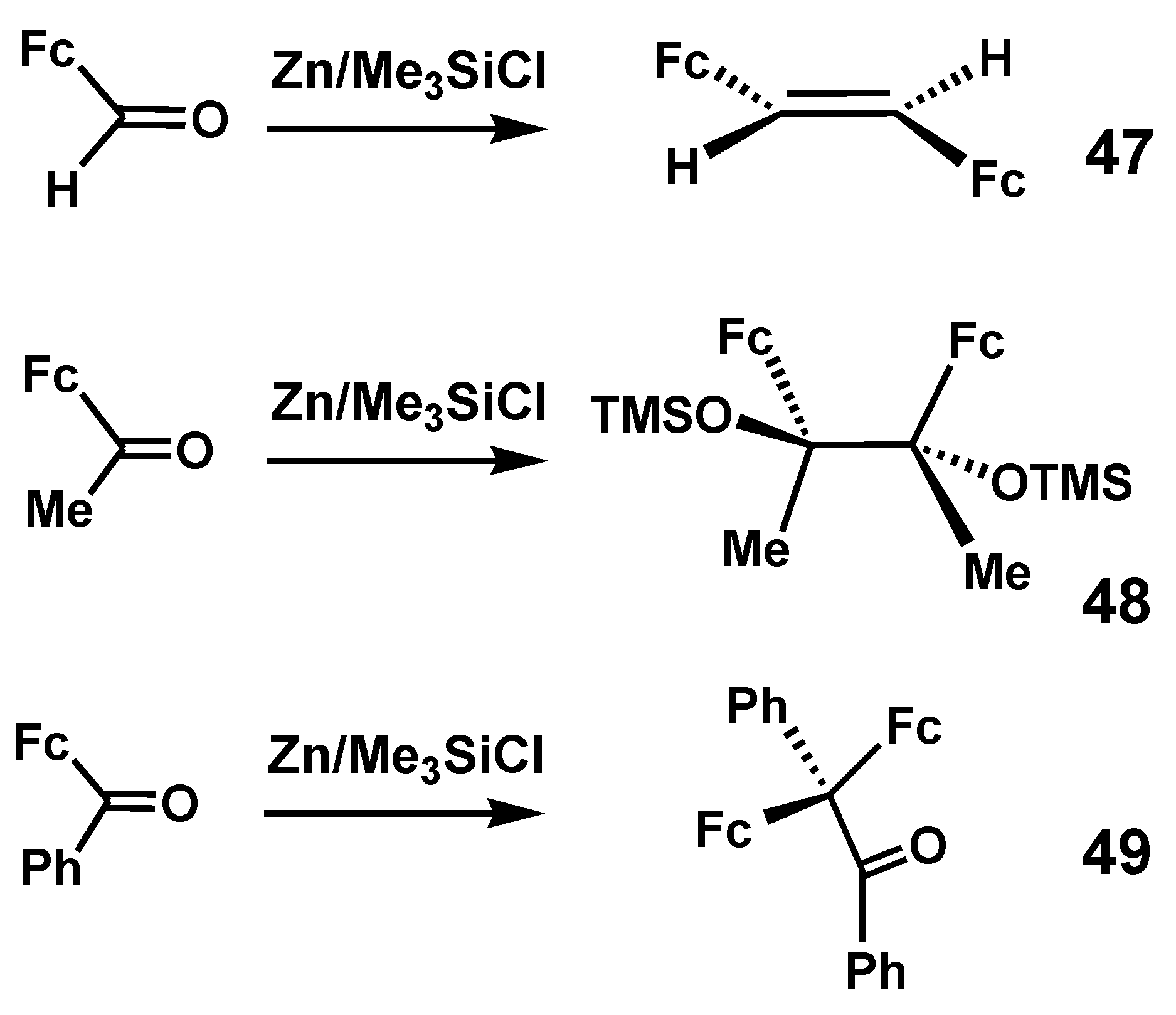
Much more recently, a comprehensive study of the modified Clemmensen reduction of formylferrocene, acetylferrocene, and benzoylferrocene was reported by Bildstein [32]. In this case, the normal reagents, Zn/HCl, were replaced by Zn/Me3SiCl in the expectation that deoxygenation to obtain the corresponding alkenes, R(Fc)C=C(Fc)R, would be enhanced by formation of Me3Si–O–SiMe3 with its energetically favorable silicon-oxygen linkages. Experimentally, however, as shown in Scheme 13, the isolated products were trans-diferrocenylethene, 47, 2,3-diferrocenyl-2,3-bis(trimethylsiloxy)butane, 48, and 2,2-diferrocenyl-1,2-diphenyl-ethan-1-one, 49, respectively. These three products were each unambiguously characterized by X-ray crystallography. It is reasonable to assume that the pinacolone, 49, must have been derived from the diphenyl analogue of the disiloxy-pinacol 48. Related work in this area, including synthetic routes to ferrocenyl cumulenes, has been fully reviewed elsewhere [33].
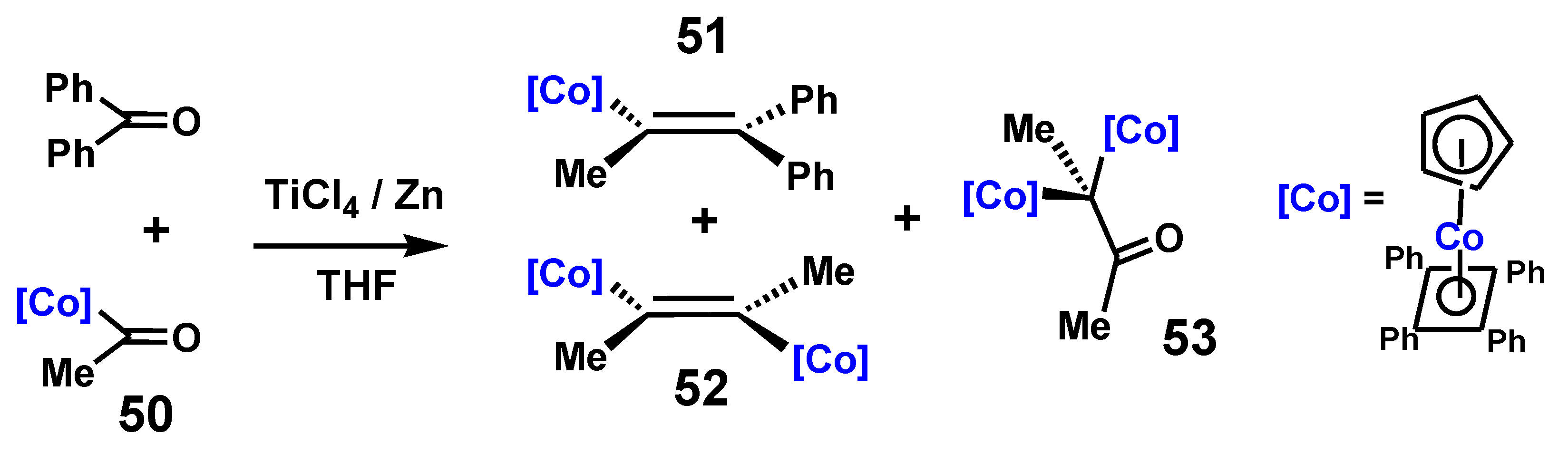
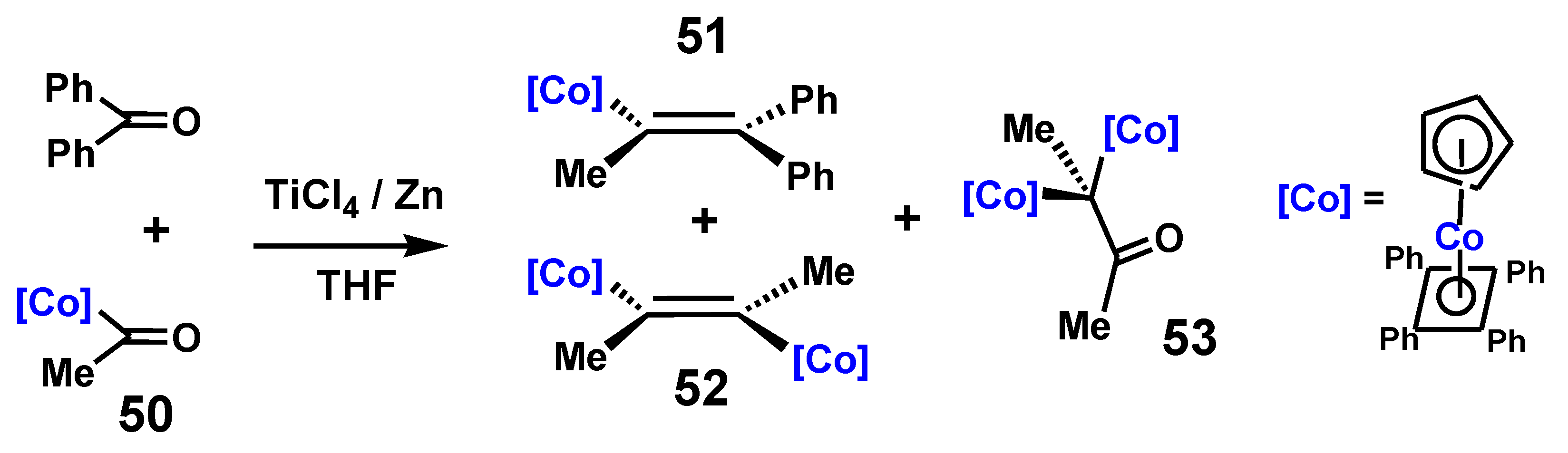
These results may be compared with those found in the course of the closely analogous McMurry reaction of (η5-acetylcyclopentadienyl)(η4-tetraphenylcyclobutadiene)cobalt, 50, with benzophenone [34]. The products observed in addition to the desired hetero-coupled alkene, 51, were trans-2,3-bis[(η5-cyclopentadienyl)(η4-tetraphenylcyclobutadienecobalt]-2-butene, 52, and 3,3-bis-[(η5-cyclopentadienyl)(η4-tetraphenylcyclobutadiene)cobalt]butan-2-one, 53, which parallel the structures of Bildstein’s iron complexes, 47 and 49. Once again, the homo-coupled alkene adopts the trans structure, and it is the cobalt sandwich moiety that has migrated to form the pinacolone, as shown in Scheme 14.
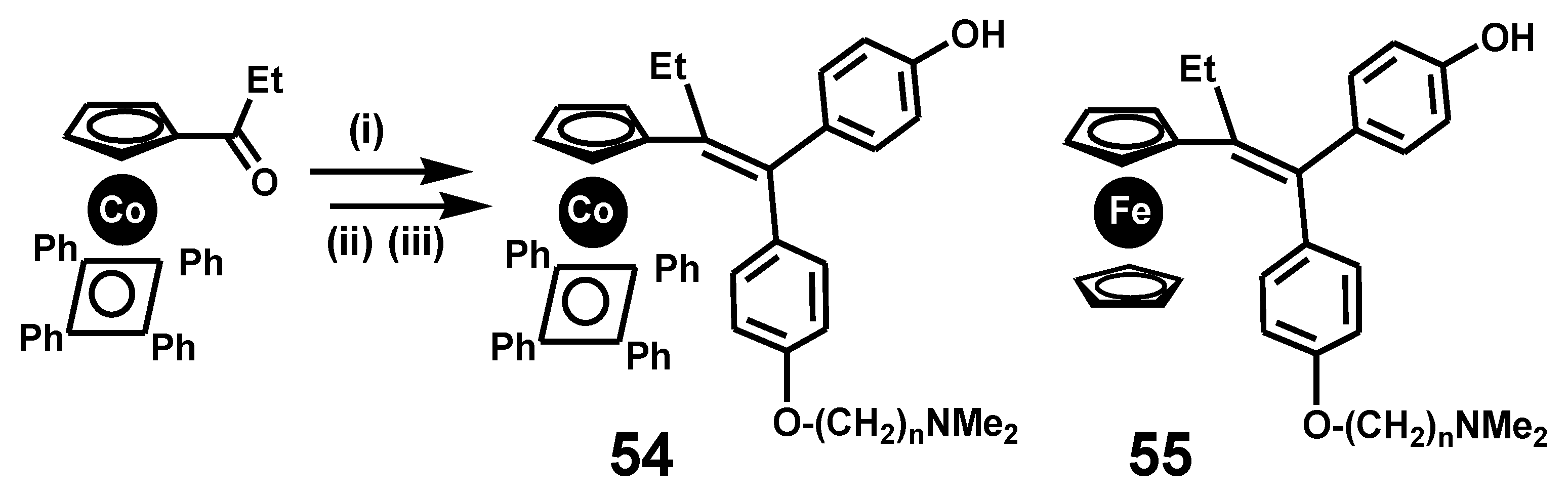
This reaction was important for the development of a synthetic route to cobaltifens, 54, (Scheme 15) [35], the cobalt analogues of ferrocifens, 55, that are now under intensive investigation for their activity against breast cancers and other related diseases [36] (see Section 7).
Following the mechanistic proposals of Bogdanović [37] and of Geise [38], it was suggested that one could visualize formation of the trans-alkene 52 from the C2-symmetric intermediate 56 (Scheme 16). In contrast, when the bulky metallocenyl groups are positioned in a cis fashion, alkene formation may be sterically hindered and instead a pinacol-to-pinacolone rearrangement via a metal-stabilized cationic structure, 57, is the preferred option (Scheme 17).
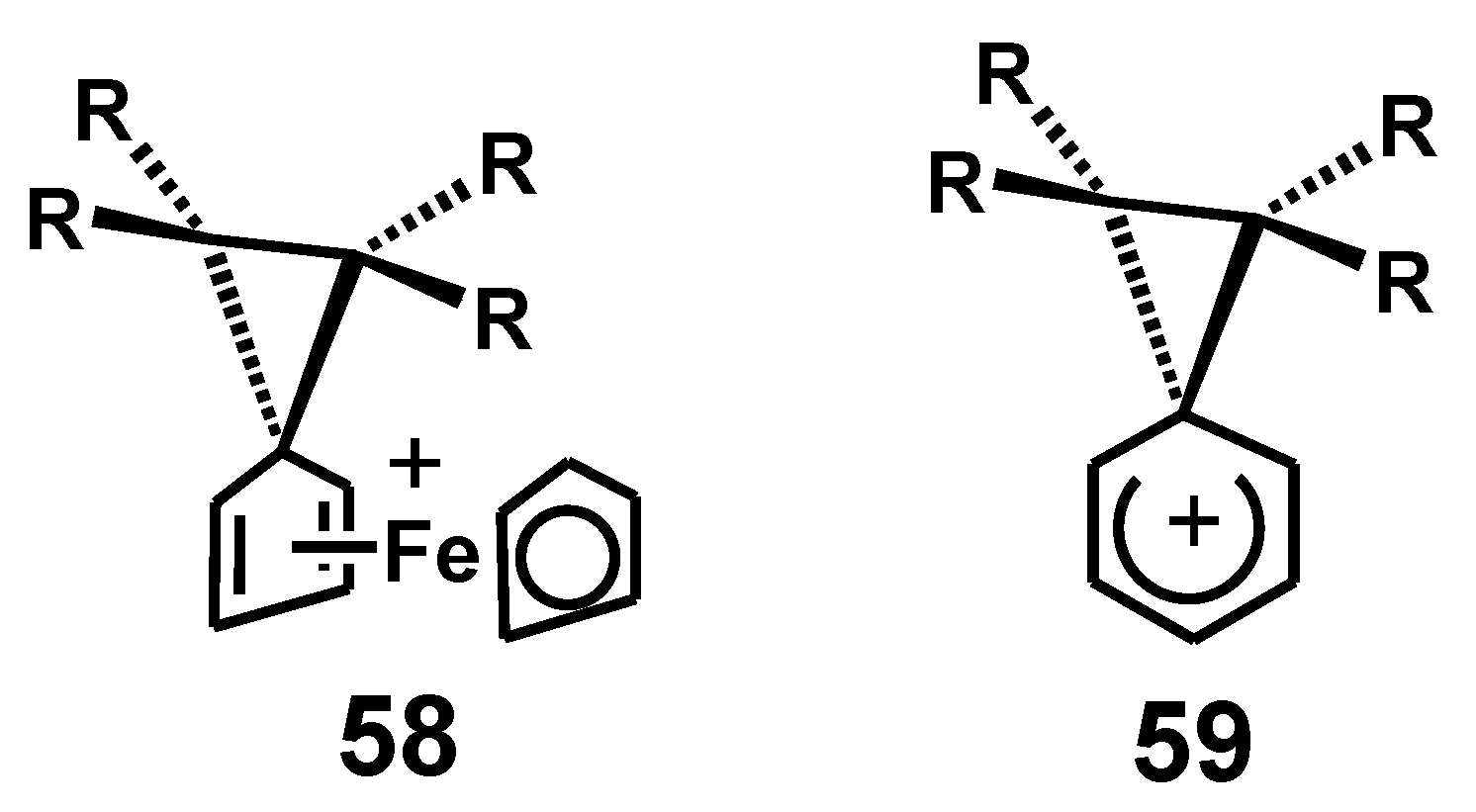
The corresponding bridging structure, 58, that facilitates a ferrocenyl migration would serve the same purpose (but markedly more efficiently) as the phenonium ion, 59, that allows delocalization of positive charge in the transition state for phenyl migrations (Figure 3).
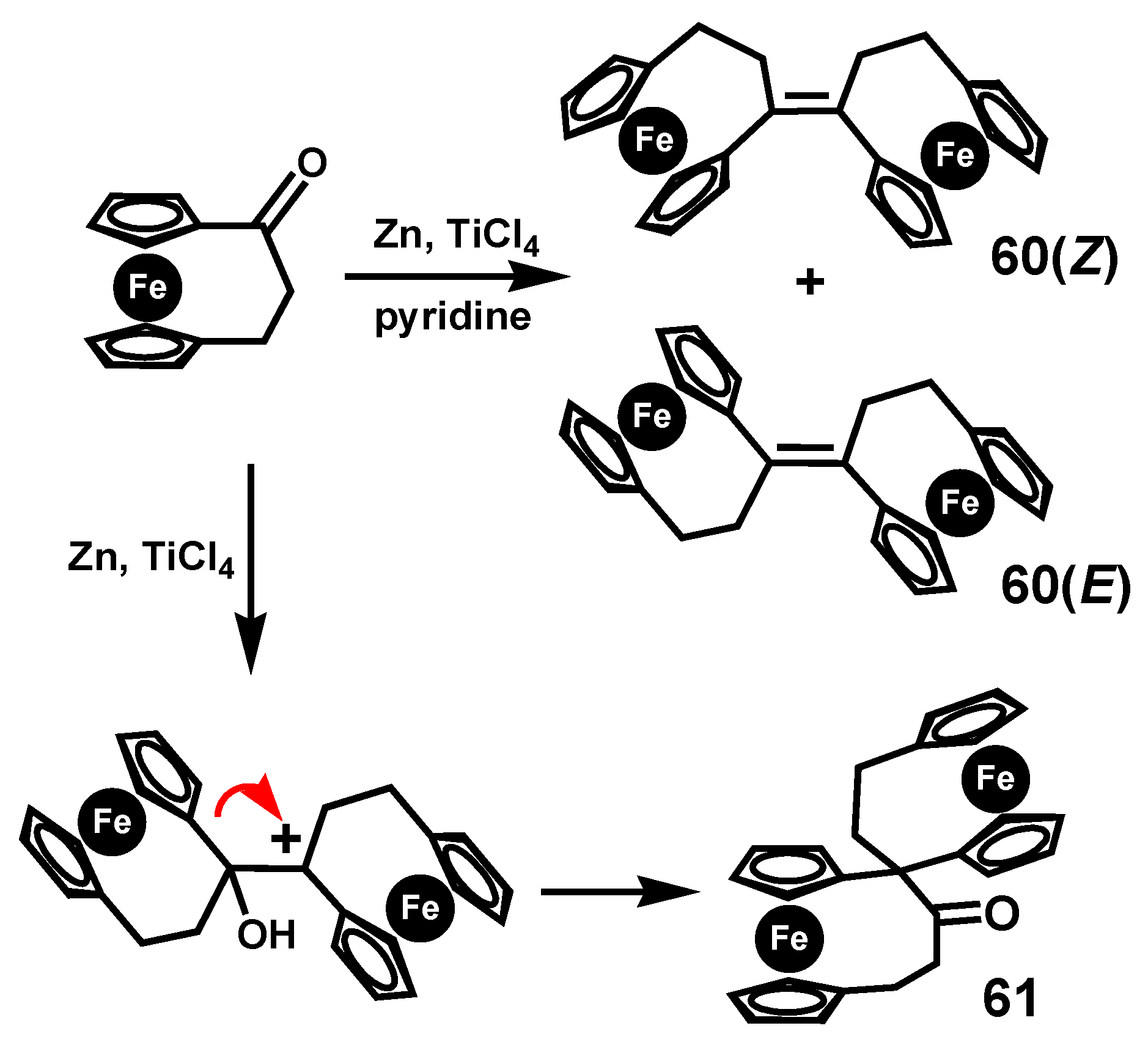
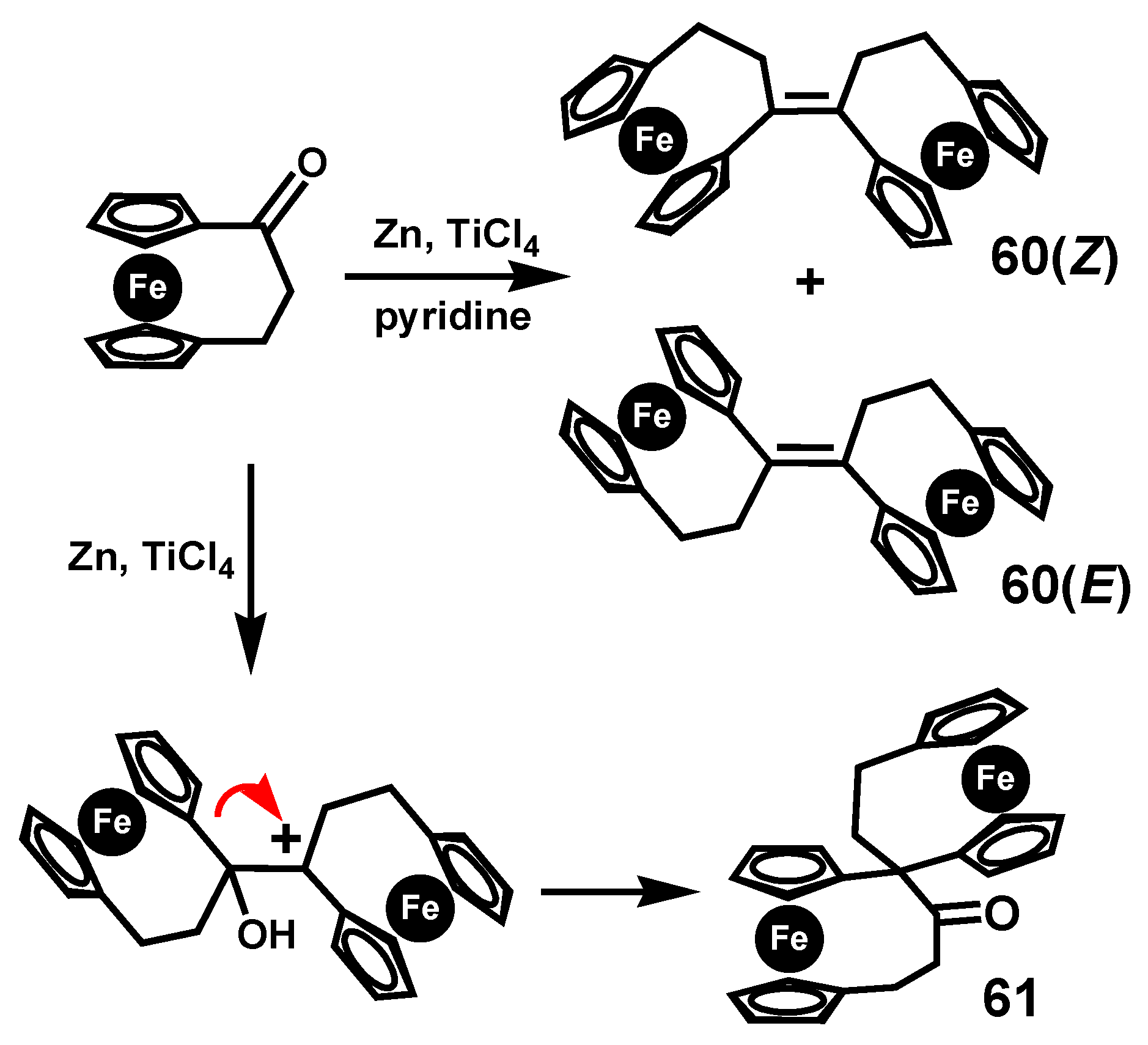
A particularly fascinating pinacol-to-pinacolone rearrangement was reported by Härter and colleagues who studied the McMurry reaction of [3]ferrocenophanone under different conditions [39]. When the standard reagent system (TiCl4/Zn) was augmented with pyridine, the homo-coupled product bis-[3]ferrocenophane-1-ylidene, 60, (as an E/Z mixture) was obtained in 70% yield (Scheme 18). In contrast, in the absence of pyridine, the major product was the spiro-linked pinacolone 61, which was unequivocally characterized by X-ray crystallography.
4.2. Wolff Rearrangements
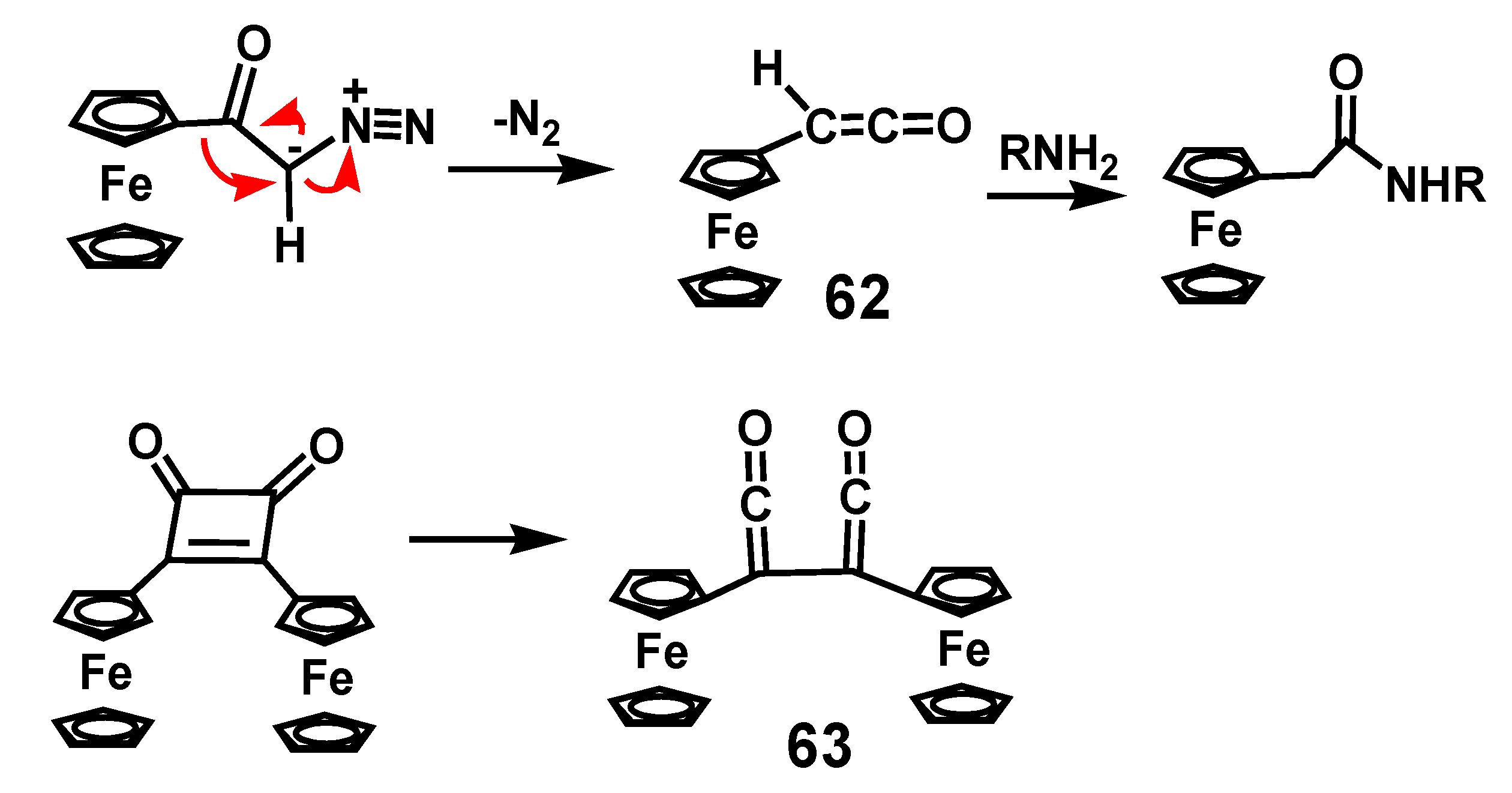
Thermal or photochemical dissociation of α-diazoketones liberates nitrogen to form the corresponding ketene. Whether the reaction involves formation of free carbenes, or proceeds via a concerted process is still the subject of discussion. Nevertheless, the ketenes produced by migration of the alkyl group onto the electron-deficient carbon are versatile species that react with nucleophiles, such as water, alcohols or amines, to form carboxylic acids, esters, or amides, but are also routinely used as partners in [2 + 2] cycloadditions [40]. Diphenylketene is a stable solid but the existence of ferrocenylketene, 62, has only been verified spectroscopically by laser flash photolysis, and was then trapped by reaction with n-butylamine [41]. Similarly, the bis-ketene 63 is detectable in the photolysis of 1,2-diferrocenyl-1-cyclobuten-3,4-dione (Scheme 19).
4.3. Rearrangements of Alkynyl-4-hydroxy-2-cyclobutenones
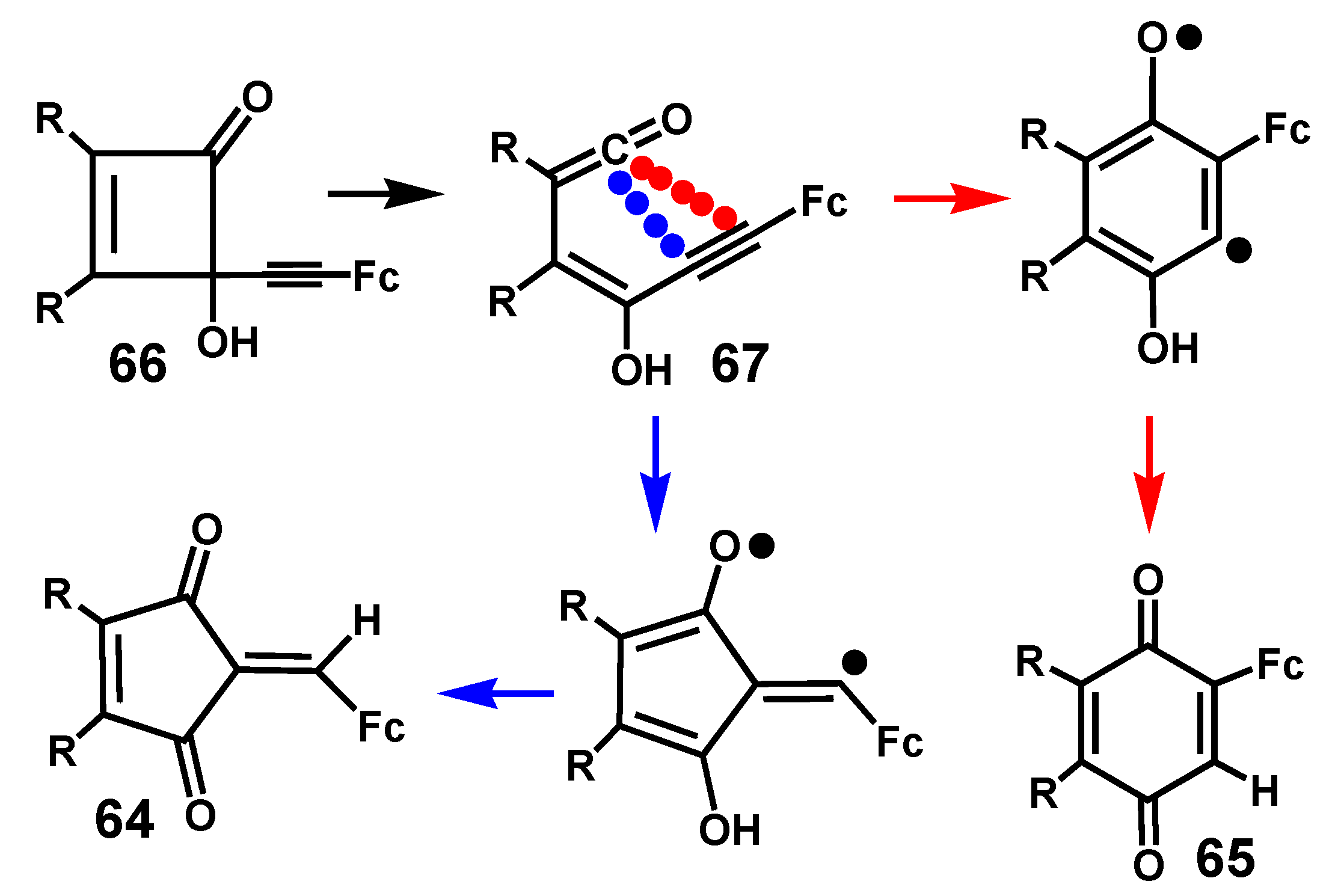
In the continuing search for novel anti-cancer agents, the 2-alkylidene-4-cyclopentene-1,3-dione framework [42] has attracted considerable recent attention. The remarkable bioactivity of ferrocenyl-containing molecules, in particular the ferrocifens [36], prompted the preparation of a series of 2-ferrocenylidene-4,5-dialkyl-4-cyclopenten-1,3-diones, 64. As shown in Scheme 20, these molecules are formed, along with their isomeric benzoquinones, 65, when 4-ferrocenyl-ethynyl-4-hydroxy-2-cyclobutenones, 66, undergo thermal rearrangement when heated at reflux in dioxane [43].
Although this reaction does not start from a diazoketone, the intermediacy of a ketene, and the role of the ferrocenyl unit in stabilizing adjacent cations or radicals, reveal some parallels to the Wolff rearrangement. Upon heating, 66 undergoes electrocyclic ring opening to form the conjugated ketene 67 that can ring-close in two different ways. In each case, the diradical so-formed then proceeds by hydrogen migration to yield the observed products.
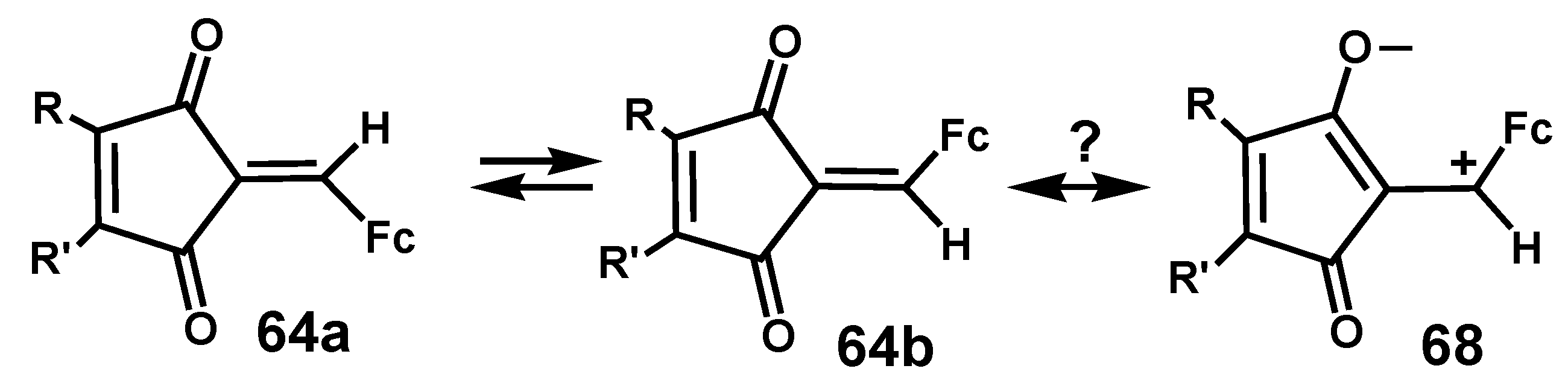
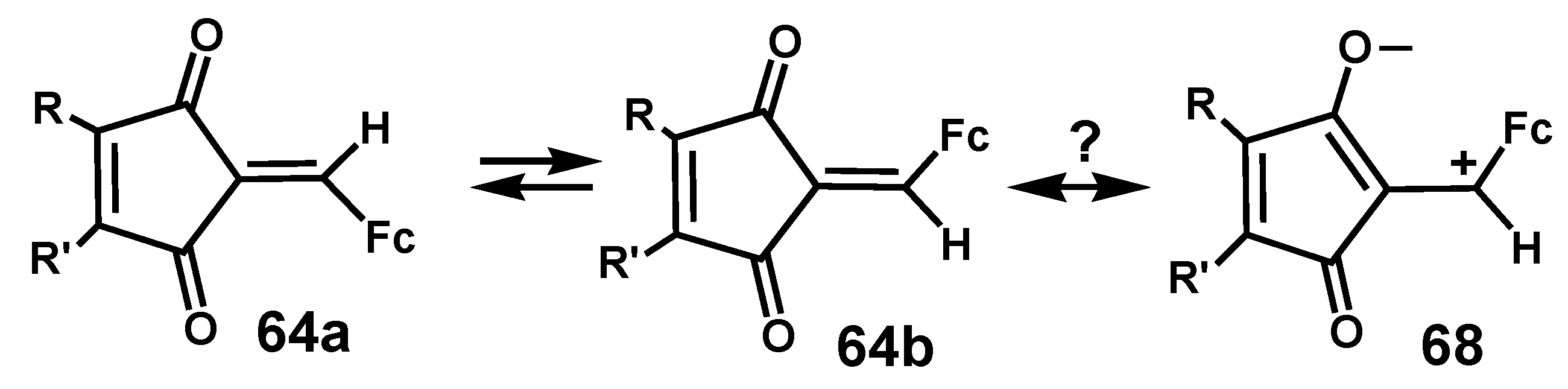
Interestingly, when R ≠ R’, the two geometric isomers 64a and 64b interconvert when heated at 125 °C, and it has been suggested that a zwitterionic contribution, 68, to the structure would facilitate rotation about the C=CHFc linkage (Scheme 21).
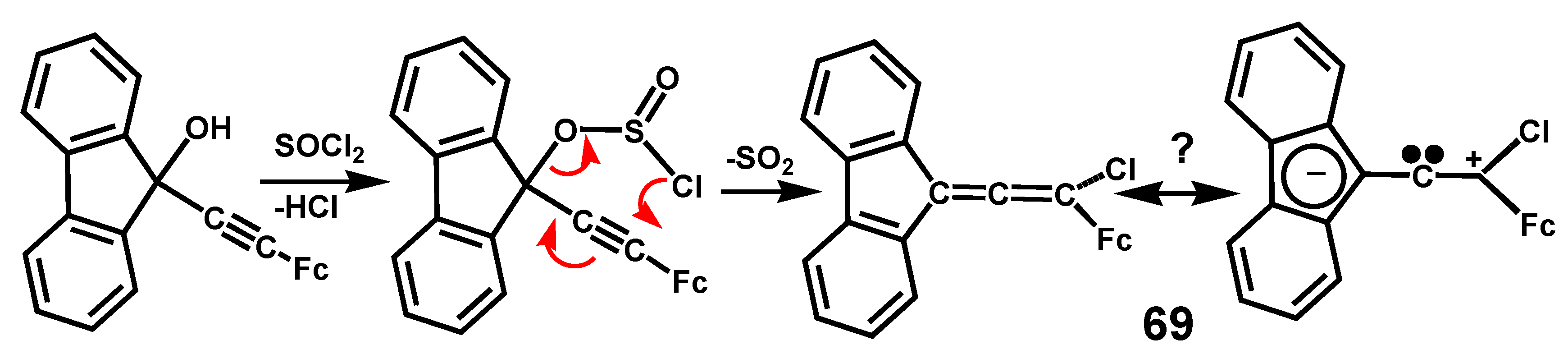
In a somewhat comparable situation, the potential push-pull ferrocenylidene-fluorenyl allene, 69, that was synthesized by Meyer-Schuster rearrangement of the precursor alkynol with thionyl chloride (Scheme 22), was investigated to test the possibility that the terminal groups might stabilize the separate charges thus generating carbene character at the allene central carbon. Although the 13C NMR spectrum of 69 revealed a peak at 198 ppm, in the normal range for the central carbon in an allene, the molecule was stable only below −20 °C and readily dimerized [44].
5. Ferrocenyl Migration onto an Electron-Deficient Nitrogen Center
5.1. Beckmann Rearrangements
Oximes are readily preparable by reaction of the precursor aldehyde or ketone with hydroxylamine hydrochloride [45]. When treated with reagents (e.g., H2SO4, PCl5, or SOCl2) that convert the hydroxyl into a leaving group, it brings about migration of the anti substituent to form a nitrilium ion, 70; typically, Beckmann rearrangement of the ferrocenyl oxime 71 and subsequent hydrolysis yielded the secondary amide 72 (Scheme 23) [46]. This specific geometric requirement contrasts with the situation in the pinacol rearrangement whereby the identity of the migrating group is controlled primarily by its migratory aptitude rather than its geometric alignment. Interestingly, several carbamate derivatives of ferrocenyl ketoximes have demonstrated acetylcholin-esterase activity [47].
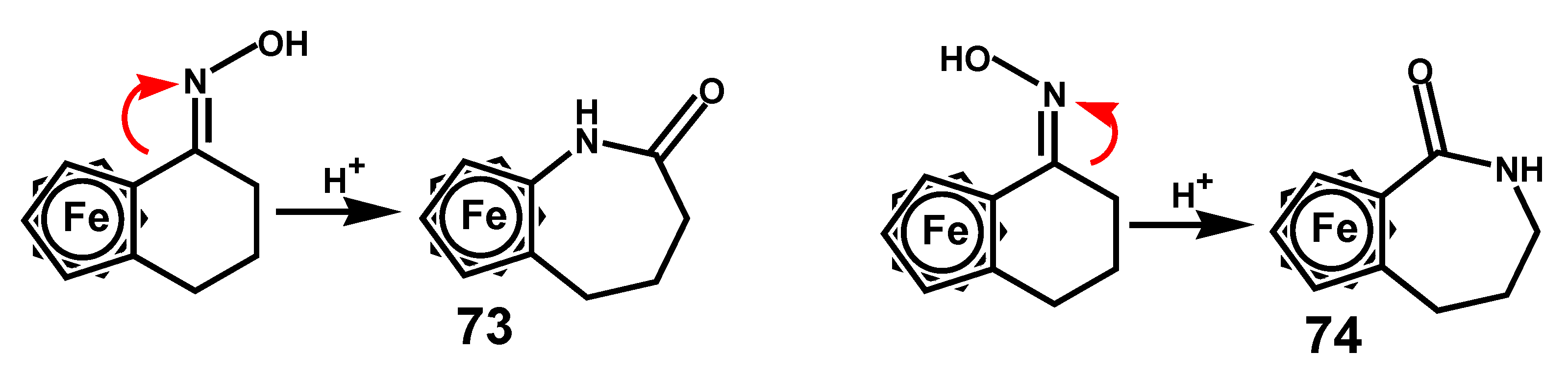
While the Beckmann reaction was extensively explored in pioneering investigations by Schlögl involving a range of alkyl- or aryl-substituted ferrocenyl oximes, probably the most explicit examples are those derived from ferrocenocyclohexanones, as in Scheme 24. In these cases, the structures of the resulting ferrocenocaprolactams, 73 and 74, are clearly controlled by the syn or anti orientation of their precursor oximes [46].
5.2. Curtius Rearrangements
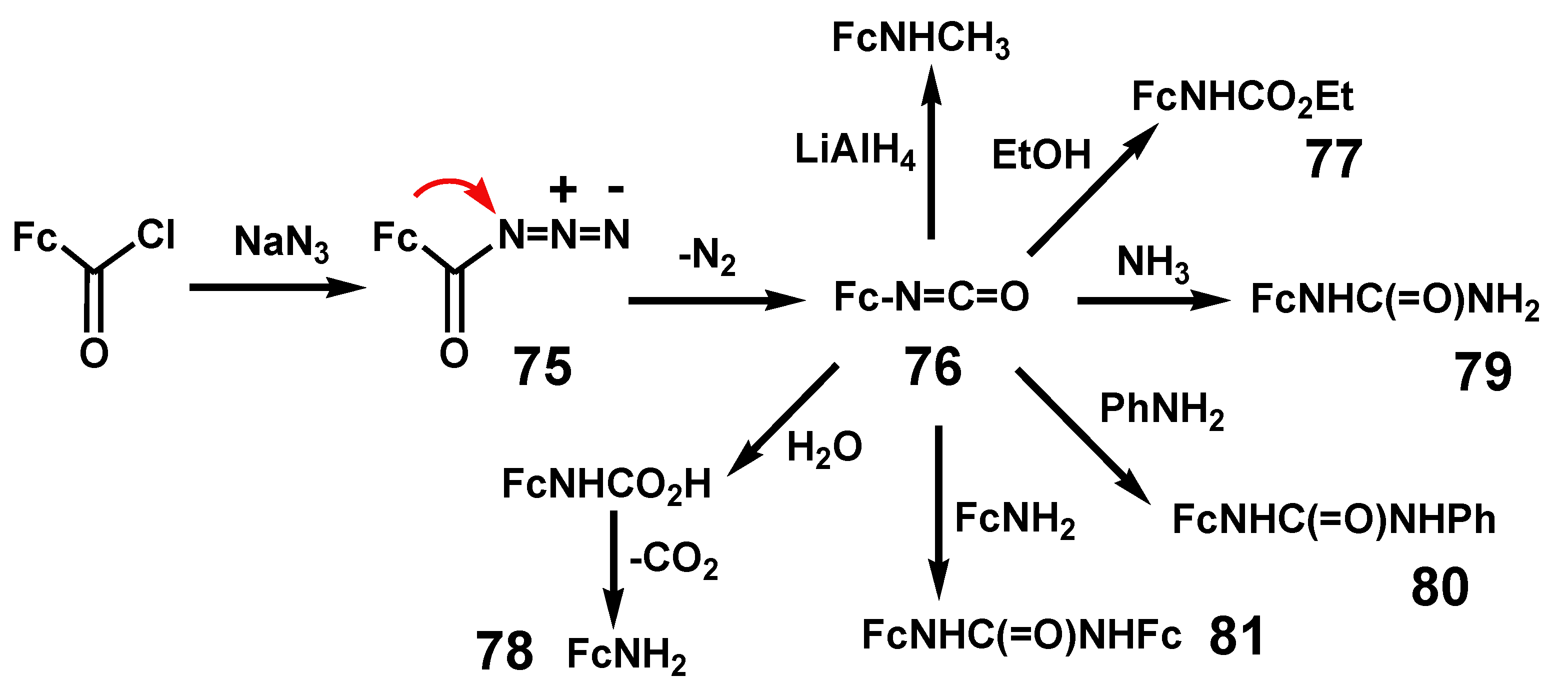
Thermolysis of acyl azides proceeds with loss of nitrogen to form isocyanates. Although they can be isolated, the reaction is often carried out in the presence of a nucleophilic solvent such as an alcohol or amine to form esters or ureas. In contrast to the Wolff rearrangement that can proceed via a carbene intermediate, the Curtius rearrangement is almost certainly a concerted process that does not involve a discrete nitrene [25]. Rearrangement of ferrocenoyl azide, 75, by thermolysis in refluxing dry toluene furnished ferrocenyl isocyanate, 76. As shown in Scheme 25, reaction with ethanol or water produces ethyl ferrocenylcarbamate, 77, or ferrocenylcarbamic acid, prior to loss of CO2 and formation of ferrocenylamine, 78. In addition, ammonia, aniline, and ferrocenylamine react with ferrocenoyl isocyanate to yield N-ferrocenylurea, 79, N-ferrocenyl-N’-phenylurea, 80, and N,N’-diferrocenylurea, 81, respectively [48].
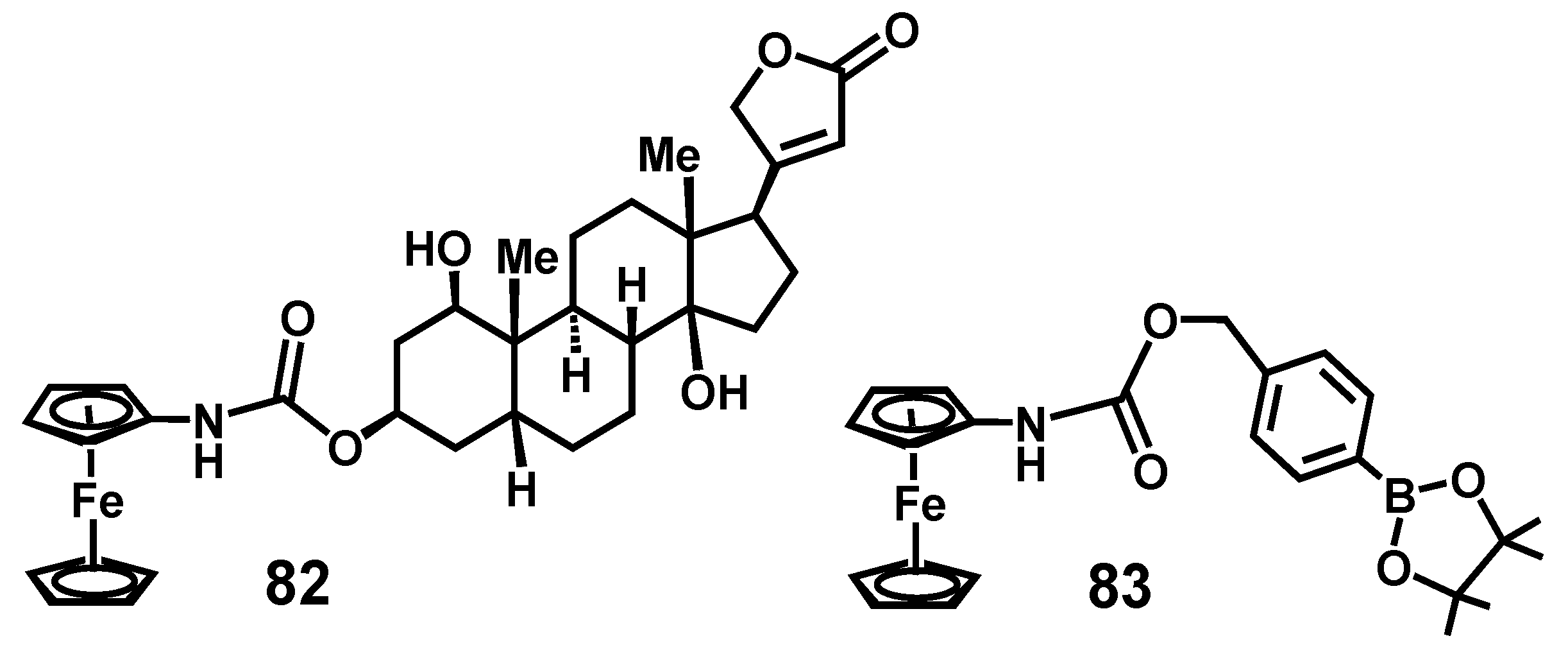
Several practical applications of this procedure have since been reported. Curtius rearrangement of ferrocenoyl azide in the presence of a number of hydroxysteroids to form carbamates, such as 82 (Figure 4), has been used in high performance liquid chromatography using electrochemical detection. Maximum sensitivity was achieved by using a silver–silver chloride reference electrode with a detection limit of 0.5 pmol. This derivatization procedure has allowed the facile characterization of the products of the bioconversion of digoxigenin [49]. Moreover, in a more recent study, it has been shown that molecules in which the ferrocenyl carbamate unit is linked to a boronic acid ester, as in 83, provide a very sensitive and rapid electrochemical detection technique for hydrogen peroxide and glucose; this can be used to follow enzyme activity and cell signaling pathways [50].
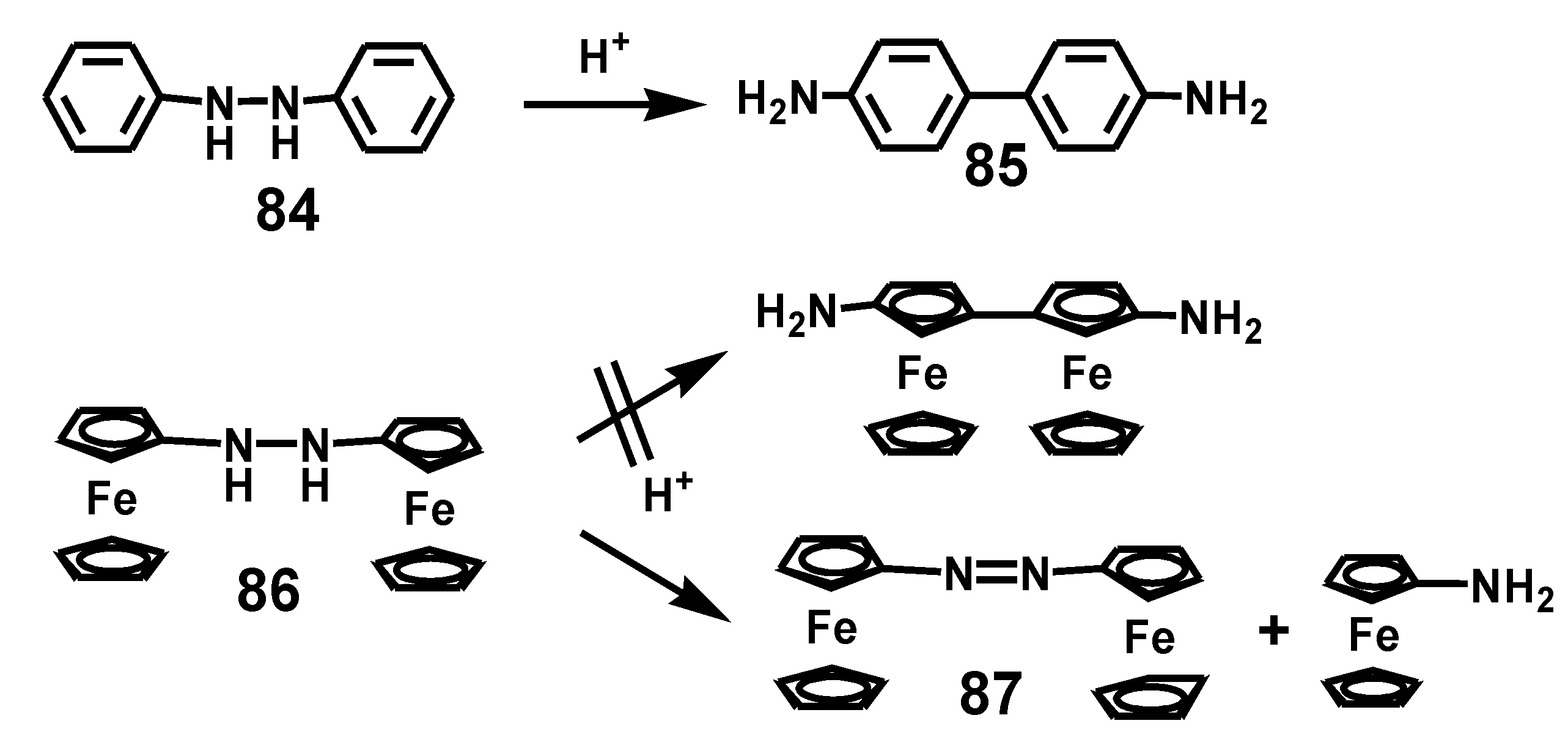
5.3. Benzidine Rearrangement
Protonation of hydrazobenzene, 84, brings about cleavage of the HN–NH linkage and coupling of the phenyl substituents, normally in the para-positions to form 4,4’-diaminobiphenyl (benzidine), 85. In the course of their pioneering studies of derivatives of ferrocene, the Nesmeyanov group attempted the benzidine rearrangement of hydrazoferrocene, 86, but to no avail (Scheme 26). The major products arose from disproportionation to form azoferrocene, 87, and ferrocenylamine [51]. When the ferrocenyl unit was instead incorporated as a substituent in hydrazobenzene, the major products upon treatment with HCl were ferrocenylazobenzene, ferrocenylaniline, and aniline, again resulting from disproportionation rather than rearrangement [52].
6. Anionic Rearrangements of Ferrocenyl Compounds
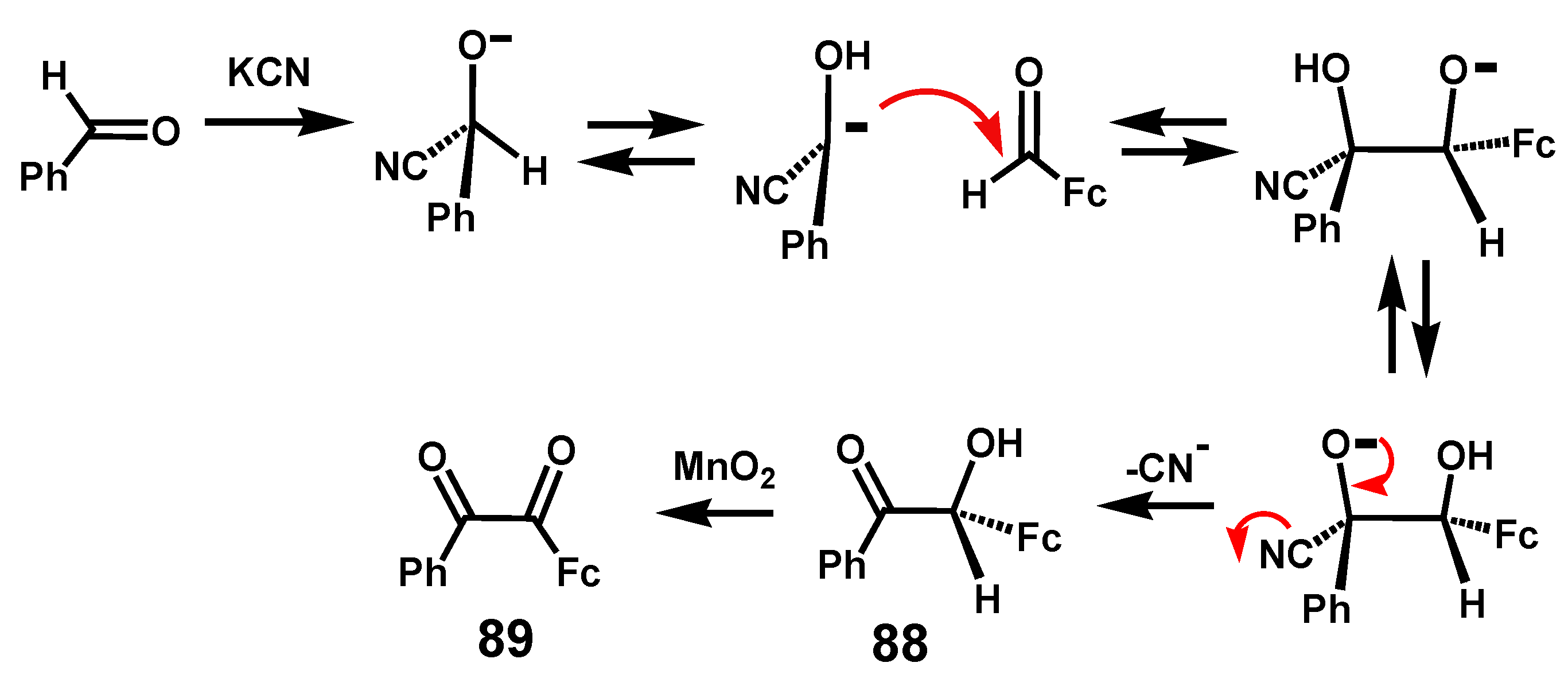
6.1. Benzoins and Benzils
The cyanide-catalyzed condensation of aromatic aldehydes, with subsequent oxidation of the benzoin so-formed, provides a convenient route to benzils [53]. However, when starting from formylferrocene, this procedure failed. Moreover, the mixed benzoin condensation of benzaldehyde and formylferrocene yields only a single product, Ph(C=O)–CH(OH)Fc, 88 [54]. As is evident from the mechanism shown in Scheme 27, this situation arises because nucleophilic attack by cyanide at the ferrocenyl carbonyl, followed by tautomerization, would generate a carbanionic site adjacent to the sandwich moiety—evidently an unfavorable and unlikely circumstance. Instead, the cyanide undergoes addition to the benzaldehyde carbonyl group. Oxidation with MnO2 yields the mixed benzil Fc(C=O)(C=O)Ph, 89 [54].
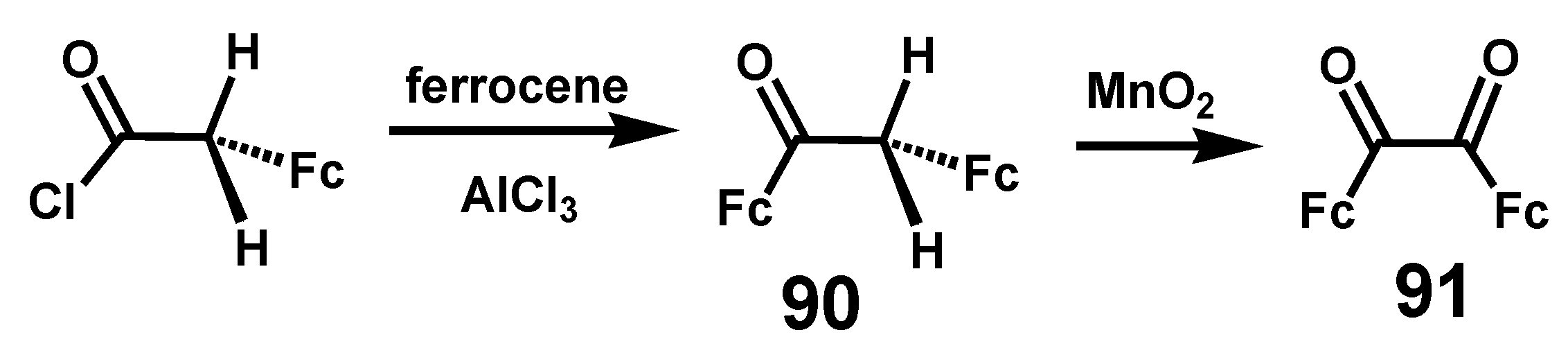
However, an indirect route to ferrocil (Scheme 28) was successfully achieved by Rinehart who found that the Friedel–Crafts acylation of ferrocene with the acid chloride of ferrocenylacetic acid yielded ferrocenylacetylferrocene (desoxyferrocoin), 90 [55]; subsequent oxidation with freshly prepared MnO2 furnished ferrocil, 91, in 86% yield [56].
In a modification of this approach, the Nesmeyanov group showed that the MnO2-mediated oxidation of 1,2-diferrocenylethane furnished trans-diferrocenylethene, 47, and small quantities of ferrocil. Satisfyingly, further oxidation of the alkene gave 91 in 40% yield [57]. Unsurprisingly, ferrocil does not undergo the benzil-to-benzilic acid rearrangement; once again, such a process would have required the migration of a ferrocenyl unit under electronically unfavorable anionic conditions.
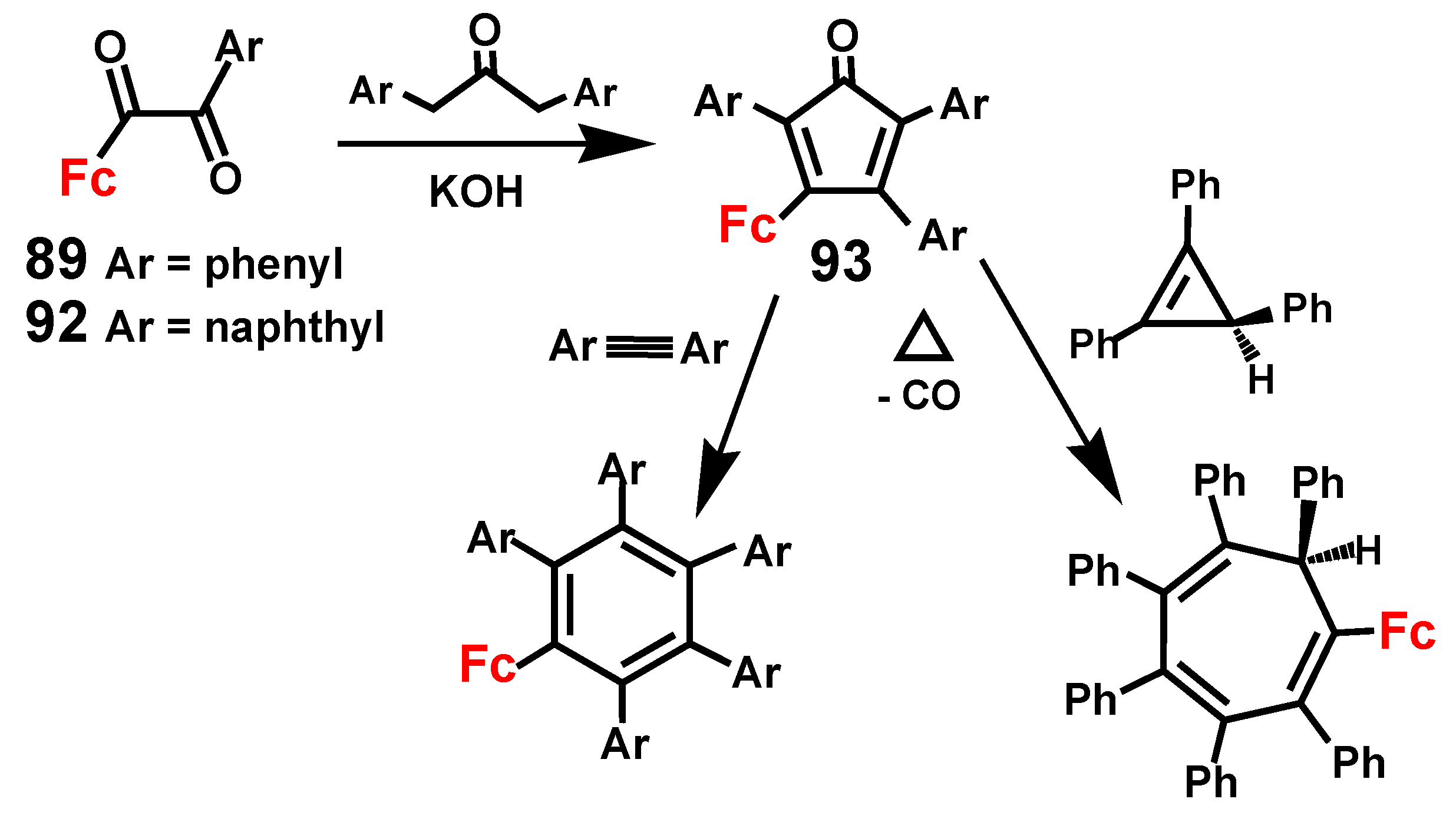
The ready availability of the mixed benzils 1-ferrocenyl-2-aryl-1,2-dioxoethane, 89 and 92, has been shown to provide a convenient route to ferrocenyl-triaryl-cyclopentadienones, 93 that are versatile precursors to the sterically hindered arenes C6R5Fc, where R = phenyl or β-naphthyl, and the cycloheptatriene C7Ph6HFc (Scheme 29) [54,58,59].
6.2. Stevens Rearrangement
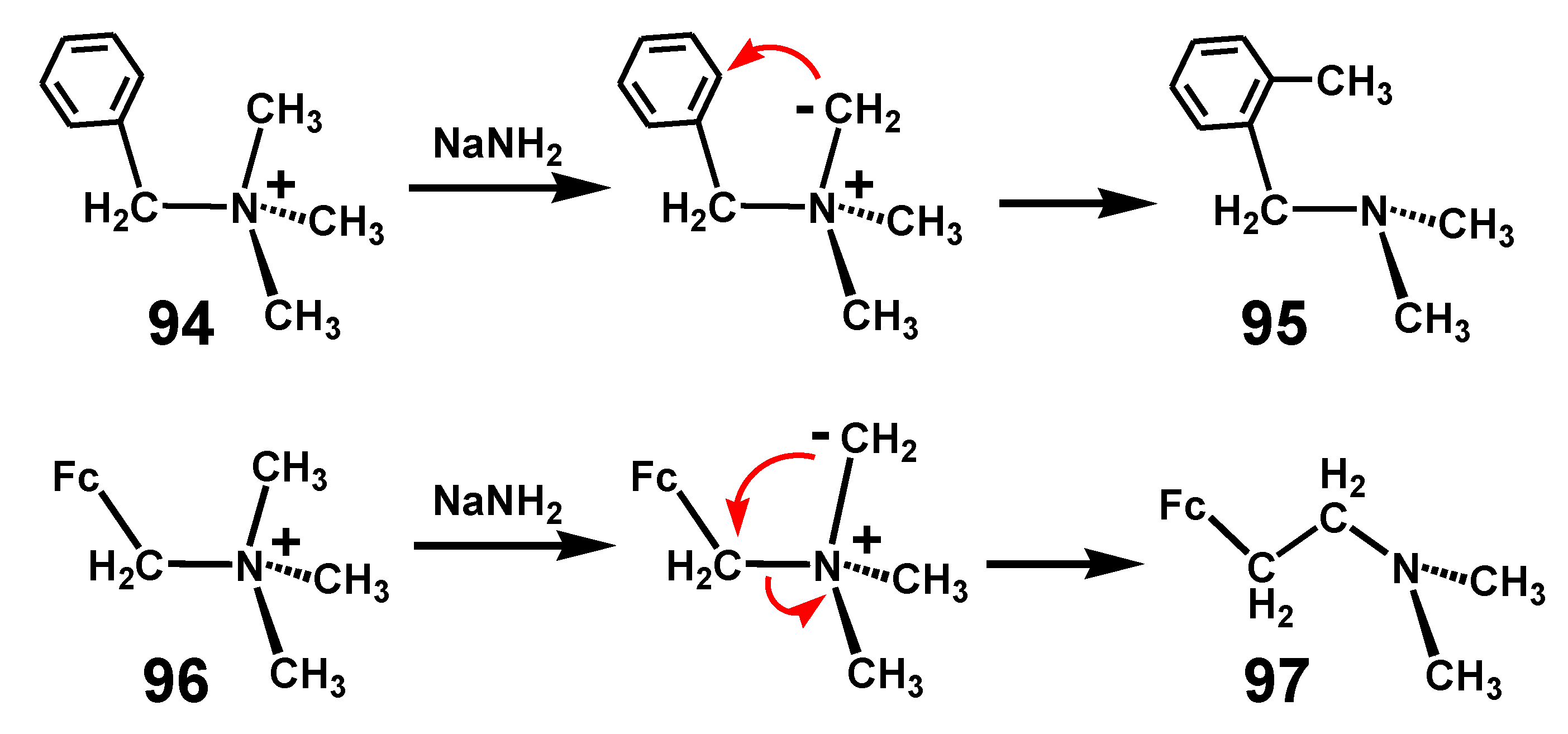
In a conventional Stevens rearrangement, deprotonation of a quaternary ammonium salt generates an ylid whereby one of the substituents undergoes migration from nitrogen. Typically, as shown in Scheme 30, under these conditions the benzyltrimethylammonium salt, 94, is converted to 2-methylbenzyldimethylamine, 95. In contrast, upon methylation to form 96, N,N-dimethyl-aminomethylferrocene undergoes a quite different isomerization process to generate N,N-dimethylaminoethylferrocene, 97 [60]. This may be rationalized on the basis that, as noted in Section 6.1, the ferrocenyl moiety is less willing than phenyl to tolerate negative charge. We note, however, that more recently, it has been suggested that the mechanism of the Stevens rearrangement may actually involve radical coupling within a solvent cage [61].
7. Ferrocenes in Bioorganometallic Chemistry
7.1. Quinone Methides
Bioorganometallic chemistry, a neologism introduced by a small group of us in the mid-1980s [62,63], focuses on the bioactivity of complexes possessing at least one metal-carbon bond. It was developed to extend the range and efficacy of metal-based systems beyond that of the widely-used platinum drugs. Although many transition-metal complexes have been studied, ferrocifens, the ferrocenyl analogues of tamoxifen, the current first-line treatment of hormone-dependent breast cancers, whereby a phenyl has been replaced by ferrocenyl, have proved particularly successful against both hormone-dependent and hormone-independent tumors [64].
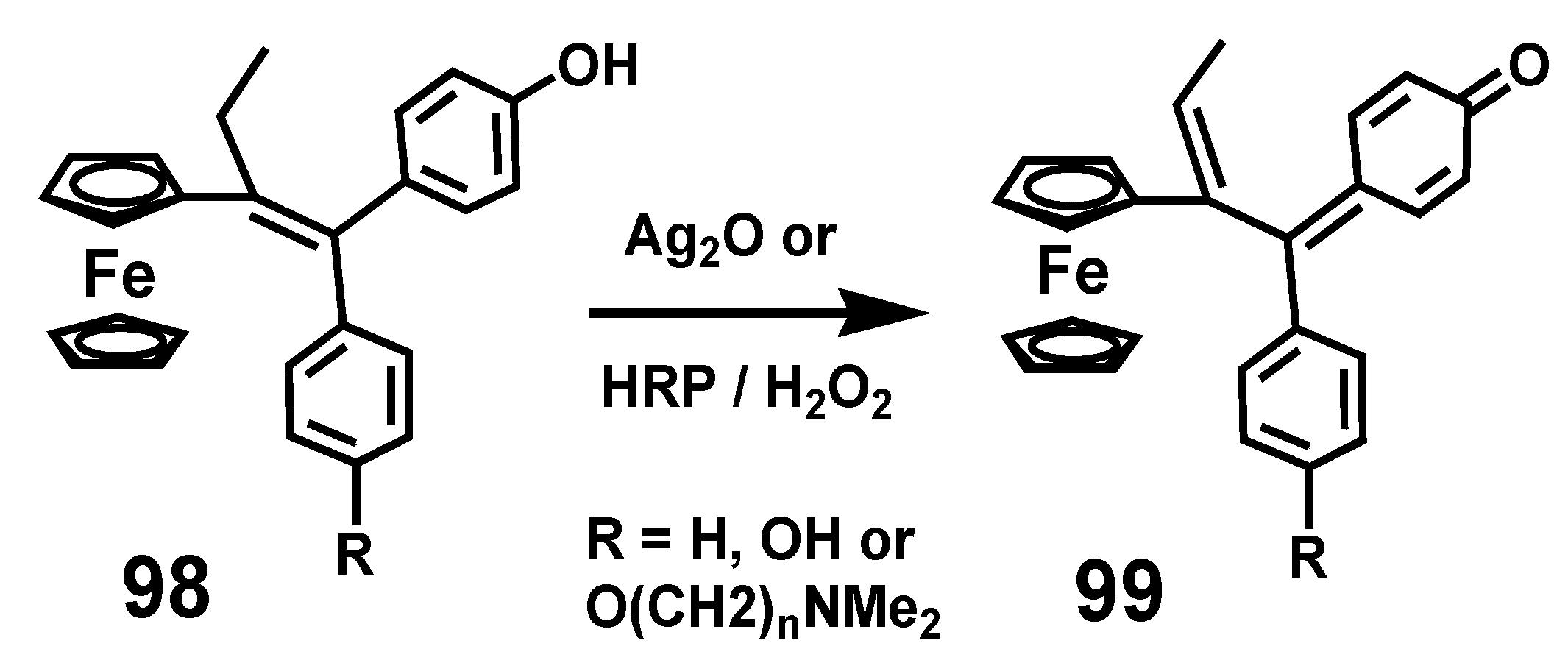
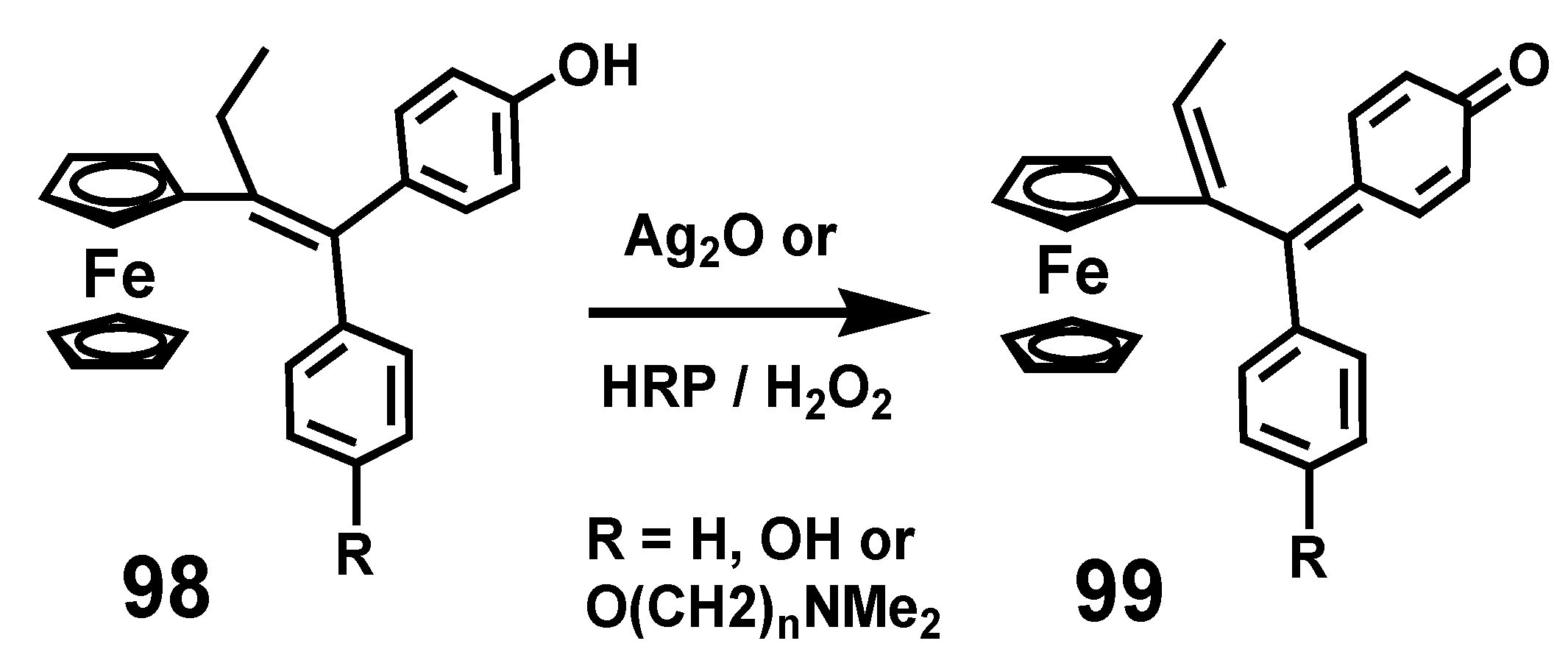
Their activity is predicated on the reversible redox activity of the ferrocenyl moiety that mediates conversion of ferrociphenols, 98, into the corresponding quinone methides, 99, that undergo Michael additions with thiols or selenols of key proteins crucial to maintaining redox balance in the cell [65,66,67]. The net result is to inhibit the regeneration of thioredoxin that defends the cell from attack by reactive oxygen species (ROS), such as hydroxyl radicals. The mode of action of thioredoxin is to trap the ROS by abstraction of hydrogen from its vicinal cysteine thiols, which then form disulfides, a process reversible only by thioredoxin reductase in conjunction with NADPH [67]. Generation of these quinone methides can also be accomplished in vitro by chemical oxidation of ferrociphenols with silver oxide, or enzymatically by using horseradish peroxidase (HRP/H2O2), as depicted in Scheme 31.
7.2. Formation of Indenes
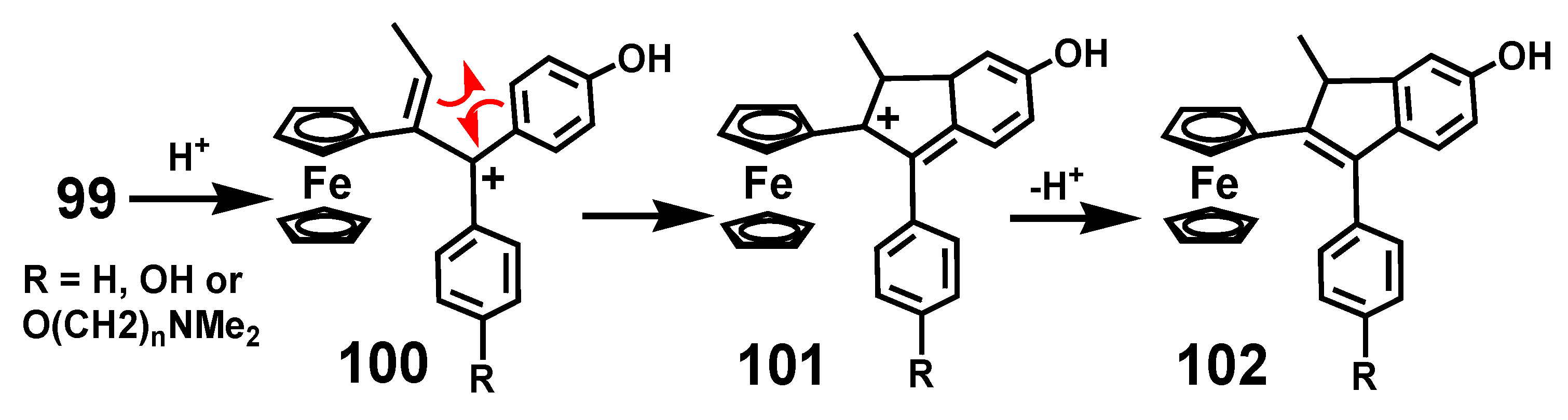
The rearrangement behavior of these bioactive ferrocenyl-containing molecules parallels that of the more conventional systems discussed already. Protonation of ferrocenyl quinone methides brings about rearomatization of the quinone ring and initially generates a doubly-benzylic carbocation, 100, that readily cyclizes so as to place the electron deficiency adjacent to the ferrocenyl substituent, as in 101, leading ultimately to formation of indene 102 (Scheme 32) [68], entirely analogous to the reactions described in Section 2.2.
7.3. Formation and Rearrangement of a Ferrocenyl-Tetrahydrofuranyl Quinone Methide
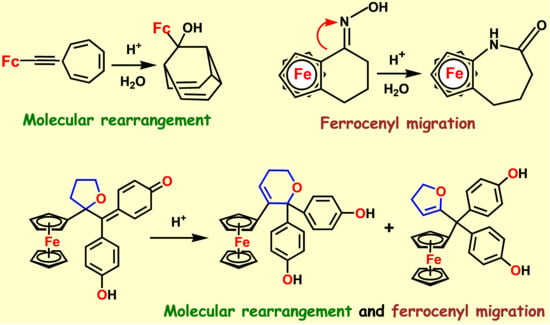
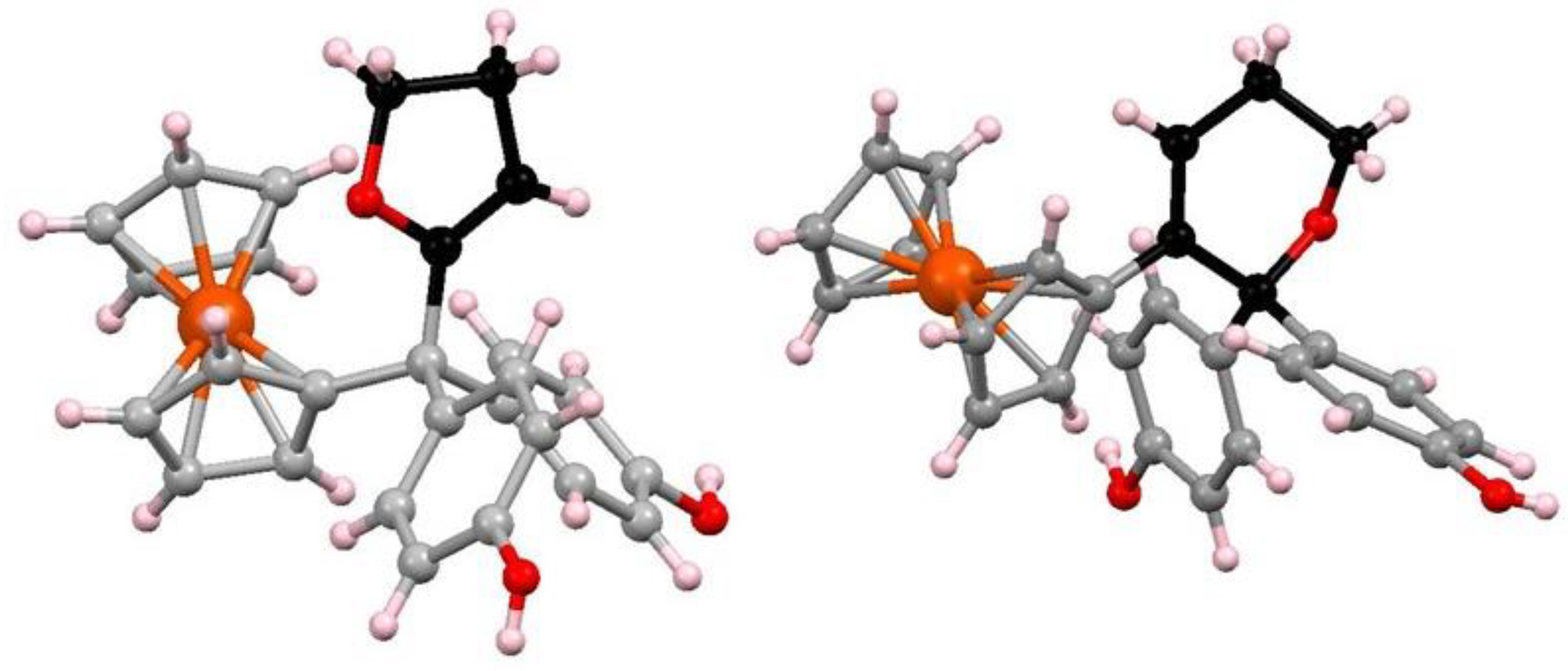
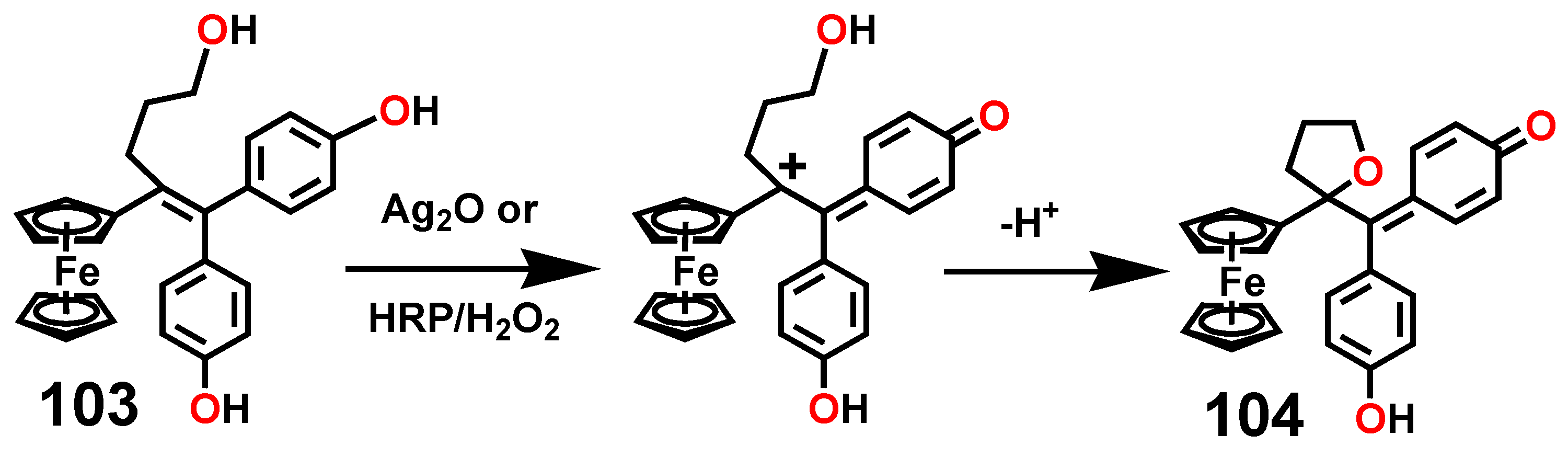
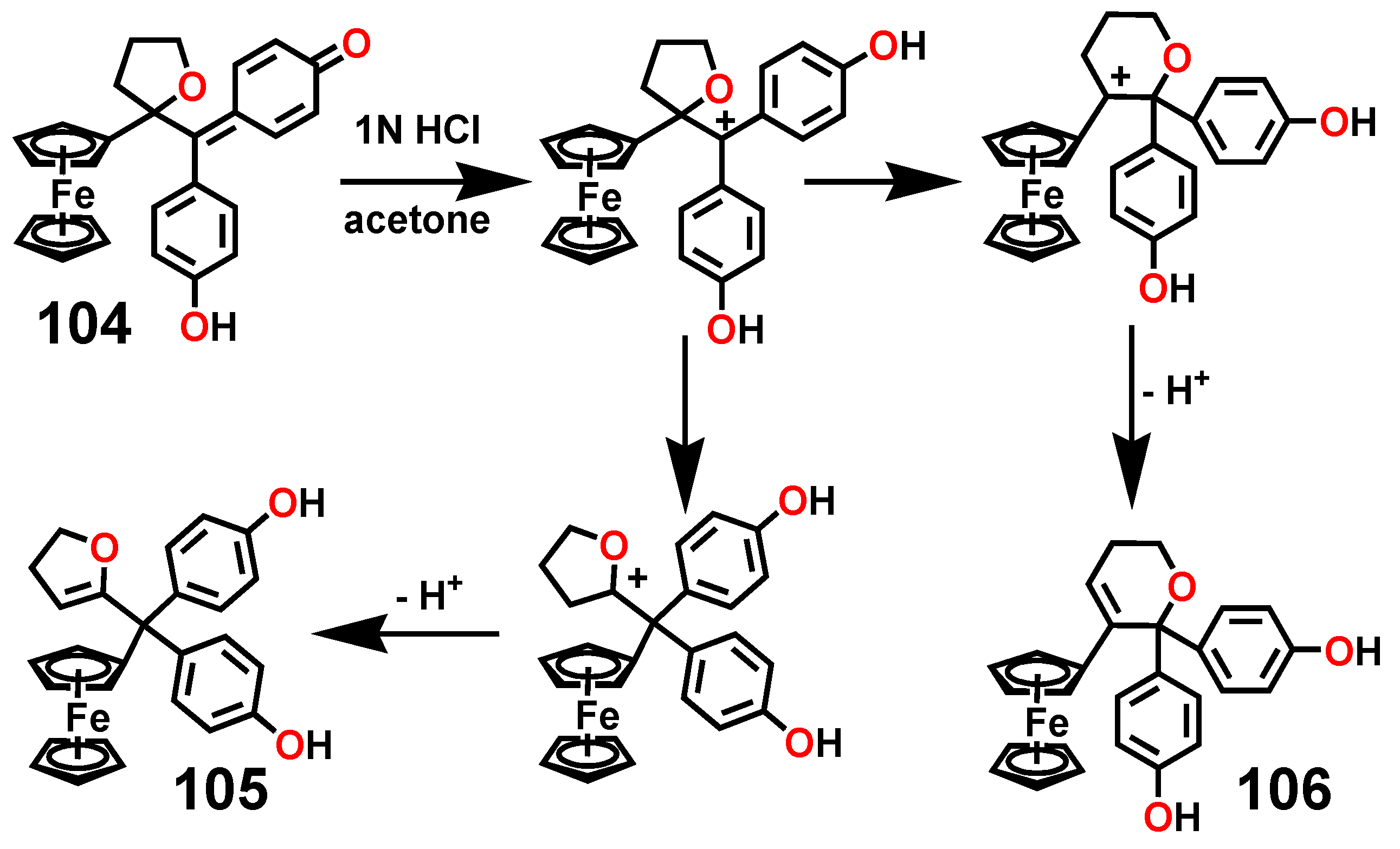
A particularly interesting transformation occurs in the enzymatic or chemical oxidation of the hydroxypropyl-ferrociphenol, 103, that yields, among other products, a spiro-bonded tetrahydrofuran derivative, 104 (Scheme 33). When protonated, this molecule undergoes both ferrocenyl migration and ring expansion to yield the dihydrofuran, 105, and the dihydropyran, 106, respectively (Scheme 34) [65]. Their X-ray crystal structures are shown as Figure 5.
7.4. Pinacol-to-Pinacolone Rearrangement of Ansa-Ferrociphenols
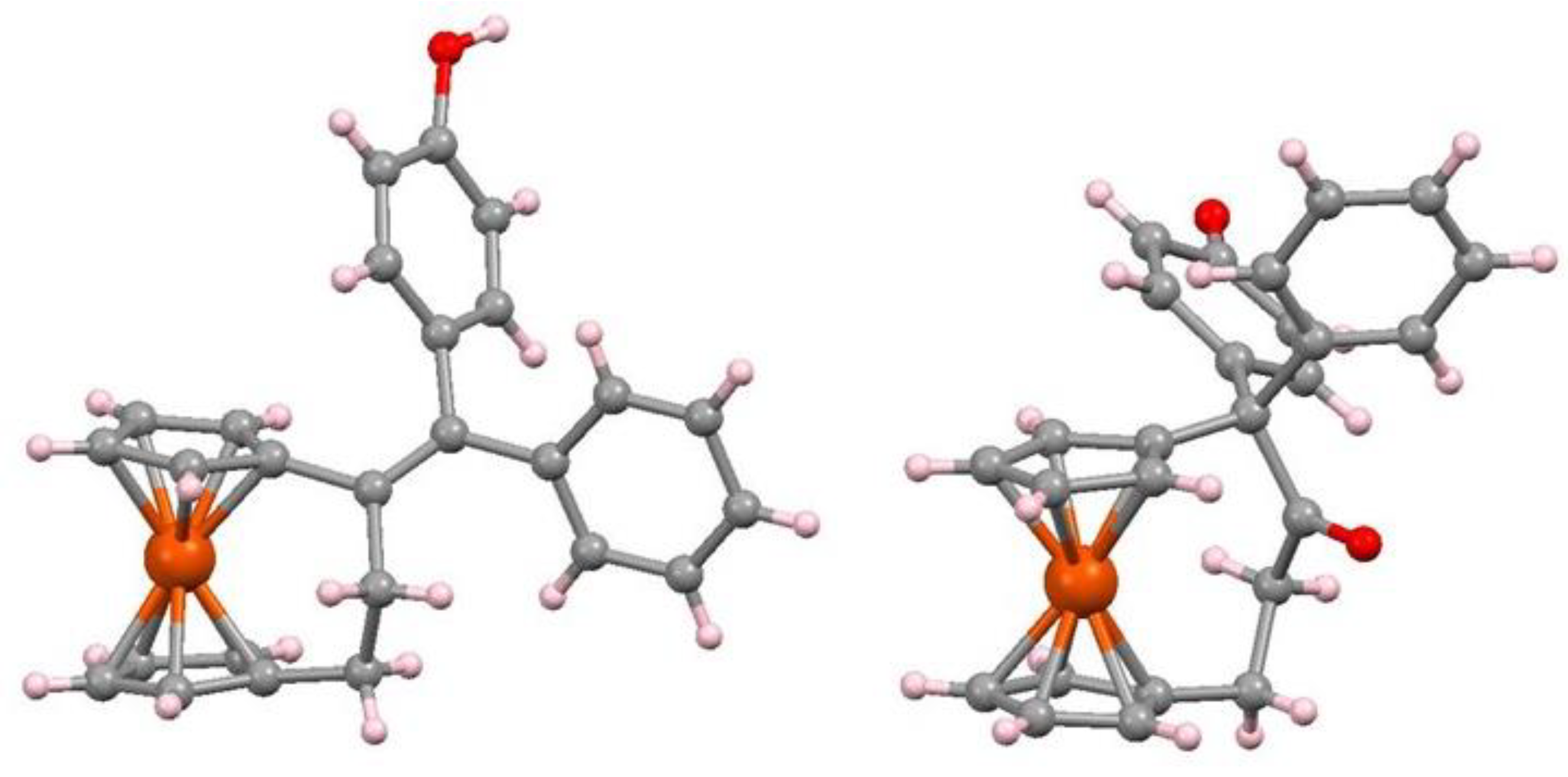
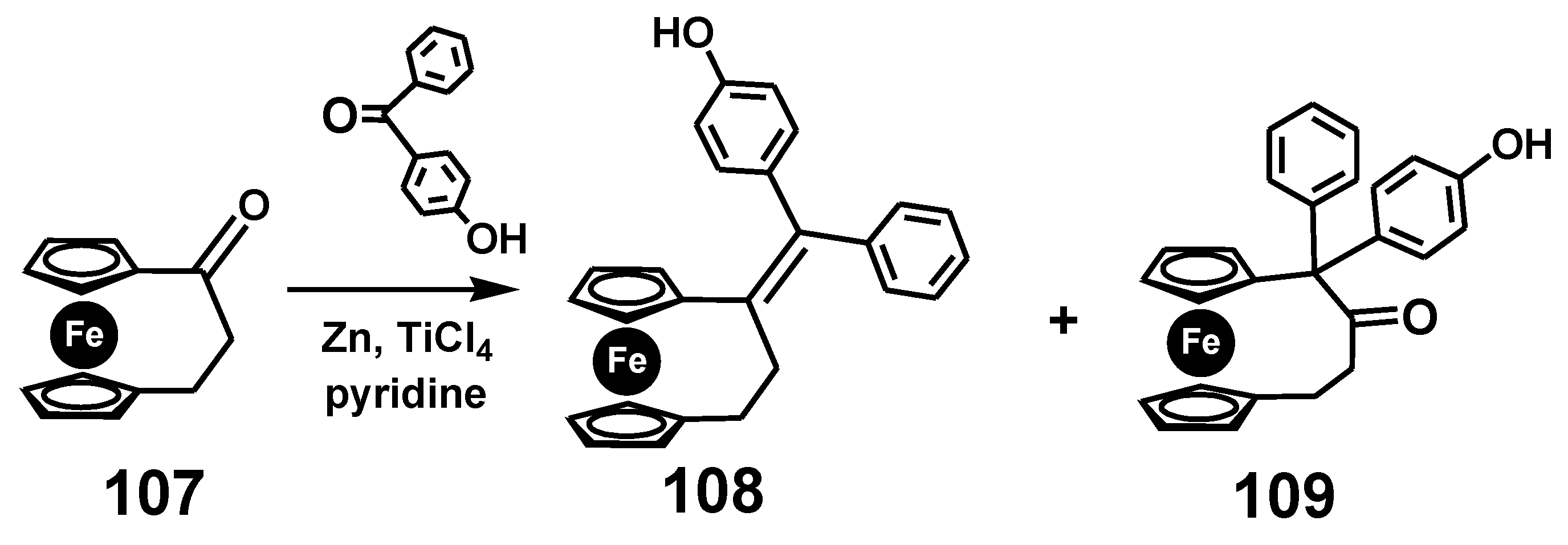
In yet another example in the bioorganometallic arena, the synthesis of an ansa-ferrociphenol from [3]ferrocenophanone, 107, by McMurry coupling yields not only the desired alkene, 108, but also the corresponding pinacolone, 109, (Scheme 35) both of whose X-ray crystal structures are shown as Figure 6.
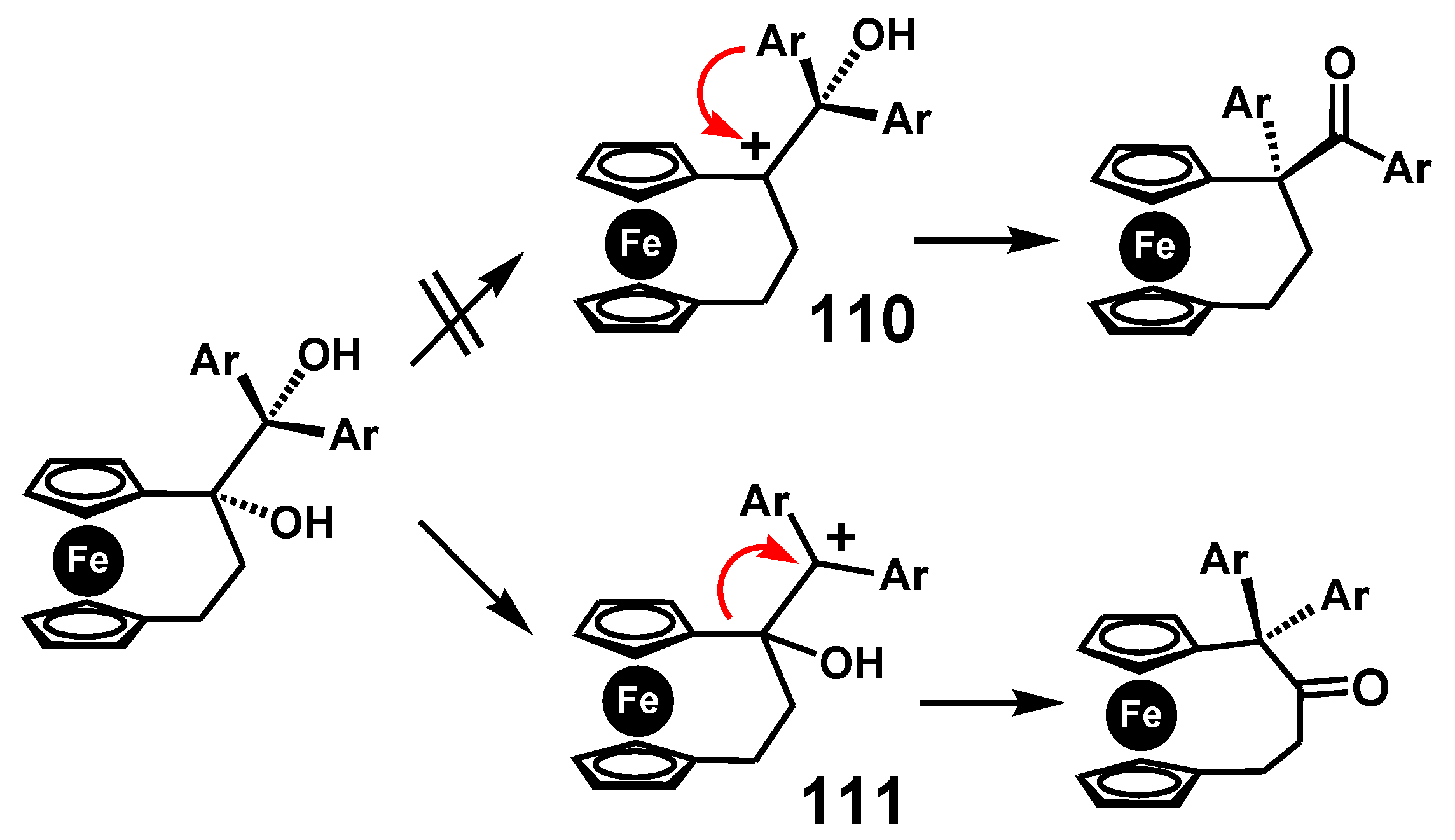
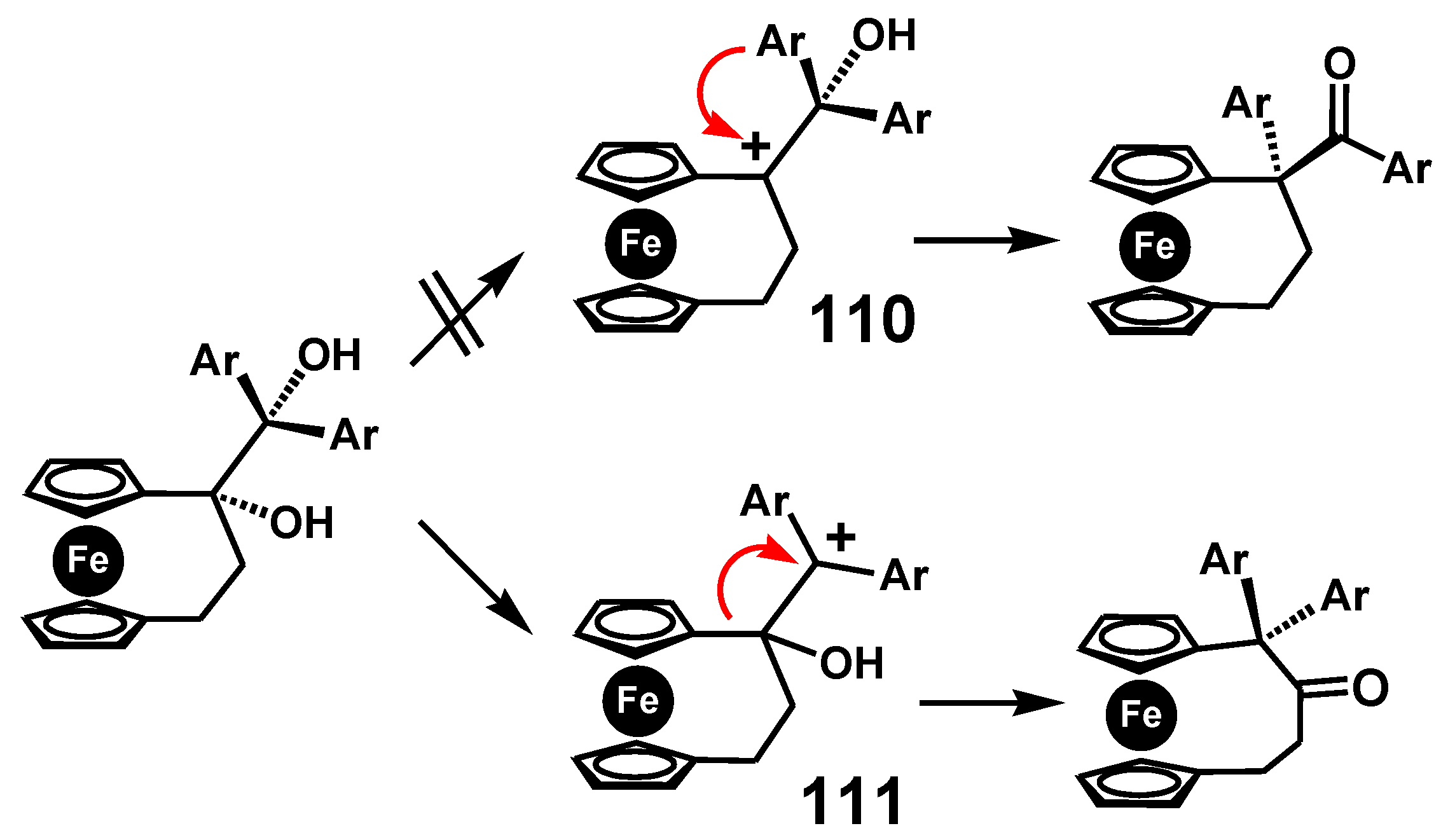
In this case, there are two possible pinacolones: one derived by cation formation adjacent to the ferrocenyl group, as in 110, the other at the doubly-benzylic position, 111, as illustrated in Scheme 36. In the event it is the latter that succeeds, it is presumably to relieve the ring strain in the three-carbon bridge thereby forming the pinacolone possessing a four-carbon link between the cyclopentadienyl rings [69]. Moreover, in this strained system, the potential carbocation, 110, adjacent to ferrocene, is unable to enhance its stability by bending towards the metal.
8. Concluding Comments
The remarkable ability of the ferrocenyl moiety to stabilize a neighboring carbocationic site manifests itself in a number of ways. In some cases, such as [(C5H5)Fe(C5H4-CPh2][BF4], the species is sufficiently stable to be isolated and structurally characterized by X-ray crystallography. In others, it directs the course of an isomerization or migration by stabilizing a transition state or intermediate by alleviating the electron deficiency and tolerating any excess positive charge on the iron atom.
It is particularly gratifying that the incorporation of ferrocenyl units into bioactive materials has been so successful. We have already discussed its role in enhancing the efficacy and range of applicability of tamoxifen by replacing a phenyl substituent by ferrocenyl in the ferrocifens for the treatment of a broad range of cancers [63]. Another spectacular advance has been the adoption of ferroquine as a replacement for chloroquine in the world-wide fight against malaria [70]. It is evident that the electronic control exerted by the ferrocenyl group continues to attract attention, and will undoubtedly lead to additional practical applications.
Funding
For those sections concerning our own work, this research was funded for many years by the Natural Sciences and Engineering Research Council of Canada (NSERC), the Petroleum Research Fund (PRF), administered by the American Chemical Society, and by Science Foundation Ireland (SFI).
Acknowledgments
The author thanks University College Dublin and the UCD School of Chemistry for additional financial support, and the Centre for Synthesis and Chemical Biology (CSCB) for the use of analytical facilities. He also thanks his long-time French colleagues Anne Vessières and Gérard Jaouen for many valuable discussions, and the reviewers for their helpful and constructive comments.
Conflicts of Interest
The author declares no conflict of interest.
References
- Richards, J.H.; Hill, E.A. Carbonium ion stabilization by metallocene nuclei. II. α-Metallocenylcarbonium ions. J. Am. Chem. Soc. 1961, 83, 3840–3846. [Google Scholar] [CrossRef]
- Lupan, S.; Kapon, M.; Cais, M.; Herbstein, F.H. Structure and properties of α,α-diferrocenylmethylium ion. Angew. Chem. Int. Ed. Engl. 1972, 11, 1025–1027. [Google Scholar] [CrossRef]
- Behrens, U. Transition metal-fulvene complexes. 14. Crystal and molecular structure of ferrocenyldiphenylcarbenium tetrafluoroborate—Fulvene-iron complex. J. Organometal. Chem. 1979, 182, 89–98. [Google Scholar] [CrossRef]
- Gleiter, R.; Seeger, R. Structure of ferrocenyl-methyl cation. Helv. Chim. Acta 1971, 54, 1217–1220. [Google Scholar] [CrossRef]
- Casper, L.A.; Osswald, S.; Anders, P.; Rosenbaum, L.-C.; Winter, R.F. Extremely electron-poor bis(diarylmethylium)-substituted ferrocenes and the first peroxoferrocenophane. Z. Anorg. Allg. Chem. 2020, 646, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Turbitt, T.D.; Watts, W.E. Stable carbonium ions. Part IV. Torsional barriers in secondary and tertiary ferrocenylalkylium ions. J. Chem. Soc. 1974, 2, 177–184. [Google Scholar] [CrossRef]
- Gleiter, R.; Bleiholder, C.; Rominger, F. α-Metallocenylmethylium ions and isoelectronic fulvene complexes of d6 to d9 metals. Structural considerations. Organometallics 2007, 26, 4850–4859. [Google Scholar] [CrossRef]
- Cais, M.; Dannenberg, J.J.; Eisenstadt, A.; Levenberg, M.L.; Richards, J.H. Nuclear magnetic resonance spectra of ferrocenyl carbonium ions. Tetrahedron Lett. 1966, 7, 1695–1701. [Google Scholar] [CrossRef]
- Traylor, T.G.; Ware, J.C. The chemistry of metallocenes. Carbonium stabilization by the ferrocenyl group. J. Am. Chem. Soc. 1967, 89, 2304–2316. [Google Scholar] [CrossRef]
- Feinberg, J.; Rosenblum, M. The structure of α-ferrocenylcarbonium ions. J. Am. Chem. Soc. 1969, 91, 4324–4325. [Google Scholar] [CrossRef]
- Hisatome, M.; Yamakawa, K. Organometallic compounds IX. The structure of α-ferrocenylcarbonium ions. Tetrahedron 1971, 27, 2101–2110. [Google Scholar] [CrossRef]
- Watts, W.E. The organic chemistry of metal-coordinated cyclopentadienyl and arene ligands. In Comprehensive Organometallic Chemistry; Wilkinson, G., Stone, F.G.A., Abel, E.W., Eds.; Pergamon: Oxford, UK, 1982; Volume 8, Chapter 59; pp. 1051–1071. [Google Scholar]
- Turbitt, T.D.; Watts, W.E. Double-shift rearrangements of secondary ferrocenylcarbonium ions. J. Organometal. Chem. 1973, 57, C78–C79. [Google Scholar] [CrossRef]
- Abram, T.S.; Turbitt, T.D.; Watts, W.E. Stable carbocations. Part 15. Double-shift rearrangements of ferrocenylcarbenium ions. J. Chem. Soc. 1977, 13, 1536–1541. [Google Scholar] [CrossRef]
- Abram, T.S.; Watts, W.E. Stable carbocations. Part 13. Intra-ionic cyclisation reactions of ferrocenyl- and phenyl-stabilised vinyl cations. J. Chem. Soc. 1977, 13, 1527–1531. [Google Scholar] [CrossRef]
- Schottenberger, H.; Buchmeiser, M.R.; Angleitner, H.; Wurst, K.; Herber, R.H. Rearrangements and dimerizations of congested ferrocenyl allyl alcohols. J. Organometal. Chem. 2000, 605, 174–183. [Google Scholar] [CrossRef]
- Gupta, H.K.; Reginato, N.; Ogini, F.O.; Brydges, S.; McGlinchey, M.J. SiCl4-ethanol as a trimerization agent for organometallics: Convenient syntheses of the symmetrically substituted arenes 1,3,5-C6H3R3 where R is (C5H4)Mn(CO)3 and (C5H4)Fe(C5H5). Can. J. Chem. 2002, 80, 1546–1554. [Google Scholar] [CrossRef]
- Štěpnička, P.; Císařová, I.; Sedláček, J.; Vohlídal, J.; Polásek, M. Synthesis of triferrocenylbenzenes by tantalum(V)-catalyzed cyclotrimerization of ethynylferrocene. The crystal structure of 1,3,5-triferrocenylbenzene. Collect. Czech. Chem. Commum. 1997, 62, 1577–1584. [Google Scholar] [CrossRef]
- Fiorentini, S.; Floris, B.; Galloni, P.; Grepioni, F.; Polito, M.; Tagliatesta, P. The metal-catalyzed cyclotrimerization of ferocenylethyne—Preparation and characterization of 1,2,4-triferrocenylbenzene. Eur. J. Org. Chem. 2006, 7, 1726–1732. [Google Scholar] [CrossRef]
- Ogini, F.O.; Ortin, Y.; Mahmoudkhani, A.H.; Cozzolino, A.F.; McGlinchey, M.J.; Vargas-Baca, I. An investigation of the formation of 1,3,5-heterosubstituted benzene rings by cyclocondensation of acetyl-substituted organometallic complexes. J. Organometal. Chem. 2008, 693, 1957–1967. [Google Scholar] [CrossRef]
- Top, S.; Jaouen, G.; Sayer, B.G.; McGlinchey, M.J. Asymmetric induction via co-cordination to Cr(CO)3: Nucleophilic attack on acyclic carbenium ions. J. Chem. Soc. Chem. Comm. 1980, 23, 1110–1112. [Google Scholar] [CrossRef]
- Top, S.; Jaouen, G.; Sayer, B.G.; McGlinchey, M.J. Rotational barriers in diphenylmethyl anions stabilized by trimethylsilyl and tricarbonylchromium(0) moieties. J. Am. Chem. Soc. 1983, 105, 6426–6429. [Google Scholar] [CrossRef]
- Klimova, E.I.; Garcia, M.M.; Klimova, T.; Ramirez, L.R.; Meleshonkova, N.N. 3-Cyclo-butyl- 3-ferrocenylcyclopropene and 3-cyclobutylidene-3-ferrocenylpropyne. Synthesis and chemical properties. Russ. Chem. Bull. 1999, 48, 2153–2157. [Google Scholar] [CrossRef]
- Méndez, D.; Klimova, E.; Klimova, T.; Hernández, O.S.; Martínez, G.M. Synthesis of ferrocenylvinylcyclopropene and its transformation into cyclopentadiene. J. Organometal. Chem. 2003, 681, 115–119. [Google Scholar] [CrossRef]
- Harwood, L.M. Polar Rearrangements; Oxford University Press: Oxford, UK, 1992; pp. 9–10, 49. [Google Scholar]
- Bachmann, W.E.; Sternberger, H.R. The pinacol-pinacolone rearrangement. V. The rearrangement of unsymmetrical aromatic pinacols. J. Am. Chem. Soc. 1934, 56, 170–173. [Google Scholar] [CrossRef]
- Weliky, N.; Gould, E.S. Studies in the ferrocene series. I. Some reactions of compounds related to monobenzoylferrocene. J. Am. Chem. Soc. 1957, 79, 2742–2746. [Google Scholar] [CrossRef]
- Pauson, P.L.; Watts, W.E. Ferrocene derivatives. Part XII. Di-and tri-ferrocenylmethane derivatives. J. Chem. Soc. 1962, 3880–3886. [Google Scholar] [CrossRef]
- Moffett, L.R., Jr. Pinacol and pinacolone derivatives of some acylferrocenes. J. Org. Chem. 1964, 29, 3726–3727. [Google Scholar] [CrossRef]
- Rausch, M.D.; Adams, D.L. The Clemmensen reduction of benzoylferrocene. J. Org. Chem. 1967, 32, 4144–4145. [Google Scholar] [CrossRef]
- Goldberg, S.I.; Bailey, W.D. Evidence for appreciable phenyl migration in the rearrangements of threo and erythro-1,2-diferroceny-1,2-diphenylethane-1,2-diols. Chem. Commun. 1969, 18, 1059–1060. [Google Scholar] [CrossRef]
- Denifl, P.; Hradsky, A.; Bildstein, B.; Wurst, K. Trimethylchlorosilane-modified Clemmensen reduction of metallocenyl ketones: Trapping and X-ray structures of aliphatic, olefinic, silylated pinacolic, and rearranged pinacolonic products. J. Organometal. Chem. 1996, 523, 79–91. [Google Scholar] [CrossRef]
- Bildstein, B. Cationic and neutral cumulene sp-carbon chains with ferrocenyl termini. Coord. Chem. Rev. 2000, 206, 369–394. [Google Scholar] [CrossRef]
- Ortin, Y.; Grealis, J.; Scully, C.; Müller-Bunz, H.; Manning, A.R.; McGlinchey, M.J. McMurry reactions of (η5-acetylcyclopentadienyl)cobalt(η4-tetraphenylcyclobutadiene) with benzophenone: Ketone couplings and a pinacol/pinacolone rearrangement. J. Organometal. Chem. 2004, 689, 4683–4690. [Google Scholar] [CrossRef]
- Nikitin, K.; Ortin, Y.; Müller-Bunz, H.; Plamont, M.-A.; Jaouen, G.; Vessières, A.; McGlinchey, M.J. Organometallic SERMS [selective estrogen receptor modulators]: Cobaltifens, the (cyclobutadiene)cobalt analogues of hydroxytamoxifen. J. Organometal. Chem. 2010, 695, 595–608. [Google Scholar] [CrossRef]
- Jaouen, G.; Vessières, A.; Top, S. Ferrocifen Type Anti Cancer Drugs. Chem. Soc. Rev. 2015, 44, 8802–8817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdanovic, B.; Bolte, A. A comparative study of the McMurry reaction utilizing [HTiCl(THF)~0.5]x, TiCl3(DME)1.5-Zn(Cu) and TiCl2·LiCl as coupling reagents. J. Organometal. Chem. 1995, 502, 109–121. [Google Scholar] [CrossRef]
- Dams, R.; Malinowski, M.; Westdorp, I.; Geise, H.Y. On the mechanism of the titanium-induced reductive couplings to olefins. J. Org. Chem. 1982, 47, 248–259. [Google Scholar] [CrossRef]
- Härter, P.; Latzel, K.; Spiegler, M.; Herdtweck, E. Migration of the ferrocenyl moiety during the McMurry coupling of ferrocenyl ketones. Polyhedron 1998, 17, 1141–1148. [Google Scholar] [CrossRef]
- Ye, T.; McKervey, M.A. Organic synthesis with α-diazo carbonyl compounds. Chem. Rev. 1994, 94, 1091–1160. [Google Scholar] [CrossRef]
- Aguilar-Aguilar, A.; Allen, A.D.; Cabrera, E.P.; Federov, A.; Fu, N.; Henry-Riyad, H.; Leuninger, J.; Schmid, U.; Tidwell, T.T.; Verma, R. Ferrocenylketene and ferrocenyl-1,2-bisketenes: Direct observation and reactivity measurements. J. Org. Chem. 2005, 70, 9556–9561. [Google Scholar] [CrossRef]
- Inayama, S.; Mamoto, K.; Shibata, T.; Hirose, T. Structure and antitumor activity relationship of 2-arylidene-4-cyclopentene-1,3-diones and 2-arylideneindan-1,3-diones. J. Med. Chem. 1976, 19, 433–436. [Google Scholar] [CrossRef]
- Zora, M.; Kokturk, M.; Eralp, T. Synthesis of 2-ferrocenylidene-4-cyclopentene-1,3-diones. Tetrahedron 2006, 62, 10344–10351. [Google Scholar] [CrossRef]
- Banide, E.V.; Ortin, Y.; Chamiot, B.; Cassidy, A.; Niehaus, J.; Moore, A.; Seward, C.M.; Müller-Bunz, H.; McGlinchey, M.J. Syntheses, structures and dimerizations of ferrocenyl-and fluorenylideneallenes: Push-pull multiple bonds? Organometallics 2008, 27, 4173–4182. [Google Scholar] [CrossRef]
- Yamakawa, K.; Hisatome, M. The stereochemistry of acylferrocene oxime derivatives. Tetrahedron 1970, 26, 4483–4489. [Google Scholar] [CrossRef]
- Schlögl, K.; Mechtler, H. Über die Umlagerung von Acylferrocen-oximen bei der Reduktion mit Lithiumalanat-Aluminiumchlorid. Monatsh. 1966, 97, 150–167. [Google Scholar] [CrossRef]
- Hetnarski, B.; Lajtha, A.; Wisniewski, H.M. On some derivatives of ferrocene, novel acetylcholinesterase inhibitors. J. Neurosci. Res. 1980, 5, 1–5. [Google Scholar] [CrossRef]
- Schlögl, K.; Seiler, H. Ferrocenyl-isocyanat. Naturwiss 1958, 45, 337. [Google Scholar] [CrossRef]
- Shimada, K.; Orii, S.; Tanaka, M.; Nambara, T. New ferrocene reagents for derivatization of alcohols in high-performance liquid chromatography with electrochemical detection. J. Chromatogr. 1986, 352, 329–335. [Google Scholar] [CrossRef]
- Goggins, S.; Apsey, E.A.; Mahon, M.F.; Frost, C.G. Ratiometric electrochemical detection of hydrogen peroxide and glucose. Org. Biomol. Chem. 2017, 15, 2458–2466. [Google Scholar] [CrossRef] [Green Version]
- Nesmeyanov, A.N.; Perevalova, E.G.; Nikitina, T.V.; Kuznetsova, N.I. Behavior of 3- and 4-ferrocenyl-hydrazobenzenes under the conditions of the benzidine rearrangement. Izv. Akad. Nauk Ser. Khim. 1965, 14, 2120–2124. [Google Scholar]
- Nesmeyanov, A.N.; Perevalova, E.G.; Nikitina, T.V.; Kuznetsova, N.I. Action of hydrochloric acid on azo derivatives of ferrocene. Izv. Akad. Nauk Ser. Khim. 1965, 14, 2124–2128. [Google Scholar] [CrossRef]
- Kuebrich, J.P.; Schowen, R.L.; Wang, M.-S.; Lupes, M.E. Mechanism of the benzoin condensation. J. Am. Chem. Soc. 1971, 93, 1214–1220. [Google Scholar] [CrossRef]
- Gupta, H.K.; Brydges, S.; McGlinchey, M.J. Diels-Alder reactions of 3-ferrocenyl-2,4,5-triphenyl-cyclopentadienone: Syntheses of C6Ph5Fc, C7Ph6FcH and [C7Ph6FcH][SbCl6]. Organometallics 1999, 18, 115–122. [Google Scholar] [CrossRef]
- Rinehart, K.L., Jr.; Michejda, C.J.; Kittle, P.A. 1,2-Diferrocenylethane from an unusual reaction. J. Am. Chem. Soc. 1959, 81, 3162–3163. [Google Scholar] [CrossRef]
- Rinehart, K.L., Jr.; Ellis, A.F.; Michejda, C.J.; Kittle, P.A. Acyl- from alkyl-ferrocenes by manganese dioxide oxidation. Ferrocobenzoquinone. J. Am. Chem. Soc. 1960, 82, 4112–4113. [Google Scholar] [CrossRef]
- Nesmeyanov, A.N.; Perevalova, E.G.; Tsiskaridze, T.T. Diferrocenoyl and diferrocenylethylene. Izv. Akad. Nauk Ser. Khim. 1966, 15, 2209–2211. [Google Scholar]
- Harrington, L.E.; Britten, J.F.; McGlinchey, M.J. Ferrocenyl-penta(β-naphthyl)benzene: Synthesis, structure and molecular dynamics. Can. J. Chem. 2003, 81, 1180–1186. [Google Scholar] [CrossRef]
- McGlinchey, M.J. Diels-Alder additions as mechanistic probes–interception of silyl-isoindenes: Organometallic derivatives of polyphenylated cycloheptatrienes and related seven-membered rings. Molecules 2020, 25, 4730. [Google Scholar] [CrossRef]
- Hauser, C.R.; Lindsay, J.K.; Lednicer, D.; Cain, C.E. Rearrangement of the methiodide of N,N-dimethyl-amino-methylferrocene by potassium amide in liquid ammonia. J. Org. Chem. 1958, 23, 358–360. [Google Scholar] [CrossRef]
- March, J. Chapter 18. Rearrangements. In Advanced Organic Chemistry, Reactions, Mechanisms and Structure, 4th ed.; John Wiley & Sons: New York, NY, USA, 1992; pp. 1100–1102. [Google Scholar]
- Top, S.; Jaouen, G.; Vessières, A.; Abjean, J.P.; Davoust, D.; Rodger, C.A.; Sayer, B.G.; McGlinchey, M.J. Chromium tricarbonyl complexes of estradiol derivatives: Differentiation of α- and β-diastereomers using one- and two-dimensional NMR spectroscopy at 500 MHz. Organometallics 1985, 4, 2143–2150. [Google Scholar] [CrossRef]
- Jaouen, G.; Beck, W.; McGlinchey, M.J. A novel field of research: Bioorganometallic chemistry, origins and founding principles. In Bioorganometallics, Biomolecules, Labeling, Medicine; Jaouen, G., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 1–37. [Google Scholar]
- Jaouen, G.; Top, S.; Vessières, A.; Leclercq, G.; McGlinchey, M.J. The first organometallic Selective Estrogen Receptor Modulators (SERMs) and their relevance to breast cancer. Curr. Med. Chem. 2004, 11, 2505–2517. [Google Scholar] [CrossRef]
- Wang, Y.; Pigeon, P.; Top, S.; McGlinchey, M.J.; Jaouen, G. Organometallic antitumour compounds: Ferrocifens as precursors to quinone methides. Angew. Chem. Int. Ed. 2015, 54, 10230–10233. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dansette, P.M.; Pigeon, P.; Top, S.; McGlinchey, M.J.; Mansuy, D.; Jaouen, G. A new generation of ferrociphenols leads to a great diversity of reactive intermediates, and exhibits remarkable antiproliferative properties. Chem. Sci. 2018, 9, 70–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vessières, A.; Wang, Y.; McGlinchey, M.J.; Jaouen, G. Multifaceted chemical behaviour of metallocene (M = Fe, Os) quinone methides. Their contribution to biology. Coord. Chem. Rev. 2020. In Press. [Google Scholar] [CrossRef]
- Richard, M.-A.; Hamels, D.; Pigeon, P.; Top, S.; Dansette, P.M.; Lee, H.Z.S.; Vessières, A.; Mansuy, D.; Jaouen, G. Oxidative metabolism of ferrocene analogues of tamoxifen: Characterization and anti-proliferative activities of the metabolites. ChemMedChem 2015, 10, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Görmen, M.; Pigeon, P.; Hillard, E.A.; Vessières, A.; Huché, M.; Richard, M.-A.; McGlinchey, M.J.; Top, S.; Jaouen, G. Synthesis and antiproliferative effects of [3]ferrocenophane transposition products and pinacols obtained from McMurry cross-coupling reactions. Organometallics 2012, 31, 5856. [Google Scholar] [CrossRef]
- Dubar, F.; Biot, C. On the molecular mechanisms of the antimalarial action of ferroquine. In Bioorganometallic Chemistry, Applications in Drug Discovery, Biocatalysis, and Imaging; Jaouen, G., Salmain, M., Eds.; Wiley-VCH: Weinheim, Germany, 2015; pp. 141–164. [Google Scholar]
Figure 1.
Delocalization of charge from the cationic α carbon onto the iron atom.

Scheme 1.
Rearrangement of 2° to 3° ferrocenyl trimethyl-carbocations.
Scheme 2.
Rearrangement of 2° to 3° ferrocenyl triphenyl-carbocations.

Scheme 3.
Indene formation by cyclization of a ferrocenyl-vinyl carbocation.
Scheme 4.
Ferrocenopentalene formation by cyclization of a ferrocenyl-vinyl carbocation.

Scheme 5.
Cyclization of a cycloheptatrienyl unit onto a ferrocenyl-vinyl carbocation.
Scheme 6.
Dimerization of a ferrocenyl-allyl cation.
Scheme 7.
Ferrocenyl-mediated retro-Nazarov ring-opening to form a hydroxytetramethyl-tetrahydropyran.
Scheme 7.
Ferrocenyl-mediated retro-Nazarov ring-opening to form a hydroxytetramethyl-tetrahydropyran.

Scheme 8.
Proposed mechanism for the cyclization of (CH3C(=O)C5H4)MLn precursors, where MLn = Fe(C5H5) or Mn(CO)3, to form 1,3,5-trisubstituted benzenes.
Scheme 8.
Proposed mechanism for the cyclization of (CH3C(=O)C5H4)MLn precursors, where MLn = Fe(C5H5) or Mn(CO)3, to form 1,3,5-trisubstituted benzenes.
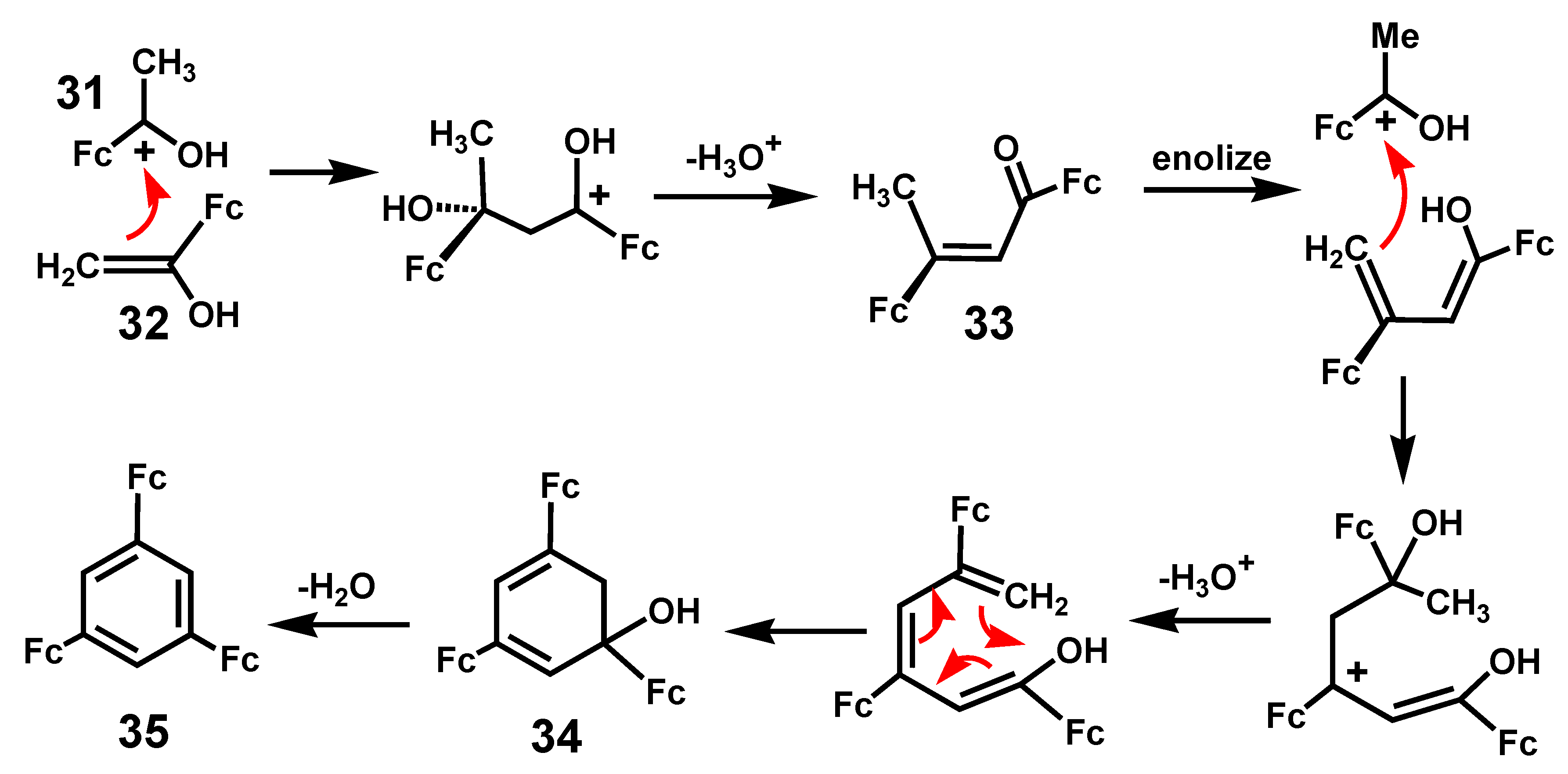
Figure 2.
Molecular structures of the mixed iron-manganese complexes 36 and 37.
Scheme 9.
Thermal rearrangement of 3-cyclobutyl-3-ferrocenylcyclopropene.

Scheme 10.
Thermal rearrangement of 3-ferrocenyl-3-(2-ferrocenyl-1-methylvinyl)cyclopropene.

Scheme 11.
Pinacol rearrangement showing methyl rather than phenyl migration.
Scheme 12.
Pinacol rearrangement showing favored migration of ferrocenyl over phenyl.
Scheme 13.
Experimentally observed products derived from Clemmensen reduction of formylferrocene, acetylferrocene, and benzoylferrocene.
Scheme 13.
Experimentally observed products derived from Clemmensen reduction of formylferrocene, acetylferrocene, and benzoylferrocene.
Scheme 14.
Experimentally observed products derived from the McMurry reaction of [(η5-MeC(=O)C5H4)Co(η4-C4Ph4)] with benzophenone.
Scheme 14.
Experimentally observed products derived from the McMurry reaction of [(η5-MeC(=O)C5H4)Co(η4-C4Ph4)] with benzophenone.
Scheme 15.
Synthetic route to cobaltifens: reagents (i) (p-HOC6H4)2C=O, Zn, TiCl4, THF; (ii) NaH, DMF, (iii) Cl(CH2)nNMe2, NEt3, THF.
Scheme 15.
Synthetic route to cobaltifens: reagents (i) (p-HOC6H4)2C=O, Zn, TiCl4, THF; (ii) NaH, DMF, (iii) Cl(CH2)nNMe2, NEt3, THF.
Scheme 16.
McMurry coupling of (C4Ph4)Co[C5H4C(=O)Me] to form the trans-alkene, 52.
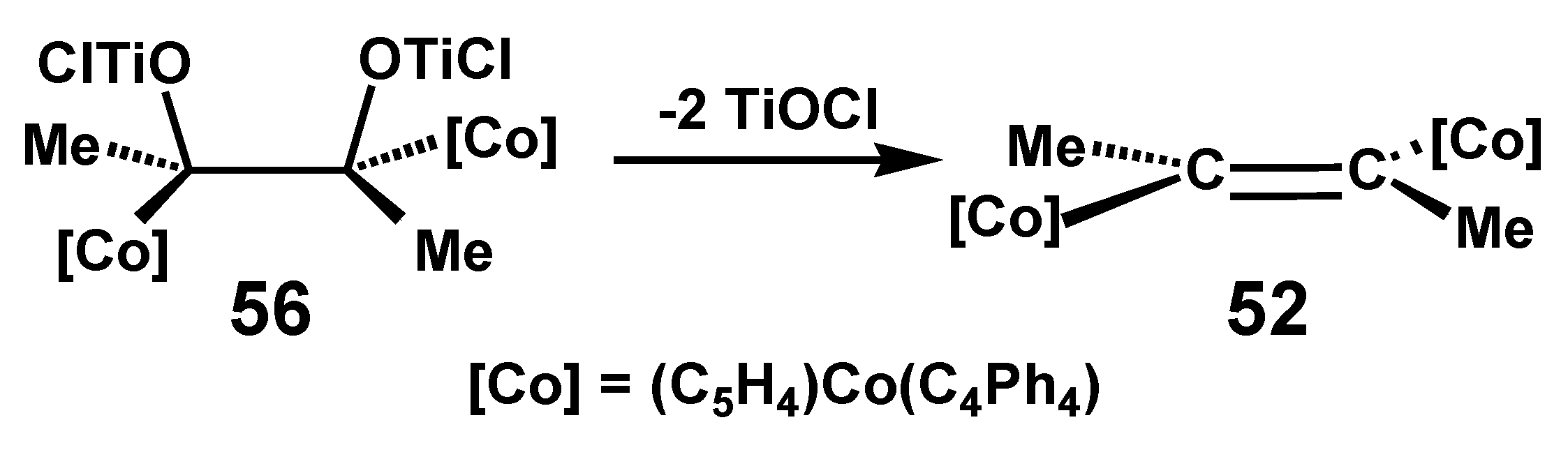
Scheme 17.
Pinacolone formation in a McMurry reaction of (C4Ph4)Co[C5H4C(=O)Me].

Figure 3.
Transition states for ferrocenyl and phenyl rearrangements whereby positive charge is delocalized onto the migrating group.
Figure 3.
Transition states for ferrocenyl and phenyl rearrangements whereby positive charge is delocalized onto the migrating group.

Scheme 18.
McMurry coupling and pinacol rearrangement of [3]ferrocenophanone.
Scheme 19.
Generation of ferrocenylketene, 62, and bis-ferrocenylketene, 63.
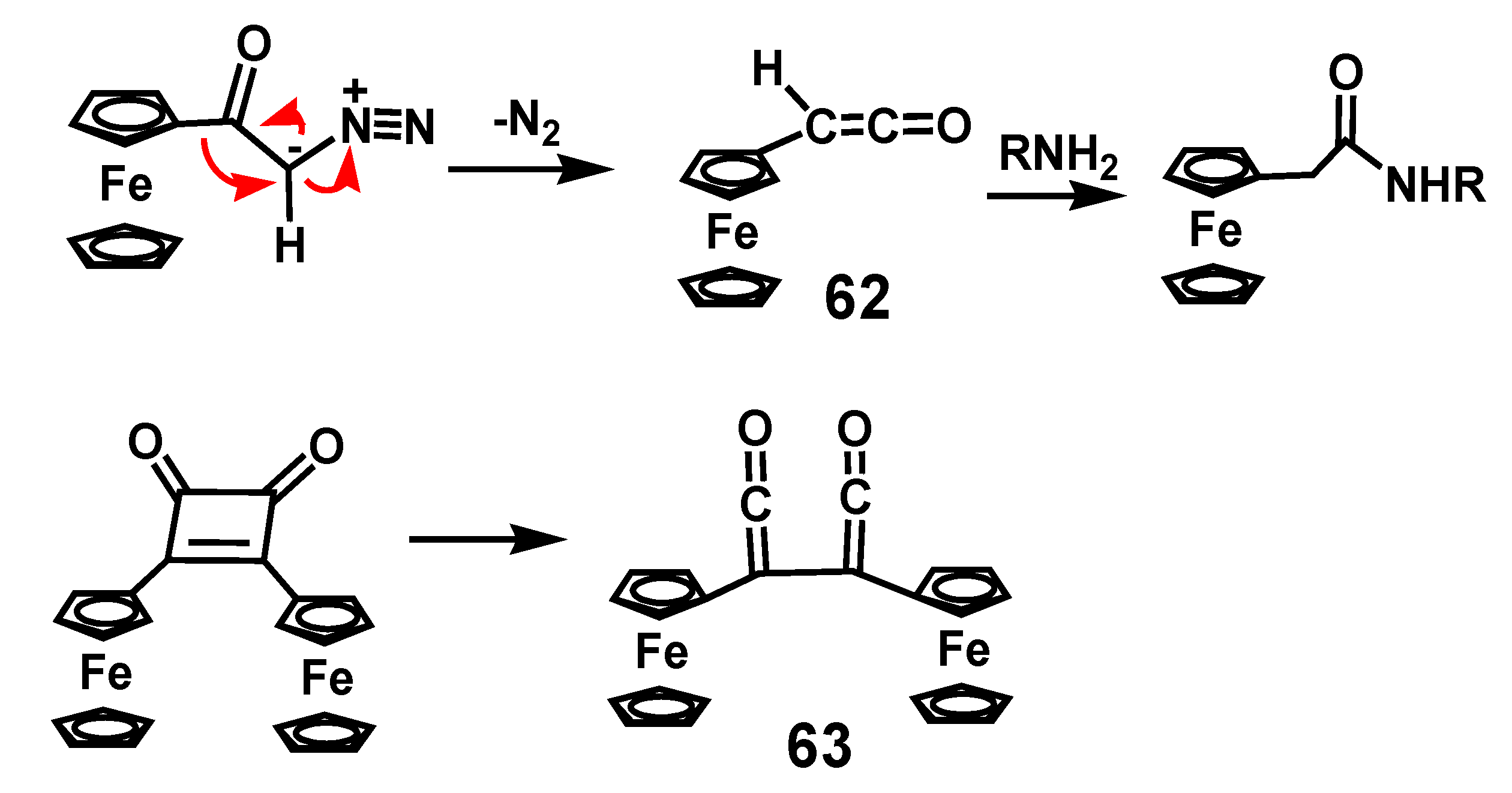
Scheme 20.
Rearrangement of 4-ferrocenylethynyl-4-hydroxy-2-cyclobutenones into 2-ferrocenylidene-4-cyclopenten-1,3-diones and 5-ferrocenyl-1,4-benzoquinones.
Scheme 20.
Rearrangement of 4-ferrocenylethynyl-4-hydroxy-2-cyclobutenones into 2-ferrocenylidene-4-cyclopenten-1,3-diones and 5-ferrocenyl-1,4-benzoquinones.

Scheme 21.
Proposed mechanism for the interconversion of 42a and 42b via a zwitterionic structure.
Scheme 22.
Meyer-Schuster rearrangement of an alkynol to form the potential push-pull allene, 69.

Scheme 23.
Beckmann rearrangement of a ferrocenyl oxime.

Scheme 24.
Beckmann rearrangement of anti and syn ferrocenocyclohexanone oximes.

Scheme 25.
Curtius rearrangement of ferrocenoyl azide to ferrocenyl isocyanate.
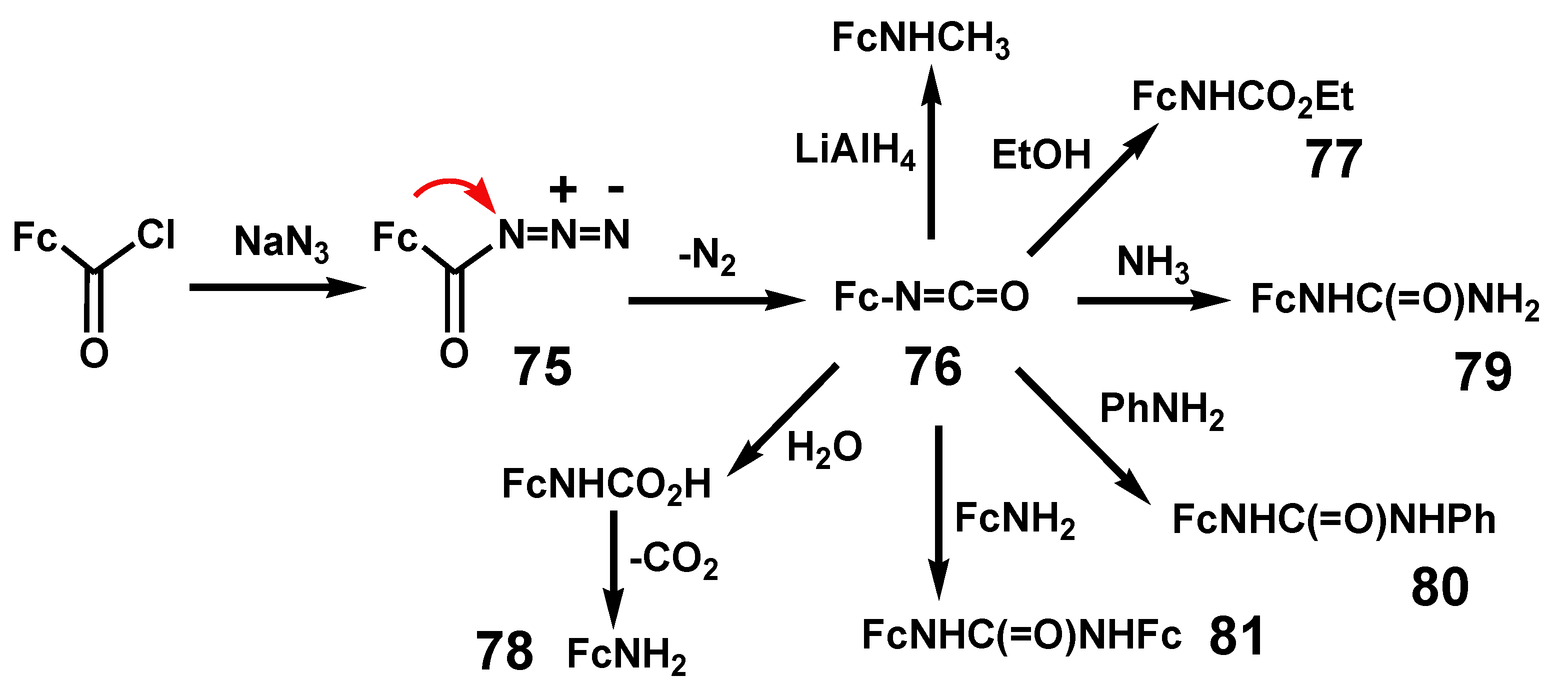
Figure 4.
Steroidal and boronic acid ester carbamates derived by reaction of alcohols with ferrocenyl isocyanate.
Figure 4.
Steroidal and boronic acid ester carbamates derived by reaction of alcohols with ferrocenyl isocyanate.

Scheme 26.
Attempted benzidine rearrangement of hydrazoferrocene.

Scheme 27.
Benzoin condensation of benzaldehyde with formylferrocene.

Scheme 28.
Synthetic route to ferrocil.

Scheme 29.
Synthetic routes to poly-arylated benzenes and cycloheptatrienes.

Scheme 30.
Different Stevens rearrangements for analogous phenyl and ferrocenyl structures.

Scheme 31.
Chemical or enzymatic conversion of ferrociphenols into quinone methides.
Scheme 32.
Protonation of a ferrocenyl quinone methide leads to formation of indenes.
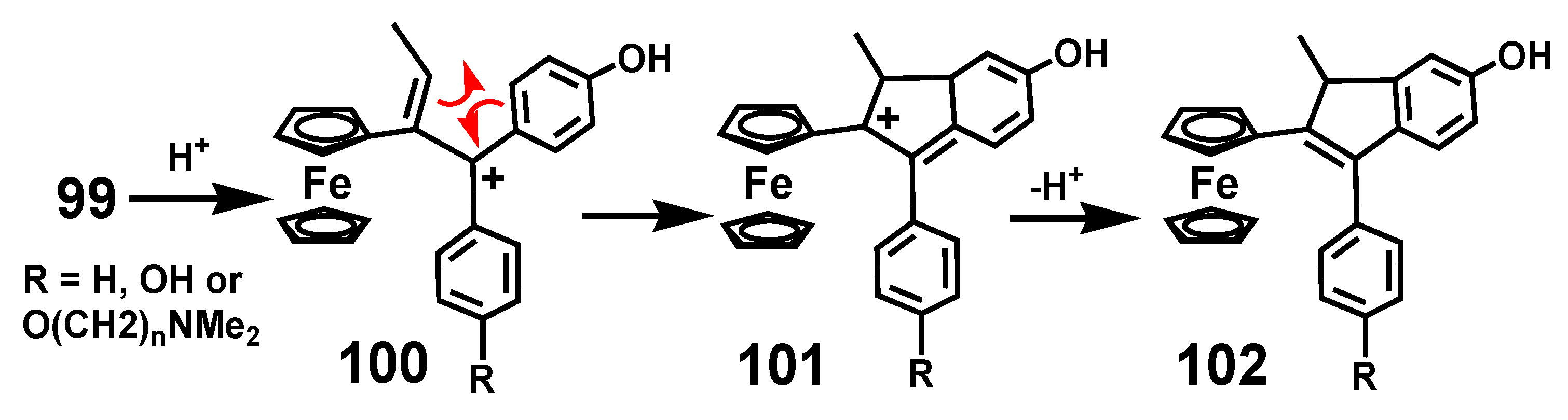
Scheme 33.
Chemical or enzymatic oxidation of the hydroxypropyl-ferrociphenol, 103, yields the tetrahydrofuranyl-quinone methide, 104.
Scheme 33.
Chemical or enzymatic oxidation of the hydroxypropyl-ferrociphenol, 103, yields the tetrahydrofuranyl-quinone methide, 104.
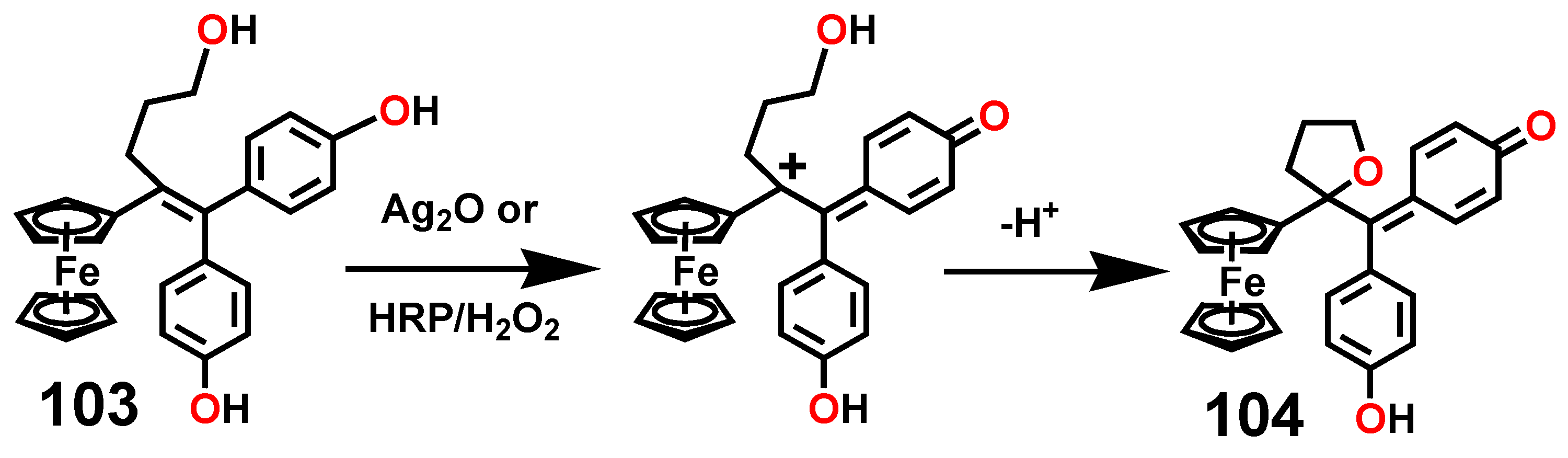
Scheme 34.
Ferrocenyl migration and ring expansion upon protonation of the tetrahydrofuranyl-quinone methide, 104.
Scheme 34.
Ferrocenyl migration and ring expansion upon protonation of the tetrahydrofuranyl-quinone methide, 104.
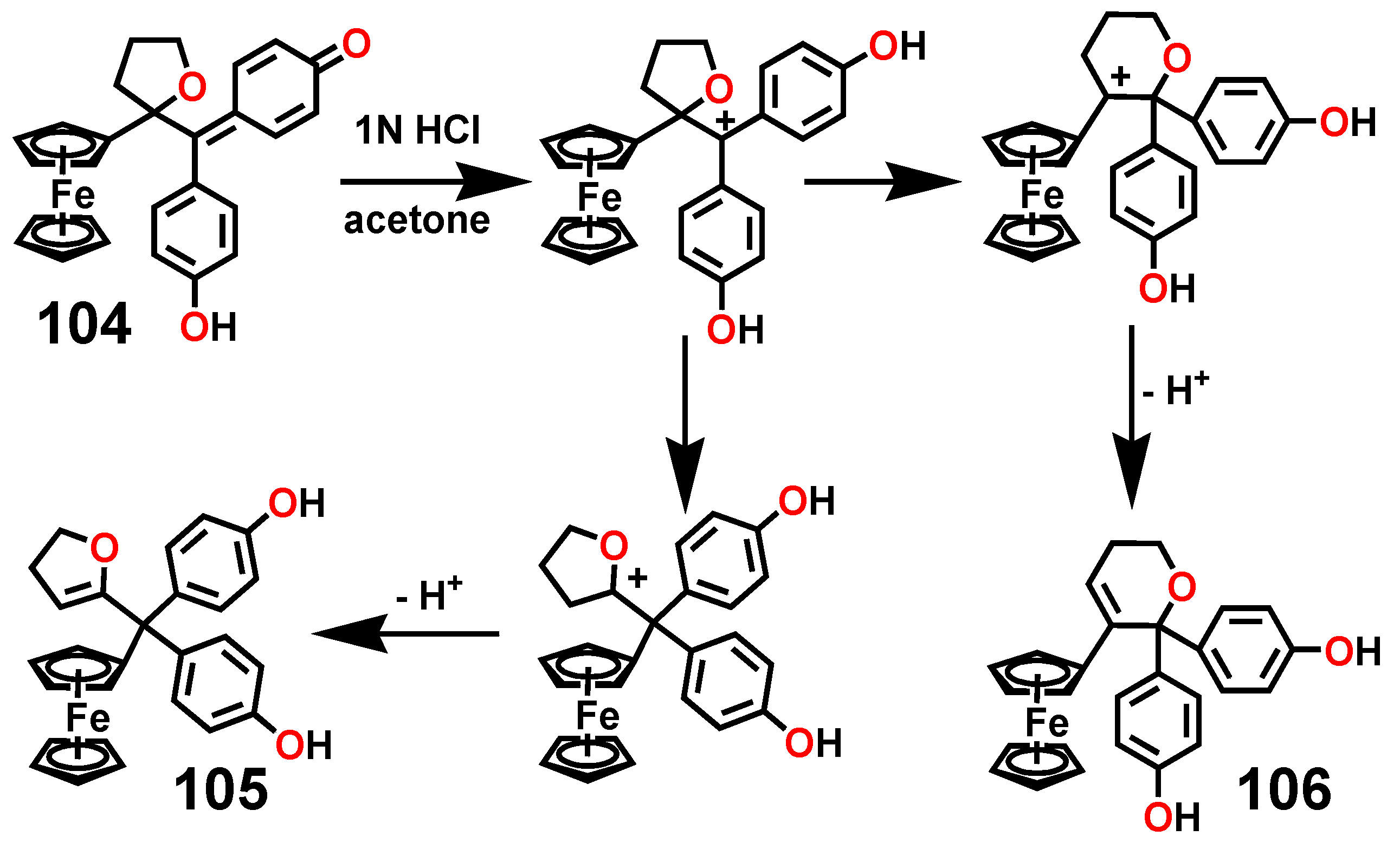
Figure 5.
Molecular structures of (left) dihydrofuran, 105, and (right) dihydropyran, 106.

Scheme 35.
Pinacol rearrangement of a [3]ferrocenophanone.

Figure 6.
Molecular structures of the ansa-ferrociphenol, 108, and the pinacolone, 109.

Scheme 36.
Carbocation formation at a doubly-benzylic site, rather that adjacent to ansa-ferrocenyl.
Scheme 36.
Carbocation formation at a doubly-benzylic site, rather that adjacent to ansa-ferrocenyl.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
McGlinchey, M.J. Ferrocenyl Migrations and Molecular Rearrangements: The Significance of Electronic Charge Delocalization. Inorganics 2020, 8, 68. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8120068
AMA Style
McGlinchey MJ. Ferrocenyl Migrations and Molecular Rearrangements: The Significance of Electronic Charge Delocalization. Inorganics. 2020; 8(12):68. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8120068
Chicago/Turabian StyleMcGlinchey, Michael J. 2020. "Ferrocenyl Migrations and Molecular Rearrangements: The Significance of Electronic Charge Delocalization" Inorganics 8, no. 12: 68. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8120068
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.