Structure: Function Studies of the Cytosolic, Mo- and NAD+-Dependent Formate Dehydrogenase from Cupriavidus necator


 and
and
Abstract
:1. Introduction
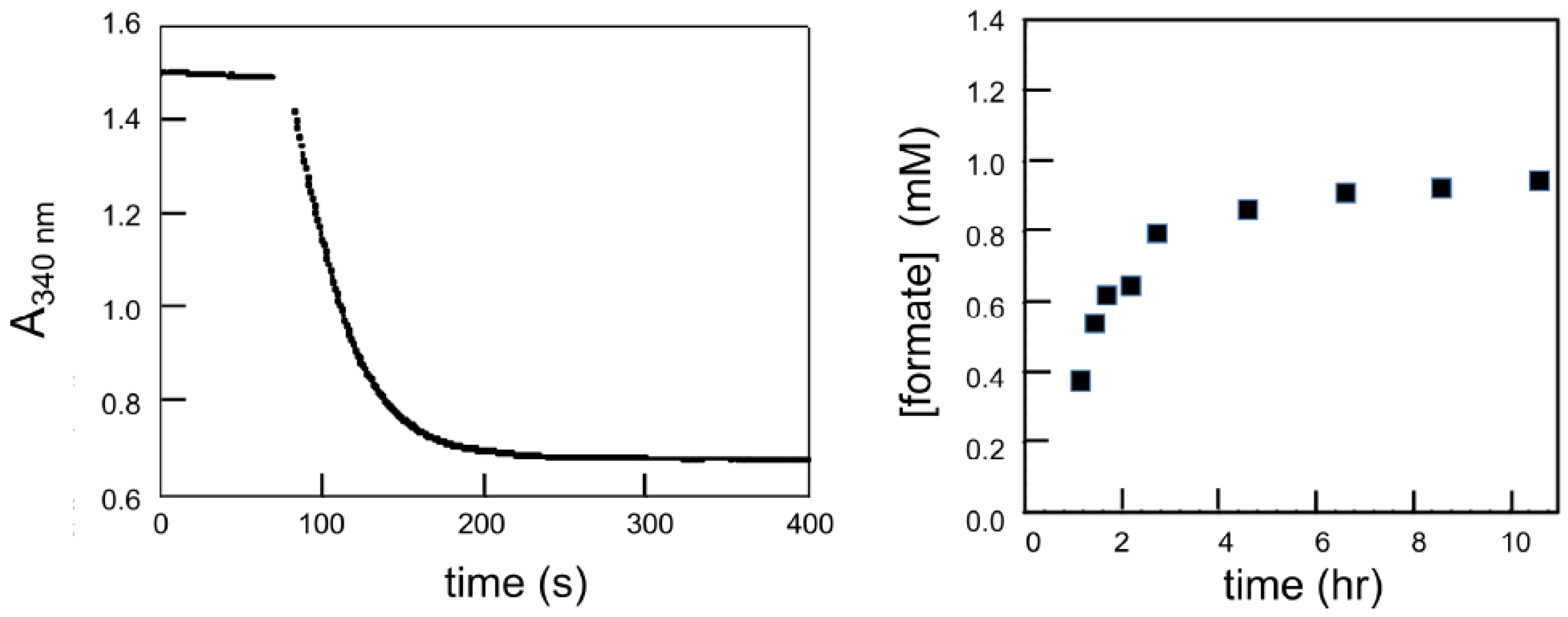
2. Catalysis of CO2 Reduction by the C. necator Formate Dehydrogenase
3. Insertion of the Molybdenum Cofactor into Formate Dehydrogenase and Other Members of the DMSO Reductase Family
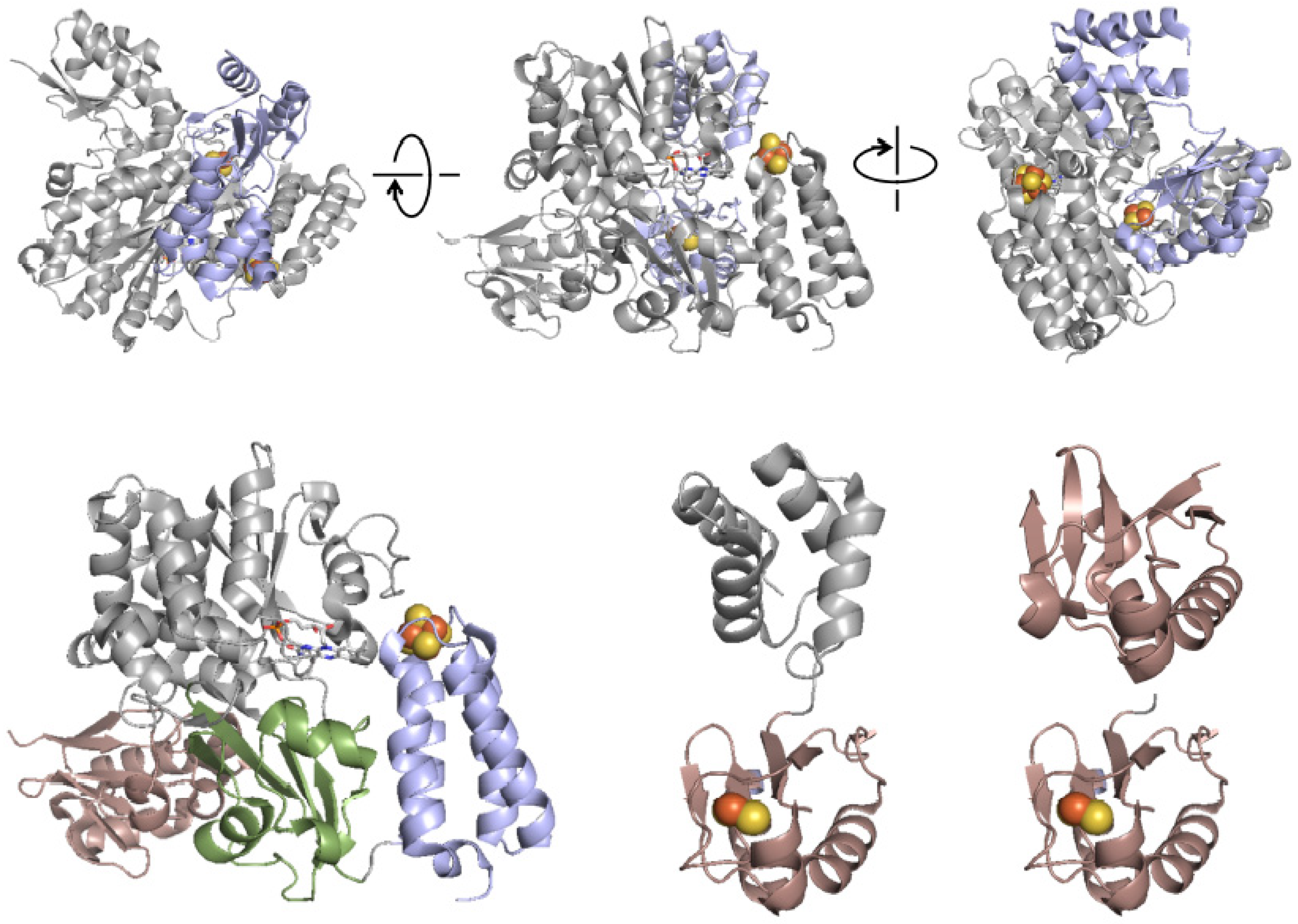
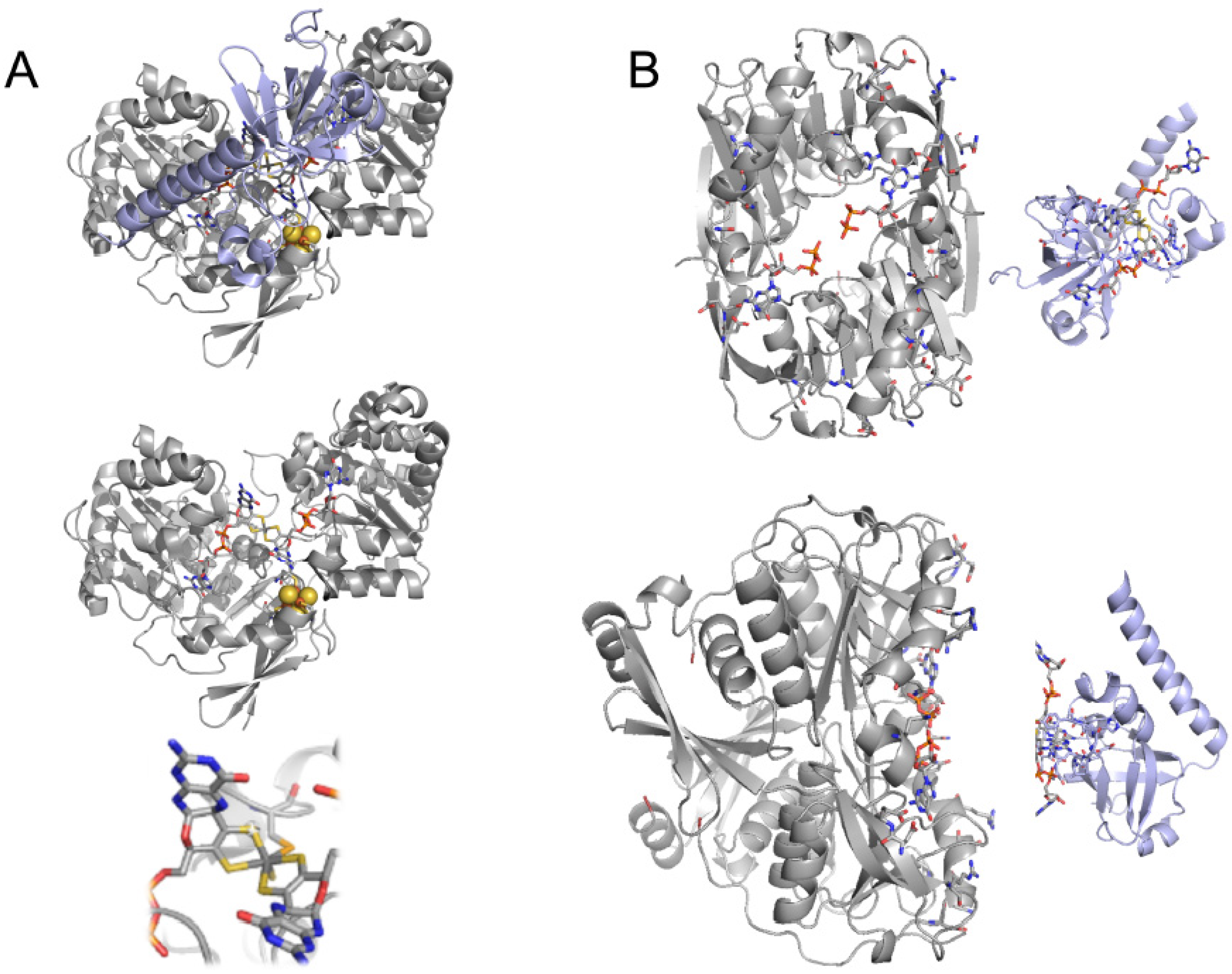
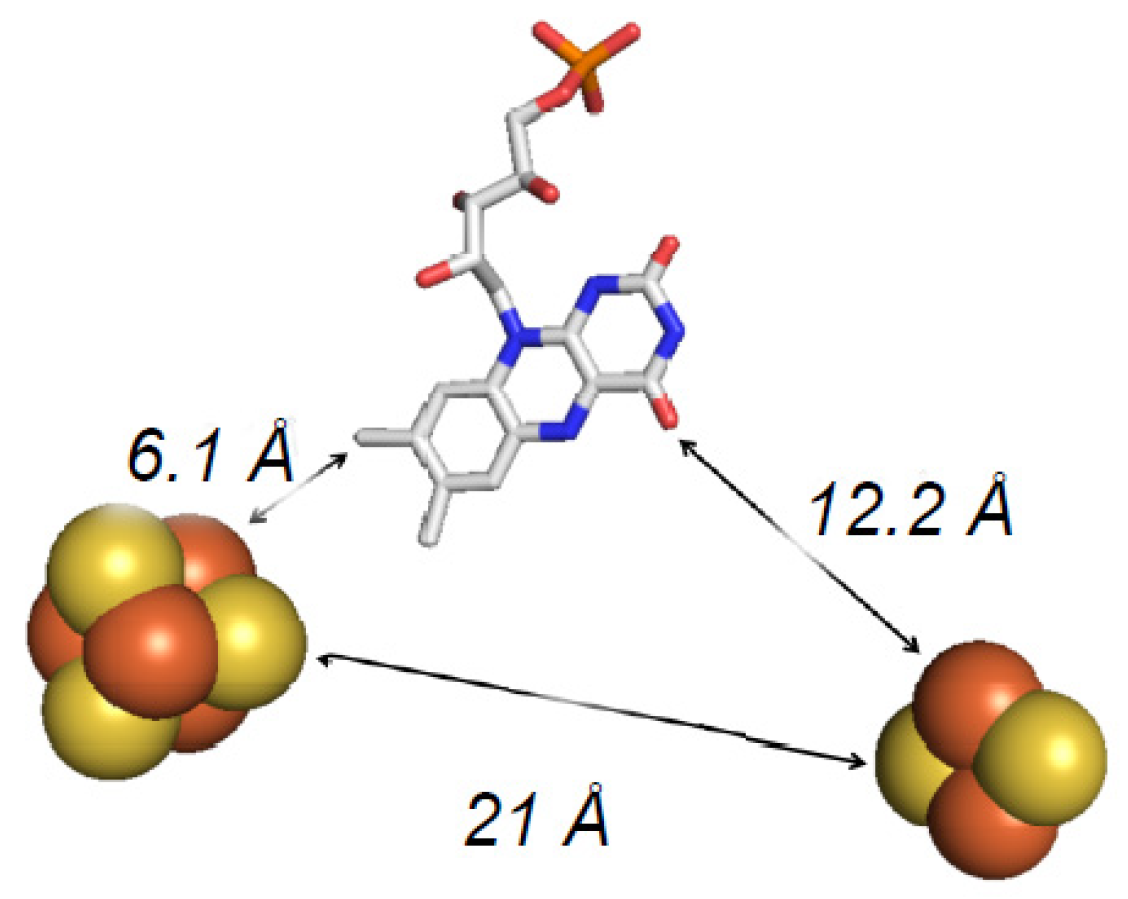
4. The Crystal Structure of FdsBG
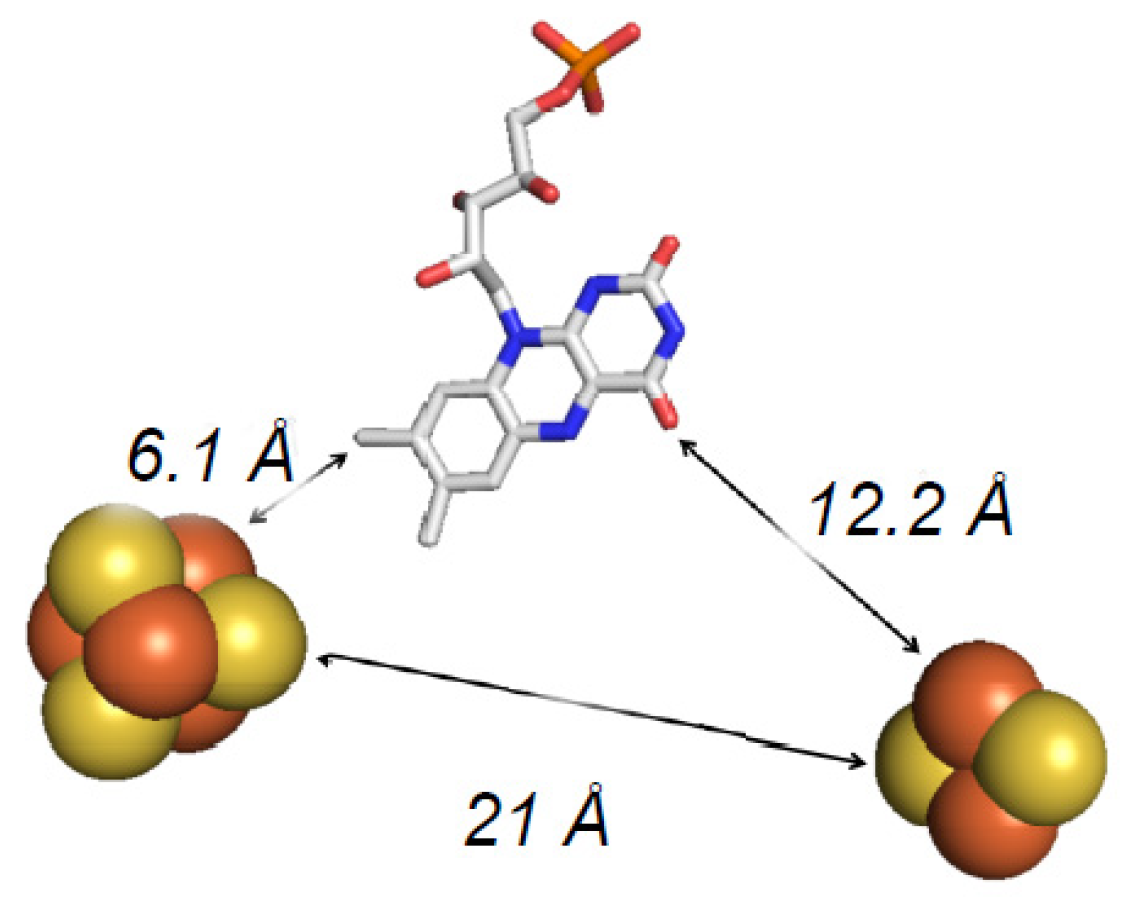
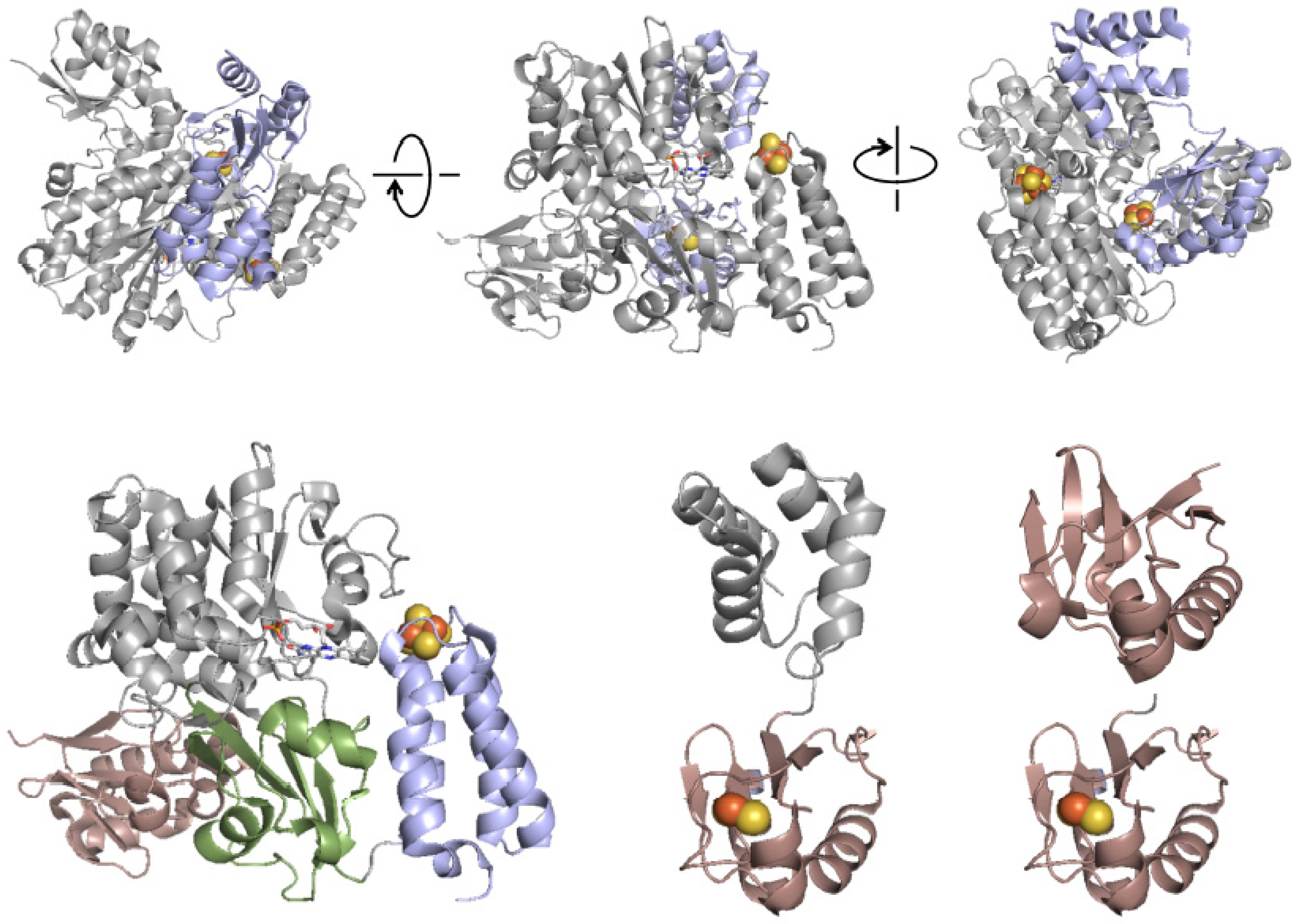
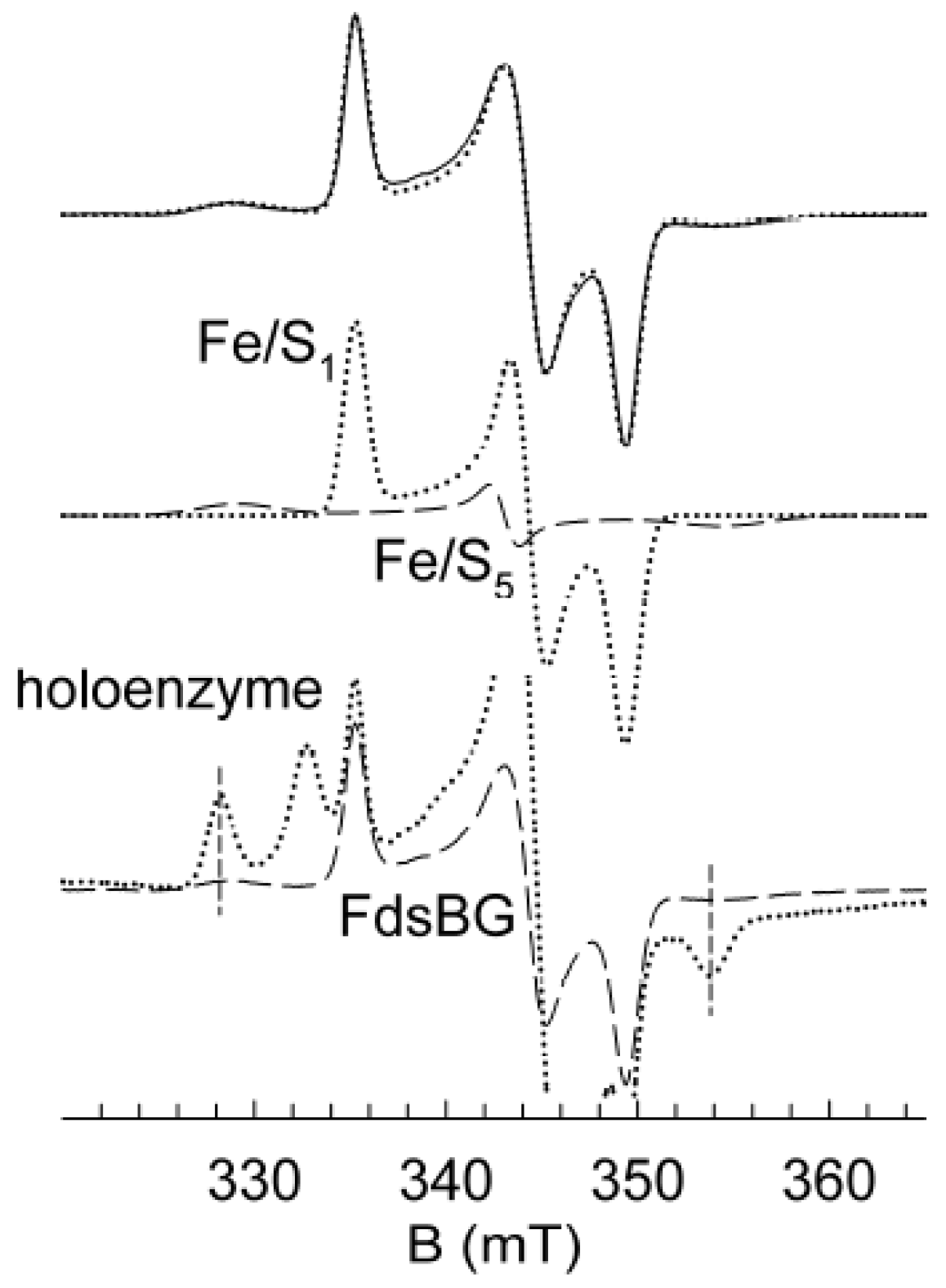
5. EPR Characterization of the Iron–Sulfur Clusters of FdsBG
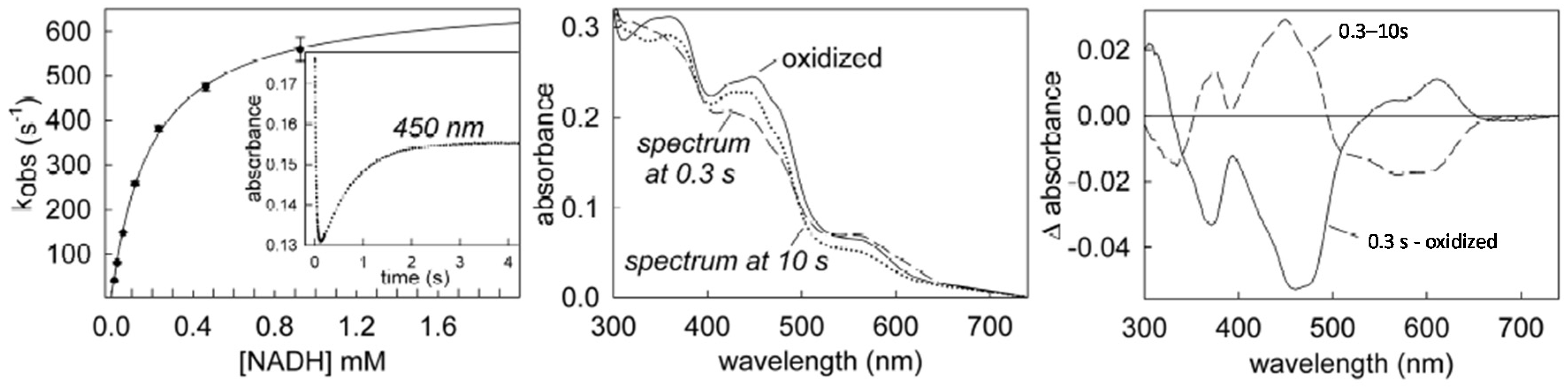
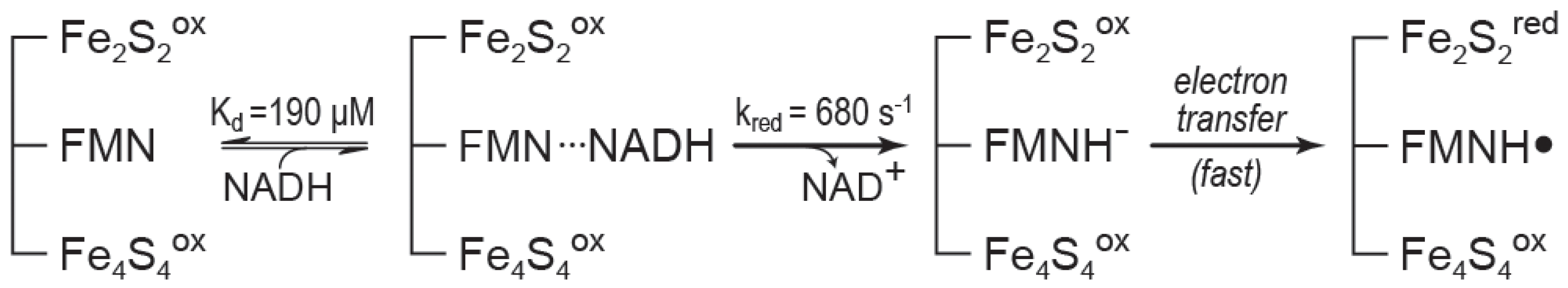
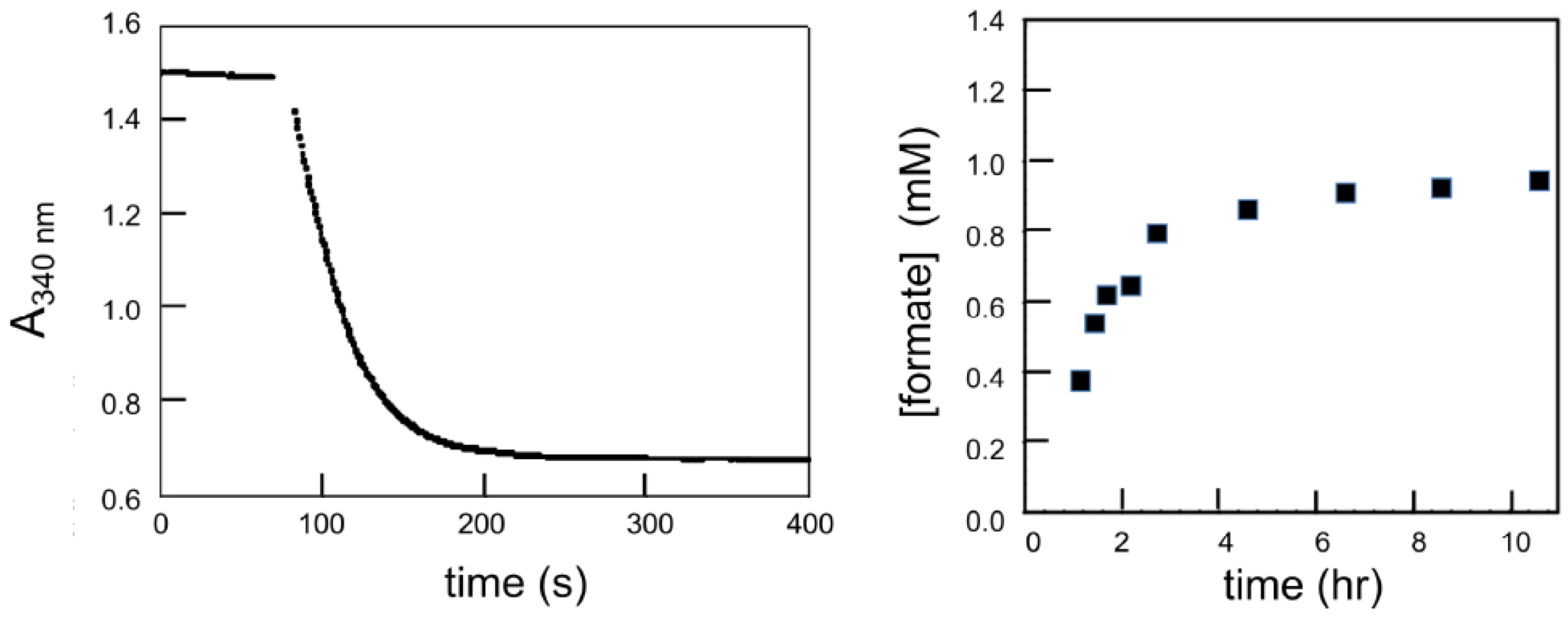
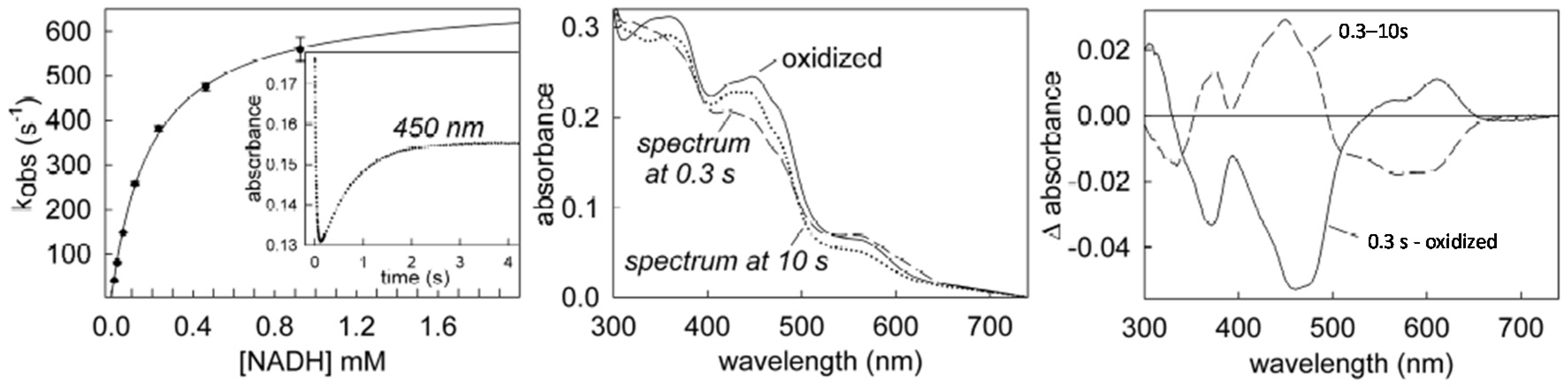
6. Rapid-Reaction Kinetics of FdsBG Reduction by NADH
7. The Thioredoxin-Like Domain of FdsB
8. Electron Transfer in FdsBG
9. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pohlmann, A.; Fricke, W.F.; Reinecke, F.; Kusian, B.; Liesegang, H.; Cramm, R.; Eitinger, T.; Ewering, C.; Potter, M.; Schwartz, E.; et al. Genome sequence of the bioplastic-producing “Knallgas” bacterium Ralstonia eutropha H16. Nat. Biotechnol. 2006, 24, 1257–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Little, G.T.; Ehsaan, M.; Arenas-Lopez, C.; Jawed, K.; Winzer, K.; Kovacs, K.; Minton, N.P. Complete genome sequence of Cupriavidus necator H16 (DSM 428). Microbiol. Resour. Announc. 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramm, R. Genomic view of energy metabolism in Ralstonia eutropha H16. J. Mol. Microbiol. Biotechnol. 2009, 16, 38–52. [Google Scholar] [CrossRef]
- Friedebold, J.; Bowien, B. Physiological and biochemical characterization of the soluble formate dehydrogenase, a molybdoenzyme from Alcaligenes eutrophus. J. Bacteriol. 1993, 175, 4719–4728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedebold, J.; Mayer, F.; Bill, E.; Trautwein, A.X.; Bowien, B. Structural and immunological studies on the soluble formate dehydrogenase from Alcaligenes eutrophus. Biol. Chem. Hoppe Seyler 1995, 376, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.I.; Bowien, B. Structural analysis of the Fds operon encoding the NAD+-linked formate dehydrogenase of Ralstonia eutropha. J. Biol. Chem. 1998, 273, 26349–26360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sazanov, L.A.; Hinchliffe, P. Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science 2006, 311, 1430–1436. [Google Scholar] [CrossRef] [Green Version]
- Berrisford, J.M.; Sazanov, L.A. Structural basis for the mecahnism of respiratory complex I. J. Biol. Chem. 2009, 284, 29773–29783. [Google Scholar] [CrossRef] [Green Version]
- Efremov, R.G.; Baradaran, R.; Sazanov, L.A. The architecture of respiratory complex I. Nature 2010, 465, 441–445. [Google Scholar] [CrossRef]
- Hille, R.; Hall, J.; Basu, P. The mononuclear molybdenum enzymes. Chem. Rev. 2014, 114, 3963–4038. [Google Scholar] [CrossRef] [Green Version]
- Boyington, J.C.; Gladyshev, V.N.; Khangulov, S.V.; Stadtman, T.C.; Sun, P.D. Crystal structure of formate dehydrogenase H: Catalysis involving Mo, molybdopterin, selenocysteine, and an Fe4S4 cluster. Science 1997, 275, 1305–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Niks, D.; Mulchandani, A.; Hille, R. Efficient reduction of CO2 by the molybdenum-containing formate dehydrogenase from Cupriavidus necator (Ralstonia eutropha). J. Biol. Chem. 2017, 292, 16872–16879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niks, D.; Duvvuru, J.; Escalona, M.; Hille, R. Spectroscopic and kinetic properties of the molybdenum-containing, NAD+-dependent Formate Dehydrogenase from Ralstonia eutropha. J. Biol. Chem. 2016, 291, 1162–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Niks, D.; Ge, X.; Liu, H.; Hille, R.; Mulchandani, A. Synthesis of formate from CO2 gas catalyzed by an O2-tolerant NAD+-dependent formate dehydrogenase and glucose dehydrogenase. Biochemistry 2019, 58, 1861–1868. [Google Scholar] [CrossRef] [PubMed]
- Thomé, R.; Gust, A.; Toci, R.; Mendel, R.; Bittner, F.; Magalon, A.; Walburger, A. A sulfurtransferase is essential for activity of formate dehydrogenase in Escherichia coli. J. Biol. Chem. 2012, 287, 4671–4678. [Google Scholar] [CrossRef] [Green Version]
- Arnoux, P.; Ruppelt, C.; Oudouhou, F.; Lavergne, J.; Siponen, M.I.; Toci, R.; Mendel, R.R.; Bittner, F.; Pignol, D.; Magalon, A.; et al. Sulphur shuttling across a chaperone during molybdenum cofactor maturation. Nat. Commun. 2015, 6, 6148. [Google Scholar] [CrossRef] [Green Version]
- Dow, J.M.; Grabel, F.; Sargent, F.; Palmer, T. Characterization of a pre-export enzyme-chaperone complex on the twin-arginine transport pathway. Biochem. J. 2013, 452, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Dow, J.M.; Grahl, S.; Ward, R.; Evans, R.; Byron, O.; Norman, D.G.; Palmer, T.; Sargent, F. Characterization of a periplasmic nitrate reductase in complex with its biosynthetic chaperone. FEBS J. 2014, 281, 246–260. [Google Scholar] [CrossRef] [Green Version]
- Young, T.; Niks, D.; Hakopian, S.; Tam, T.K.; Yu, X.; Hille, R.; Blaha, G.M. Crystallographic and kinetic analyses of the FdsBG subcomplex of the cytosolic formate dehydrogenase from Cupriavidus necator. J. Biol. Chem. 2020, 295, 6570–6585. [Google Scholar] [CrossRef] [Green Version]
- Schulte, M.; Frick, K.; Gnandt, E.; Jurkovic, S.; Burschel, S.; Labatzke, R.; Aierstock, K.; Fiegen, D.; Wohlwend, D.; Gerhardt, S.; et al. A mechanism to prevent production of reactive oxygen species by Escherichia coli respiratory complex I. Nat. Commun. 2019, 10, 2551. [Google Scholar] [CrossRef] [Green Version]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef]
- Hinchliffe, P.; Sazanov, L.A. Organization of iron-sulfur clusters in respiratory complex I. Science 2005, 309, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Shomura, Y.; Taketa, M.; Nakashima, H.; Tai, H.; Nakagawa, H.; Ikeda, Y.; Ishii, M.; Igarashi, Y.; Nishihara, H.; Yoon, K.S.; et al. Structural basis of the redox switches in the NAD(+)-reducing soluble [NiFe]-hydrogenase. Science 2017, 357, 928–932. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, T.; Ohnishi, S.T.; Salerno, J.C. Five decades of research on mitochondrial NADH-quinone oxidoreductase (complex I). Biol. Chem. 2018, 399, 1249–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reda, T.; Barker, C.D.; Hirst, J. Reduction of the iron-sulfur clusters in mitochondrial NADH:ubiquinone oxidoreductase (complex I) by EuII-DTPA, a very low potential reductant. Biochemistry 2008, 47, 8885–8893. [Google Scholar] [CrossRef]
- Edmondson, D.E.; Tollin, G. Semiquinone formation in flavo- and metalloflavoproteins. Top. Curr. Chem. 1983, 108, 109–138. [Google Scholar] [PubMed]
- Edmondson, D.E.; Ackrell, B.A.; Kearney, E.B. Identification of neutral and anionic 8 alpha-substituted flavin semiquinones in flavoproteins by electron spin resonance spectroscopy. Arch. Biochem. Biophys. 1981, 208, 69–74. [Google Scholar] [CrossRef]
- Palmer, G.; Mueller, F.; Massey, V. Electron Paramagnetic Resonance Studies on Flavoprotein Radicals. In Third International Symposium of Flavins and Flavoproteins; Kamin, H., Ed.; University Park Press: Baltimore, MD, USA, 1971; pp. 123–140. [Google Scholar]
- Bliven, S.; Lafita, A.; Parker, A.; Capitani, G.; Duarte, J.M. Automated evaluation of quaternary structures from protein crystals. PLoS Comput. Biol. 2018, 14, e1006104. [Google Scholar] [CrossRef] [Green Version]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Yano, T.; Sled, V.D.; Ohnishi, T.; Yagi, T. Expression and characterization of the flavoprotein subcomplex composed of 50-kDa (NQO1) and 25-kDa (NQO2) subunits of the proton-translocating NADH-quinone oxidoreductase of Paracoccus denitrificans. J. Biol. Chem. 1996, 271, 5907–5913. [Google Scholar] [CrossRef] [Green Version]
- Galante, Y.M.; Hatefi, Y. Purification and molecular and enzymic properties of mitochondrial NADH dehydrogenase. Arch. Biochem. Biophys. 1979, 192, 559–568. [Google Scholar] [CrossRef]
- Lauterbach, L.; Idris, Z.; Vincent, K.A.; Lenz, O. Catalytic properties of the isolated diaphorase fragment of the NAD-reducing [NiFe]-hydrogenase from Ralstonia eutropha. PLoS ONE 2011, 6, e25939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radon, C.; MIttelStádt, G.; Duffus, B.R.; Bürger, J.; Hartmann, T.; Mielke, T.; Teutloff, C.; Leimkühler, S.; Wendler, P. Cryo-EM structures reveal intricate Fe-S cluster arrangement and charging in Rhodobacter capsulatus formate dehydrogenase. Nat. Commun. 2020, 11, 1912. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.; Ermler, U.; Shima, S. The methanogenic CO2 redcuing-and-fixing enzyme is bifunctional and contains 46 [4Fe-4S] clusters. Science 2016, 354, 114–117. [Google Scholar] [CrossRef]
- Spatzao, T.; Schlesier, J.; Burger, E.-M.; Sippel, D.; Zhang, L.; Andrade, S.L.A.; Rees, D.C.; Einsle, O. Nitrogenase FeMoCo investigated by spatially resolved anomalous dispersion refinement. Nat. Commun. 2016, 7, 10902. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward | Reverse | |
|---|---|---|
| kcat | 201 s−1 | 10 s−1 |
| Kmformate | 130 µM | – |
| KmNAD+ | 310 µM | – |
| KmCO2 | – | 2700 µM |
| KmNADH | – | 46 µM |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hille, R.; Young, T.; Niks, D.; Hakopian, S.; Tam, T.K.; Yu, X.; Mulchandani, A.; Blaha, G.M. Structure: Function Studies of the Cytosolic, Mo- and NAD+-Dependent Formate Dehydrogenase from Cupriavidus necator. Inorganics 2020, 8, 41. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8070041
Hille R, Young T, Niks D, Hakopian S, Tam TK, Yu X, Mulchandani A, Blaha GM. Structure: Function Studies of the Cytosolic, Mo- and NAD+-Dependent Formate Dehydrogenase from Cupriavidus necator. Inorganics. 2020; 8(7):41. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8070041
Chicago/Turabian StyleHille, Russ, Tynan Young, Dimitri Niks, Sheron Hakopian, Timothy K. Tam, Xuejun Yu, Ashok Mulchandani, and Gregor M. Blaha. 2020. "Structure: Function Studies of the Cytosolic, Mo- and NAD+-Dependent Formate Dehydrogenase from Cupriavidus necator" Inorganics 8, no. 7: 41. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8070041