Photophysical and Biological Properties of Iridium Tetrazolato Complexes Functionalised with Fatty Acid Chains

 ,
,  , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
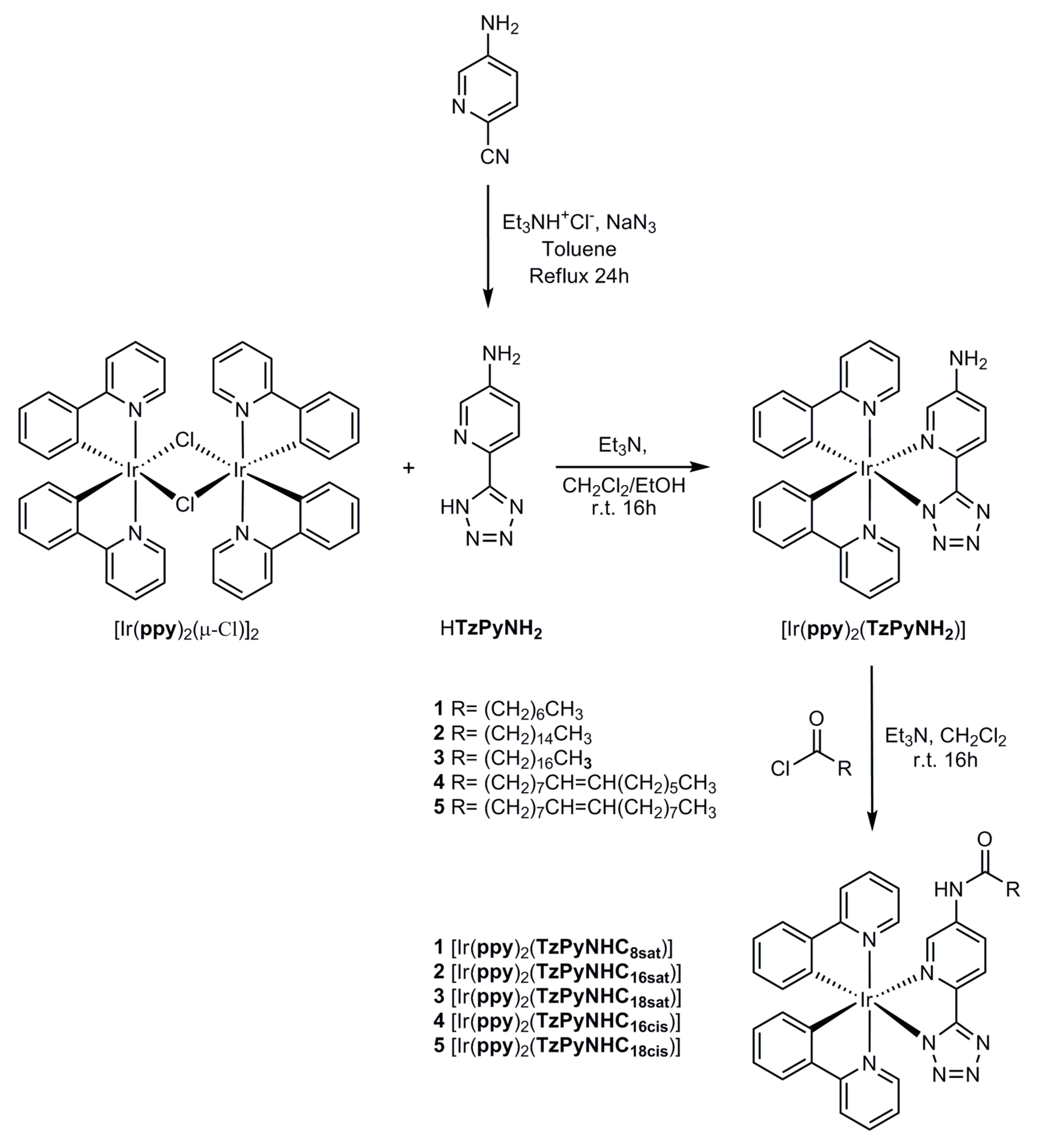
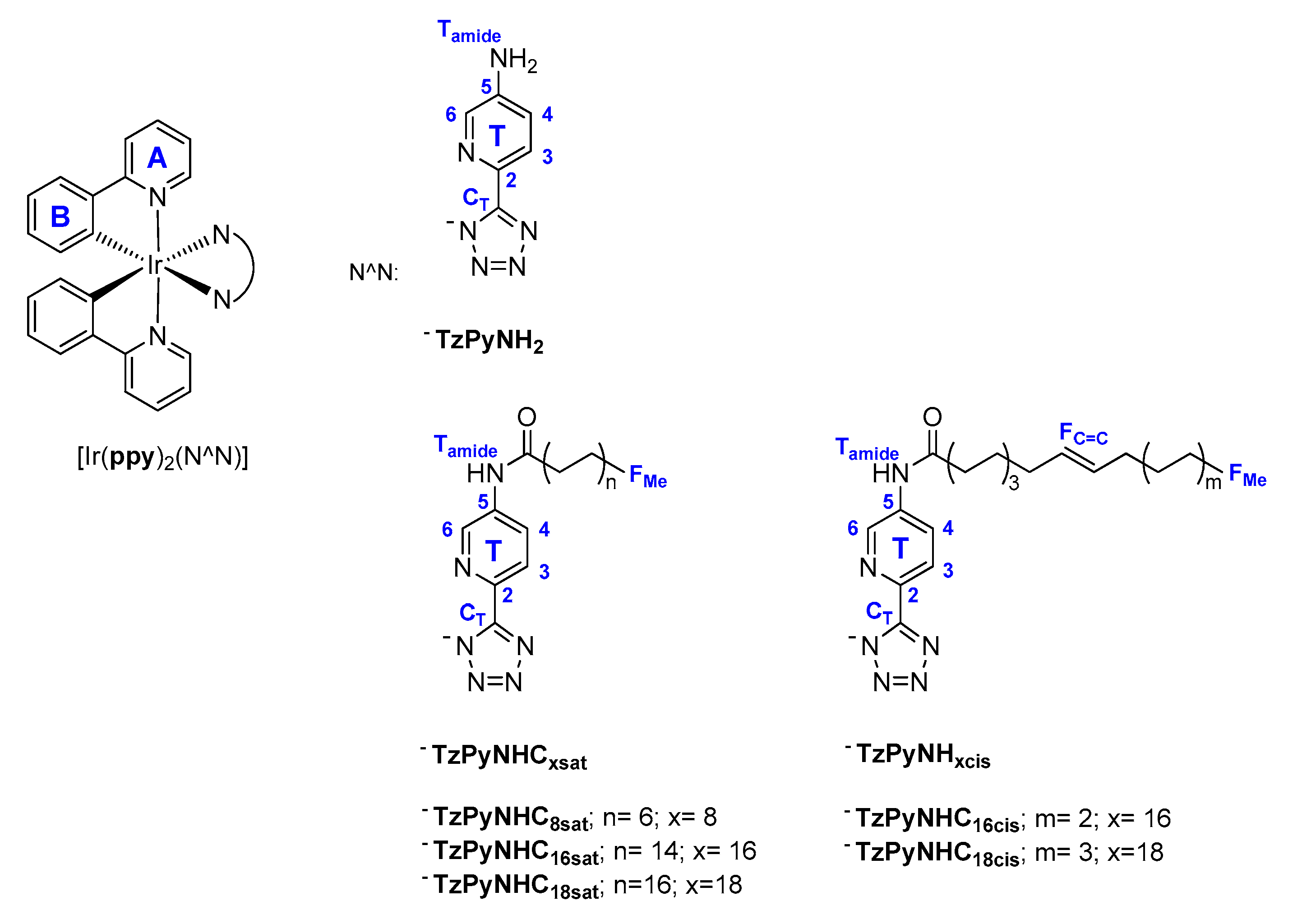
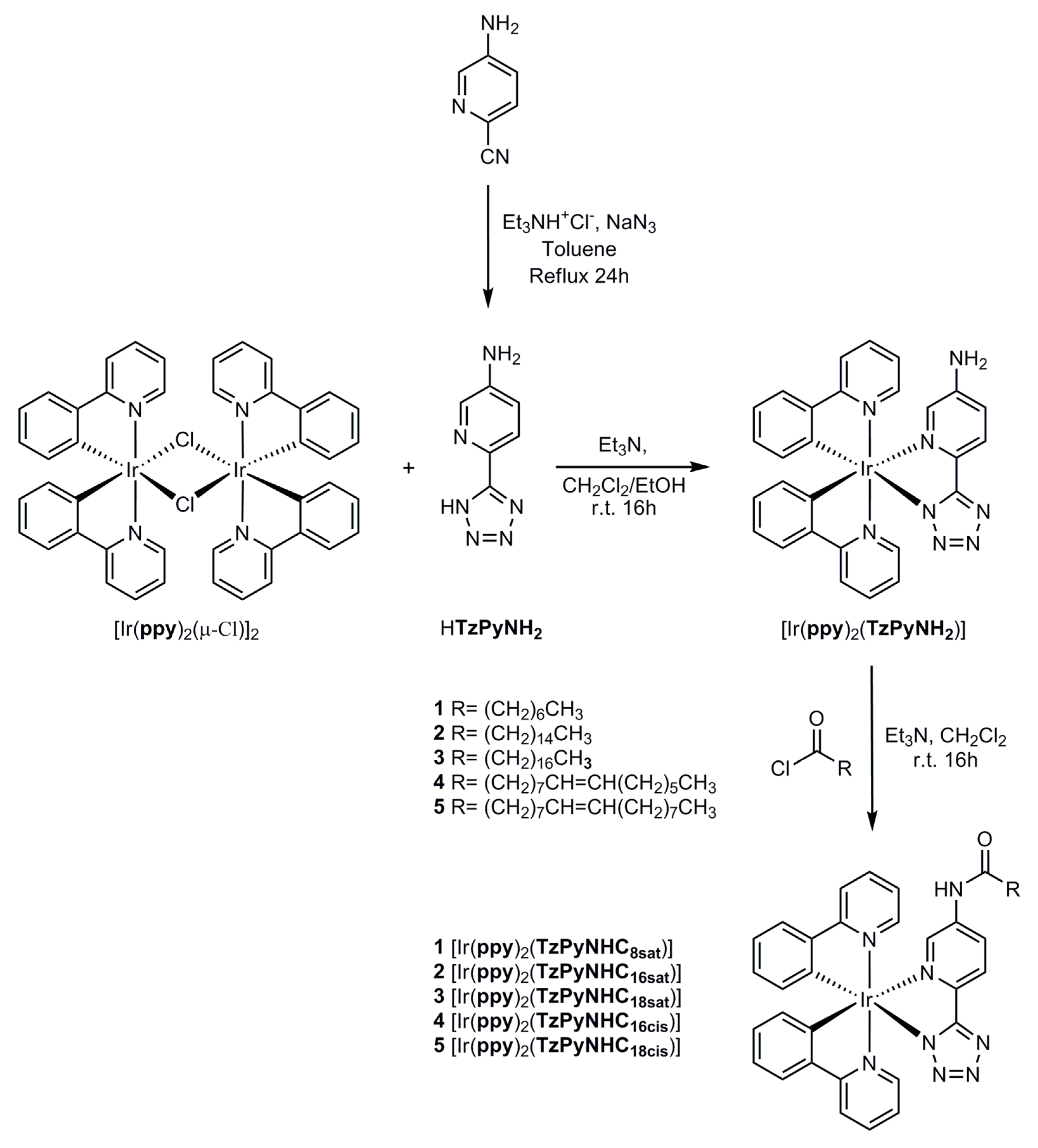
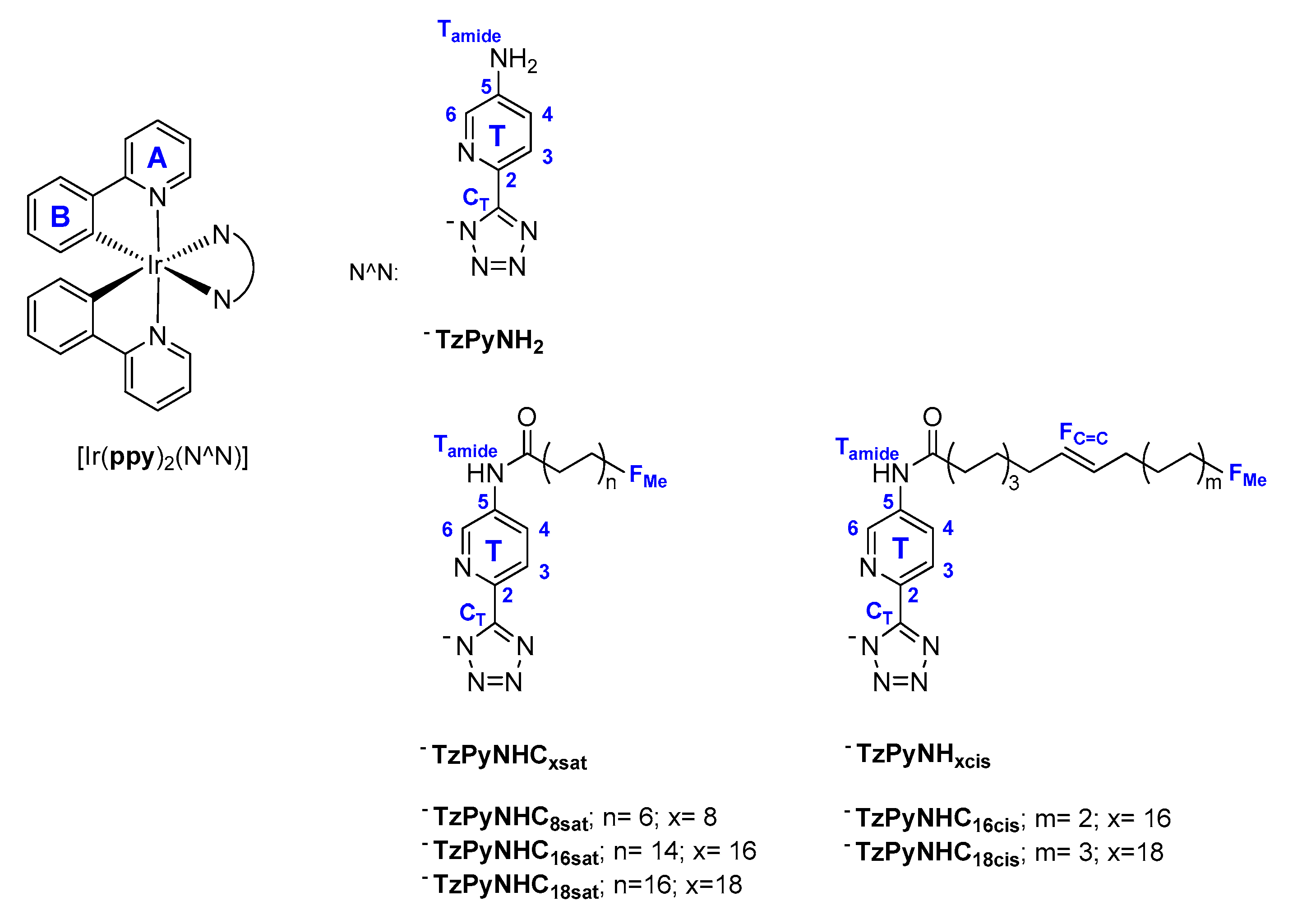
2.1. Synthesis of the Ligands and Iridium Complexes
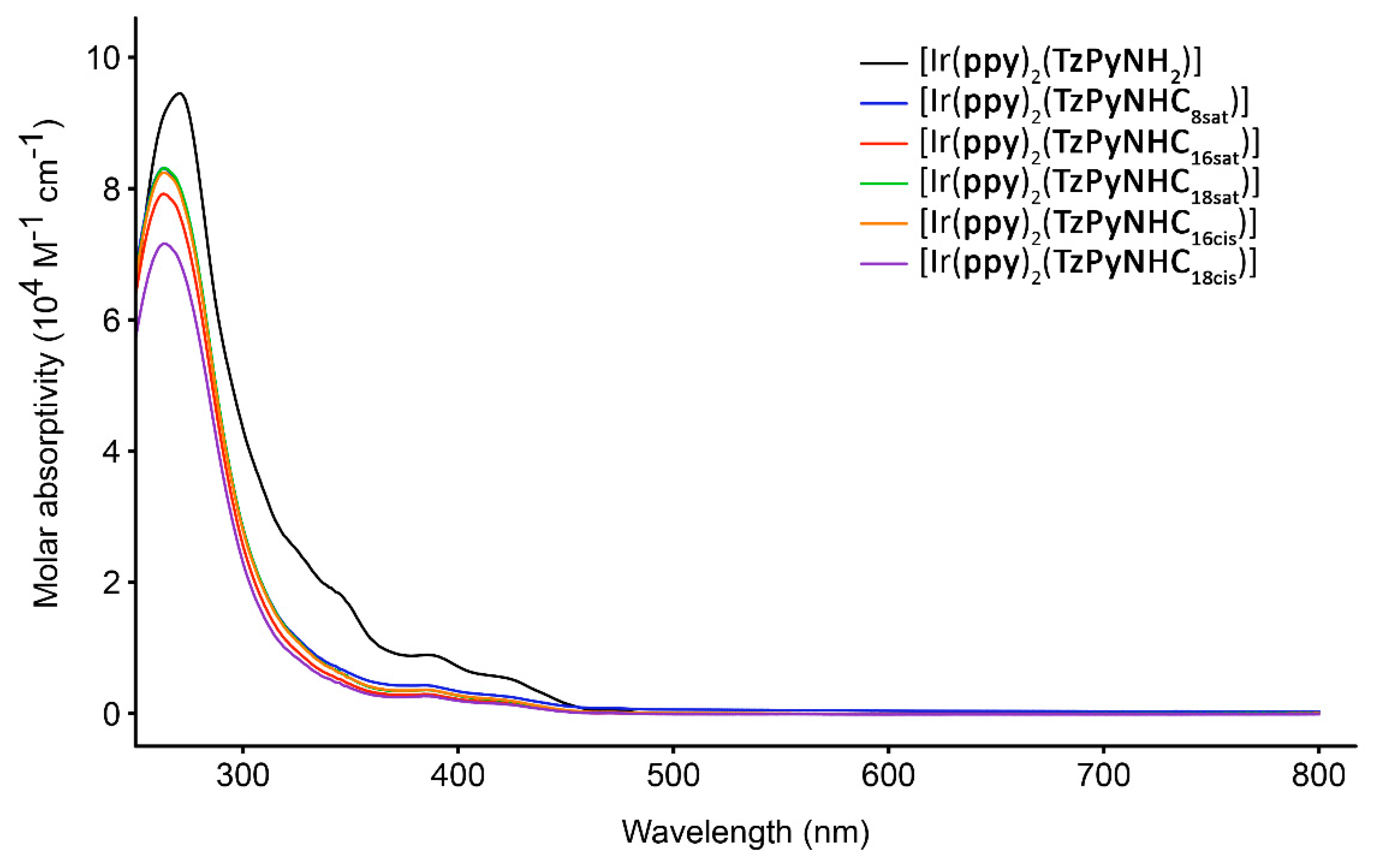
2.2. Photophysical Properties
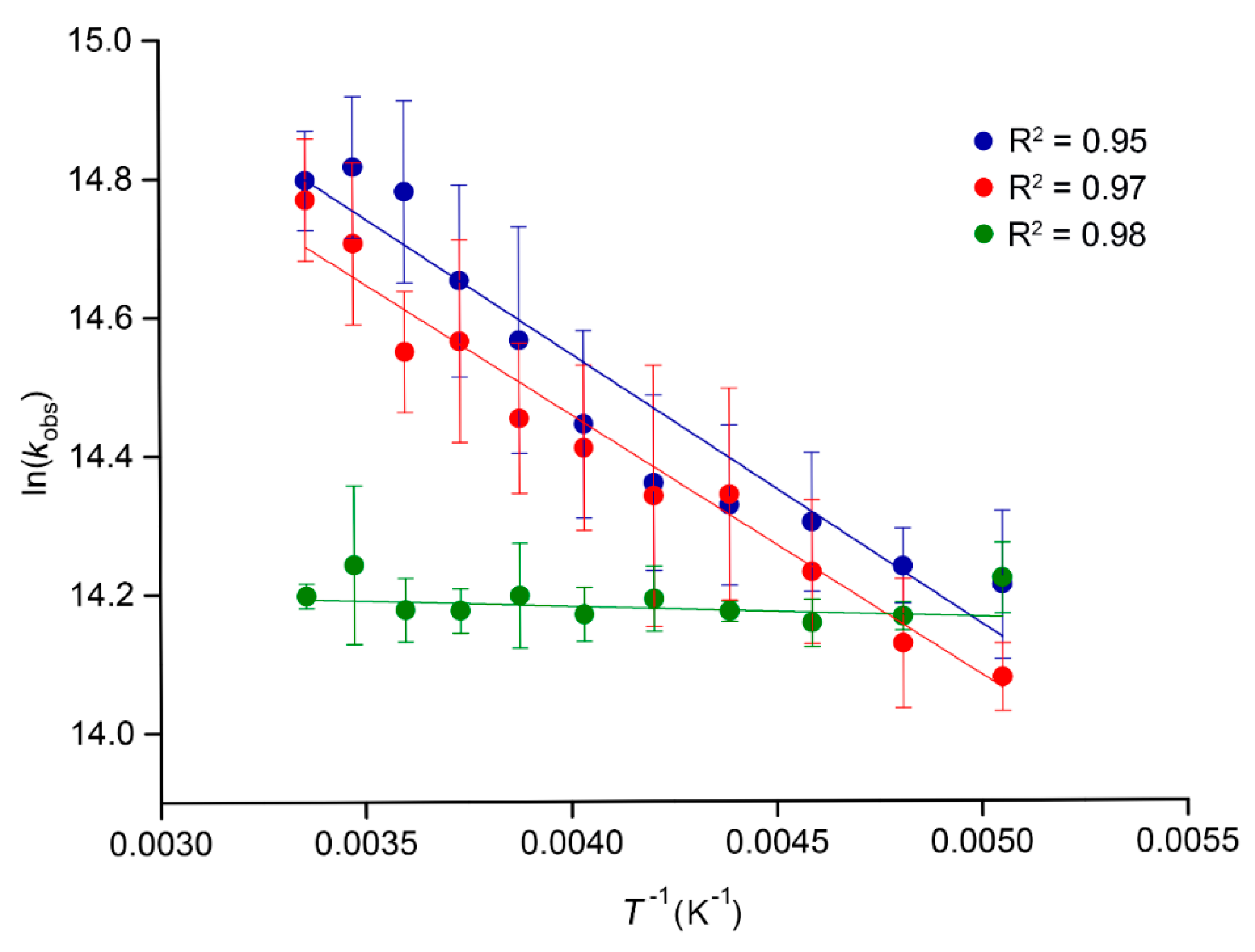
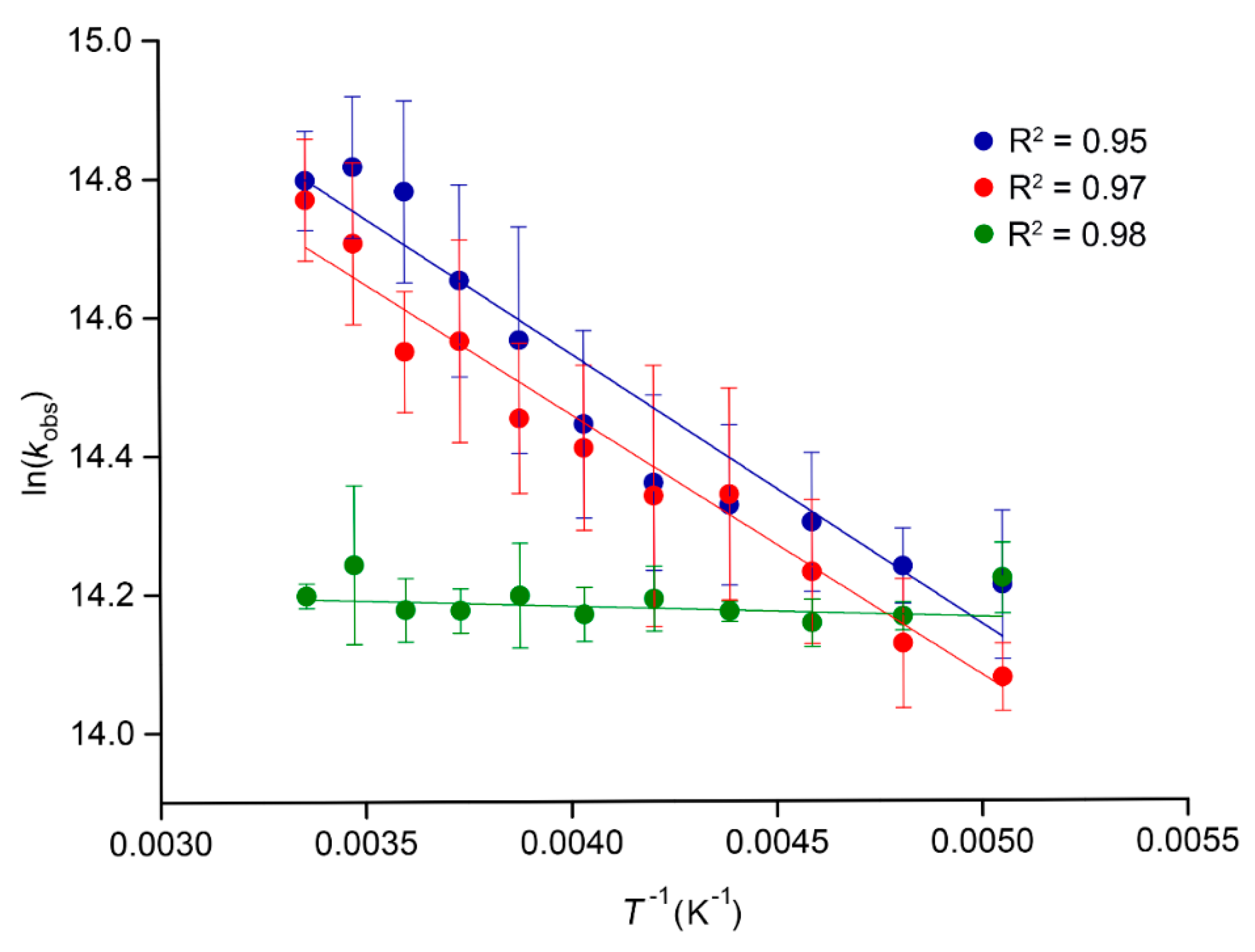
2.3. Temperature-Dependent Emission Lifetimes
2.4. Lipophilicity
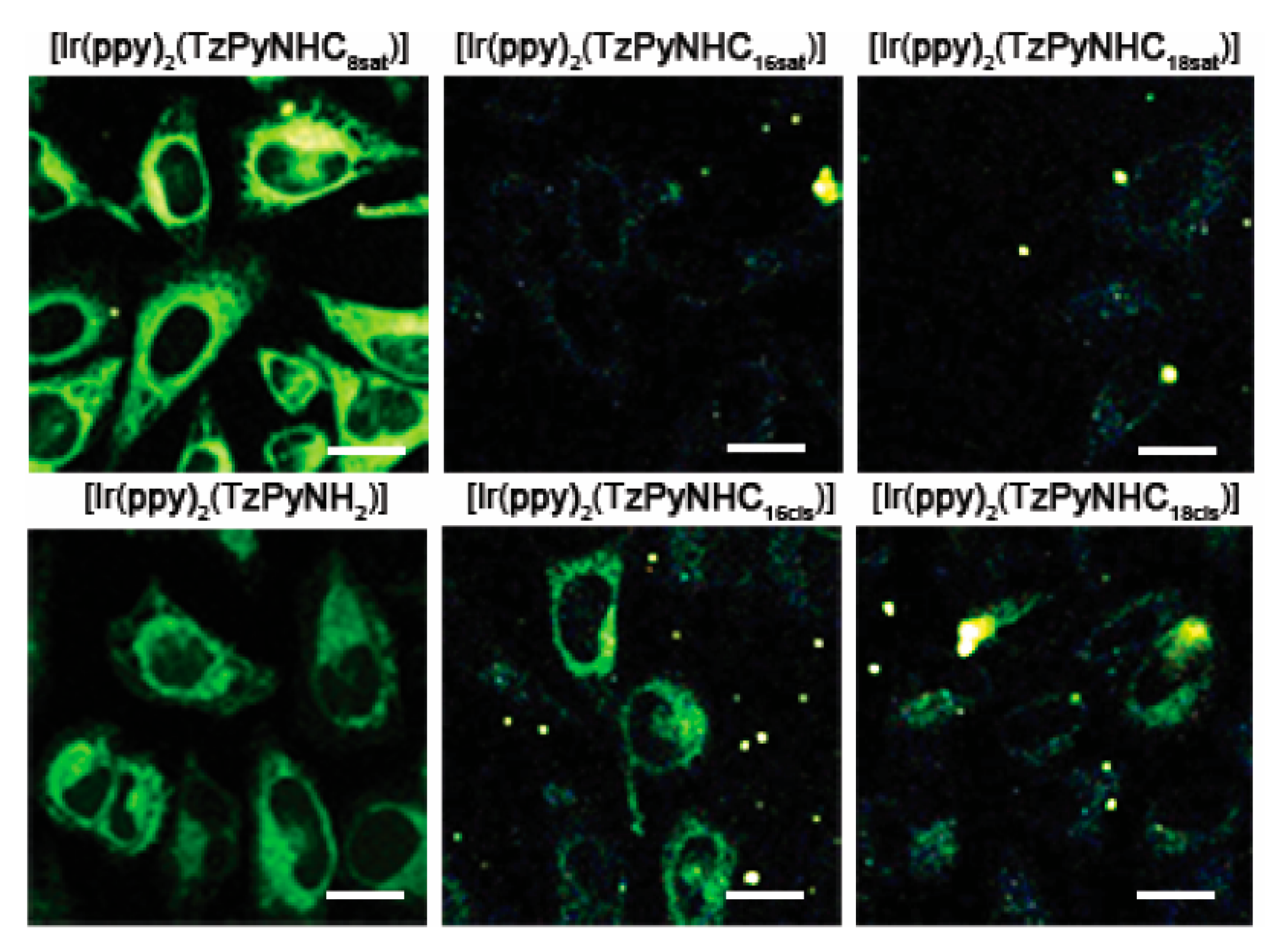
2.5. Cellular Internalisation and Localisation
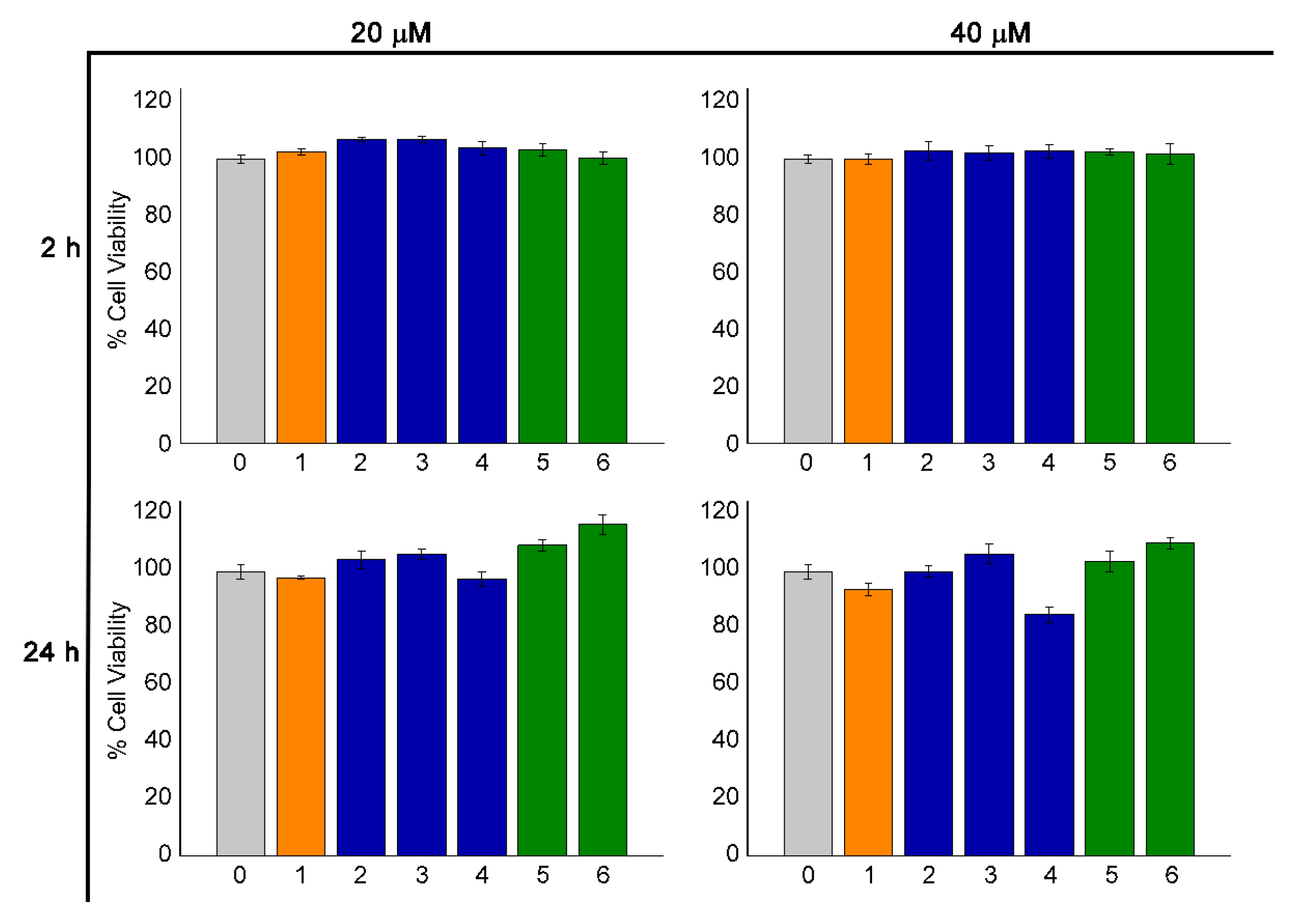
2.6. Cell Viability Assay
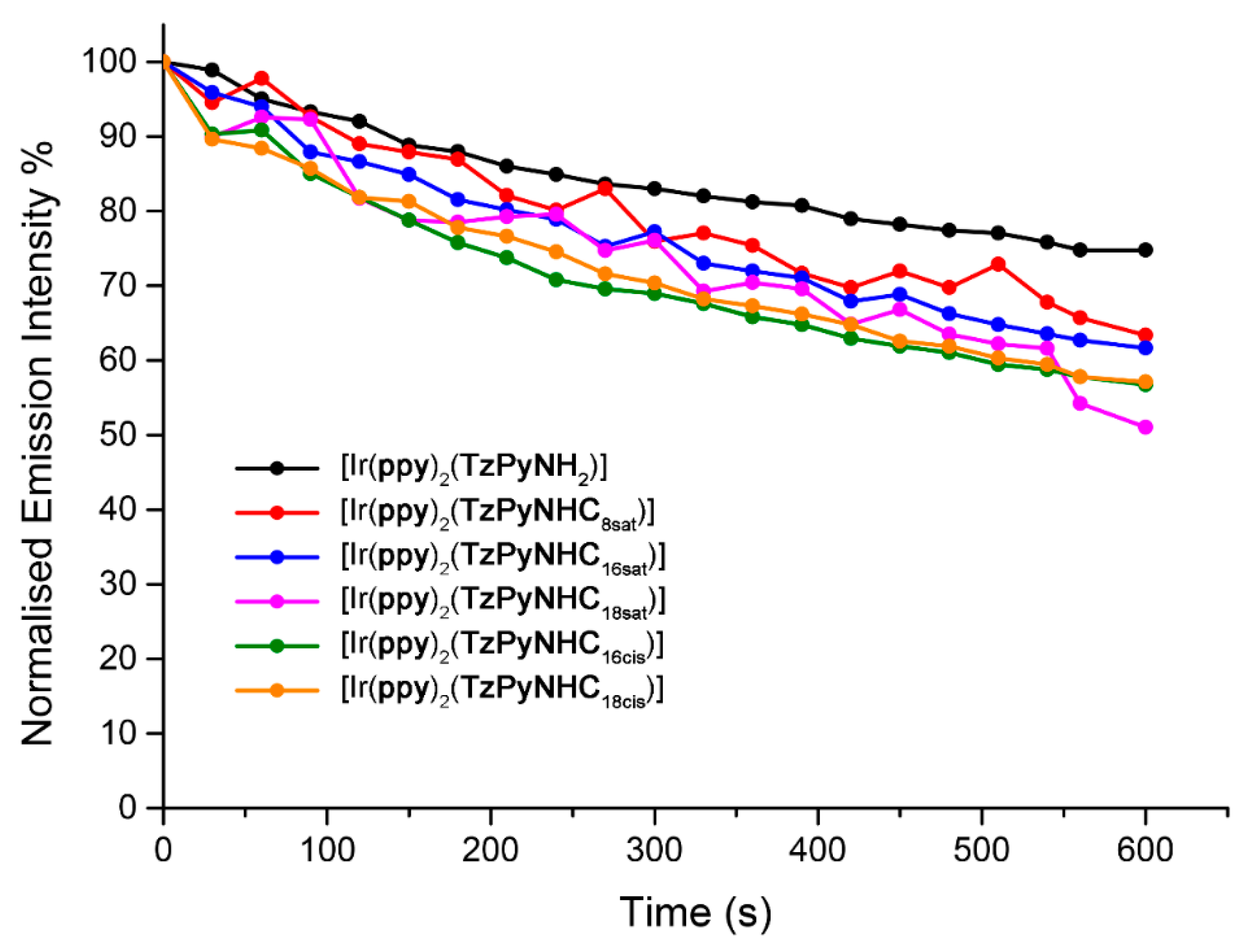
2.7. Photostability
3. Materials and Methods
3.1. General Considerations
3.2. Photophysical Measurements
3.3. Temperature-Dependent Emission Lifetimes
3.4. Lipophilicity
3.5. Cell Staining
3.6. Cell Viability
3.7. Photobleaching
3.8. Confocal Microscopy
3.9. Synthesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kolanowski, J.L.; Liu, F.; New, E.J. Fluorescent probes for the simultaneous detection of multiple analytes in biology. Chem. Soc. Rev. 2018, 47, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Kolanowski, J.L.; New, E.J. Reversible Fluorescent Probes for Biological Redox States. Angew. Chem. Int. Ed. 2016, 55, 1602–1613. [Google Scholar] [CrossRef]
- Depaoli, M.R.; Bischof, H.; Eroglu, E.; Burgstaller, S.; Ramadani-Muja, J.; Rauter, T.; Schinagl, M.; Waldeck-Weiermair, M.; Hay, J.C.; Graier, W.F.; et al. Live cell imaging of signaling and metabolic activities. Pharmacol. Ther. 2019, 202, 98–119. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; He, Y.; Sharma, A.; Kim, J.; Pan, W.H.; Yang, Z.G.; Li, J.; Yan, W.; Liu, L.W.; Qu, J.L.; et al. Fluorescent Probes for Nanoscopic Imaging of Mitochondria. Chem 2019, 5, 1697–1726. [Google Scholar] [CrossRef]
- Fernandez-Moreira, V.; Thorp-Greenwood, F.L.; Coogan, M.P. Application of d6 transition metal complexes in fluorescence cell imaging. Chem. Commun. 2010, 46, 186–202. [Google Scholar] [CrossRef] [PubMed]
- Lo, K.K.W. Luminescent Rhenium(I) and Iridium(III) Polypyridine Complexes as Biological Probes, Imaging Reagents, and Photocytotoxic Agents. Acc. Chem. Res. 2015, 48, 2985–2995. [Google Scholar] [CrossRef] [PubMed]
- Heffern, M.C.; Matosziuk, L.M.; Meade, T.J. Lanthanide Probes for Bioresponsive Imaging. Chem. Rev. 2014, 114, 4496–4539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coogan, M.P.; Fernandez-Moreira, V. Progress with, and prospects for, metal complexes in cell imaging. Chem. Commun. 2014, 50, 384–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnard, P.J.; Wedlock, L.E.; Baker, M.V.; Berners-Price, S.J.; Joyce, D.A.; Skelton, B.W.; Steer, J.H. Luminescence studies of the intracellular distribution of a dinuclear gold(I) N-heterocyclic carbene complex. Angew. Chem. Int. Ed. 2006, 45, 5966–5970. [Google Scholar] [CrossRef]
- Botchway, S.W.; Charnley, M.; Haycock, J.W.; Parker, A.W.; Rochester, D.L.; Weinstein, J.A.; Williams, J.A.G. Time-resolved and two-photon emission imaging microscopy of live cells with inert platinum complexes. Proc. Natl. Acad. Sci. USA 2008, 105, 16071–16076. [Google Scholar] [CrossRef] [Green Version]
- Mauro, M.; Aliprandi, A.; Septiadi, D.; Kehra, N.S.; De Cola, L. When self-assembly meets biology: Luminescent platinum complexes for imaging applications. Chem. Soc. Rev. 2014, 43, 4144–4166. [Google Scholar] [CrossRef] [PubMed]
- New, E.J.; Congreve, A.; Parker, D. Definition of the uptake mechanism and sub-cellular localisation profile of emissive lanthanide complexes as cellular optical probes. Chem. Sci. 2010, 1, 111–118. [Google Scholar] [CrossRef]
- Lee, L.C.C.; Leung, K.K.; Lo, K.K.W. Recent development of luminescent rhenium(I) tricarbonyl polypyridine complexes as cellular imaging reagents, anticancer drugs, and antibacterial agents. Dalton Trans. 2017, 46, 16357–16380. [Google Scholar] [CrossRef] [PubMed]
- Jarman, P.J.; Noakes, F.; Fairbanks, S.; Smitten, K.; Griffiths, I.K.; Saeed, H.K.; Thomas, J.A.; Smythe, C. Exploring the Cytotoxicity, Uptake, Cellular Response, and Proteomics of Mono- and Dinuclear DNA Light-Switch Complexes. J. Am. Chem. Soc. 2019, 141, 2925–2937. [Google Scholar] [CrossRef]
- Pettinari, R.; Marchetti, F.; Pettinari, C.; Condello, F.; Petrini, A.; Scopelliti, R.; Riedel, T.; Dyson, P.J. Organometallic rhodium(III) and iridium(III) cyclopentadienyl complexes with curcumin and bisdemethoxycurcumin co-ligands. Dalton Trans. 2015, 44, 20523–20531. [Google Scholar] [CrossRef] [Green Version]
- Colombo, A.; Fontani, M.; Dragonetti, C.; Roberto, D.; Williams, J.A.G.; di Perrotolo, R.S.; Casagrande, F.; Barozzi, S.; Polo, S. A Highly Luminescent Tetrahydrocurcumin Ir-III Complex with Remarkable Photoactivated Anticancer Activity. Chem. Eur. J. 2019, 25, 7948–7952. [Google Scholar] [CrossRef]
- Mari, C.; Panigati, M.; D’Alfonso, L.; Zanoni, I.; Donghi, D.; Sironi, L.; Collini, M.; Maiorana, S.; Baldoli, C.; D’Alfonso, G.; et al. Luminescent Conjugates between Dinuclear Rhenium Complexes and Peptide Nucleic Acids (PNA): Synthesis, Photophysical Characterization, and Cell Uptake. Organometallics 2012, 31, 5918–5928. [Google Scholar] [CrossRef]
- Ranieri, A.M.; Caporale, C.; Fiorini, V.; Hubbard, A.; Rigby, P.; Stagni, S.; Watkin, E.; Ogden, M.I.; Hackett, M.J.; Massi, M. Complementary Approaches to Imaging Subcellular Lipid Architectures in Live Bacteria Using Phosphorescent Iridium Complexes and Raman Spectroscopy. Chem. Eur. J. 2019, 25, 10566–10570. [Google Scholar] [CrossRef]
- Bader, C.A.; Sorvina, A.; Simpson, P.V.; Wright, P.J.; Stagni, S.; Plush, S.E.; Massi, M.; Brooks, D.A. Imaging nuclear, endoplasmic reticulum and plasma membrane events in real time. Febs Lett. 2016, 590, 3051–3060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadowski, M.C.; Mcpherson, S.J.; Rockstroh, A.; Soekmadji, C.; Tevz, G.; Gunter, J.H.; Jeet, V.; Nelson, C.C. Comprehensive profiling of androgen signalling and energy and lipid metabolism highlights heterogeneity of prostate cancer cell lines. Bju Int. 2016, 118, 34. [Google Scholar]
- Bader, C.A.; Carter, E.A.; Safitri, A.; Simpson, P.V.; Wright, P.; Stagni, S.; Massi, M.; Lay, P.A.; Brooks, D.A.; Plush, S.E. Unprecedented staining of polar lipids by a luminescent rhenium complex revealed by FTIR microspectroscopy in adipocytes. Mol. Biosyst. 2016, 12, 2064–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Gui, C.; Zhao, E.; Wang, J.; Li, X.; Qin, A.; Zhao, Z.; Yu, Z.; Tang, B.Z. Specific Fluorescence Probes for Lipid Droplets Based on Simple AIEgens. ACS Appl. Mater. Interfaces 2016, 8, 10193–10200. [Google Scholar] [CrossRef] [PubMed]
- Caporale, C.; Bader, C.A.; Sorvina, A.; MaGee, K.D.M.; Skelton, B.W.; Gillam, T.A.; Wright, P.J.; Raiteri, P.; Stagni, S.; Morrison, J.L.; et al. Investigating Intracellular Localisation and Cytotoxicity Trends for Neutral and Cationic Iridium Tetrazolato Complexes in Live Cells. Chem. Eur. J. 2017, 23, 15666–15679. [Google Scholar] [CrossRef] [Green Version]
- Koguro, K.; Oga, T.; Mitsui, S.; Orita, R. Novel Synthesis of 5-Substituted Tetrazoles from Nitriles. Synthesis 1998, 1998, 910–914. [Google Scholar] [CrossRef]
- Stagni, S.; Colella, S.; Palazzi, A.; Valenti, G.; Zacchini, S.; Paolucci, F.; Marcaccio, M.; Albuquerque, R.Q.; De Cola, L. Essential role of the ancillary ligand in the color tuning of iridium tetrazolate complexes. Inorg. Chem. 2008, 47, 10509–10521. [Google Scholar] [CrossRef]
- Wissner, A.; Grudzinskas, C.V. Reaction of Tert-Butyldimethylsilyl Esters with Oxalyl Chloride-Dimethylformamide: Preparation of Carboxylic Acid Chlorides under Neutral Conditions. J. Org. Chem. 1978, 43, 3972–3974. [Google Scholar] [CrossRef]
- Beniwal, M.; Jain, N. Review Article on Vilsmeier Haack Reaction and Its. Eur. J. Biomed. Pharm. Sci. 2015, 2, 1340–1374. [Google Scholar]
- Cherney, A.H.; Kadunce, N.T.; Reisman, S.E. Catalytic asymmetric reductive acyl cross-coupling: Synthesis of enantioenriched acyclic α,α-disubstituted ketones. J. Am. Chem. Soc. 2013, 135, 7442–7445. [Google Scholar] [CrossRef] [Green Version]
- Nakamaru, K. Synthesis, luminescence quantum yields, and lifetimes of trischelated ruthenium(II) mixed-ligand complexes including 3, 3’-dimethyl-2, 2’-bipyridyl. Bull. Chem. Soc. Jpn. 1982, 55, 2697–2705. [Google Scholar] [CrossRef] [Green Version]
- Umamahesh, B.; Karthikeyan, N.S.; Sathiyanarayanan, K.I.; Malicka, J.M.; Cocchi, M. Tetrazole iridium(III) complexes as a class of phosphorescent emitters for high-efficiency OLEDs. J. Mater. Chem. C 2016, 4, 10053–10060. [Google Scholar] [CrossRef]
- Lamansky, S.; Djurovich, P.; Murphy, D.; Abdel-Razzaq, F.; Lee, H.E.; Adachi, C.; Burrows, P.E.; Forrest, S.R.; Thompson, M.E. Highly phosphorescent bis-cyclometalated iridium complexes: Synthesis, photophysical characterization, and use in organic light emitting diodes. J. Am. Chem. Soc. 2001, 123, 4304–4312. [Google Scholar] [CrossRef]
- Flamigni, L.; Barbieri, A.; Sabatini, C.; Ventura, B.; Barigelletti, F. Photochemistry and photophysics of coordination compounds: Iridium. Top. Curr. Chem. 2007, 171, 143–203. [Google Scholar] [CrossRef]
- Mauro, M.; De Paoli, G.; Otter, M.; Donghi, D.; D’Alfonso, G.; De Cola, L. Aggregation induced colour change for phosphorescent iridium(III) complex-based anionic surfactants. Dalton Trans. 2011, 40, 12106–12116. [Google Scholar] [CrossRef]
- Leung, S.-K.; Liu, H.-W.; Lo, K.K.-W. Functionalization of luminescent cyclometalated iridium(III) polypyridine complexes with a fluorous moiety: Photophysics, protein-binding, bioconjugation, and cellular uptake properties. Chem. Commun. 2011, 47, 10548–10550. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, C.; Schmidt, R. Physical Mechanisms of Generation and Deactivation of Singlet Oxygen. Chem. Rev. 2003, 103, 1685–1757. [Google Scholar] [CrossRef] [PubMed]
- Abdel-shafi, A.A.; Worrall, D.R. Mechanism of the excited singlet and triplet states quenching by molecular oxygen in acetonitrile. J. Photochem. Photobiol. 2005, 172, 170–179. [Google Scholar] [CrossRef]
- Caspar, J.V.; Meyer, T.J. Application of the energy gap law to nonradiative, excited-state decay. J. Phys. Chem. 1983, 87, 952–957. [Google Scholar] [CrossRef]
- Koike, K.; Okoshi, N.; Hori, H.; Takeuchi, K.; Ishitani, O.; Tsubaki, H.; Clark, I.P.; George, M.W.; Johnson, F.P.A.; Turner, J.J. Mechanism of the photochemical ligand substitution reactions of fac-[Re(bPY)(CO)3(PR3)]+ complexes and the properties of their triplet ligand-field excited states. J. Am. Chem. Soc. 2002, 124, 11448–11455. [Google Scholar] [CrossRef]
- Costa, R.D.; Monti, F.; Accorsi, G.; Barbieri, A.; Bolink, H.J.; Orti, E.; Armaroli, N. Photophysical Properties of Charged Cyclometalated Ir(III) Complexes: A Joint Theoretical and Experimental Study. Inorg. Chem. 2011, 50, 7229–7238. [Google Scholar] [CrossRef]
- Huber, W.; Linder, R.; Niesel, J.; Schatzschneider, U.; Spingler, B.; Kunz, P.C. A comparative study of tricarbonylmanganese photoactivatable CO releasing molecules (PhotoCORMs) by using the myoglobin assay and time-resolved IR spectroscopy. Eur. J. Inorg. Chem. 2012, 3140–3146. [Google Scholar] [CrossRef]
- He, L.; Li, Y.; Tan, C.-P.; Ye, R.-R.; Chen, M.-H.; Cao, J.-J.; Ji, L.-N.; Mao, Z.-W. Cyclometalated iridium(III) complexes as lysosome-targeted photodynamic anticancer and real-time tracking agents. Chem. Sci. 2015, 6, 5409–5418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.Y.; Li, S.P.-Y.; Zhu, N.; Or, L.W.-S.; Cheung, M.S.-H.; Lam, Y.-W.; Lo, K.K.-W. Structure, photophysical and electrochemical properties, biomolecular interactions, and intracellular uptake of luminescent cyclometalated iridium(III) dipyridoquinoxaline complexes. Inorg. Chem. 2010, 49, 2530–2540. [Google Scholar] [CrossRef] [PubMed]
- Steunenberg, P.; Ruggi, A.; Van Den Berg, N.S.; Buckle, T.; Kuil, J.; Van Leeuwen, F.W.B.; Velders, A.H. Phosphorescence imaging of living cells with amino acid-functionalized tris(2-phenylpyridine)iridium(III) complexes. Inorg. Chem. 2012, 51, 2105–2114. [Google Scholar] [CrossRef]
- Law, W.H.-T.; Lee, L.C.-C.; Louie, M.-W.; Liu, H.-W.; Ang, T.W.-H.; Lo, K.K.-W. Phosphorescent cellular probes and uptake indicators derived from cyclometalated iridium(III) bipyridine complexes appended with a glucose or galactose entity. Inorg. Chem. 2013, 52, 13029–13041. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.-J.; Tan, C.-P.; Chen, M.-H.; Wu, N.; Yao, D.-Y.; Liu, X.-G.; Ji, L.-N.; Mao, Z.-W. Targeting cancer cell metabolism with mitochondria-immobilized phosphorescent cyclometalated iridium(III) complexes. Chem. Sci. 2017, 8, 631–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, K.K.W.; Lee, P.K.; Lau, J.S.Y. Synthesis, characterization, and properties of luminescent organoiridium(III) polypyridine complexes appended with an alkyl chain and their interactions with lipid bilayers, surfactants, and living cells. Organometallics 2008, 27, 2998–3006. [Google Scholar] [CrossRef]
- Sansee, A.; Meksawangwong, S.; Chainok, K.; Franz, K.J.; Gál, M.; Pålsson, L.O.; Puniyan, W.; Traiphol, R.; Pal, R.; Kielar, F. Novel aminoalkyl tris-cyclometalated iridium complexes as cellular stains. Dalton Trans. 2016, 45, 17420–17430. [Google Scholar] [CrossRef]
- Owen, T.; Butler, A. Metallosurfactants of bioinorganic interest: Coordination- induced self assembly. Coord. Chem. Rev. 2011, 225, 678–687. [Google Scholar] [CrossRef] [Green Version]
- Lo, K.K.-W.; Law, W.H.-T.; Chan, J.C.-Y.; Liu, H.-W.; Zhang, K.Y. Photophysical and cellular uptake properties of novel phosphorescent cyclometalated iridium(III) bipyridine d-fructose complexes. Metallomics 2013, 5, 808–812. [Google Scholar] [CrossRef]
- Li, L.; Szmacinski, H.; Lakowicz, J.R. Synthesis and luminescence spectral characterization of long-lifetime lipid metal-ligand probes. Anal. Biochem. 1997, 244, 80–85. [Google Scholar] [CrossRef]
- Li, L.; Szmacinski, H.; Lakowicz, J.R. Long-lifetime lipid probe containing a luminescent metal-ligand complex. Biospectroscopy 1997, 3, 155–159. [Google Scholar] [CrossRef]
- Balakrishnan, G.; Rajendran, T.; Senthil Murugan, K.; Sathish Kumar, M.; Sivasubramanian, V.K.; Ganesan, M.; Mahesh, A.; Thirunalasundari, T.; Rajagopal, S. Interaction of rhenium(I) complex carrying long alkyl chain with Calf Thymus DNA: Cytotoxic and cell imaging studies. Inorg. Chim. Acta 2015, 434, 51–59. [Google Scholar] [CrossRef]
- Kumar, S.V.; Scottwell, S.O.; Waugh, E.; McAdam, C.J.; Hanton, L.R.; Brooks, H.J.L.; Crowley, J.D. Antimicrobial Properties of Tris(homoleptic) Ruthenium(II) 2-Pyridyl-1,2,3-triazole “Click” Complexes against Pathogenic Bacteria, Including Methicillin-Resistant Staphylococcus aureus (MRSA). Inorg. Chem. 2016, 55, 9767–9777. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, P.; Qiu, K.; Huang, J.; Chen, Y.; Ji, L.; Chao, H. Mitochondrial Dynamics Tracking with Two-Photon Phosphorescent Terpyridyl Iridium(III) Complexes. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, W.; Zuo, J.; Ji, L.; Chao, H. Dinuclear iridium(III) complexes as phosphorescent trackers to monitor mitochondrial dynamics. J. Mater. Chem. B 2015, 3, 3306–3314. [Google Scholar] [CrossRef]
- Huang, H.; Yang, L.; Zhang, P.; Qiu, K.; Huang, J.; Chen, Y.; Diao, J.J.; Liu, J.; Ji, L.; Long, J.; et al. Real-time tracking mitochondrial dynamic remodeling with two-photon phosphorescent iridium(III) complexes. Biomaterials 2016, 83, 321–331. [Google Scholar] [CrossRef]
- Qiu, K.; Huang, H.; Liu, B.; Liu, Y.; Huang, Z.; Chen, Y.; Ji, L.; Chao, H. Long-Term Lysosomes Tracking with a Water-Soluble Two-Photon Phosphorescent Iridium(III) Complex. ACS Appl. Mater. Interfaces 2016, 8, 12702–12710. [Google Scholar] [CrossRef]
- Lv, W.; Yang, T.; Yu, Q.; Zhao, Q.; Zhang, K.Y.; Liang, H.; Liu, S.; Li, F.; Huang, W. A Phosphorescent Iridium(III) Complex-Modified Nanoprobe for Hypoxia Bioimaging Via Time-Resolved Luminescence Microscopy. Adv. Sci. 2015, 2, 1500107. [Google Scholar] [CrossRef]
- Sprouse, S.; King, K.A.; Spellane, P.J.; Watts, R.J. Photophysical Effects of Metal-Carbon c Bonds in Ortho-Metalated Complexes of Ir(III) and Rh(III). J. Am. Chem. Soc. 1984, 106, 6647–6653. [Google Scholar] [CrossRef]
- Hedberg, Y.; Gustafsson, J.; Karlsson, H.L.; Moller, L.; Wallinder, I.O. Bioaccessibility, bioavailability and toxicity of commercially relevant iron- and chromium-based particles: In vitro studies with an inhalation perspective. Part. Fibre Toxicol. 2010, 7. [Google Scholar] [CrossRef] [Green Version]
- Demas, J.N.; Crosby, G.A. The Measurement of Photoluminescence Quantum Yields. A Review. J. Phys. Chem. 1971, 75, 991–1024. [Google Scholar]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef]
- Rampersad, S.N. Multiple Applications of Alamar Blue as an Indicator of Metabolic Function and Cellular Health in Cell Viability Bioassays. Sens. Basel 2012, 12, 12347–12360. [Google Scholar] [CrossRef] [PubMed]
- Hamid, R.; Rotshteyn, Y.; Rabadi, L.; Parikh, R.; Bullock, P. Comparison of alamar blue and MTT assays for high through-put screening. Toxicol Vitro 2004, 18, 703–710. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Solvent a | λabs (nm) (104 ε [M−1 cm−1]) | λem (nm) | τaer τdeaer (ns) b | Φaer c Φdeaer c |
|---|---|---|---|---|---|
| [Ir(ppy)2 (TzPyNH2)] | CH2Cl2 | 270 (9.45), 346 (1.79), 387 (0.89), 423 (0.53) | 484, 514 | 101 1032 | 0.005 0.028 |
| H2O | 273 (5.30), 388 (0.91), 422 (0.56) | 484, 508 | 30 (3), 466 (97) | 0.012 | |
| [Ir(ppy)2 (TzPyNHC8sat)] | CH2Cl2 | 264 (8.30), 364 (0.46), 420 (0.26) | 500, 514 | 83 393 | 0.006 0.067 |
| H2O | 287 (4.17), 393 (1.56), 426 (1.20) | 493, 520 | 55 (81), 277 (19) | 0.004 | |
| [Ir(ppy)2 (TzPyNHC16sat)] | CH2Cl2 | 264 (7.91), 386 (0.29), 423 (0.15) | 500, 530 | 80 374 | 0.006 0.067 |
| H2O | 280 (2.63), 392 (0.45), 433 (0.28) | 495, 526 | 53 (43), 197 (57) | 0.009 | |
| [Ir(ppy)2 (TzPyNHC18sat)] | CH2Cl2 | 264 (8.31), 386 (0.36), 422 (0.20) | 502, 526 | 83 383 | 0.007 0.060 |
| H2O | 281 (5.27), 393 (2.22), 452 (1.77) | 498, 530 | 195 (57), 999 (43) | 0.024 | |
| [Ir(ppy)2 (TzPyNHC16cis)] | CH2Cl2 | 264 (8.24), 386 (0.36), 421 (0.21) | 496, 548 | 75 356 | 0.007 0.054 |
| H2O | 284 (6.38), 397 (1.33), 435 (0.84) | 496, 548 | 70 (25), 946 (75) | 0.009 | |
| [Ir(ppy)2 (TzPyNHC18cis)] | CH2Cl2 | 264 (7.16), 386 (0.26), 418 (0.15) | 493, 537 | 75 360 | 0.008 0.060 |
| H2O | 282 (6.04), 394 (1.28), 431 (0.85) | 496, 547 | 34 (59), 339 (41) | 0.005 |
| Complexes | Log D7.4 |
|---|---|
| [Ir(ppy)2(TzPyNH2)] | 1.99 ± 0.04 |
| [Ir(ppy)2(TzPyNHC8sat)] | 2.66 ± 0.07 |
| [Ir(ppy)2(TzPyNHC16sat)] | 2.84 ± 0.08 |
| [Ir(ppy)2(TzPyNHC18sat)] | 2.63 ± 0.06 |
| [Ir(ppy)2(TzPyNHC16cis)] | 2.31 ± 0.06 |
| [Ir(ppy)2(TzPyNHC18cis)] | 2.26 ± 0.06 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caporale, C.; Ranieri, A.M.; Paternoster, S.; Bader, C.A.; Falasca, M.; Plush, S.E.; Brooks, D.A.; Stagni, S.; Massi, M. Photophysical and Biological Properties of Iridium Tetrazolato Complexes Functionalised with Fatty Acid Chains. Inorganics 2020, 8, 23. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8040023
Caporale C, Ranieri AM, Paternoster S, Bader CA, Falasca M, Plush SE, Brooks DA, Stagni S, Massi M. Photophysical and Biological Properties of Iridium Tetrazolato Complexes Functionalised with Fatty Acid Chains. Inorganics. 2020; 8(4):23. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8040023
Chicago/Turabian StyleCaporale, Chiara, Anna Maria Ranieri, Silvano Paternoster, Christie A. Bader, Marco Falasca, Sally E. Plush, Douglas A. Brooks, Stefano Stagni, and Massimiliano Massi. 2020. "Photophysical and Biological Properties of Iridium Tetrazolato Complexes Functionalised with Fatty Acid Chains" Inorganics 8, no. 4: 23. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8040023