A Mixed-Valence Tetra-Nuclear Nickel Dithiolene Complex: Synthesis, Crystal Structure, and the Lability of Its Nickel Sulfur Bonds



Abstract
:
1. Introduction
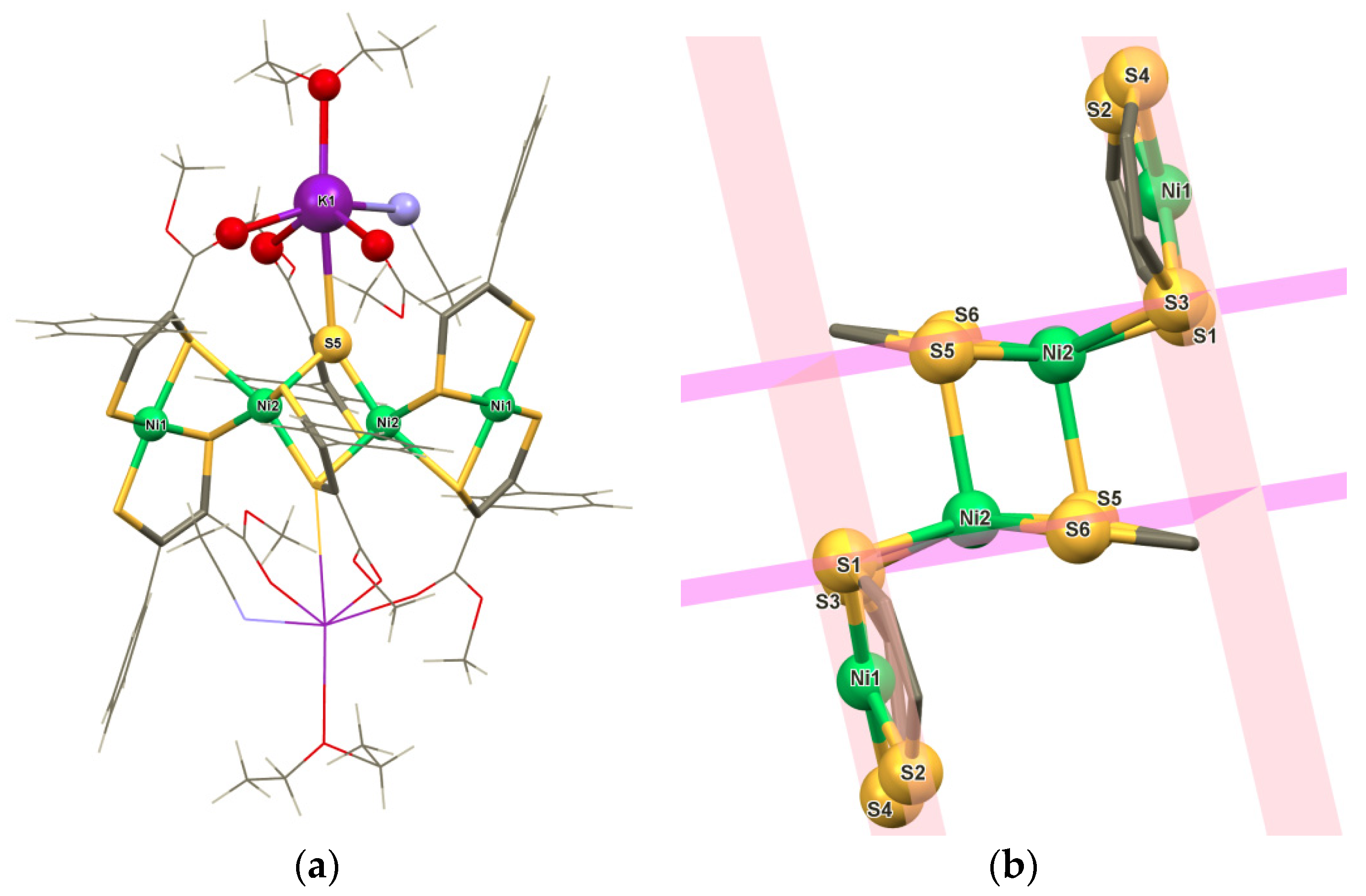
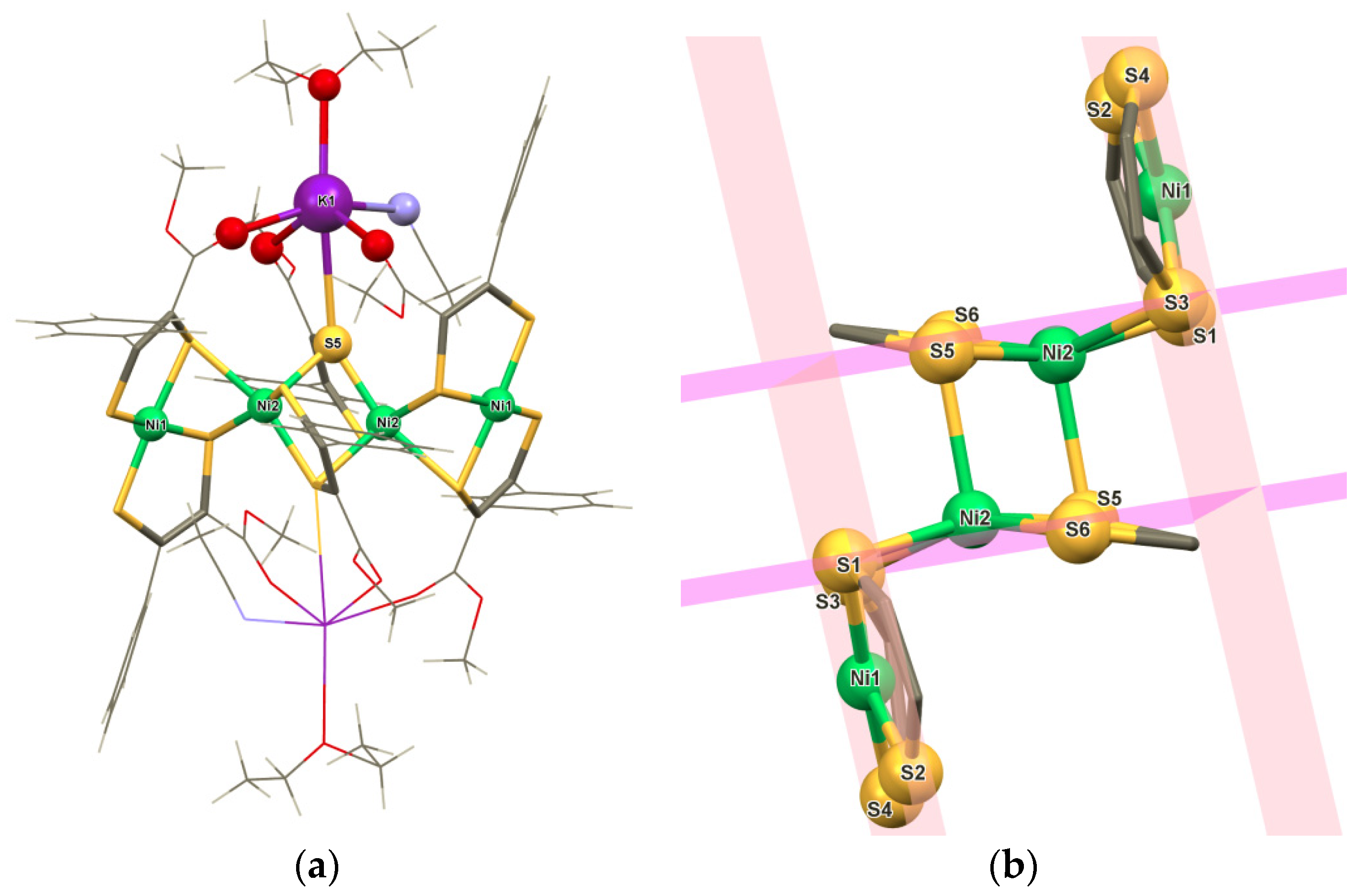
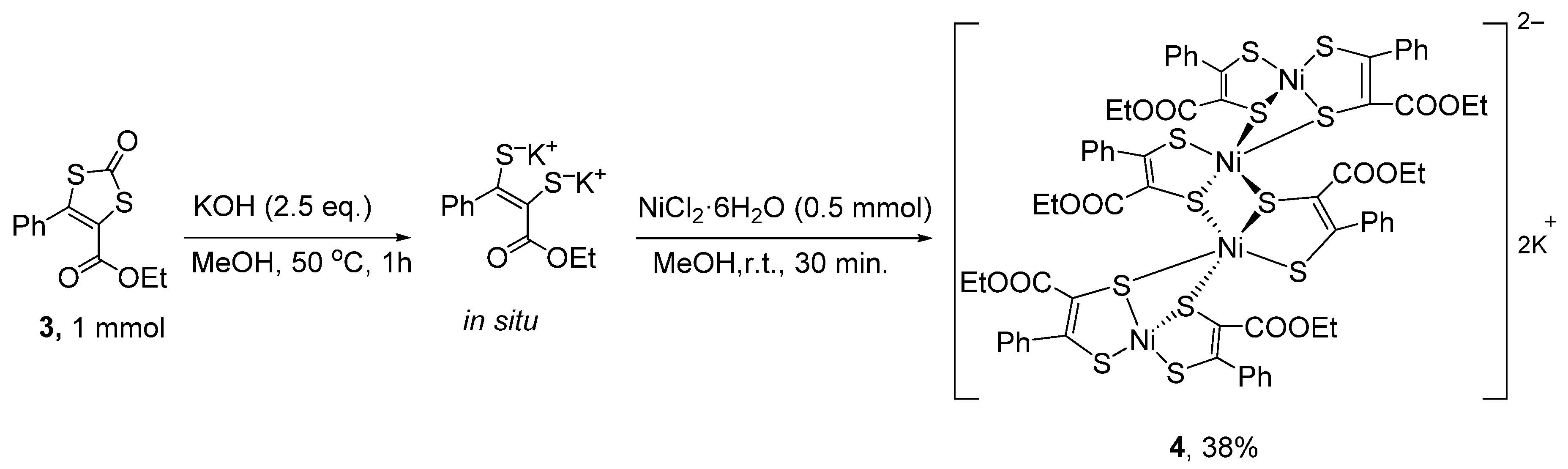
2. Results and Discussion
3. Materials and Methods
3.1. Physical Measurements
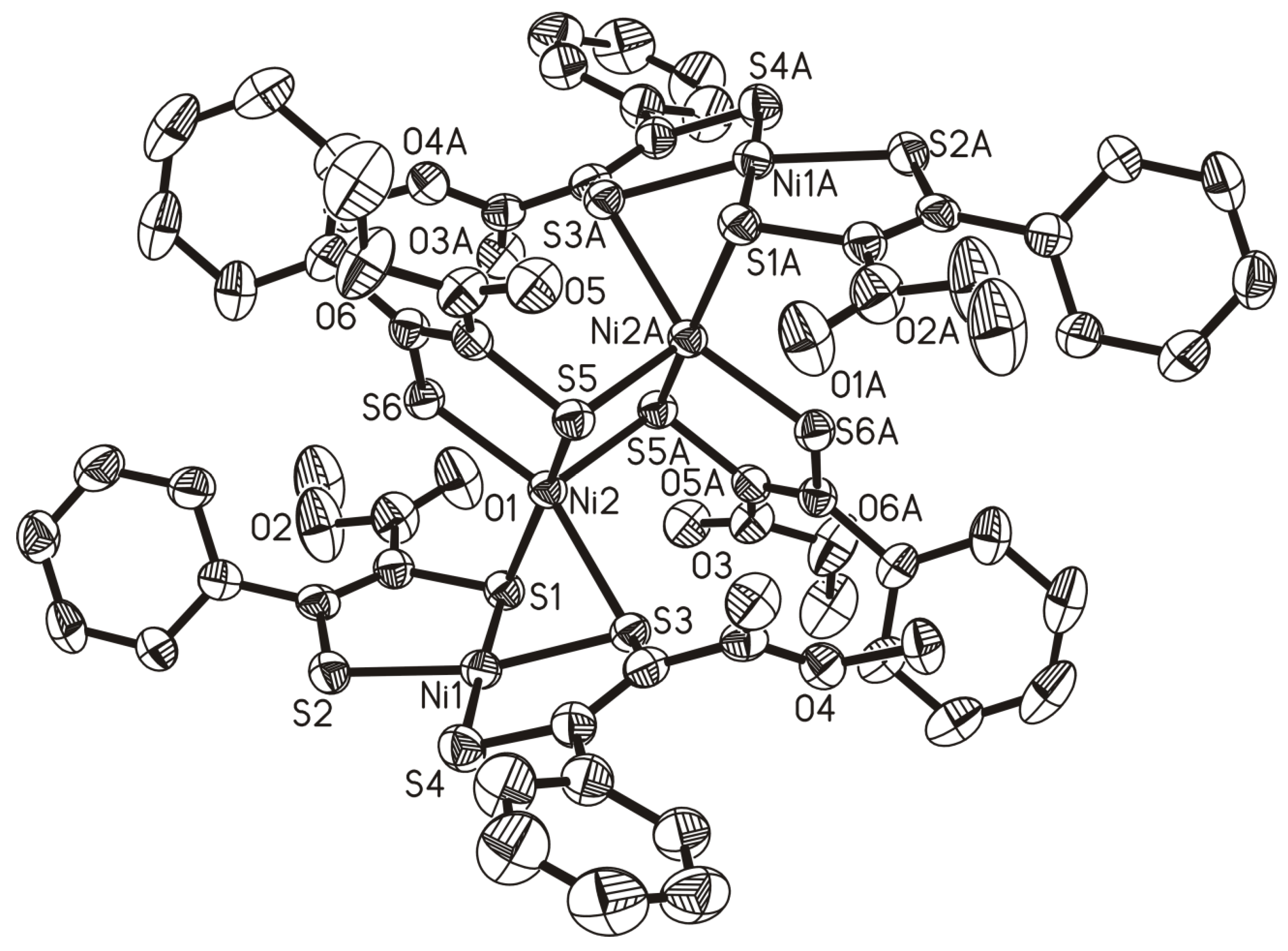
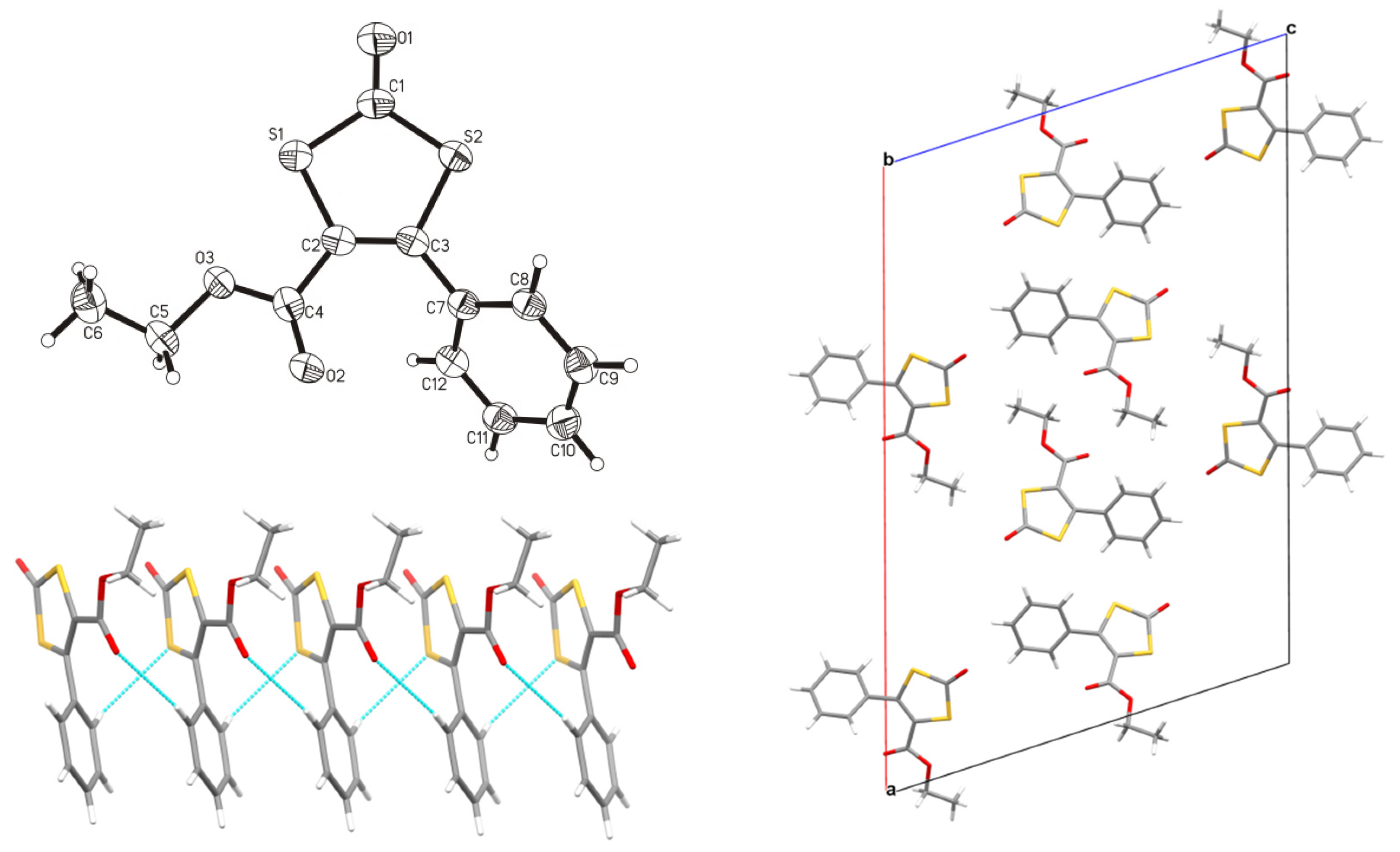
3.2. X-Ray Crystallography
3.3. Syntheses
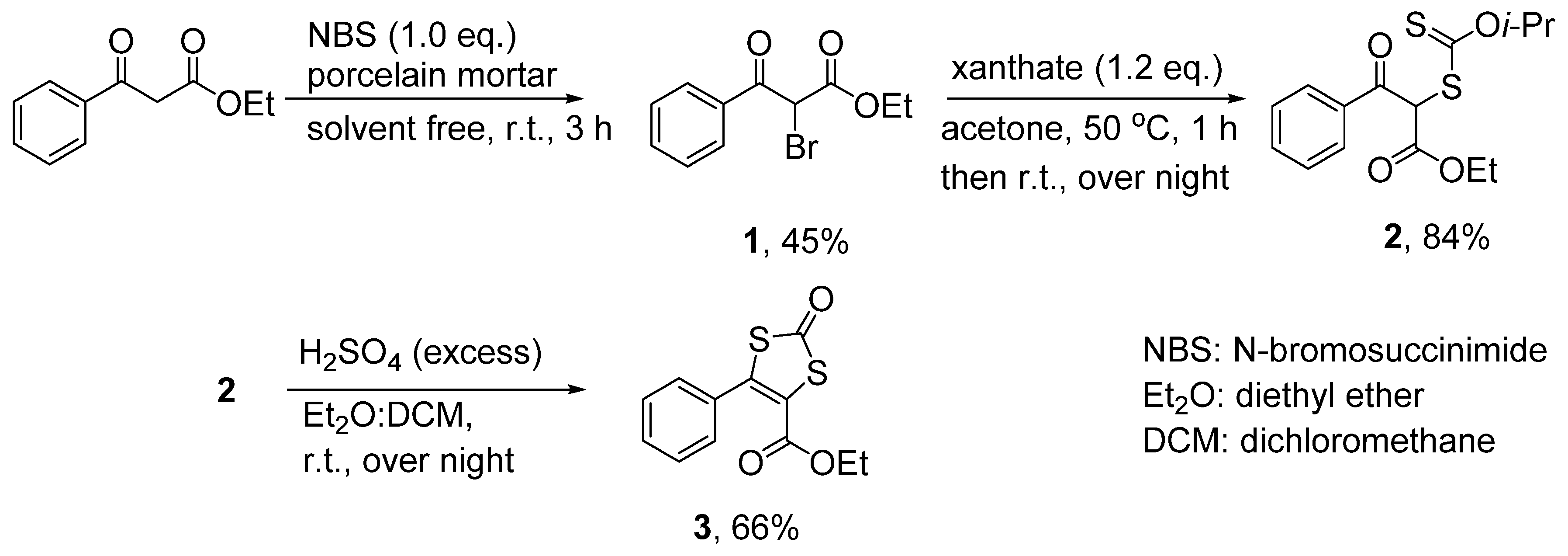
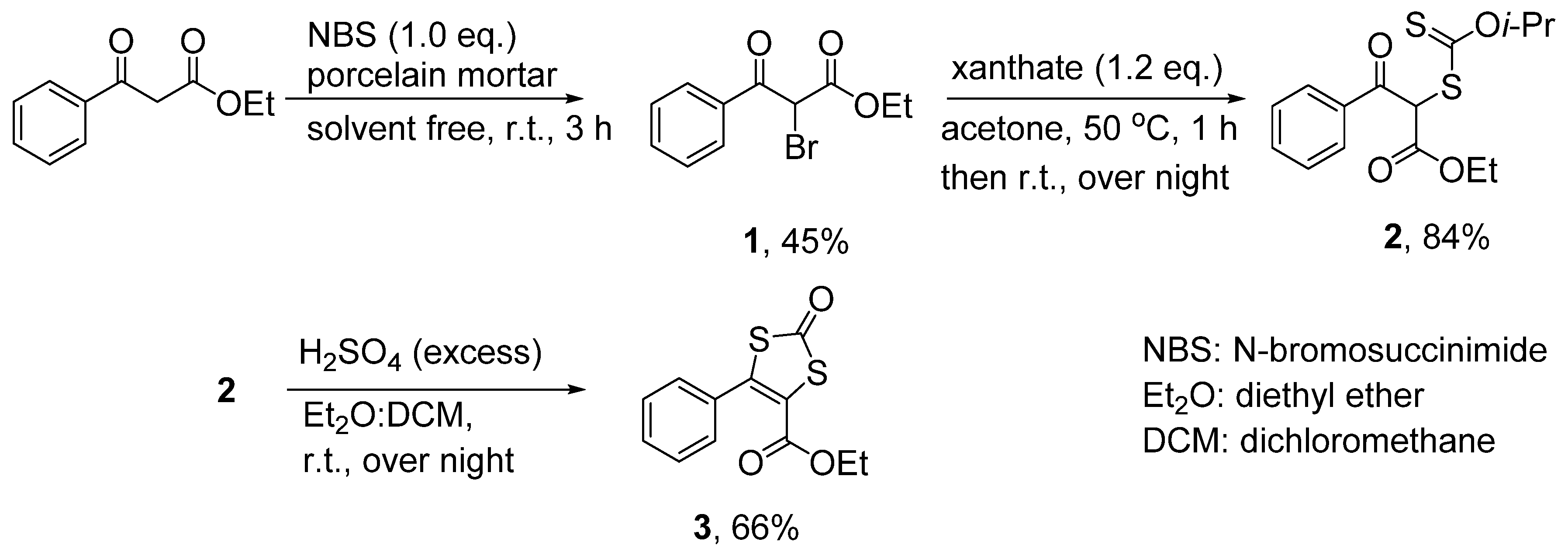
3.3.1. Ethyl 2-bromo-3-oxo-3-phenylpropanoate (1)
3.3.2. Ethyl 2-(isopropoxycarbonothioylthio)-3-oxo-3-phenylpropanoate (2)
3.3.3. 4-Ethylcarboxylate-5-phenyl-1,3-dithiole-2-one (3)
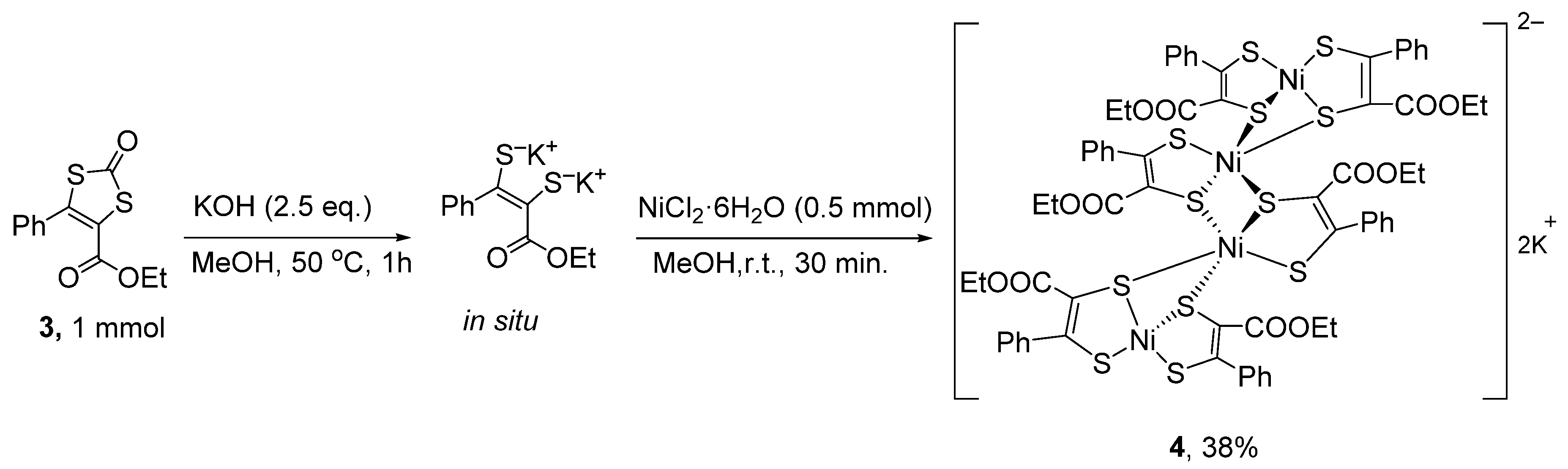
3.3.4. K2[Ni4(ecpdt)6] ·2CH3CN·2Et2O (4·2CH3CN·2Et2O)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schrauzer, G.N.; Mayweg, V.P. Preparation, Reactions, and Structure of Bisdithio-α-diketone Complexes of Nickel, Palladium, and Platinum. J. Am. Chem. Soc. 1965, 87, 1483. [Google Scholar] [CrossRef]
- Schrauzer, G.N.; Mayweg, V. Reaction of Diphenylacetylene with Nickel Sulfides. J. Am. Chem. Soc. 1962, 84, 3221. [Google Scholar] [CrossRef]
- Eisenberg, R.; Stiefel, E.I.; Rosenberg, R.C.; Gray, H.B. Six-Coordinate Trigonal-Prismatic Complexes of First-Row Transition Metals1. J. Am. Chem. Soc. 1966, 88, 2874–2876. [Google Scholar] [CrossRef]
- Stiefel, E.I.; Eisenberg, R.; Rosenberg, R.C.; Gray, H.B. Characterization and Electronic Structures of Six-Coordinate Trigonal-Prismatic Complexes. J. Am. Chem. Soc. 1966, 88, 2956–2966. [Google Scholar] [CrossRef]
- Balch, A.L.; Dance, I.G.; Holm, R.H. Characterization of dimeric dithiolene complexes. J. Am. Chem. Soc. 1968, 90, 1139–1145. [Google Scholar] [CrossRef]
- Mueller-Westerhoff, U.T.; Vance, B.; Yoon, D.I. The synthesis of dithiolene dyes with strong near-IR absorption. Tetrahedron 1991, 47, 909–932. [Google Scholar] [CrossRef]
- Winter, C.S.; Oliver, S.N.; Manning, R.J.; Rush, J.D.; Hill, C.A.S.; Underhill, A.E. Non-linear optical studies of nickel dithiolene complexes. J. Mater. Chem. 1992, 2, 443–447. [Google Scholar] [CrossRef]
- Oku, H.; Ueyama, N.; Kondo, M.; Nakamura, A. Oxygen atom transfer systems in which the (m-oxo)dimolybdenum(V) complex formation does not occur: Syntheses, structures, and reactivities of monooxomolybdenum(IV) benzenedithiolato complexes as models of molybdenum oxidoreductases. Inorg. Chem. 1994, 33, 209–216. [Google Scholar] [CrossRef]
- Davies, E.S.; Beddoes, R.L.; Collison, D.; Dinsmore, A.; Docrat, A.; Joule, J.A.; Wilson, C.R.; Garner, C.D. Synthesis of oxomolybdenum bis(dithiolene) complexes related to the cofactor of the oxomolybdoenzymes. J. Chem. Soc. Dalton Trans. 1997, 3985–3995. [Google Scholar] [CrossRef]
- Donahue, J.P.; Goldsmith, C.R.; Nadiminti, U.; Holm, R.H. Synthesis, Structures, and Reactivity of Bis(dithiolene)molybdenum(IV,VI) Complexes Related to the Active Sites of Molybdoenzymes. J. Am. Chem. Soc. 1998, 120, 12869–12881. [Google Scholar] [CrossRef]
- Holm, R.H.; Solomon, E.I.; Majumdar, A.; Tenderholt, A. Comparative molecular chemistry of molybdenum and tungsten and its relation to hydroxylase and oxotransferase enzymes. Coord. Chem. Rev. 2011, 255, 993–1015. [Google Scholar] [CrossRef]
- Periyasamy, G.; Burton, N.A.; Hillier, I.H.; Vincent, M.A.; Disley, H.; McMaster, J.; Garner, C.D. The dithiolene ligand—Innocent or ‘non-innocent’? A theoretical and experimental study of some cobalt-dithiolene complexes. Faraday Discuss. 2007, 135, 469–488. [Google Scholar] [CrossRef]
- Adams, H.; Morris, M.J.; Robertson, C.C.; Tunnicliffe, H.C.I. Synthesis of Mono- and Diiron Dithiolene Complexes as Hydrogenase Models by Dithiolene Transfer Reactions, Including the Crystal Structure of [{Ni(S2C2Ph2)}6]. Organometallics 2019, 38, 665–676. [Google Scholar] [CrossRef]
- Maroney, M.J.; Ciurli, S. Nonredox Nickel Enzymes. Chem. Rev. 2014, 114, 4206–4228. [Google Scholar] [CrossRef] [Green Version]
- Can, M.; Armstrong, F.A.; Ragsdale, S.W. Structure, Function, and Mechanism of the Nickel Metalloenzymes, CO Dehydrogenase, and Acetyl-CoA Synthase. Chem. Rev. 2014, 114, 4149–4174. [Google Scholar] [CrossRef]
- Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef]
- Van Gastel, M.; Shaw, J.L.; Blake, A.J.; Flores, M.; Schröder, M.; McMaster, J.; Lubitz, W. Electronic Structure of a Binuclear Nickel Complex of Relevance to [NiFe] Hydrogenase. Inorg. Chem. 2008, 47, 11688–11697. [Google Scholar] [CrossRef]
- Choudhury, S.B.; Pressler, M.A.; Mirza, S.A.; Day, R.O.; Maroney, M.J. Structure and Redox Chemistry of Analogous Nickel Thiolato and Selenolato Complexes: Implications for the Nickel Sites in Hydrogenases. Inorg. Chem. 1994, 33, 4831–4839. [Google Scholar] [CrossRef]
- Mondragón, A.; Flores-Alamo, M.; Martínez-Alanis, P.R.; Aullón, G.; Ugalde-Saldívar, V.M.; Castillo, I. Electrocatalytic Proton Reduction by Dimeric Nickel Complex of a Sterically Demanding Pincer-type NS2 Aminobis(thiophenolate) Ligand. Inorg. Chem. 2015, 54, 619–627. [Google Scholar] [CrossRef]
- Song, L.-C.; Lu, Y.; Cao, M.; Yang, X.-Y. Reactions of dinuclear Ni2 complexes [Ni(RNPyS4)]2 (RNPyS4 = 2,6-bis(2-mercaptophenylthiomethyl)-4-R-pyridine) with Fe(CO)3(BDA) (BDA = benzylidene acetone) leading to heterodinuclear NiFe and mononuclear Fe complexes related to the active sites of [NiFe]- and [Fe]-hydrogenases. RSC Adv. 2016, 6, 39225–39233. [Google Scholar]
- Schrauzer, G.N.; Mayweg, V.P.; Heinrich, W. Coordination Compounds with Delocalized Ground States. α-Dithiodiketone-Substituted Group VI Metal Carbonyls and Related Compounds. J. Am. Chem. Soc. 1966, 88, 5174–5179. [Google Scholar] [CrossRef]
- Schrauzer, G.N. Coordination compounds with delocalized ground states. Transition metal derivatives of dithiodiketones and ethylene-1,2-dithiolates (metal dithienes). Acc. Chem. Res. 1969, 2, 72–80. [Google Scholar] [CrossRef]
- Lim, B.S.; Donahue, J.P.; Holm, R.H. Synthesis and Structures of Bis(dithiolene)molybdenum Complexes Related to the Active Sites of the DMSO Reductase Enzyme Family. Inorg. Chem. 2000, 39, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Obanda, A.; Martinez, K.; Schmehl, R.H.; Mague, J.T.; Rubtsov, I.V.; MacMillan, S.N.; Lancaster, K.M.; Sproules, S.; Donahue, J.P. Expanding the Scope of Ligand Substitution from [M(S2C2Ph2] (M = Ni2+, Pd2+, Pt2+) To Afford New Heteroleptic Dithiolene Complexes. Inorg. Chem. 2017, 56, 10257–10267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goddard, C.A.; Holm, R.H. Synthesis and Reactivity Aspects of the Bis(dithiolene) Chalcogenide Series [WIVQ(S2C2R2)2]2− (Q = O, S, Se). Inorg. Chem. 1999, 38, 5389–5398. [Google Scholar] [CrossRef]
- Lim, B.S.; Fomitchev, D.V.; Holm, R.H. Nickel Dithiolenes Revisited: Structures and Electron Distribution from Density Functional Theory for the Three-Member Electron-Transfer Series [Ni(S2C2Me2)2]0,1−,2−. Inorg. Chem. 2001, 40, 4257–4262. [Google Scholar] [CrossRef]
- Ghosh, A.C.; Schulzke, C. Selectively detecting Hg2+—A “mercury quick test” with bis-(coumarin–dithiolene) niccolate. Inorg. Chim. Acta 2016, 445, 149–154. [Google Scholar] [CrossRef]
- Kean, C.L.; Pickup, P.G. A low band gap conjugated metallopolymer with nickel bis(dithiolene) crosslinks. Chem. Commun. 2001, 815–816. [Google Scholar] [CrossRef]
- Papavassiliou, G.C.; Anyfantis, G.C.; Mousdis, G.A. Neutral Metal 1,2-Dithiolenes: Preparations, Properties and Possible Applications of Unsymmetrical in Comparison to the Symmetrical. Crystals 2012, 2, 762–811. [Google Scholar] [CrossRef] [Green Version]
- Anyfantis, G.C.; Papavassiliou, G.C.; Assimomytis, N.; Terzis, A.; Psycharis, V.; Raptopoulou, C.P.; Kyritsis, P.; Thoma, V.; Koutselas, I.B. Some unsymmetrical nickel 1,2-dithiolene complexes as candidate materials for optics and electronics. Solid State Sci. 2008, 10, 1729–1733. [Google Scholar] [CrossRef]
- Cassoux, P. Molecular (super)conductors derived from bis-dithiolate metal complexes. Coord. Chem. Rev. 1999, 185–186, 213–232. [Google Scholar] [CrossRef]
- Kato, R. Conducting Metal Dithiolene Complexes: Structural and Electronic Properties. Chem. Rev. 2004, 104, 5319–5346. [Google Scholar] [CrossRef] [PubMed]
- Kusamoto, T.; Nishihara, H. Zero-, one- and two-dimensional bis(dithiolato)metal complexes with unique physical and chemical properties. Coord. Chem. Rev. 2019, 380, 419–439. [Google Scholar] [CrossRef]
- Lieffrig, J.; Jeannin, O.; Auban-Senzier, P.; Fourmigué, M. Chiral Conducting Salts of Nickel Dithiolene Complexes. Inorg. Chem. 2012, 51, 7144–7152. [Google Scholar] [CrossRef]
- Neves, A.I.S.; Santos, I.C.; Pereira, L.C.J.; Rovira, C.; Ruiz, E.; Belo, D.; Almeida, M. Ni-2,3-thiophenedithiolate Anions in New Architectures: An In-Line Mixed-Valence Ni Dithiolene (Ni4–S12) Cluster. Eur. J. Inorg. Chem. 2011, 2011, 4807–4815. [Google Scholar] [CrossRef]
- Jeannin, O.; Clérac, R.; Fourmigué, M. Order−Disorder Transition Coupled with Magnetic Bistability in the Ferricinium Salt of a Radical Nickel Dithiolene Complex. J. Am. Chem. Soc. 2006, 128, 14649–14656. [Google Scholar] [CrossRef]
- Deplano, P.; Pilia, L.; Espa, D.; Mercuri, M.L.; Serpe, A. Square-planar d8 metal mixed-ligand dithiolene complexes as second order nonlinear optical chromophores: Structure/property relationship. Coord. Chem. Rev. 2010, 254, 1434–1447. [Google Scholar] [CrossRef]
- Basu, P.; Nigam, A.; Mogesa, B.; Denti, S.; Nemykin, V.N. Synthesis, characterization, spectroscopy, electronic and redox properties of a new nickel dithiolene system. Inorg. Chim. Acta 2010, 363, 2857–2864. [Google Scholar] [CrossRef]
- Alves, H.; Simão, D.; Cordeiro Santos, I.; Gama, V.; Teives Henriques, R.; Novais, H.; Almeida, M. A Series of Transition Metal Bis(dicyanobenzenedithiolate) Complexes [M(dcbdt)2] (M = Fe, Co, Ni, Pd, Pt, Cu, Au and Zn). Eur. J. Inorg. Chem. 2004, 1318–1329. [Google Scholar] [CrossRef]
- Ghosh, A.C.; Weisz, K.; Schulzke, C. Selective Capture of Ni2+ Ions by Naphthalene- and Coumarin-Substituted Dithiolenes. Eur. J. Inorg. Chem. 2016, 2016, 208–218. [Google Scholar] [CrossRef]
- Perochon, R.; Piekara-Sady, L.; Jurga, W.; Clerac, R.; Fourmigue, M. Amphiphilic paramagnetic neutral gold dithiolene complexes. Dalton Trans. 2009, 3052–3061. [Google Scholar] [CrossRef]
- Perochon, R.; Poriel, C.; Jeannin, O.; Piekara-Sady, L.; Fourmigué, M. Chiral, Neutral, and Paramagnetic Gold Dithiolene Complexes Derived from Camphorquinone. Eur. J. Inorg. Chem. 2009, 2009, 5413–5421. [Google Scholar] [CrossRef]
- McLauchlan, C.C.; Ibers, J.A. Synthesis and Characterization of the Silver Maleonitrilediselenolates and Silver Maleonitriledithiolates [K([2.2.2]-cryptand)]4[Ag4(Se2C2(CN)2)4], [Na([2.2.2]-cryptand)]4[Ag4(S2C2(CN)2)4]·0.33MeCN, [NBu4]4[Ag4(S2C2(CN)2)4], [K([2.2.2]-cryptand)]3[Ag(Se2C2(CN)2)2]·2MeCN, and [Na([2.2.2]-cryptand)]3[Ag(S2C2(CN)2)2]. Inorg. Chem. 2001, 40, 1809–1815. [Google Scholar]
- Watanabe, E.; Fujiwara, M.; Yamaura, J.-I.; Kato, R. Synthesis and properties of novel donor-type metal–dithiolene complexes based on 5,6-dihydro-1,4-dioxine-2,3-dithiol (edo) ligand. J. Mater. Chem. 2001, 11, 2131–2141. [Google Scholar] [CrossRef]
- Llusar, R.; Vicent, C. Trinuclear molybdenum cluster sulfides coordinated to dithiolene ligands and their use in the development of molecular conductors. Coord. Chem. Rev. 2010, 254, 1534–1548. [Google Scholar] [CrossRef]
- Castillo, O.; Delgado, E.; Gómez-García, C.J.; Hernández, D.; Hernández, E.; Martín, A.; Martínez, J.I.; Zamora, F. Group 10 Metal Benzene-1,2-dithiolate Derivatives in the Synthesis of Coordination Polymers Containing Potassium Countercations. Inorg. Chem. 2017, 56, 11810–11818. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, J.R.; Christou, G.; Huffman, J.C.; Folting, K. The synthesis, structure and spectroscopic properties of the di- and tri-nuclear Ni(II) thiolate complexes. Polyhedron 1987, 6, 863–870. [Google Scholar] [CrossRef]
- Breitzer, J.G.; Rauchfuss, T.B. Studies on α-C3S52− (dmit2−) and its dinuclear Ni(II) complex: Spectroscopic and structural characterization. Polyhedron 2000, 19, 1283–1291. [Google Scholar] [CrossRef]
- Sheng, T.; Zhang, W.; Gao, X.; Lin, P. Two new nickel-dmit-based molecular conductors based on heteroleptic polymetallic complexes: Synthesis, structures and electrical properties. Chem. Commun. 1998, 263–264. [Google Scholar] [CrossRef]
- Ahmadi, M.; Fischer, C.; Ghosh, A.C.; Schulzke, C. An Asymmetrically Substituted Aliphatic Bis-Dithiolene Mono-Oxido Molybdenum(IV) Complex With Ester and Alcohol Functions as Structural and Functional Active Site Model of Molybdoenzymes. Front. Chem. 2019, 7, 486. [Google Scholar] [CrossRef] [Green Version]
- Chrysochos, N.; Ahmadi, M.; Wahlefeld, S.; Rippers, Y.; Zebger, I.; Mroginski, M.A.; Schulzke, C. Comparison of molybdenum and rhenium oxo bis-pyrazine-dithiolene complexes—In search of an alternative metal centre for molybdenum cofactor models. Dalton Trans. 2019, 48, 2701–2714. [Google Scholar] [CrossRef]
- Ghosh, A.C.; Samuel, P.P.; Schulzke, C. Synthesis, characterization and oxygen atom transfer reactivity of a pair of Mo(IV)O− and Mo(VI)O2− enedithiolate complexes—A look at both ends of the catalytic transformation. Dalton Trans. 2017, 46, 7523–7533. [Google Scholar] [CrossRef]
- Holleman, A.; Wiberg, N.; Krieger-Hauwede, M. Anorganische Chemie Band1: Grundlagen und Hauptgruppenelemente, 103rd ed.; Walter de Gruyter: Berlin, Germany; Boston, MA, USA, 2017; Volume 1. [Google Scholar]
- Eisenberg, R.; Gray, H.B. Noninnocence in Metal Complexes: A Dithiolene Dawn. Inorg. Chem. 2011, 50, 9741–9751. [Google Scholar] [CrossRef]
- Williams, R.; Billig, E.; Waters, J.H.; Gray, H.B. The Toluenedithiolate and Maleonitriledithiolate Square-Matrix Systems. J. Am. Chem. Soc. 1966, 88, 43–50. [Google Scholar] [CrossRef]
- Yang, J.; Enemark, J.H.; Kirk, M.L. Metal–Dithiolene Bonding Contributions to Pyranopterin Molybdenum Enzyme Reactivity. Inorganics 2020, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Volbeda, A.; Martin, L.; Cavazza, C.; Matho, M.; Faber, B.W.; Roseboom, W.; Albracht, S.P.J.; Garcin, E.; Rousset, M.; Fontecilla-Camps, J.C. Structural differences between the ready and unready oxidized states of [NiFe] hydrogenases. J. Biol. Inorg. Chem. 2005, 10, 239–249. [Google Scholar] [CrossRef]
- Higuchi, Y.; Yagi, T.; Yasuoka, N. Unusual ligand structure in Ni–Fe active center and an additional Mg site in hydrogenase revealed by high resolution X-ray structure analysis. Structure 1997, 5, 1671–1680. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, Y.; Ogata, H.; Miki, K.; Yasuoka, N.; Yagi, T. Removal of the bridging ligand atom at the Ni–Fe active site of [NiFe] hydrogenase upon reduction with H2, as revealed by X-ray structure analysis at 1.4 Å resolution. Structure 1999, 7, 549–556. [Google Scholar] [CrossRef]
- Ogata, H.; Kellers, P.; Lubitz, W. The Crystal Structure of the [NiFe] Hydrogenase from the Photosynthetic Bacterium Allochromatium vinosum: Characterization of the Oxidized Enzyme (Ni-A State). J. Mol. Biol. 2010, 402, 428–444. [Google Scholar] [CrossRef]
- Marques, M.C.; Tapia, C.; Gutiérrez-Sanz, O.; Ramos, A.R.; Keller, K.L.; Wall, J.D.; De Lacey, A.L.; Matias, P.M.; Pereira, I.A.C. The direct role of selenocysteine in [NiFeSe] hydrogenase maturation and catalysis. Nat. Chem. Biol. 2017, 13, 544–550. [Google Scholar] [CrossRef]
- Volbeda, A.; Charon, M.-H.; Piras, C.; Hatchikian, E.C.; Frey, M.; Fontecilla-Camps, J.C. Crystal structure of the nickel–iron hydrogenase from Desulfovibrio gigas. Nature 1995, 373, 580–587. [Google Scholar] [CrossRef]
- Moubarak, S.; Elghobashi-Meinhardt, N.; Tombolelli, D.; Mroginski, M.A. Probing the Structure of [NiFeSe] Hydrogenase with QM/MM Computations. Appl. Sci. 2020, 10, 781. [Google Scholar] [CrossRef] [Green Version]
- Schlaepfer, C.W.; Nakamoto, K. Infrared spectra and normal-coordinate analysis of 1,2-dithiolate complexes with nickel. Inorg. Chem. 1975, 14, 1338–1344. [Google Scholar] [CrossRef]
- Bui, T.-T.; Thiebaut, O.; Grelet, E.; Achard, M.-F.; Garreau-de Bonneval, B.; Moineau-Chane Ching, K.I. Discotic Nickel Bis(dithiolene) Complexes—Synthesis, Optoelectrochemical and Mesomorphic Properties. Eur. J. Inorg. Chem. 2011, 2011, 2663–2676. [Google Scholar] [CrossRef]
- Kirk, M.L.; McNaughton, R.L.; Helton, M.E. The Electronic Structure and Spectroscopy of Metallo-Dithiolene Complexes. In Dithiolene Chemistry; Stiefel, E.I., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004; Volume 52, pp. 111–212. [Google Scholar]
- Ray, K.; Weyhermüller, T.; Neese, F.; Wieghardt, K. Electronic Structure of Square Planar Bis(benzene-1,2-dithiolato)metal Complexes [M(L)2]z (z = 2−, 1−, 0; M = Ni, Pd, Pt, Cu, Au): An Experimental, Density Functional, and Correlated ab Initio Study. Inorg. Chem. 2005, 44, 5345–5360. [Google Scholar] [CrossRef]
- Izutsu, K. Potentiometry in Nonaqueous Solutions. In Electrochemistry in Nonaqueous Solutions; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; Chapter 6; pp. 171–208. [Google Scholar] [CrossRef]
- Evans, D.F. 400. The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J. Chem. Soc. 1959, 2003–2005. [Google Scholar] [CrossRef]
- Evans, D.F.; James, T.A. Variable-temperature magnetic-susceptibility measurements of spin equilibria for iron(III) dithiocarbamates in solution. J. Chem. Soc. Dalton Trans. 1979, 723–726. [Google Scholar] [CrossRef]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crsyt. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Ahmadi, M. Phosphate Substituted Dithiolene Complexes as Models for the Active Site of Molybdenum Dependent Oxidoreductases. Universität Greifswald, Greifswald, Germany, 2019. Available online: https://epub.ub.uni-greifswald.de/frontdoor/index/index/start/1/rows/10/sortfield/score/sortorder/desc/searchtype/simple/query/Mohsen+Ahmadi/docId/3279 (accessed on 1 March 2020).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Lengths | Bond Angles | ||
|---|---|---|---|
| Ni(1)–Ni(2) | 2.6747(11) | S(1)–Ni(1)–S(2) | 91.82(6) |
| Ni(2)–Ni(2A) | 2.9187(16) | S(1)–Ni(1)–S(3) | 84.52(6) |
| Ni(1)–S(1) | 2.1487(17) | S(1)–Ni(1)–S(4) | 176.32(7) |
| Ni(1)–S(2) | 2.1482(17) | S(1)–Ni(2)–S(3) | 77.09(6) |
| Ni(1)–S(3) | 2.1725(17) | S(1)–Ni(2)–S(5) | 164.00(6) |
| Ni(1)–S(4) | 2.1464(18) | S(1)–Ni(2)–S(5A) | 95.06(6) |
| Ni(2)–S(1) | 2.3125(16) | S(1)–Ni(2)–S(6) | 92.80(6) |
| Ni(2)–S(3) | 2.3509(16) | S(3)–Ni(2)–S(5) | 94.06(6) |
| Ni(2)–S(5) | 2.1758(16) | S(3)–Ni(2)–S(5A) | 102.43(5) |
| Ni(2)–S(5A) | 2.3554(16) | S(3)–Ni(2)–S(6) | 156.26(6) |
| Ni(2)–S(6) | 2.1793(16) | S(5)–Ni(2)–S(5A) | 99.91(6) |
| C(1)–C(4) | 1.366(8) | S(5)–Ni(2)–S(1) | 90.24(6) |
| C(11)–C(14) | 1.364(8) | Ni(1)–Ni(2)–S(5) | 113.74(5) |
| C(21)–C(24) | 1.369(8) | Ni(1)–Ni(2)–S(5A) | 136.58(5) |
| Ni(1)–Ni(2)–Ni(2A) | 152.41(4) | ||
| 3 | 4·2CH3CN·2Et2O | ||
|---|---|---|---|
| C(2)–C(3) | 1.353(5) | C(1)–C(4) | 1.366(8) |
| C(2)–S(1) (ester side) | 1.756(4) | C(1)–S(1) (ester side) | 1.745(6) |
| C(3)–S(2) (phenyl side) | 1.747(4) | C(4)–S(2) (phenyl side) | 1.711(6) |
| C(11)–C(14) | 1.364(8) | ||
| C(11)–S(3) (ester side) | 1.749(6) | ||
| C(14)–S(4) (phenyl side) | 1.729(6) | ||
| C(21)–C(24) | 1.369(8) | ||
| C(21)–S(5) (ester side) | 1.748(6) | ||
| C(24)–S(6) (phenyl side) | 1.712(6) | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmadi, M.; Correia, J.; Chrysochos, N.; Schulzke, C. A Mixed-Valence Tetra-Nuclear Nickel Dithiolene Complex: Synthesis, Crystal Structure, and the Lability of Its Nickel Sulfur Bonds. Inorganics 2020, 8, 27. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8040027
Ahmadi M, Correia J, Chrysochos N, Schulzke C. A Mixed-Valence Tetra-Nuclear Nickel Dithiolene Complex: Synthesis, Crystal Structure, and the Lability of Its Nickel Sulfur Bonds. Inorganics. 2020; 8(4):27. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8040027
Chicago/Turabian StyleAhmadi, Mohsen, Jevy Correia, Nicolas Chrysochos, and Carola Schulzke. 2020. "A Mixed-Valence Tetra-Nuclear Nickel Dithiolene Complex: Synthesis, Crystal Structure, and the Lability of Its Nickel Sulfur Bonds" Inorganics 8, no. 4: 27. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8040027