Tungstoenzymes: Occurrence, Catalytic Diversity and Cofactor Synthesis


1
Faculty of Biology-Microbiology, Albert-Ludwigs-Universität Freiburg, 79104 Freiburg, Germany
2
Faculty of Biology, Philipps-Universität Marburg, 35037 Marburg, Germany
*
Author to whom correspondence should be addressed.
Inorganics 2020, 8(8), 44; https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8080044
Submission received: 30 June 2020
/
Revised: 24 July 2020
/
Accepted: 28 July 2020
/
Published: 31 July 2020
(This article belongs to the Special Issue Transition Metals in Catalysis: The Functional Relationship of Fe–S Clusters and Molybdenum or Tungsten Cofactor-Containing Enzyme Systems)
Abstract
:Tungsten is the heaviest element used in biological systems. It occurs in the active sites of several bacterial or archaeal enzymes and is ligated to an organic cofactor (metallopterin or metal binding pterin; MPT) which is referred to as tungsten cofactor (Wco). Wco-containing enzymes are found in the dimethyl sulfoxide reductase (DMSOR) and the aldehyde:ferredoxin oxidoreductase (AOR) families of MPT-containing enzymes. Some depend on Wco, such as aldehyde oxidoreductases (AORs), class II benzoyl-CoA reductases (BCRs) and acetylene hydratases (AHs), whereas others may incorporate either Wco or molybdenum cofactor (Moco), such as formate dehydrogenases, formylmethanofuran dehydrogenases or nitrate reductases. The obligately tungsten-dependent enzymes catalyze rather unusual reactions such as ones with extremely low-potential electron transfers (AOR, BCR) or an unusual hydration reaction (AH). In recent years, insights into the structure and function of many tungstoenzymes have been obtained. Though specific and unspecific ABC transporter uptake systems have been described for tungstate and molybdate, only little is known about further discriminative steps in Moco and Wco biosynthesis. In bacteria producing Moco- and Wco-containing enzymes simultaneously, paralogous isoforms of the metal insertase MoeA may be specifically involved in the molybdenum- and tungsten-insertion into MPT, and in targeting Moco or Wco to their respective apo-enzymes. Wco-containing enzymes are of emerging biotechnological interest for a number of applications such as the biocatalytic reduction of CO2, carboxylic acids and aromatic compounds, or the conversion of acetylene to acetaldehyde.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Tungsten and molybdenum are transition metals of the sixth group and occur in nature predominantly in form of their oxyanions—tungstate (WO42−) and molybdate (MoO42−). While their average abundance in the earth’s crust is highly similar, their bioavailability may differ in various aqueous environments. Both metals are present in biological systems in ligation to the so-called metallopterin (or metal binding pterin, MPT, initially introduced as molybdopterin), which occurs as a three-ring pyranopterin compound in most known molybdo- or tungstoenzymes. The entire metal cofactors are referred to as Moco or Wco, respectively [1,2,3,4,5,6]. There are many variants of MPT-derived cofactors with regard to the metal inserted, the presence of additional ligands of the metal, the number of pyranopterin molecules bound per metal (one or two), and the attachment of additional nucleotides to the MPT core. According to this structural diversity and the underlying amino acid sequence similarities, MPT-containing enzymes can be divided into four unrelated families: (i) xanthine oxidase (XO), (ii) sulfite oxidase (SO), (iii) dimethyl sulfoxide reductase (DMSOR), and (iv) aldehyde oxidoreductase (AOR) families. Wco-containing enzymes belong to the DMSOR and AOR families, whereas Moco is predominantly found in members of the XO, SO and DMSOR families. In the AOR family, tungsten is always bound by two MPTs, referred to as W-bis-MPT; in DMSOR family members, tungsten or molybdenum is coordinated by two MPT guanine dinucleotide (MGD) moieties, referred to as W-/Mo-bis-MGD cofactor (Figure 1). All tungstoenzymes contain at least one Fe-S cluster next to Wco, and especially the multi-subunit enzymes harbor a number of additional redox-active cofactors such as Fe-S clusters, flavins or hemes.
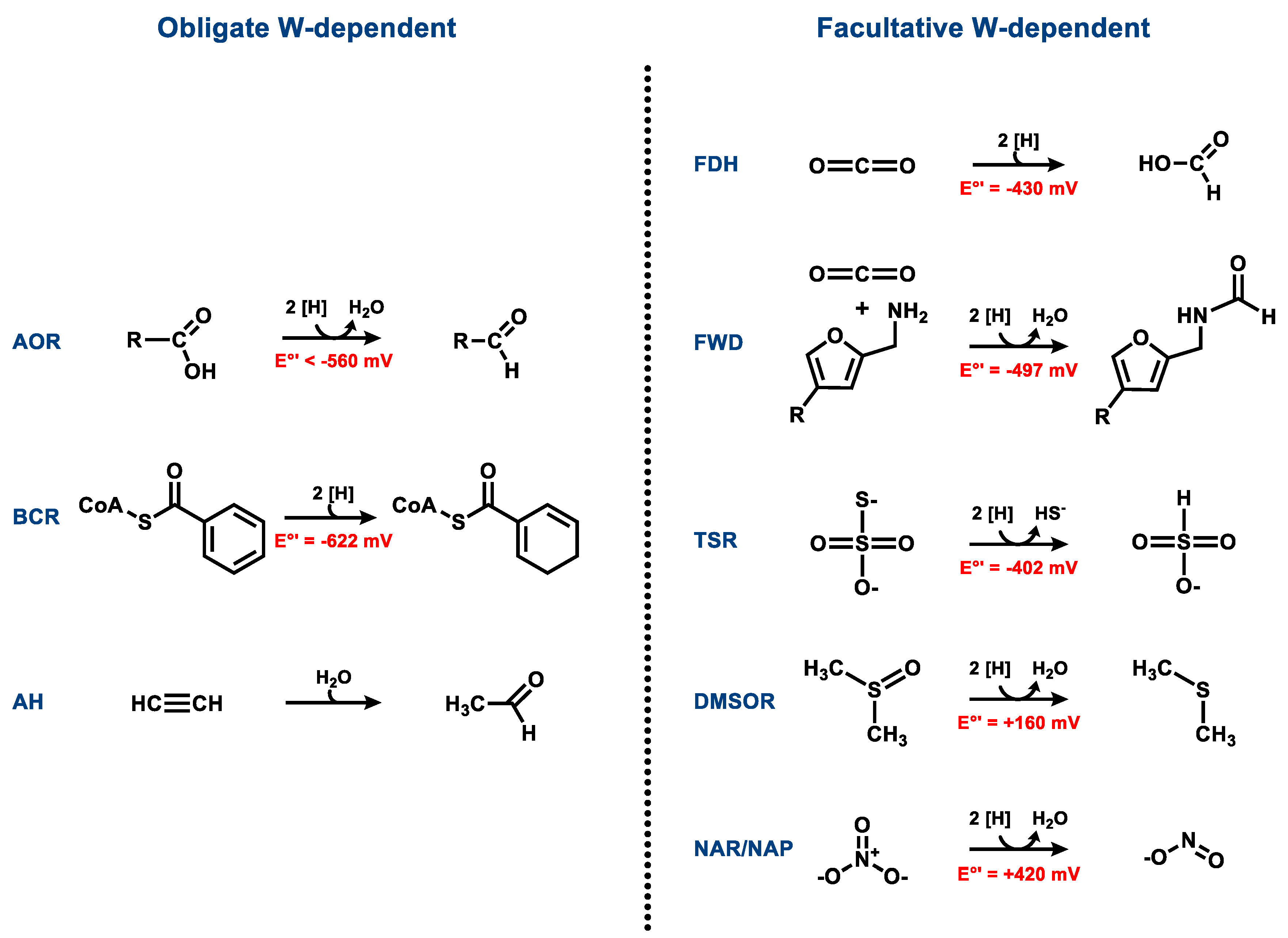
The majority of Moco- or Wco-containing enzymes catalyze hydroxy- or oxo-transfer reactions such as water-dependent hydroxylations or hydrations via water-, hydroxyl-, or oxo-intermediates bound to the metal in the catalytic course. There are also examples of MPT-dependent enzymes catalyzing hydride or hydrogen atom transfers (e.g., formate dehydrogenase or class II benzoyl-CoA reductase) or sulfur atom transfer reactions (polysulfide reductase). With the exception of acetylene hydratase, all Wco- and Moco-containing enzymes catalyze redox reactions (Figure 2). It is evident that most naturally occurring tungstoenzymes are involved in low-potential (E°’ < −400 mV) electron transfer reactions. This finding can be rationalized by the generally lower redox potential of the biologically relevant redox transitions of W(IV/V/VI) vs. Mo(IV/V/VI). Though acetylene hydratase does not catalyze a redox reaction, the enzyme depends on a low-potential redox activation (see below).
Based on the highly similar physicochemical properties of molybdate and tungstate, the latter was initially considered to act as a general inhibitor of Moco-dependent enzymes, which has been observed in many cases if the replacement of molybdenum by tungsten is enforced by high tungstate concentrations (for an excellent review on this aspect, see [7]). As an example, a recent study used tungstate to inhibit Moco-dependent enzymes from facultatively anaerobic bacteria involved in gut disorders [8]. In facultatively tungsten-dependent enzymes that occur with either of the two metals, the bioavailability of tungstate or molybdate may govern which of the two metals is incorporated into bis-MPT-containing enzymes.
Today, it has become evident that Wco is an essential cofactor of many archaeal and bacterial enzymes that have been discussed in previous reviews with various focuses [7,9,10,11,12]. Here, we first aim to provide a state-of-the-art overview of the occurrence and function of obligately and facultatively tungsten-containing enzymes. We then discuss processes that may be involved in discriminating between molybdenum and tungsten during uptake, Mo/W-MPT synthesis and the incorporation into their respective apo-enzymes. This aspect is of particular importance for the emerging number of organisms that simultaneously produce Wco- and Moco-dependent enzymes.
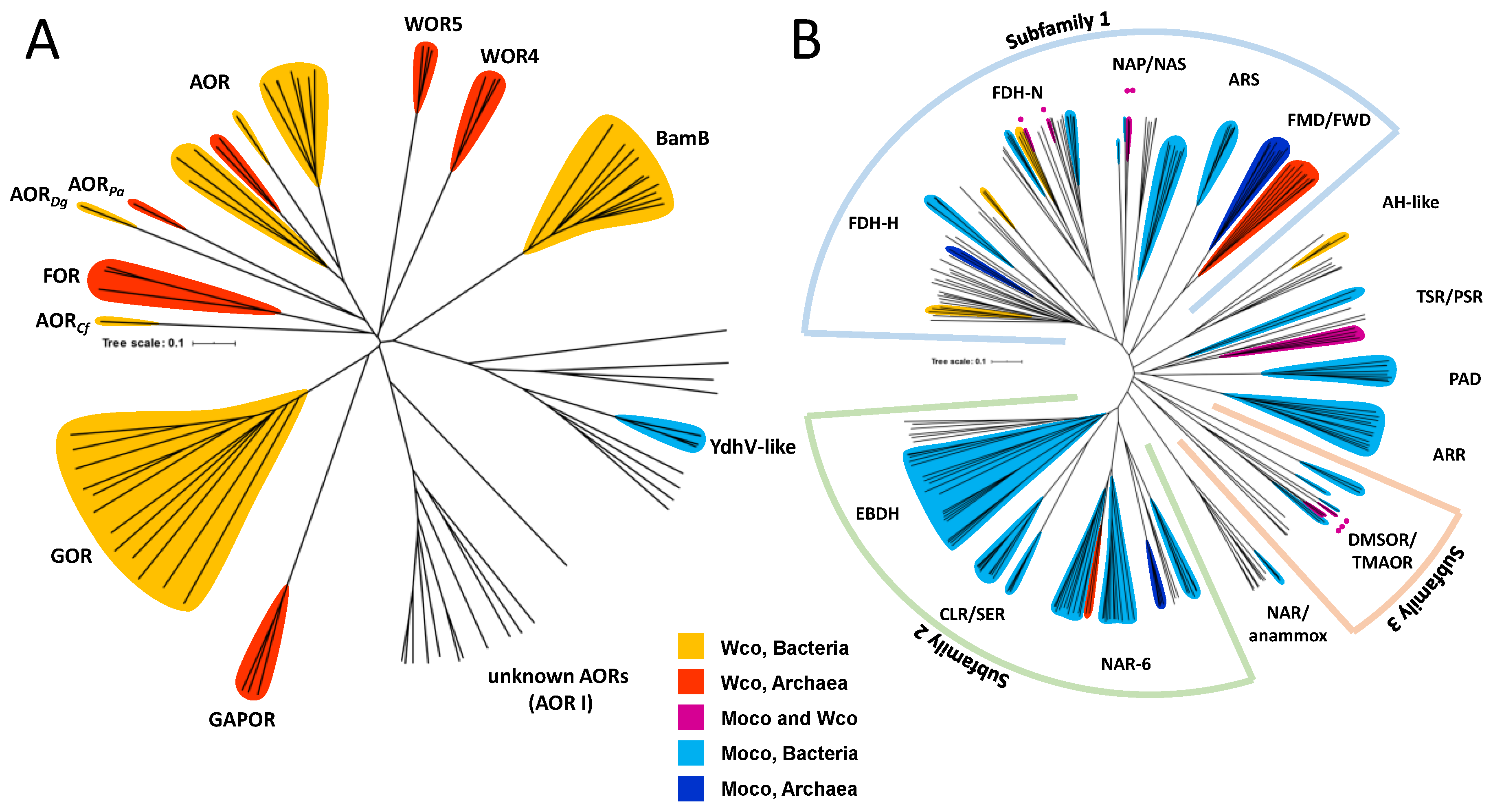
2. Affiliation of Tungsten Enzymes in the AOR and DMSOR Enzyme Families
The presence of Wco is restricted to members of a few clades of the DMSOR and AOR enzyme families. The biochemically characterized tungstoenzymes of the AOR family are affiliated to five clades of AORs (AOR, FOR, GAPOR, GOR, and WOR5) and one clade of the active site subunit BamB of class II benzoyl-CoA reductases (Figure 3A). In addition, the enzyme family contains one clade of archaeal enzymes and two of bacterial enzymes, whose biological functions are yet unknown (WOR4, YdhV and AOR1). Enzymes of the YdhV clade are unique in containing Moco instead of Wco, whereas WOR4 has been identified as a Wco-containing protein. The metal content of the enzymes of the AOR1 clade is unknown, since these have been only identified from genome sequences and not on the protein level. AORs of the XO family [13,14] are solely Moco-dependent and therefore not further discussed in this review.
The DMSOR family is subdivided into three subfamilies and several additional clades, and the tungstoenzymes of this family are affiliated to at least eight clades, which mostly contain either Moco- or Wco-dependent members (in case of formate or formylmethanofuran dehydrogenases, and nitrate-, DMSO-, or thiosulfate reductases [15]). Only the acetylene hydratase clade seems to be predominantly W-dependent, but this is based on only a single biochemically characterized enzyme, and it is unknown whether all these enzymes or closely related clades share the same metal preference (Figure 3B).
3. Obligately W-Containing Enzymes
3.1. Aldehyde Oxidoreductases
Most known clades of obligately tungsten-dependent enzymes show activities as aldehyde oxidoreductases (Figure 2). Four of the five enzymes of the AOR family encoded in the genome of the archaeon Pyrococcus furiosus have been characterized as hyperthermophilic and extremely O2-sensitive aldehyde oxidoreductases of various specificities. Together with the tungsten-dependent oxidoreductase WOR4, which was shown to contain Wco and a [3Fe-4S] cluster, but did not show any detectable activity [16], these enzymes define five separate phylogenetic clades of the AOR enzyme family (Figure 3A), namely the subfamilies of archaeal AOR, formaldehyde oxidoreductase (FOR), glyceraldehyde-3-phosphate oxidoreductases (GAPOR), and tungsten-dependent oxidoreductases (WOR4 and WOR5). Attempts to replace tungsten in some of these proteins with molybdenum or vanadium led either to proteins lacking any metal or to molybdenum-containing, inactive variants, proving the necessity of the Wco for their activities [17,18]. Additional members of the enzyme family emerged recently from obligately or facultatively anaerobic bacteria, namely bacterial AOR and GAPOR (GOR), which represent sister clades of the archaeal AOR and GAPOR, respectively (Figure 3A).
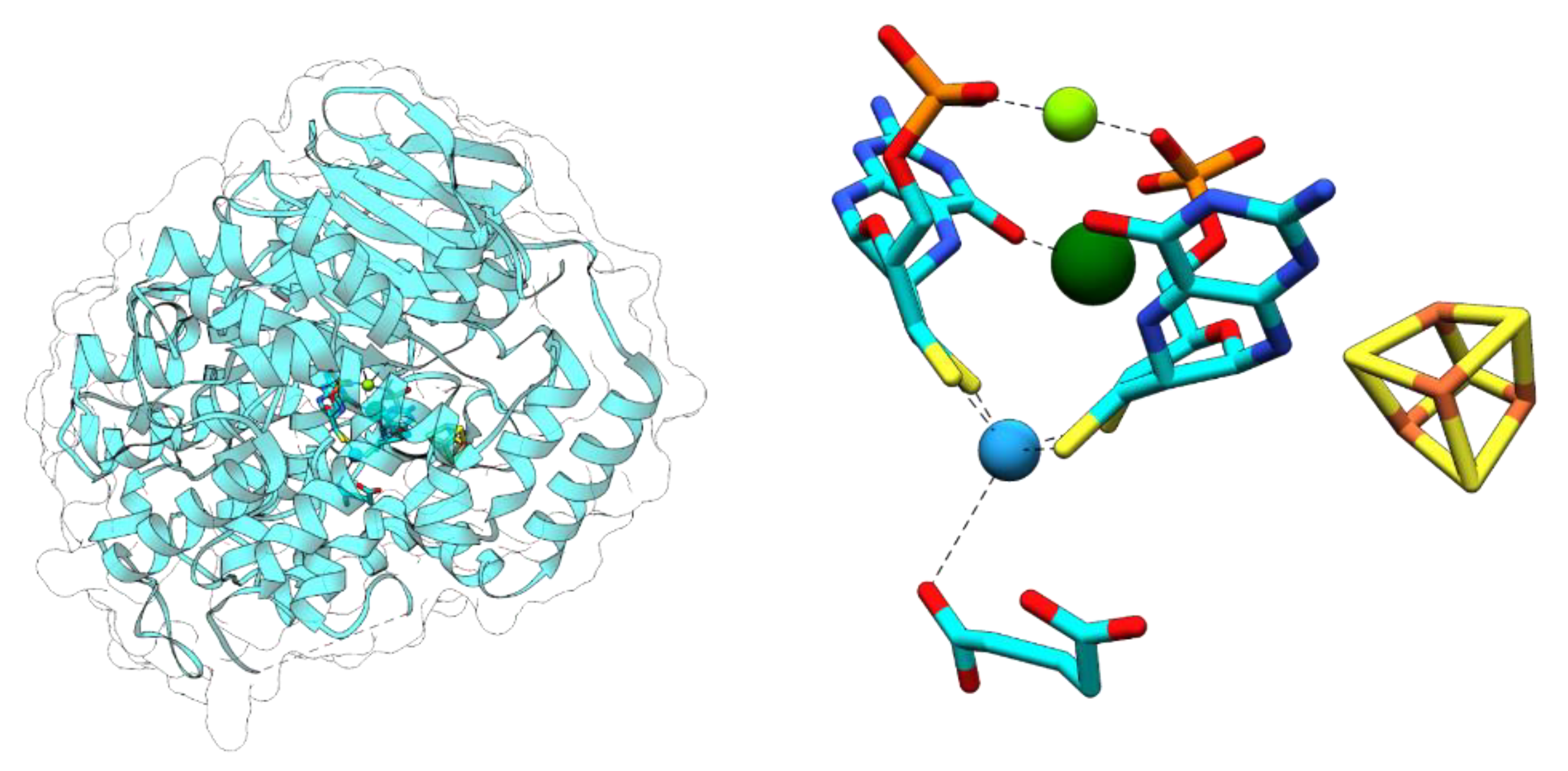
Archaeal and bacterial AORs (sensu stricto) represent the most abundant and best-studied clades exhibiting aldehyde oxidoreductase activity. Both contain a subunit of a similar size harboring the W-bis-MPT cofactor with an additional Mg2+ ion bridging the phosphate groups of the two cofactors, as well as an additional [4Fe-4S] cluster [19,20,21] (Figure 4). The archaeal AORs, as well as some AORs from strictly anaerobic bacteria, occur as homodimers with a bridging iron atom [20], and utilize ferredoxin as a physiological electron acceptor. In contrast, AORs from Aromatoleum aromaticum and other facultatively anaerobic denitrifying bacteria, as well as from the strictly anaerobic Moorella thermoacetica, consist of three different subunits in an α2β2γ2 composition. The additional electron-transferring small subunit with four [4Fe-4S] clusters and the medium-size FAD-containing subunit allows for the use of a broader electron acceptor range, including NAD+ [21]. All characterized enzymes of the archaeal or bacterial AOR clades oxidize a variety of aldehydes to their respective carboxylic acids. Their main physiological function was proposed to be the detoxification of aldehydes that accumulate in different metabolic pathways. Though known for decades, there is still little knowledge of the reaction mechanisms of AORs and the internal electron transfer events involved, largely because of the difficult genetic accessibility of the respective host organisms. Remarkably, AORs also catalyze the reverse reaction, namely the reduction of non-activated acids to the corresponding aldehydes at E°’ ≈ −560 mV, albeit at a rate lower than 1% compared to those of aldehyde oxidation. This property is reflected in the alternative name “carbonic acid reductase” (CAR) for the enzyme from M. thermoacetica. By utilizing this property, the metabolism of fermentative archaea has been artificially shifted from the production of acetic acid to ethanol. In spite of the very low observed reverse in vitro activities of AORs, the reaction seems to work well in whole-cell systems [22]. A similar potential of AORs in the biotechnologically interesting utilization of syngas as a substrate for acetogenic bacteria has been suggested [22,23].
FORs are known from various hyperthermophilic archaea and account together with AOR and GAPOR for the most abundant tungstoenzymes in these organisms. Unlike other AORs, FORs show only low specific activities with short chain aldehydes. They turn over formaldehyde with reasonable rates but show very high Km-values for almost all substrates. A possible exception is glutardialdehyde with the lowest Km-value of the tested substrates, but the data available do not allow to propose a rational physiological function of FOR [24]. The X-ray structure of the FOR for P. furiosus confirms its composition as a homotetramer without a bridging iron atom, as in AORs (Figure 4). Instead, FOR contains an additional Ca2+ ion ligated to one of the MPT cofactors, which is attributed to a structural function. The binding of W-bis-MPT and the [4Fe-4S] cluster is highly similar in AOR and FOR, and the FOR structure with a bound glutarate as a substrate mimic led to the speculation that diacid semialdehydes may serve as potential physiological substrates [25].
Two further clades comprise GAPOR from archaea and GOR from bacteria, which are involved in glucose metabolism via a modified glycolytic pathway. They specifically couple the oxidation of glyceraldehyde-3-phosphate to 3-phosphoglycerate without 1,3-bisphosphoglycerate as an intermediate to the reduction of ferredoxin. GAPORs are monomeric enzymes containing Zn2+ as an additional metal [26,27].
Finally, two further clades of tungsten-containing AOR family members of unknown physiological relevance are represented by WOR4 and WOR5 from P. furiosus. The former showed no aldehyde-oxidizing activity with any substrate. Furthermore, it is the only enzyme of the family containing a [3Fe-4S] cluster, consistent with the loss of one of the cysteine ligands [16], the latter was found to oxidize a similar aldehyde as AOR [28]. Orthologous genes putatively encoding AOR-like enzymes are found in many hyperthermophilic archaea and anaerobic bacteria, and some of them have been biochemically characterized, such as AORs and FORs from Thermococcus litoralis, AORs from T. paralvinellae, Clostridium formicoaceticum, Eubacterium acidaminophilum, Desulfovibrio gigas, Methanobacterium thermoautotrophicum, and GAPOR from Pyrobaculum aerophilum [7,29,30,31].
3.2. Class II Benzoyl-CoA Reductases
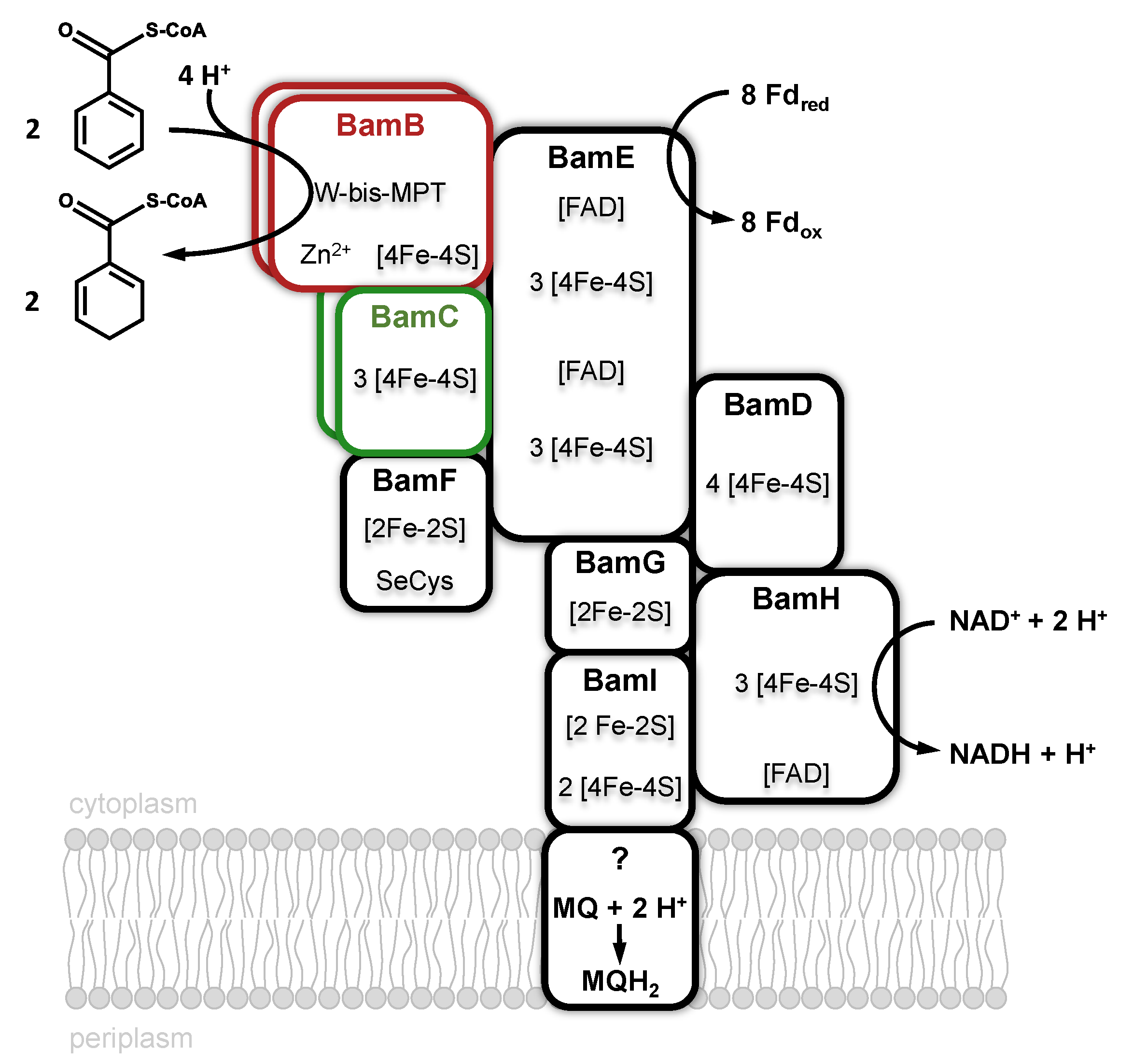
Benzoyl-CoA-reductases (BCRs) are key enzymes for aromatic compound degradation in anaerobic prokaryotes [32,33,34,35]. They catalyze the reduction of the central intermediate benzoyl-CoA to cyclohexa-1,5-diene-1-carboxyl-CoA (dienoyl-CoA) at a redox potential of E’° = −622 mV [36]. Two phylogenetically unrelated classes of BCRs have been identified that both catalyze the same reaction. Class I BCRs are present in facultative anaerobes such as the denitrifying bacterium Thauera aromatica [37]. They employ [4Fe-4S] clusters as the only cofactors and use reduced ferredoxin as an electron donor. Class I BCRs couple the thermodynamically unfavorable reduction of benzoyl-CoA to the stoichiometric hydrolysis of ATP. Obligately anaerobic bacteria employ an ATP-independent, Wco-containing one megadalton class II BCR complex [38]. Class II BCRs have been purified from the Fe(III)-respiring Geobacter metallireducens [39] and the sulfate-respiring Desulfosarcina cetonica [40], both are composed of eight subunits (Bam[(BC)2DEFGHI]2, Bam = benzoic acid metabolism). With four W-bis-MPTs, four Zn, >50 Fe-S clusters, four selenocysteines, and six FADs, class II BCRs represent one of the most complex metalloenzyme machineries known (Figure 5). BamB harbors the Wco-binding active site subunit and can be purified along with the electron transferring BamC subunit [41]. The remaining BamDEFGHI subunits are proposed to be involved in the endergonic electron transfer to Wco, driven by a flavin-based electron bifurcation [39]. In this process, the endergonic electron transfer from a donor to a low-potential acceptor, here benzoyl-CoA, is driven by the exergonic reduction of a second high-potential acceptor using the same donor [42]. The anticipated high-potential acceptor(s) of class II BCRs are still unclear, but recent studies suggest that class II BCRs transfer electrons to menaquinone, as evident from the membrane-association of the complex from G. metallireducens [39].
The BamB subunit belongs to the AOR enzyme family and contains Wco, Zn2+ and a [4Fe-4S] cluster as cofactors. Bioinformatic analyses suggest that all strictly anaerobic bacteria employ a tungsten-containing BamB for benzoyl-CoA reductions during growth with aromatic substrates [34]. Initial studies with Desulfococcus multivorans [43] and G. metallireducens [38] indicated that growth with aromatics depends on molybdenum and selenium, however, the isolated Bam(BC)2 contained stoichiometric amounts of tungsten, whereas molybdenum was virtually absent [41]. This finding suggests that traces of tungstate in the molybdate stock solution were sufficient to produce an active, Wco-containing class II BCR. A TupABC transporter is induced during growth with aromatics that guarantees selective tungstate uptake from the medium [44].
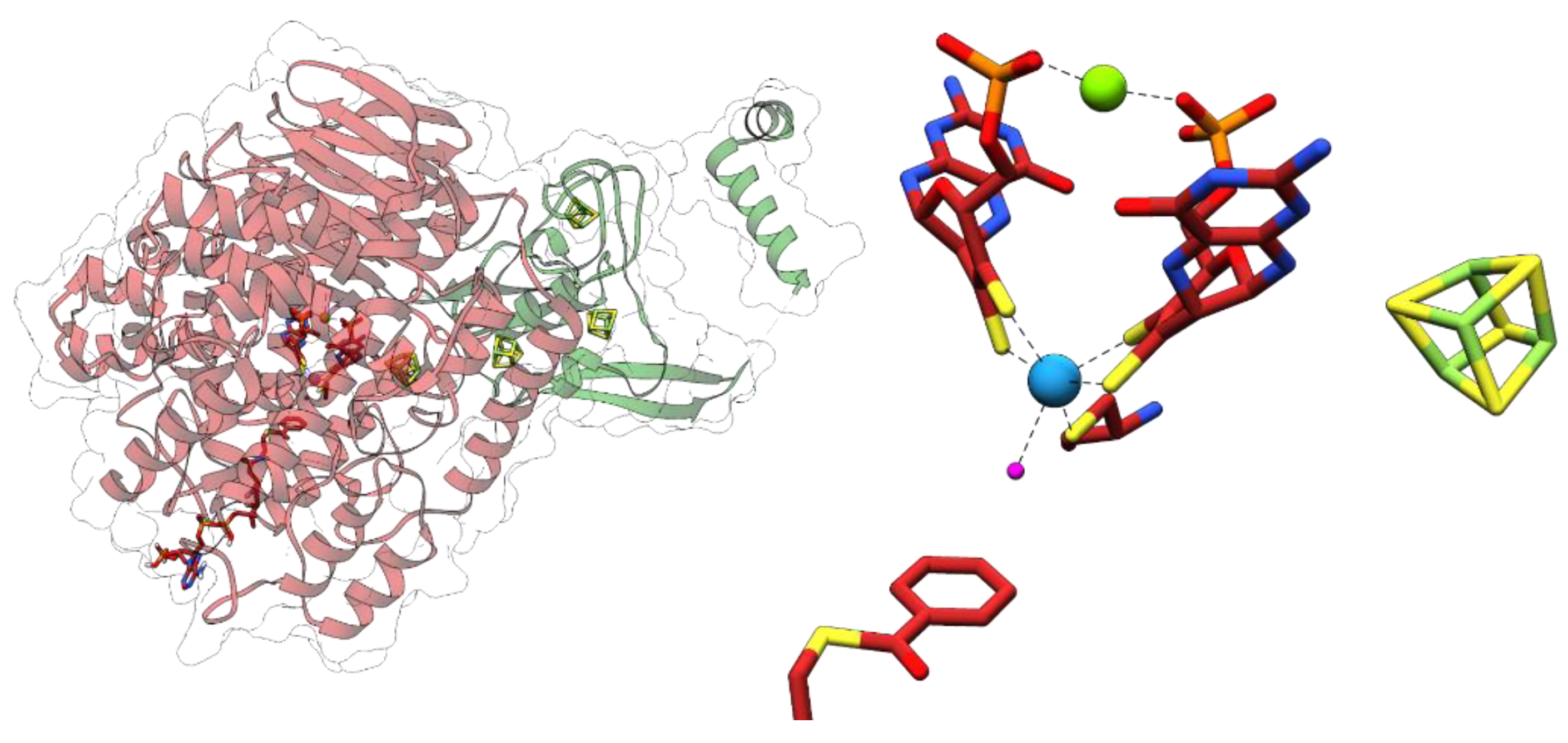
The 1.9 Å resolution X-ray structure of Bam(BC)2 crystals revealed that Wco is accommodated in a highly hydrophobic pocket where it is coordinated by four dithiolene sulfurs of W-bis-MPT, a thiolate of a conserved cysteine (Cys322) and a sixth non-proteinogenic ligand [45] (Figure 6). Its identity could so far not be resolved unambiguously by spectroscopic methods, whereas computational studies favor a water ligand [46,47]. Neither the substrate nor the product are directly bound to tungsten. The spatial separation of the potential proton donor His260 and the W-atom with the aromatic ring positioned in-between provides the molecular basis for the anticipated biological Birch reduction of aromatic rings [48]. Continuum electrostatic and quantum-mechanical/molecular mechanics calculations suggest that benzoyl-CoA reductions are initiated by a hydrogen atom transfer from a W(IV) species via bound water yielding a W(V)-(OH−) species and a substrate radical intermediate [46,47]. These studies also suggested that the second electron derives from the pyranopterin cofactor, rather than from W(V). Proton transfers from an invariant histidine (His260) likely assist this step. A BamB catalysis is one of the rare examples of an MPT-dependent enzyme that does not involve an oxo- or hydroxyl-transfer. The unusual properties of class II BCRs among MPT-enzymes may be explained by the extremely low redox potential of the substrate/product couple that affords a radical biochemistry.
The Birch reduction of aromatic rings is widely used in synthetic chemistry for many applications [49]. Due to its negative attributes, such as its dependence on alkali metals, and ammonia and cryogenic reaction conditions, alternative procedures are being studied such as the use of photo- [50] and electrocatalytic [51] methodologies. In this light, biocatalytic BCRs may be attractive for future Birch reduction applications under mild and environmentally friendly conditions.
3.3. Acetylene Hydratases
Anaerobic acetylene degradation by Pelobacter acetylenicus [52] is initiated by the hydration and tautomerization to acetaldehyde, catalyzed by acetylene hydratase (AH). So far, only the enzyme from P. acetylenicus has been isolated and studied, and a high resolution crystal structure is available (Figure 7) [53,54,55,56]. The oxygen-sensitive enzyme belongs to the DMSOR enzyme family and contains a W-bis-MGD and a [4Fe-4S] cluster [53]. When AH was isolated from P. acetylenicus grown under tungsten-depletion (2 nm) and a 1000-fold excess of molybdate, a molybdenum-containing variant of AH was identified (45–50% metal occupation) that converted acetylene at 10% of the rate of the Wco-containing enzyme [57]. Because of these highly unphysiological expression conditions, we consider AH here as an obligately tungsten-containing enzyme, but further research on the viability of Mo-containing AH variants and their occurrence is required. Almost all studies were carried out with Wco-containing AH.
AH is unique among all Moco/Wco enzymes in terms of catalyzing a non-redox reaction, and it is the only known enzyme that acts on acetylene as a physiological substrate. Notably, acetylene inhibits many key enzymes of anaerobic metabolism, with the exception of nitrogenase, which reduces acetylene to ethylene [58,59]. AH itself is inhibited by acetylene analogues such as cyanide, carbon monoxide, nitrous oxide or substituted acetylenes [53,54]. Though the reaction catalyzed by AH is redox neutral, AH requires a strong reductant like titanium(III)-citrate or sodium dithionite to be catalytically active in vitro [53,56]. The mandatory reductive activation of AH is assigned to the reduction of Wco to the W(IV) state, though the exact redox potential of the Wco is unknown [53,55]. An intact [4Fe-4S] cluster appears to be dispensable for catalysis [56] as supported by the observation that degradation to a [3Fe-4S] cluster did not affect AH activity [53].
The mechanism of AH has been subject to many discussions, and two types of mechanistic scenarios were suggested with different roles for the catalytically active W(IV): (i) a first-shell mechanism, where tungsten coordinates acetylene and primes the substrate for a nucleophilic attack by water (or hydroxide), and (ii) a second-shell mechanism that relies on the polarization of tungsten-bound water, followed by an electrophilic attack on the C≡C bond of acetylene [55]. The second-shell mechanism was favored by density-functional theory calculations based on a high resolution crystal structure, but no spectroscopic evidence was obtained to corroborate this mechanism [55]. Based on calculations for the active site mimics of AH, another study concluded that the direct coordination of acetylene by tungsten was favored over the binding of water [60]. It was reported that an inorganic model complex (NEt4)2[WIVO(mnt)2], where mnt = malonitrile, was able to catalyze the same reaction as AH, whereas the W(VI) analogue failed [61]. While these results could not be reproduced, recent developments and thorough characterizations of other biomimetic tungsten complexes strongly support a mechanism of acetylene binding directly to tungsten (first-shell mechanism) [62,63]. Such a metal-acetylene coordination is known from other organometallic molybdenum-/tungsten-complexes [64].
Acetaldehyde is an important building block for many chemicals and pharmaceuticals. Its industrial production from acetylene is typically achieved in the presence of a mercury catalyst-containing mercuric chloride that exhibits adverse effects on human health and the environment [65]. Thus, enzymatic solutions for this industrial process are of potential interest.
4. Enzymes Containing Either Tungsten or Molybdenum
A number of isofunctional enzymes of the DMSOR family exist in nature that may contain either Moco or Wco. This apparent promiscuity may be explained by an adaptation to molybdate or tungstate enriched/depleted environments rather than to substantial differences in enzyme function. In some organisms, sets of genes exist that specifically code either for Moco- and/or for Wco-dependent variants of MPT-dependent enzymes. The induction of the individual gene clusters in response to varying molybdate/tungstate concentrations has been described in some cases. The molybdenum- or tungsten-containing variants may differ in their activity or affinity to the individual substrates. Owing to the lower redox potential of the biologically relevant W(IV/V/VI) vs. Mo(IV/V/VI) states, Wco-containing variants appear to be catalytically more efficient for reactions occurring at low redox potentials and Moco for those at more positive redox potentials.
4.1. Formate Dehydrogenases
The widely abundant tungsten- or molybdenum-dependent FDHs catalyze the reversible conversion of formate to CO2 at E°’ = −430 mV with various electron donors/acceptors [66,67,68,69,70]. Metal-dependent FDHs are involved in formate-dependent respiration, syntrophy and methanogenesis, acetogenesis, methylotrophy or in fermentations as components of formate hydrogen lyase complexes. Unlike most other Moco/Wco enzymes, an FDH catalysis does not involve an oxygen atom transfer reaction. Though the reaction mechanism of FDHs is still under debate, a hydride transfer from formate to a sulfido-ligand at Mo(VI)/W(VI) appears to be the most plausible as it circumvents the difficult deprotonation of the Cα proton of formate [68,71,72,73]. Here, we briefly summarize the occurrence and function of Wco-dependent FDHs and refer to recent in-depth reviews of the diversity, structure and function of metal-containing FDHs [66,67,68].
Many FDHs depend on molybdenum, and elevated tungstate concentrations in the medium yield inactive Mo-dependent variants as reported for FDHs from E. coli [74] or Methanobacterium formicium [75]. On the other side, a number of FDHs have been isolated with a clear preference for tungsten over molybdenum, whereas only a few FDHs appear to be active with either of the two metals. Wco-containing FDHs are affiliated to two enzyme clades, the membrane-bound FDH-N-like enzymes and the soluble or membrane-associated FDH-H-like enzymes, whose active sites are oriented towards the periplasm or the cytoplasm, respectively. Cytoplasmic Wco-containing FDHs have initially been described in a number of acetogenic clostridia such as Moorella thermoaceticum [76], Clostridium formicoaceticum [77] or C. carboxidivorans [78], where they catalyze the NAD(P)H-dependent reduction of CO2. A tungsten-containing FDH of Peptoclostridium (formerly Eubacterium) acidaminophilum serves either as an electron donor system for amino acid fermentations or as component of a formate hydrogen lyase complex [79]. A tungsten-dependent FDH of this clade is also involved in the oxidation of reduced C1 compounds to CO2 with dioxygen as an electron acceptor in the aerobic methylotrophic Methylobacterium extorquens [80]. This finding indicates that the use of Wco is not restricted to anaerobic organisms. Two tungsten-specific FDHs, an FDH-H and a FDH-N type enzyme, were reported from Syntrophobacter fumaroxidans during syntrophic propionate fermentations, where they play a role in electron transfers to the methanogenic partner [81]. Further Wco-containing FDHs of the FDH-N clade are known from Desulfovibrio species and other sulfate reducing bacteria [82,83,84,85]. One of these species, D. alaskensis, is known to incorporate either Moco or Wco into the same FDH, depending on the tungstate and molybdate concentrations in the medium [83,85]. In contrast, D. vulgaris Hildenborough produces a specific Moco-dependent FDH during growth with molybdate and an FDH active with either metal during growth with tungstate [84]. A similar case of an active FDH-N type enzyme with either Moco or Wco in differently grown cells was reported for Campylobacter jejuni [86], which contains only one set of FDH encoding genes.
Biocatalysts that sequester CO2 are of increasing general interest. In this light, FDHs that reduce CO2 to formate as a viable energy source are of a potential biotechnological use. A tungsten-containing FDH has been adsorbed to an electrode and successfully used for electrocatalytical CO2 reduction [87]. Furthermore, an enzyme complex of FDH and hydrogenase (formate hydrogen lyase) reversibly interconverts H2 + CO2 to formate demonstrating that formate may be used as an energy storage/transport compound [88,89]. Finally, the solar-driven CO2 reduction by FDHs in combination with artificial and/or biological photosystems has been demonstrated [90,91].
4.2. Formylmethanofuran Dehydrogenases
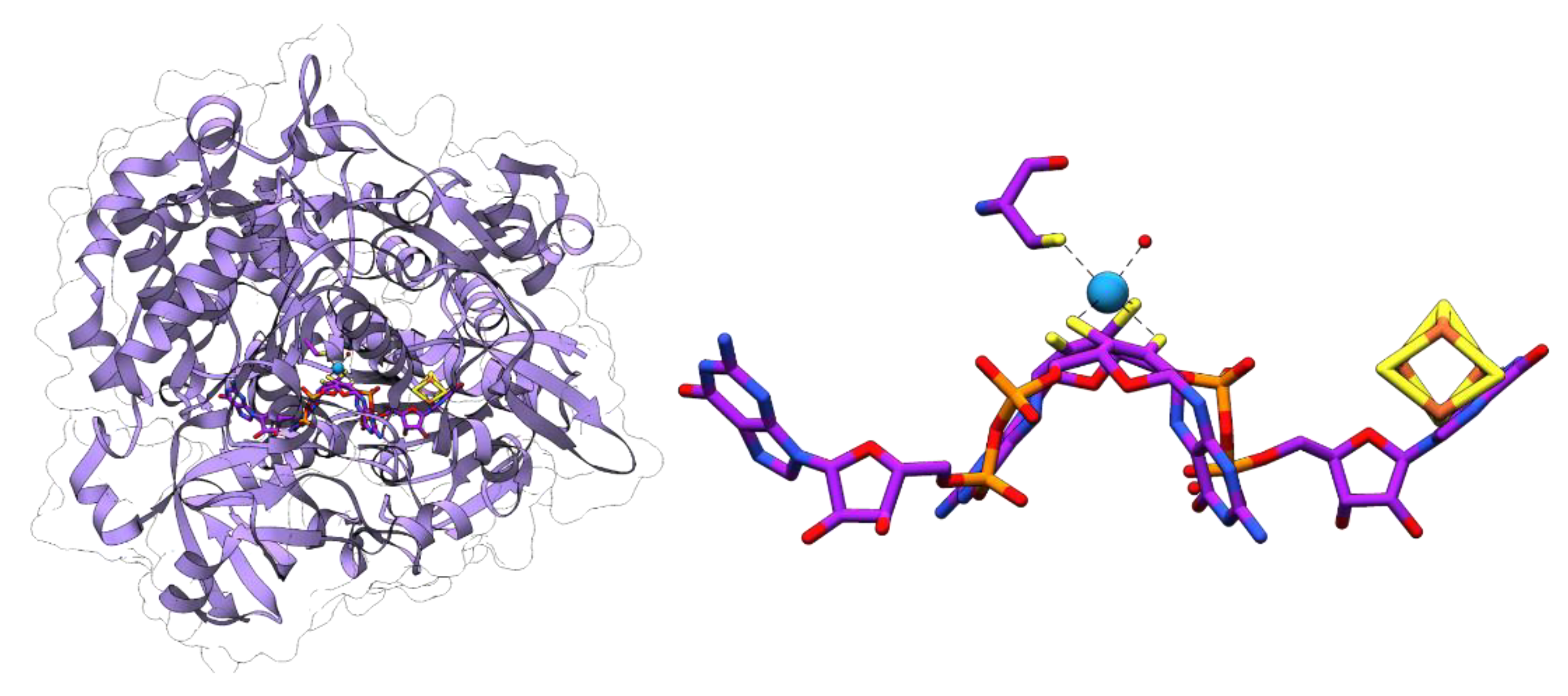
Formylmethanofuran dehydrogenase catalyzes the first step of methanogenesis that is related to the reaction catalyzed by FDHs: the reduction of CO2 yielding formate is bound in an activated form at the methanofuran cofactor [69]. Methanogens have been found to contain isoenzymes being either specific for tungsten or molybdenum [92,93]. The genome of Methanososarcina acetivorans encodes four putative formylmethanofuran dehydrogenases. Two of these are proposed to be specific for tungsten or molybdenum, respectively, with one of the tungsten-dependent isoenzymes being specifically required for growth with carbon monoxide [94]. The crystal structure of the tungsten-containing formylmethanofuran dehydrogenase from Methanothermobacter wolfei revealed that the enzyme harbors two active sites being separated by a 43 Å tunnel which allows the directed diffusion of the formate from the first active site (the Wco center) to the second one, the formylmethanofuran forming center [95].
4.3. Respiratory Nitrate Reductases
Two versions of respiratory nitrate reductases belong to the DMSOR enzyme family: the membrane bound enzymes (NarGHI) and the periplasmic ones (NAP). The active site subunit of the former, NarG, is largely conserved among all nitrate-respiring bacteria and archaea and usually contains Moco in the active site subunit. Tungstate usually acts as an inhibitor of the assembly of Moco into NarG, NAP or other respiratory Moco-containing enzymes [8,74]. However, Paracoccus pantotrophus is able to grow under denitrifying conditions in the presence of 100 µm tungstate, and indirect evidence was obtained that tungsten was incorporated into its periplasmic nitrate reductase NAP [96]. Furthermore, the nitrate-respiring, hyperthermophilic archaea Pyrobaculum aerophilum and Aeropyrum pernix grow at temperatures and environments where tungstate is enriched and molybdate is depleted. Under these special growth conditions, a tungsten-containing NarGHI-type enzyme was isolated from of P. aerophilum with a two-fold lower turnover number than the Mo-containing enzyme [97].
4.4. Dimethyl Sulfoxide and Trimethylamine N-Oxide Reductases
DMSOR and trimethylaminoxide reductase (TMAOR) are both required to use DMSO or TMAO as terminal electron acceptors in oxygen-independent respiratory chains. In the DMSOR from Rhodobacter capsulatus and R. sphaeroides, the Mo bound to the bis-Mo-MGD cofactor was substituted with tungsten, and the resulting Wco-containing enzyme reduced DMSO even at a higher rate, but was inactive in terms of catalyzing the reverse dimethyl sulfide oxidation [15,98]. This finding is rationalized by the generally lower redox potential of Wco in comparison to Moco [99]. Likewise, TMAOR from E. coli was also active when molybdenum was substituted by tungsten even with a slightly increased catalytic efficiency [100].
4.5. Thiosulfate Reductases
A recent report shows that a thiosulfate reductase (TSR) of the DMSOR family is involved in thiosulfate respiration in the archaeon P. aerophilum. The enzyme was recombinantly produced in P. furiosus and contained either Mo or W, dependent on the growth conditions [101]. The molybdenum-containing enzyme showed a ten-fold higher conversion rate and a twice as high affinity to thiosulfate than the tungsten-containing one. This study represents the first case of producing an active Moco- or Wco-containing enzyme in P. furiosus.
5. Tungsten Uptake and Assembly of the Wco
5.1. Tungsten Uptake
Tungsten is taken up by prokaryotes in form of the tungstate oxyanion (WO42−) by three high-affinity ATP-binding cassette (ABC)-type systems: the highly tungstate-specific TupABC system, and the ModABC/WtpABC systems that transport either tungstate or molybdate [102]. They all consist of a periplasmic binding protein (component A), a transmembrane channel (component B), and an ATPase subunit (component C). The ModABC system with similar affinities to molybdate and tungstate in the low to high nM range is highly abundant in bacteria and archaea and allows for the specific transport of molybdate and tungstate even in the presence of high concentrations of similar other oxyanions (e.g., 28 mm sulfate in marine environments). The WtpABC transporter is present in archaea that lack TupABC or ModABC systems, such as P. furiosus, and is rather distantly related to these uptake systems. The binding protein WtpA shows by far the highest affinity for tungstate with Kd = 19 pm [103]. The TupABC system has originally been studied in P. acidaminophilum [104,105], and later in C. jejuni [106,107] and D. alaskensis [108,109]. The encoding TupABC genes are widely abundant in bacterial and archaeal genomes [110]. The structural basis by which TupA discriminates between tungstate and molybdate, considering their highly similar atomic radii, is still unknown.
5.2. Metallopterin Cofactor Synthesis
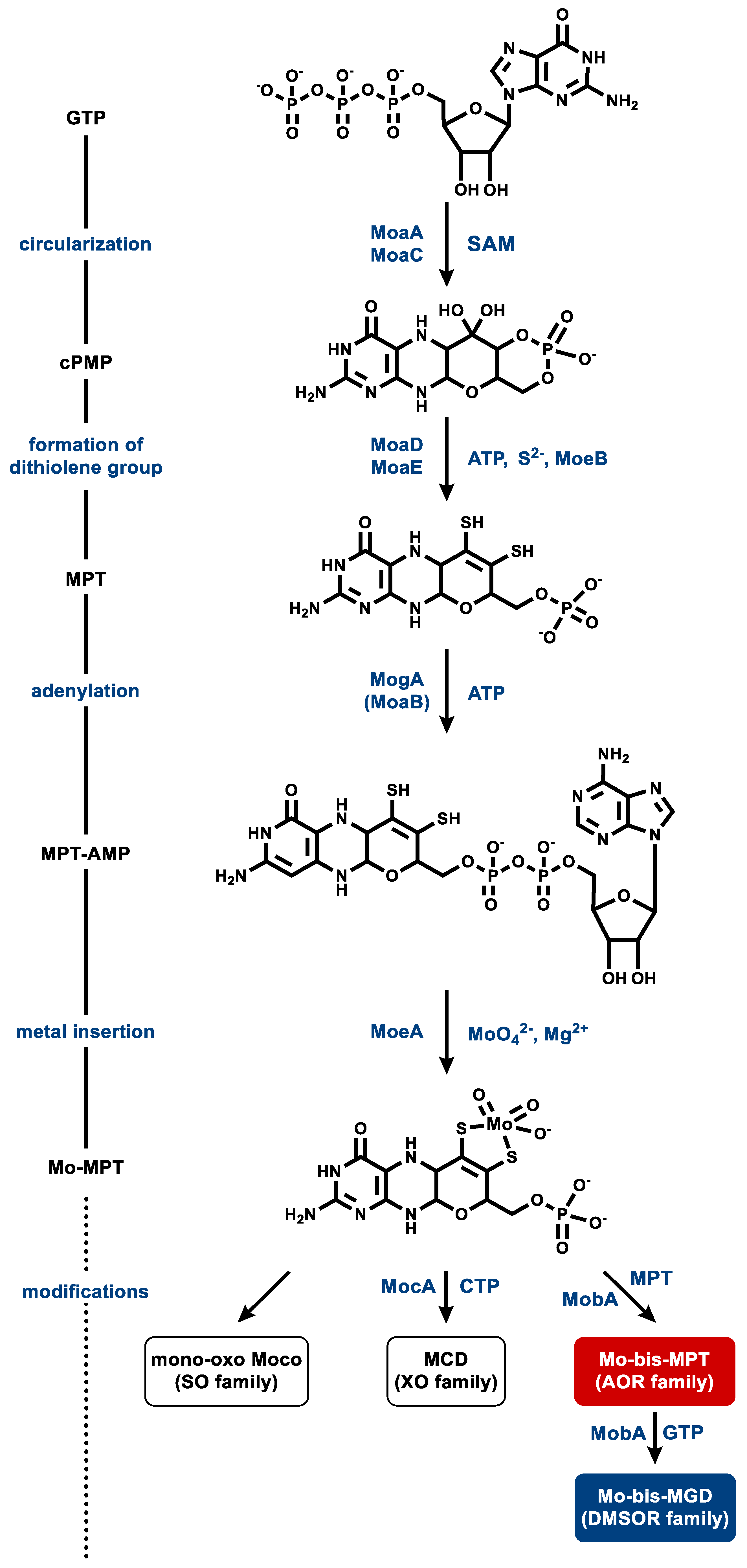
Most knowledge of MPT biosynthesis derives from research on Moco (for initial and recent reviews on Moco synthesis see [4,5,111,112,113]). As almost all genes required for Moco synthesis are present in the genomes of prokaryotes that synthesize Wco-dependent enzymes, it is generally accepted that MPT synthesis is catalyzed by similar enzymes and identical intermediates [114]. In contrast, very little is known about the steps that discriminate between molybdenum and tungsten during the insertion of the metals into MPT. Here, we first briefly summarize knowledge of Moco synthesis in the prokaryotic model organism E. coli, followed by the presentation of initial insights and hypotheses about Wco synthesis. There, we will address the special challenges of simultaneously synthesizing Moco- and Wco-dependent enzymes.
Moco biosynthesis proceeds via a four-step process: (i) conversion of guanosine-5′-triphosphate (GTP) to the four-ringed cyclic intermediate pyranopterin monophosphate (cPMP), (ii) introduction of the two thiols of the dithiolene group, (iii) adenylation to MPT-adenosine monophosphate (AMP), and (iv) metal insertion and release of AMP; the latter two steps are often merged as a single one (Figure 8) [4,5,111,113]. The formation of cPMP (or “precursor Z”) from GTP occurs in two steps involving complex molecule rearrangements. The radical S-adenosylmethionine (SAM) enzyme MoaA catalyzes an initial remodeling of the carbon backbone of GTP and afterwards, MoaC completes the synthesis of cPMP with the concomitant release of the pyrophosphate moiety of the original GTP [115,116].
In the second step, two sulfur atoms are inserted into cPMP at the C1′ and C2′ position, resulting in MPT formation. This step is catalyzed by MoaD and MoaE, which form a heterotetrameric complex, also referred to as MPT synthase. Two MoaEs bind one cPMP each, while MoaD transfers one sulfur atom at a time from a thiocarboxylate group on its conserved C-terminal glycine residue, producing a hemisulfurated intermediate in the process [117,118,119,120]. MoaD itself receives its thiogylcyl modification by the sulfurylase MoeB. MoaD forms a complex with MoeB, which catalyses the Mg2+- and ATP-dependent adenylation of the C-terminal glycine residue of MoaD. This activated MoaD is then sulfurated by the cysteine desulfurase IscS [121,122].
In the third step, MPT is adenylated in an ATP-dependent reaction by MogA (in bacteria) or MoaB (in archaea), yielding MPT-AMP. This activated intermediate is transferred to MoeA, where molybdate is bound to the dithiolene sulfurs of MPT, accompanied by hydrolysis and the release of the AMP moiety [114,123,124]. The product is referred to as Mo-MPT, which represents the Moco found in SO family members, and which serves as a building block for the cofactors of the three other families of MPT-containing enzymes. To generate the cofactor for the XO-family enzymes, MocA catalyzes the cytidylation of MPT with CTP while releasing pyrophosphate. The generated MPT cytosine dinucleotide cofactor is then further modified by the addition of an equatorial sulfido-ligand in the active site of the enzymes [13,125]. Ligation of a molybdenum and a second MPT yields Mo-bis-MPT, which is found in the AOR family member YdhV [126]. The Mo-bis-MGD cofactor of the DMSOR family enzymes is synthesized by the attachment of two GMP moieties from GTP [127,128].
Wco synthesis is considered to involve similar or even the same enzymes for the steps up to metal insertion. So far, the adenylation of MPT by MoaB in the course of Wco synthesis was shown in the hyperthermophilic P. furiosus [114]. No other enzyme specifically involved in Wco synthesis has been studied so far.
5.3. Strategies for the Selective Insertion of Molybdenum/Tungsten into Target Proteins
The highly similar physicochemical properties of molybdate and tungstate immediately lead to the question regarding which mechanism guarantees the correct transfer of both oxyanions into their target proteins. In particular, organisms producing enzymes that are only active with either of the two metals depend on a highly selective metal insertion system. We distinguish here between three different scenarios: (i) organisms that exclusively produce either Wco- or Moco-dependent enzymes, (ii) organisms that contain genes for both type of enzymes, but only produce either Moco- or Wco-enzymes under certain conditions, and (iii) organisms that produce Moco- and Wco-dependent enzymes simultaneously. In principle, selectivity may be achieved during molybdate/tungstate uptake, Mo-MPT/W-MPT formation, or the insertion of the mature W-bis-MPT/bis-Mo-MPT into the respective target proteins.
In the first scenario, the selective uptake of either molybdate or tungstate would already be sufficient to guarantee selectivity. Specific tungstate uptake may be achieved by the periplasmic binding protein TupA from the TupABC transporter [105], or WtpA from P. furiosus, which show much higher affinities for tungstate vs. molybdate [103]. However, no transporter system exhibiting a comparable selectivity for molybdate over tungstate has been described so far, which explains the frequently observed antagonistic effect of tungstate for Moco-dependent enzymatic processes.
In the second scenario, the selective uptake may also play a role but would require at least two selective uptake systems for either of the two metal oxyanions, which may be induced under certain conditions. In addition, MoeAs have been proposed earlier as selectivity generating enzymes that may be specific for either molybdate or tungstate during metal insertion into MPT-AMP [12,85,114]. This proposal was based on the finding that at least two versions of MoeA encoding genes were identified in genomes of archaea [12] or in D. alaskensis [85]. The latter produces either Moco or Wco containing versions of the same FDH, depending on the molybdate/tungsten concentrations in the medium. In D. vulgaris Hildenborough, the production of Wco- or Moco-containing FDH isoenzymes is regulated by the molybdate/tungstate availability at the transcriptional level [84]. These results are in line with a metal-dependent induction of either tungsten- or molybdenum-specific pairs of MoeA/apo-FDH paralogues.
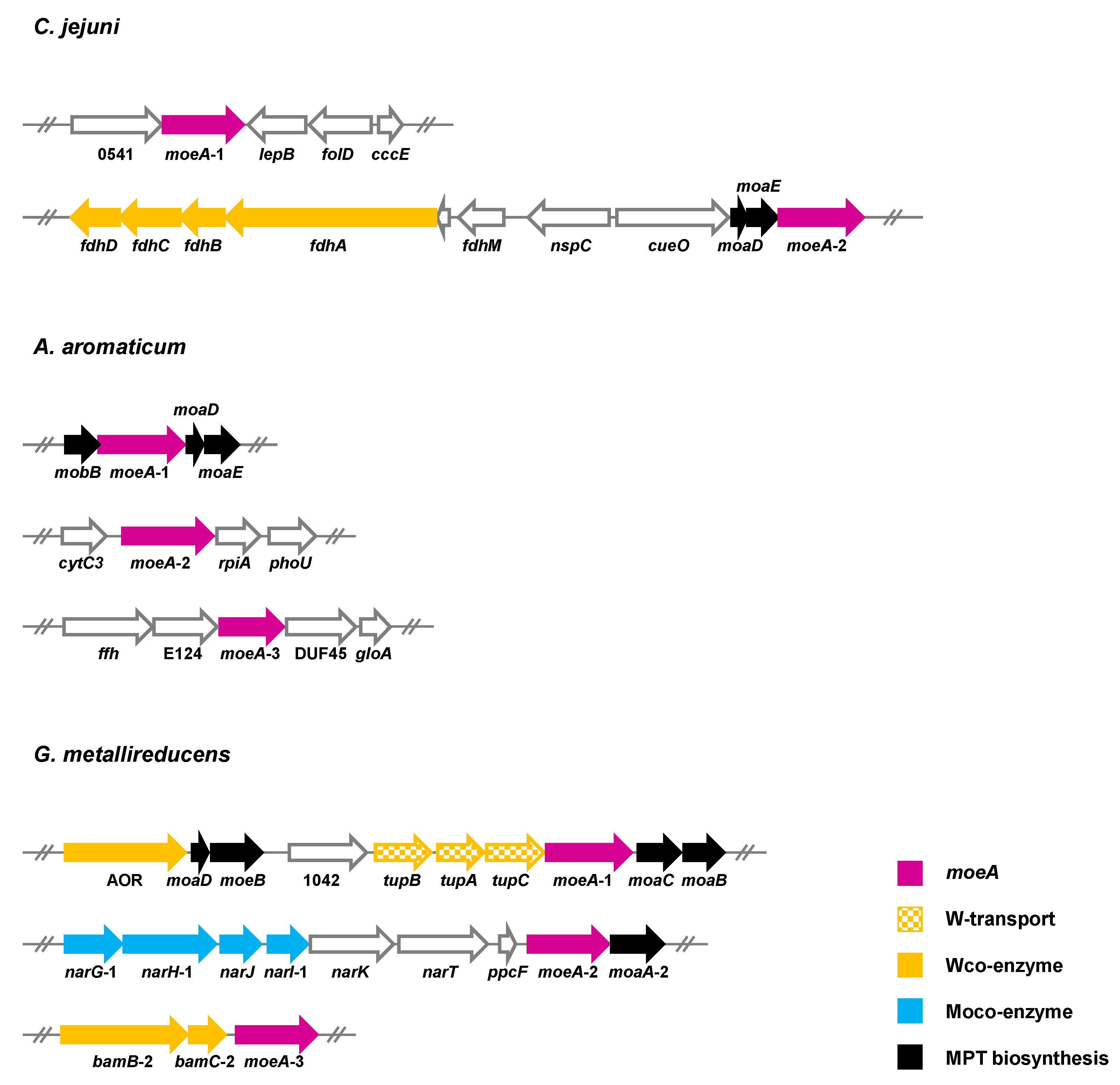
Such a mechanism is not feasible when Moco- and Wco-depending enzymes have to be produced in parallel (scenario iii). In recent years, a number of bacteria were identified that simultaneously produce obligately Wco- and Moco-dependent enzymes, and they all contain at least two moeA copies (Figure 9). C. jejuni produces a tungsten-containing FDH under certain growth conditions that may be coupled with molybdenum-dependent nitrate reductase in a menaquinone-dependent respiratory chain [86]. In A. aromaticum, a tungsten-dependent AOR and two molybdenum-dependent enzymes, phenylacetyl-CoA dehydrogenase and nitrate reductase, are produced during growth with phenylalanine under denitrifying conditions [21]. When G. metallireducens grows with p-cresol or 4-hydroxybenzoate under ammonifying conditions, it produces one strictly Wco-dependent class II benzoyl-CoA reductase [38,41], and two Moco-containing enzymes from different families: the 4-hydroxybenzoyl-CoA reductase of the XO family [129,130] and nitrate reductase of the DMSOR family [44]. While C. jejuni contains two paralogous moeA genes [106], even three paralogues are found in the genomes of A. aromatoleum and G. metallireducens. In the latter bacterium, the genetic context of the three moeA genes indicates the specificity for molybdenum or tungsten (Figure 9). In particular, moeA-1 is located adjacent to the genes encoding the tungstate-dependent TupABC transporter, and moeA-3 is adjacent to paralogues of the bamBC genes, which putatively code for an isoenzyme of tungsten-dependent class II benzoyl-CoA reductase. Thus, it is tempting to speculate that moeA-1 and moeA-3 encode a tungstate-specific metal insertase. In contrast, moeA-2 is located in a gene cluster together with genes encoding molybdenum-dependent NarGHI-type nitrate reductase. Thus, moeA-2 likely encodes a molybdenum-specific enzyme. These findings support the idea that the simultaneous production of Moco- and Wco-dependent enzymes is accomplished by MoeA isoenzymes that are selective for either molybdate or tungstate. Though this assumption appears plausible, any experimental evidence for it is lacking.
Finally, the question rises as to how the Mo-MPT/W-MPT cofactors formed by specific MoeAs are specifically processed and targeted into the individual apo-enzymes. Specific targeting excludes that Mo-MPT/W-MPT formed are released from individual MoeAs because the modifying enzymes that bind the Mo-MPT/W-MPT in the next step will hardly distinguish between the molybdenum- or tungsten-containing versions. In tungstoenzymes, the metal is always bound to two pyranopterins (bis-MPT), which affords the conversion of MoeA-bound W-MPT to W-bis-MPT in a similar manner as the MobA-catalyzed process known for molybdenum enzymes of the DMSOR family [128]. Thus, the reported complex formation between MoeA and MobA [131] appears to be essential for maintaining the selectivity of the individual bis-MPT intermediates formed. It is conceivable that individual MoeA/MobA complexes specifically interact with the respective apo-enzymes either before or after the GTP-dependent W-bis-MGD formation, including further modifying enzymes such as FdhD. The latter is involved in sulfuration of the Mo-/W-bis-MPT of FDHs and in its insertion into apo-FDHs [132,133,134]. MoeAs, specific for either tungsten or molybdenum, may enable or disable such additional interactions. Finally, the formation of supercomplexes between all the components involved in the conversion of the last common MPT-AMP intermediate to the mature cofactors inserted in individual target proteins could substantially facilitate selective Moco/Wco targeting.
6. Conclusions
Recent research has revealed a higher abundance and catalytic diversity of tungstoenzymes than previously anticipated. While tungsten was originally mostly associated with AORs from hyperthermophilic archaea and some clostridial FDHs, a number of tungstoenzymes with novel functions have been discovered in a large variety of microorganisms comprising fermenting, sulfate-reducing, metal oxide-reducing, denitrifying, aerobic and pathogenic bacteria. Tungstoenzymes are of emerging biotechnological interest due to their involvement in the low-potential reduction processes of CO2, carboxylic acids or aromatic rings, or in the industrially important conversion of acetylene to acetaldehyde. Thus, tungsten appears to be the bio-metal of choice for a number of challenging enzymatic reactions. While knowledge of the structure and function of tungstoenzymes has continuously been increasing over the past two decades, there is still a high research demand concerning the biosynthesis and incorporation of Wco. In particular, the mechanisms that discriminate between tungstate- and molybdate-insertion, and the selective targeting of Mo- or W-bis-MPTs to the individual apo-proteins, are still unknown. Considering the high similarities of molybdate and tungstate, the observed high selectivity for either of the two metals affords sophisticated molecular solutions. Insights into the molecular basis of this selectivity will help to explain how enzymes that strictly rely on tungsten are specifically loaded with this cofactor even in the presence of high molybdate concentrations in the medium. They will also provide a rationale as to why whole-cells may be inhibited by tungstate due to the antagonistic effects of tungstate and molybdate. Bacteria that simultaneously produce tungsto- and molybdoenzymes will serve as valuable model organisms to address the question of selectivity for tungstate vs. molybdate in the future.
Author Contributions
Phylogenetic analyses and visualization: J.H., C.S.S.; structural analysis: M.W.; visualization of reaction schemes and genetic organization: C.S.S., M.B.; project supervision: M.B.; the manuscript was written and revised through contributions of all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the German Research Foundation (DFG), grants BO 1565 15-1 and HE 2190 11-1, the collaborative research center SFB 1381, project ID 403222702 (M.B.) and the Synmikro center Marburg (J.H.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hille, R. The molybdenum oxotransferases and related enzymes. Dalt. Trans. 2013, 42, 3029–3042. [Google Scholar] [CrossRef] [PubMed]
- Hille, R.; Hall, J.; Basu, P. The mononuclear molybdenum enzymes. Chem. Rev. 2014, 114, 3963–4038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leimkühler, S.; Iobbi-Nivol, C. Bacterial molybdoenzymes: Old enzymes for new purposes. FEMS Microbiol. Rev. 2015, 40, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Mendel, R.R.; Leimkühler, S. The biosynthesis of the molybdenum cofactors. J. Biol. Inorg. Chem. 2015, 20, 337–347. [Google Scholar] [CrossRef]
- Leimkühler, S. The biosynthesis of the molybdenum cofactors in Escherichia coli. Environ. Microbiol. 2020, 22, 2007–2026. [Google Scholar] [CrossRef] [Green Version]
- Hille, R.; Schulzke, C.; Kirk, M.L. Molybdenum and Tungsten Enzymes: Spectroscopic and Theoretical Investigations; Metallobiology; Royal Society of Chemistry: Cambridge, UK, 2016; ISBN 978-1-78262-878-1. [Google Scholar]
- Andreesen, J.R.; Makdessi, K. Tungsten, the surprisingly positively acting heavy metal element for prokaryotes. Ann. N. Y. Acad. Sci. 2008, 1125, 215–229. [Google Scholar] [CrossRef]
- Zhu, W.; Winter, M.G.; Byndloss, M.X.; Spiga, L.; Duerkop, B.A.; Hughes, E.R.; Büttner, L.; de Lima Romão, E.; Behrendt, C.L.; Lopez, C.A.; et al. Precision editing of the gut microbiota ameliorates colitis. Nature 2018, 553, 208–211. [Google Scholar] [CrossRef]
- Hille, R. Molybdenum and tungsten in biology. Trends Biochem. Sci. 2002, 27, 360–367. [Google Scholar] [CrossRef]
- Pushie, M.J.; Cotelesage, J.J.; George, G.N. Molybdenum and tungsten oxygen transferases-structural and functional diversity within a common active site motif. Metallomics 2014, 6, 15–24. [Google Scholar] [CrossRef]
- Romão, M.J. Molybdenum and tungsten enzymes: A crystallographic and mechanistic overview. Dalt. Trans. 2009, 4053–4068. [Google Scholar] [CrossRef]
- Bevers, L.E.; Hagedoorn, P.L.; Hagen, W.R. The bioinorganic chemistry of tungsten. Coord. Chem. Rev. 2009, 253, 269–290. [Google Scholar] [CrossRef]
- Neumann, M.; Mittelstädt, G.; Iobbi-Nivol, C.; Saggu, M.; Lendzian, F.; Hildebrandt, P.; Leimkühler, S. A periplasmic aldehyde oxidoreductase represents the first molybdopterin cytosine dinucleotide cofactor containing molybdo-flavoenzyme from Escherichia coli. FEBS J. 2009, 276, 2762–2774. [Google Scholar] [CrossRef] [PubMed]
- Romão, M.J.; Archer, M.; Moura, I.; Moura, J.J.; LeGall, J.; Engh, R.; Schneider, M.; Hof, P.; Huber, R. Crystal structure of the xanthine oxidase-related aldehyde oxido-reductase from D. Gigas. Science 1995, 270, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, J.; Niks, D.; Hille, R. Kinetic and spectroscopic characterization of tungsten-substituted DMSO reductase from Rhodobacter sphaeroides. J. Biol. Inorg. Chem. 2018, 23, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Adams, M.W.W. Characterization of a fourth tungsten-containing enzyme from the hyperthermophilic archaeon Pyrococcus furiosus. J. Bacteriol. 2002, 184, 6952–6956. [Google Scholar] [CrossRef] [Green Version]
- Mukund, S.; Adams, M.W. Molybdenum and vanadium do not replace tungsten in the catalytically active forms of the three tungstoenzymes in the hyperthermophilic archaeon Pyrococcus furiosus. J. Bacteriol. 1996, 178, 163–167. [Google Scholar] [CrossRef] [Green Version]
- Sevcenco, A.-M.; Bevers, L.E.; Pinkse, M.W.H.; Krijger, G.C.; Wolterbeek, H.T.; Verhaert, P.D.E.M.; Hagen, W.R.; Hagedoorn, P.-L. Molybdenum incorporation in tungsten aldehyde oxidoreductase enzymes from Pyrococcus furiosus. J. Bacteriol. 2010, 192, 4143–4152. [Google Scholar] [CrossRef] [Green Version]
- Mukund, S.; Adams, M.W. Characterization of a novel tungsten-containing formaldehyde ferredoxin oxidoreductase from the hyperthermophilic archaeon, Thermococcus litoralis. A role for tungsten in peptide catabolism. J. Biol. Chem. 1993, 268, 13592–13600. [Google Scholar]
- Chan, M.K.; Mukund, S.; Kletzin, A.; Adams, M.W.; Rees, D.C. Structure of a hyperthermophilic tungstopterin enzyme, aldehyde ferredoxin oxidoreductase. Science 1995, 267, 1463–1469. [Google Scholar] [CrossRef]
- Arndt, F.; Schmitt, G.; Winiarska, A.; Saft, M.; Seubert, A.; Kahnt, J.; Heider, J. Characterization of an aldehyde oxidoreductase from the mesophilic bacterium Aromatoleum aromaticum Ebn1, a member of a new subfamily of tungsten-containing enzymes. Front. Microbiol. 2019, 10, 71. [Google Scholar] [CrossRef] [Green Version]
- Basen, M.; Schut, G.J.; Nguyen, D.M.; Lipscomb, G.L.; Benn, R.A.; Prybol, C.J.; Vaccaro, B.J.; Poole, F.L.; Kelly, R.M.; Adams, M.W.W. Single gene insertion drives bioalcohol production by a thermophilic archaeon. Proc. Natl. Acad. Sci. USA 2014, 111, 17618–17623. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.W.; Lipscomb, G.L.; Nguyen, D.M.; Crowley, A.T.; Schut, G.J.; Scott, I.; Kelly, R.M.; Adams, M.W.W. Ethanol production by the hyperthermophilic archaeon Pyrococcus furiosus by expression of bacterial bifunctional alcohol dehydrogenases. Microb. Biotechnol. 2017, 10, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Mukund, S.; Schut, G.J.; Dunn, D.M.; Weiss, R.; Adams, M.W. Purification and molecular characterization of the tungsten-containing formaldehyde ferredoxin oxidoreductase from the hyperthermophilic archaeon Pyrococcus furiosus: The third of a putative five-member tungstoenzyme family. J. Bacteriol. 1999, 181, 1171–1180. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Faham, S.; Roy, R.; Adams, M.W.; Rees, D.C. Formaldehyde ferredoxin oxidoreductase from Pyrococcus furiosus: The 1.85 Å resolution crystal structure and its mechanistic implications. J. Mol. Biol. 1999, 286, 899–914. [Google Scholar] [CrossRef] [PubMed]
- Mukund, S.; Adams, M.W.W. Glyceraldehyde-3-phosphate ferredoxin oxidoreductase, a novel tungsten-containing enzyme with a potential glycolytic role in the hyperthermophilic archaeon Pyrococcus furiosus. J. Biol. Chem. 1995, 270, 8389–8392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, I.M.; Rubinstein, G.M.; Poole, F.L.; Lipscomb, G.L.; Schut, G.J.; Williams-Rhaesa, A.M.; Stevenson, D.M.; Amador-Noguez, D.; Kelly, R.M.; Adams, M.W.W. The thermophilic biomass-degrading bacterium Caldicellulosiruptor bescii utilizes two enzymes to oxidize glyceraldehyde 3-phosphate during glycolysis. J. Biol. Chem. 2019, 294, 9995–10005. [Google Scholar] [CrossRef]
- Bevers, L.E.; Bol, E.; Hagedoorn, P.-L.; Hagen, W.R. WOR5, a novel tungsten-containing aldehyde oxidoreductase from Pyrococcus furiosus with a broad substrate specificity. J. Bacteriol. 2005, 187, 7056–7061. [Google Scholar] [CrossRef] [Green Version]
- Kletzin, A.; Adams, M.W. Tungsten in biological systems. FEMS Microbiol. Rev. 1996, 18, 5–63. [Google Scholar] [CrossRef]
- Hagedoorn, P.L.; Chen, T.; Schröder, I.; Piersma, S.R.; De Vries, S.; Hagen, W.R. Purification and characterization of the tungsten enzyme aldehyde:ferredoxin oxidoreductase from the hyperthermophilic denitrifier Pyrobaculum aerophilum. J. Biol. Inorg. Chem. 2005, 10, 259–269. [Google Scholar] [CrossRef]
- Reher, M.; Gebhard, S.; Schönheit, P. Glyceraldehyde-3-phosphate ferredoxin oxidoreductase (GAPOR) and nonphosphorylating glyceraldehyde-3-phosphate dehydrogenase (GAPN), key enzymes of the respective modified Embden-Meyerhof pathways in the hyperthermophilic crenarchaeota Pyrobaculum aerophilum. FEMS Microbiol. Lett. 2007, 273, 196–205. [Google Scholar]
- Heider, J.; Boll, M.; Breese, K.; Breinig, S.; Ebenau-Jehle, C.; Feil, U.; Gad’on, N.; Laempe, D.; Leuthner, B.; Mohamed, M.E.S.; et al. Differential induction of enzymes involved in anaerobic metabolism of aromatic compounds in the denitrifying bacterium Thauera aromatica. Arch. Microbiol. 1998, 170, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, G.; Boll, M.; Heider, J. Microbial degradation of aromatic compounds-From one strategy to four. Nat. Rev. Microbiol. 2011, 9, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Löffler, C.; Kuntze, K.; Vazquez, J.R.; Rugor, A.; Kung, J.W.; Böttcher, A.; Boll, M. Occurrence, genes and expression of the W/Se-containing class II benzoyl-coenzyme A reductases in anaerobic bacteria. Environ. Microbiol. 2011, 13, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Boll, M.; Löffler, C.; Morris, B.E.L.; Kung, J.W. Anaerobic degradation of homocyclic aromatic compounds via arylcarboxyl-coenzyme A esters: Organisms, strategies and key enzymes. Environ. Microbiol. 2014, 16, 612–627. [Google Scholar] [CrossRef]
- Kung, J.W.; Baumann, S.; von Bergen, M.; Müller, M.; Hagedoorn, P.-L.; Hagen, W.R.; Boll, M. Reversible biological birch reduction at an extremely low redox potential. J. Am. Chem. Soc. 2010, 132, 9850–9856. [Google Scholar] [CrossRef]
- Boll, M.; Fuchs, G. Benzoyl-coenzyme A reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism. Eur. J. Biochem. 1995, 234, 921–933. [Google Scholar] [CrossRef]
- Wischgoll, S.; Heintz, D.; Peters, F.; Erxleben, A.; Sarnighausen, E.; Reski, R.; Van Dorsselaer, A.; Boll, M. Gene clusters involved in anaerobic benzoate degradation of Geobacter metallireducens. Mol. Microbiol. 2005, 58, 1238–1252. [Google Scholar] [CrossRef]
- Huwiler, S.G.; Löffler, C.; Anselmann, S.E.L.; Stärk, H.-J.; von Bergen, M.; Flechsler, J.; Rachel, R.; Boll, M. One-megadalton metalloenzyme complex in Geobacter metallireducens involved in benzene ring reduction beyond the biological redox window. Proc. Natl. Acad. Sci. USA 2019, 116, 2259–2264. [Google Scholar] [CrossRef] [Green Version]
- Anselmann, S.E.L.; Löffler, C.; Stärk, H.; Jehmlich, N.; Bergen, M.; Brüls, T.; Boll, M. The class II benzoyl-coenzyme A reductase complex from the sulfate-reducing Desulfosarcina cetonica. Environ. Microbiol. 2019, 21, 4241–4252. [Google Scholar] [CrossRef] [Green Version]
- Kung, J.W.; Löffler, C.; Dorner, K.; Heintz, D.; Gallien, S.; Van Dorsselaer, A.; Friedrich, T.; Boll, M. Identification and characterization of the tungsten-containing class of benzoyl-coenzyme A reductases. Proc. Natl. Acad. Sci. USA 2009, 106, 17687–17692. [Google Scholar] [CrossRef] [Green Version]
- Buckel, W.; Thauer, R.K. Flavin-based electron bifurcation, a new mechanism of biological energy coupling. Chem. Rev. 2018, 118, 3862–3886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, F.; Rother, M.; Boll, M. Selenocysteine-containing proteins in anaerobic benzoate metabolism of Desulfococcus multivorans. J. Bacteriol. 2004, 186, 2156–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heintz, D.; Gallien, S.; Wischgoll, S.; Ullmann, A.K.; Schaeffer, C.; Kretzschmar, A.K.; Van Dorsselaer, A.; Boll, M. Differential membrane proteome analysis reveals novel proteins involved in the degradation of aromatic compounds in Geobacter metallireducens. Mol. Cel Proteom. 2009, 8, 2159–2169. [Google Scholar] [CrossRef] [Green Version]
- Weinert, T.; Huwiler, S.G.; Kung, J.W.; Weidenweber, S.; Hellwig, P.; Stärk, H.-J.; Biskup, T.; Weber, S.; Cotelesage, J.J.H.; George, G.N.; et al. Structural basis of enzymatic benzene ring reduction. Nat. Chem. Biol. 2015, 11, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Culka, M.; Huwiler, S.G.; Boll, M.; Ullmann, G.M. Breaking benzene aromaticity-computational insights into the mechanism of the tungsten-containing benzoyl-CoA reductase. J. Am. Chem. Soc. 2017, 139, 14488–14500. [Google Scholar] [CrossRef]
- Qian, H.X.; Liao, R.Z. QM/MM Study of tungsten-dependent benzoyl-coenzyme a reductase: Rationalization of regioselectivity and predication of W vs Mo selectivity. Inorg. Chem. 2018, 57, 10667–10678. [Google Scholar] [CrossRef]
- Birch, A.J. Reduction by dissolving metals. Nature 1946, 158. [Google Scholar] [CrossRef]
- Birch, A.J. The Birch reduction in organic synthesis. Pure Appl. Chem. 1996, 68, 553–556. [Google Scholar] [CrossRef]
- Chatterjee, A.; König, B. Birch-type photoreduction of arenes and heteroarenes by sensitized electron transfer. Angew. Chemie. Int. Ed. 2019, 58, 14289–14294. [Google Scholar] [CrossRef] [Green Version]
- Peters, B.K.; Rodriguez, K.X.; Reisberg, S.H.; Beil, S.B.; Hickey, D.P.; Kawamata, Y.; Collins, M.; Starr, J.; Chen, L.; Udyavara, S.; et al. Scalable and safe synthetic organic electroreduction inspired by Li-ion battery chemistry. Science 2019, 363, 838–845. [Google Scholar] [CrossRef]
- Schink, B. Fermentation of acetylene by an obligate anaerobe, Pelobacter acetylenicus sp. nov. Arch. Microbiol. 1985, 142, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Meckenstock, R.U.; Krieger, R.; Ensign, S.; Kroneck, P.M.H.; Schink, B. Acetylene hydratase of Pelobacter acetylenicus. Molecular and spectroscopic properties of the tungsten iron-sulfur enzyme. Eur. J. Biochem. 1999, 264, 176–182. [Google Scholar] [CrossRef]
- Rosner, B.M.; Schink, B. Purification and characterization of acetylene hydratase of Pelobacter acetylenicus, a tungsten iron-sulfur protein. J. Bacteriol. 1995, 177, 5767–5772. [Google Scholar] [CrossRef] [Green Version]
- Seiffert, G.B.; Ullmann, G.M.; Messerschmidt, A.; Schink, B.; Kroneck, P.M.H.; Einsle, O. Structure of the non-redox-active tungsten/[4Fe:4S] enzyme acetylene hydratase. Proc. Natl. Acad. Sci. USA 2007, 104, 3073–3077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- tenBrink, F.; Schink, B.; Kroneck, P.M.H. Exploring the active site of the tungsten, iron-sulfur enzyme acetylene hydratase. J. Bacteriol. 2011, 193, 1229–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boll, M.; Einsle, O.; Ermler, U.; Kroneck, P.M.H.P.M.H.; Ullmann, G.M.M. Structure and function of the unusual tungsten enzymes acetylene hydratase and class ii benzoyl-coenzyme a reductase. J. Mol. Microbiol. Biotechnol. 2016, 26, 119–137. [Google Scholar] [CrossRef]
- Hyman, M.R.; Daniel, A. Acetylene inhibition of metalloenzymes. Anal. Biochem. 1988, 173, 207–220. [Google Scholar] [CrossRef]
- Stewart, W.D.; Fitzgerald, G.P.; Burris, R.H. In situ studies on N2 fixation using the acetylene reduction technique. Proc. Natl. Acad. Sci. USA 1967, 58, 2071–2078. [Google Scholar] [CrossRef] [Green Version]
- Antony, S.; Bayse, C.A. Theoretical studies of models of the active site of the tungstoenzyme acetylene hydratase. Organometallics 2009, 28, 4938–4944. [Google Scholar] [CrossRef]
- Yadav, J.; Das, S.K.; Sarkar, S. A functional mimic of the new class of tungstoenzyme, acetylene hydratase. J. Am. Chem. Soc. 1997, 119, 4315–4316. [Google Scholar] [CrossRef]
- Schreyer, M.; Hintermann, L. Is the tungsten(IV) complex (NEt4)2[WO(mnt)2] a functional analogue of acetylene hydratase? Beilstein J. Org. Chem. 2017, 13, 2332–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidovič, C.; Peschel, L.M.; Buchsteiner, M.; Belaj, F.; Mösch-Zanetti, N.C. Structural mimics of acetylene hydratase: Tungsten complexes capable of intramolecular nucleophilic attack on acetylene. Chem. Eur. J. 2019, 25, 14267–14272. [Google Scholar] [CrossRef] [PubMed]
- Templeton, J.L.; Ward, B.C.; Chen, G.J.J.; Mcdonald, J.W.; Newton, W.E. Oxotungsten(IV)-acetylene complexes: Synthesis via intermetal oxygen atom transfer and nuclear magnetic resonance studies. Inorg. Chem. 1981, 20, 1248–1253. [Google Scholar] [CrossRef]
- Schobert, H. Production of acetylene and acetylene-based chemicals from coal. Chem. Rev. 2014, 114, 1743–1760. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, T.; Schwanhold, N.; Leimkühler, S. Assembly and catalysis of molybdenum or tungsten-containing formate dehydrogenases from bacteria. Biochim. Biophys. Acta Proteins Proteom. 2015, 1854, 1090–1100. [Google Scholar] [CrossRef]
- Maia, L.B.; Moura, J.J.G.; Moura, I. Molybdenum and tungsten-dependent formate dehydrogenases. J. Biol. Inorg. Chem. 2015, 20, 287–309. [Google Scholar] [CrossRef]
- Niks, D.; Hille, R. Molybdenum- and tungsten-containing formate dehydrogenases and formylmethanofuran dehydrogenases: Structure, mechanism, and cofactor insertion. Protein Sci. 2019, 28, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Vorholt, J.A.; Thauer, R.K. Molybdenum and tungsten enzymes in C1 metabolism. Met. Ions Biol. Syst. 2002, 39, 571–619. [Google Scholar]
- Moura, J.J.G.; Brondino, C.D.; Trincão, J.; Romão, M.J. Mo and W bis-MGD enzymes: Nitrate reductases and formate dehydrogenases. J. Biol. Inorg. Chem. 2004, 9, 791–799. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, T.; Schrapers, P.; Utesch, T.; Nimtz, M.; Rippers, Y.; Dau, H.; Mroginski, M.A.; Haumann, M.; Leimkühler, S. The Molybdenum Active Site of Formate Dehydrogenase Is Capable of Catalyzing C-H Bond Cleavage and Oxygen Atom Transfer Reactions. Biochemistry 2016, 55, 2381–2389. [Google Scholar]
- Niks, D.; Duvvuru, J.; Escalona, M.; Hille, R. Spectroscopic and kinetic properties of the molybdenum-containing, NAD+-dependent formate dehydrogenase from Ralstonia eutropha. J. Biol. Chem. 2016, 291, 1162–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maia, L.B.; Fonseca, L.; Moura, I.; Moura, J.J.G. Reduction of carbon dioxide by a molybdenum-containing formate dehydrogenase: A kinetic and mechanistic study. J. Am. Chem. Soc. 2016, 138, 8834–8846. [Google Scholar] [CrossRef]
- Enoch, H.G.; Lester, R.L. Effects of molybdate, tungstate, and selenium compounds on formate dehydrogenase and other enzyme systems in Escherichia coli. J. Bacteriol. 1972, 110, 1032–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, H.D.; Patel, P.S.; Ferry, J.G. Effect of molybdenum and tungsten on synthesis and composition of formate dehydrogenase in Methanobacterium formicicum. J. Bacteriol. 1988, 170, 3384–3389. [Google Scholar] [CrossRef] [Green Version]
- Ljungdahl, L.G.; Andreesen, J.R. Formate Dehydrogenase, a selenium-tungsten enzyme from Clostridium thermoaceticum. Methods Enzymol. 1978, 53, 360–372. [Google Scholar]
- Strobl, G.; Feicht, R.; White, H.; Lottspeich, F.; Simon, H. The tungsten-containing aldehyde oxidoreductase from Clostridium thermoaceticum and its complex with a viologen-accepting NADPH oxidoreductase. Biol. Chem. Hoppe. Seyler 1992, 373, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Alissandratos, A.; Kim, H.K.; Matthews, H.; Hennessy, J.E.; Philbrook, A.; Easton, C.J. Clostridium carboxidivorans strain P7T recombinant formate dehydrogenase catalyzes reduction of CO2 to formate. Appl. Environ. Microbiol. 2013, 79, 741–744. [Google Scholar] [CrossRef] [Green Version]
- Graentzdoerffer, A.; Rauh, D.; Pich, A.; Andreesen, J.R. Molecular and biochemical characterization of two tungsten-and selenium-containing formate dehydrogenases from Eubacterium acidaminophilum that are associated with components of an iron-only hydrogenase. Arch. Microbiol. 2003, 179, 116–130. [Google Scholar] [CrossRef]
- Laukel, M.; Chistoserdova, L.; Lidstrom, M.E.; Vorholt, J.A. The tungsten-containing formate dehydrogenase from Methylobacterium extorquens AM1: Purification and properties. Eur. J. Biochem. 2003, 270, 325–333. [Google Scholar] [CrossRef] [Green Version]
- De Bok, F.A.M.; Hagedoorn, P.L.; Silva, P.J.; Hagen, W.R.; Schiltz, E.; Fritsche, K.; Stams, A.J.M. Two W-containing formate dehydrogenases (CO2-reductases) involved in syntrophic propionate oxidation by Syntrophobacter fumaroxidans. Eur. J. Biochem. 2003, 270, 2476–2485. [Google Scholar] [CrossRef]
- Almendra, M.J.; Brondino, C.D.; Gavel, O.; Pereira, A.S.; Tavares, P.; Bursakov, S.; Duarte, R.; Caldeira, J.; Moura, J.J.G.; Moura, I. Purification and characterization of a tungsten-containing formate dehydrogenase from Desulfovibrio gigas. Biochemistry 1999, 38, 16366–16372. [Google Scholar] [CrossRef] [PubMed]
- Brondino, C.D.; Passeggi, M.C.G.; Caldeira, J.; Almendra, M.J.; Feio, M.J.; Moura, J.J.G.; Moura, I. Incorporation of either molybdenum or tungsten into formate dehydrogenase from Desulfovibrio alaskensis NCIMB 13491; EPR assignment of the proximal iron-sulfur cluster to the pterin cofactor in formate dehydrogenases from sulfate-reducing bacteria. J. Biol. Inorg. Chem. 2004, 9, 145–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, S.M.; Pimentel, C.; Valente, F.M.A.; Rodrigues-Pousada, C.; Pereira, I.A.C. Tungsten and molybdenum regulation of formate dehydrogenase expression in Desulfovibrio vulgaris Hildenborough. J. Bacteriol. 2011, 193, 2909–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mota, C.S.; Valette, O.; González, P.J.; Brondino, C.D.; Moura, J.J.G.; Moura, I.; Dolla, A.; Rivas, M.G. Effects of molybdate and tungstate on expression levels and biochemical characteristics of formate dehydrogenases produced by Desulfovibrio alaskensis NCIMB 13491. J. Bacteriol. 2011, 193, 2917–2923. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.J.; Kelly, D.J. The function, biogenesis and regulation of the electron transport chains in Campylobacter jejuni: New insights into the bioenergetics of a major food-borne pathogen. Adv. Microb. Physiol. 2019, 74, 239–329. [Google Scholar]
- Reda, T.; Plugge, C.M.; Abram, N.J.; Hirst, J. Reversible interconversion of carbon dioxide and formate by an electroactive enzyme. Proc. Natl. Acad. Sci. USA 2008, 105, 10654–10658. [Google Scholar] [CrossRef] [Green Version]
- Schuchmann, K.; Müller, V. Direct and reversible hydrogenation of CO2 to formate by a bacterial carbon dioxide reductase. Science 2013, 342, 1382–1385. [Google Scholar] [CrossRef]
- Sokol, K.P.; Robinson, W.E.; Oliveira, A.R.; Zacarias, S.; Lee, C.Y.; Madden, C.; Bassegoda, A.; Hirst, J.; Pereira, I.A.C.; Reisner, E. Reversible and selective interconversion of hydrogen and carbon dioxide into formate by a semiartificial formate hydrogenlyase mimic. J. Am. Chem. Soc. 2019, 141, 17498–17502. [Google Scholar] [CrossRef] [Green Version]
- Sokol, K.P.; Robinson, W.E.; Oliveira, A.R.; Warnan, J.; Nowaczyk, M.M.; Ruff, A.; Pereira, I.A.C.; Reisner, E. Photoreduction of CO2 with a formate dehydrogenase driven by photosystem ii using a semi-artificial z-scheme architecture. J. Am. Chem. Soc. 2018, 140, 16418–16422. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.; Robinson, W.E.; Oliveira, A.R.; Heidary, N.; Kornienko, N.; Warnan, J.; Pereira, I.A.C.; Reisner, E. Interfacing formate dehydrogenase with metal oxides for the reversible electrocatalysis and solar-driven reduction of carbon dioxide. Angew. Chemie. Int. Ed. 2019, 58, 4601–4605. [Google Scholar] [CrossRef] [Green Version]
- Hochheimer, A.; Hedderich, R.; Thauer, R.K. The formylmethanofuran dehydrogenase isoenzymes in Methanobacterium wolfei and Methanobacterium thermoautotrophicum: Induction of the molybdenum isoenzyme by molybdate and constitutive synthesis of the tungsten isoenzyme. Arch. Microbiol. 1998, 170, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Bertram, P.A.; Schmitz, R.A.; Linder, D.; Thauer, R.K. Tungstate can substitute for molybdate in sustaining growth of Methanobacterium thermoautotrophicum—Identification and characterization of a tungsten isoenzyme of formylmethanofuran dehydrogenase. Arch. Microbiol. 1994, 161, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Matschiavelli, N.; Rother, M. Role of a putative tungsten-dependent formylmethanofuran dehydrogenase in Methanosarcina acetivorans. Arch. Microbiol. 2015, 197, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.; Ermler, U.; Shima, S. The methanogenic CO2 reducing-and-fixing enzyme is bifunctional and contains 46 [4Fe-4S] clusters. Science 2016, 354, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Gates, A.J.; Hughes, R.O.; Sharp, S.R.; Millington, P.D.; Nilavongse, A.; Cole, J.A.; Leach, E.R.; Jepson, B.; Richardson, D.J.; Butler, C.S. Properties of the periplasmic nitrate reductases from Paracoccus pantotrophus and Escherichia coli after growth in tungsten-supplemented media. FEMS Microbiol. Lett. 2003, 220, 261–269. [Google Scholar] [CrossRef] [Green Version]
- De Vries, S.; Momcilovic, M.; Strampraad, M.J.F.; Whitelegge, J.P.; Baghai, A.; Schröder, I. Adaptation to a high-tungsten environment: Pyrobaculum aerophilum contains an active tungsten nitrate reductase. Biochemistry 2010, 49, 9911–9921. [Google Scholar]
- Stewart, L.J.; Bailey, S.; Bennett, B.; Charnock, J.M.; Garner, C.D.; McAlpine, A.S. Dimethylsulfoxide reductase: An enzyme capable of catalysis with either molybdenum or tungsten at the active site. J. Mol. Biol. 2000, 299, 593–600. [Google Scholar] [CrossRef]
- Johnson, M.K.; Rees, D.C.; Adams, M.W.W. Tungstoenzymes. Chem. Rev. 1996, 96, 2817–2839. [Google Scholar] [CrossRef]
- Buc, J.; Santini, C.-L.; Giordani, R.; Czjzek, M.; Wu, L.-F.; Giordano, G. Enzymatic and physiological properties of the tungsten-substituted molybdenum TMAO reductase from Escherichia coli. Mol. Microbiol. 1999, 32, 159–168. [Google Scholar] [CrossRef]
- Haja, D.K.; Wu, C.H.; Poole, F.L.; Sugar, J.; Williams, S.G.; Jones, A.K.; Adams, M.W.W. Characterization of thiosulfate reductase from Pyrobaculum aerophilum heterologously produced in Pyrococcus furiosus. Extremophiles 2020, 24, 53–62. [Google Scholar] [CrossRef]
- Aguilar-Barajas, E.; Díaz-Pérez, C.; Ramírez-Díaz, M.I.; Riveros-Rosas, H.; Cervantes, C. Bacterial transport of sulfate, molybdate, and related oxyanions. BioMetals 2011, 24, 687–707. [Google Scholar] [CrossRef]
- Bevers, L.E.; Hagedoorn, P.L.; Krijger, G.C.; Hagen, W.R. Tungsten transport protein A (WtpA) in Pyrococcus furiosus: The first member of a new class of tungstate and molybdate transporters. J. Bacteriol. 2006, 188, 6498–6505. [Google Scholar] [CrossRef] [Green Version]
- Makdessi, K.; Fritsche, K.; Pich, A.; Andreesen, J.R. Identification and characterization of the cytoplasmic tungstate/molybdate-binding protein (Mop) from Eubacterium acidaminophilum. Arch. Microbiol. 2004, 181, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Makdessi, K.; Andreesen, J.R.; Pich, A. Tungstate uptake by a highly specific ABC transporter in Eubacterium acidaminophilum. J. Biol. Chem. 2001, 276, 24557–24564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smart, J.P.; Cliff, M.J.; Kelly, D.J. A role for tungsten in the biology of Campylobacter jejuni: Tungstate stimulates formate dehydrogenase activity and is transported via an ultra-high affinity ABC system distinct from the molybdate transporter. Mol. Microbiol. 2009, 74, 742–757. [Google Scholar] [CrossRef] [PubMed]
- Taveirne, M.E.; Sikes, M.L.; Olson, J.W. Molybdenum and tungsten in Campylobacter jejuni: Their physiological role and identification of separate transporters regulated by a single ModE-like protein. Mol. Microbiol. 2009, 74, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Otrelo-Cardoso, A.R.; Nair, R.R.; Correia, M.A.S.; Rivas, M.G.; Santos-Silva, T. TupA: A tungstate binding protein in the periplasm of Desulfovibrio alaskensis G20. Int. J. Mol. Sci. 2014, 15, 11783–11798. [Google Scholar] [CrossRef] [Green Version]
- Otrelo-Cardoso, A.R.; Nair, R.R.; Correia, M.A.S.; Cordeiro, R.S.C.; Panjkovich, A.; Svergun, D.I.; Santos-Silva, T.; Rivas, M.G. Highly selective tungstate transporter protein TupA from Desulfovibrio alaskensis G20. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Gladyshev, V.N. Molybdoproteomes and evolution of molybdenum utilization. J. Mol. Biol. 2008, 379, 881–899. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, K.V. Biosynthesis and processing of the molybdenum cofactors. Biochem. Soc. Trans. 1997, 25, 757–761. [Google Scholar] [CrossRef] [Green Version]
- Leimkühler, S. Shared function and moonlighting proteins in molybdenum cofactor biosynthesis. Biol. Chem. 2017, 398, 1009–1026. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, G.; Mendel, R.R.; Ribbe, M.W. Molybdenum cofactors, enzymes and pathways. Nature 2009, 460, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Bevers, L.E.; Hagedoorn, P.L.; Santamaria-Araujo, J.A.; Magalon, A.; Hagen, W.R.; Schwarz, G. Function of MoaB proteins in the biosynthesis of the molybdenum and tungsten cofactors. Biochemistry 2008, 47, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Wuebbens, M.M.; Rajagopalan, K.V. Structural characterization of a molybdopterin precursor. J. Biol. Chem. 1993, 268, 13493–13498. [Google Scholar] [PubMed]
- Hänzelmann, P.; Schindelin, H. Binding of 5′-GTP to the C-terminal FeS cluster of the radical S-adenosylmethionine enzyme MoaA provides insights into its mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 6829–6834. [Google Scholar] [CrossRef] [Green Version]
- Gutzke, G.; Fischer, B.; Mendel, R.R.; Schwarz, G. Thiocarboxylation of molybdopterin synthase provides evidence for the mechanism of dithiolene formation in metal-binding pterins. J. Biol. Chem. 2001, 276, 36268–36274. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, M.J.; Wuebbens, M.M.; Rajagopalan, K.V.; Schindelin, H. Crystal structure of molybdopterin synthase and its evolutionary relationship to ubiquitin activation. Nat. Struct. Biol. 2001, 8, 42–46. [Google Scholar] [CrossRef]
- Wuebbens, M.M.; Rajagopalan, K.V. Mechanistic and mutational studies of Escherichia coli molybdopterin synthase clarify the final step of molybdopterin biosynthesis. J. Biol. Chem. 2003, 278, 14523–14532. [Google Scholar] [CrossRef] [Green Version]
- Pitterle, D.M.; Johnson, J.L.; Rajagopalan, K.V. In vitro synthesis of molybdopterin from precursor Z using purified converting factor. Role of protein-bound sulfur in formation of the dithiolene. J. Biol. Chem. 1993, 268, 13506–13509. [Google Scholar]
- Lake, M.W.; Wuebbens, M.M.; Rajagopalan, K.V.; Schindelin, H. Mechanism of ubiquitin activation revealed by the structure of a bacterial MoeB-MoaD complex. Nature 2001, 414, 325–329. [Google Scholar] [CrossRef]
- Leimkühler, S.; Rajagopalan, K.V. A Sulfurtransferase is required in the transfer of cysteine sulfur in the in vitro synthesis of molybdopterin from precursor z in Escherichia coli. J. Biol. Chem. 2001, 276, 22024–22031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llamas, A.; Mendel, R.R.; Schwarz, G. Synthesis of adenylated molybdopterin: An essential step for molybdenum insertion. J. Biol. Chem. 2004, 279, 55241–55246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, M.S.; Johnson, J.L.; Rajagopalan, K. V Molybdenum cofactor biosynthesis in Escherichia coli mod and mog mutants. J. Bacteriol. 1996, 178, 4310–4312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, O.; Rajagopalan, K.V. Molybdopterin in carbon monoxide oxidase from carboxydotrophic bacteria. J. Bacteriol. 1984, 157, 643–648. [Google Scholar] [CrossRef] [Green Version]
- Reschke, S.; Duffus, B.R.; Schrapers, P.; Mebs, S.; Teutloff, C.; Dau, H.; Haumann, M.; Leimkühler, S. Identification of YdhV as the first molybdoenzyme binding a Bis-Mo-MPT cofactor in Escherichia coli. Biochemistry 2019, 58, 2228–2242. [Google Scholar]
- Johnson, J.L.; Bastian, N.R.; Rajagopalan, K. V Molybdopterin guanine dinucleotide: A modified form of molybdopterin identified in the molybdenum cofactor of dimethyl sulfoxide reductase from Rhodobacter sphaeroides forma specialis denitrificans. Proc. Natl. Acad. Sci. USA 1990, 87, 3190–3194. [Google Scholar] [CrossRef] [Green Version]
- Reschke, S.; Sigfridsson, K.G.V.; Kaufmann, P.; Leidel, N.; Horn, S.; Gast, K.; Schulzke, C.; Haumann, M.; Leimkühler, S. Identification of a bis-molybdopterin intermediate in molybdenum cofactor biosynthesis in Escherichia coli. J. Biol. Chem. 2013, 288, 29736–29745. [Google Scholar] [CrossRef] [Green Version]
- Johannes, J.; Bluschke, A.; Jehmlich, N.; Von Bergen, M.; Boll, M. Purification and characterization of active-site components of the putative p-cresol methylhydroxylase membrane complex from Geobacter metallireducens. J. Bacteriol. 2008, 190, 6493–6500. [Google Scholar] [CrossRef] [Green Version]
- Peters, F.; Heintz, D.; Johannes, J.; Van Dorsselaer, A.; Boll, M. Genes, enzymes, and regulation of para-cresol metabolism in Geobacter metallireducens. J. Bacteriol. 2007, 189, 4729–4738. [Google Scholar] [CrossRef] [Green Version]
- Neumann, M.; Stöcklein, W.; Leimkühler, S. Transfer of the molybdenum cofactor synthesized by Rhodobacter capsulatus MoeA to XdhC and MobA. J. Biol. Chem. 2007, 282, 28493–28500. [Google Scholar] [CrossRef] [Green Version]
- Thomé, R.; Gust, A.; Toci, R.; Mendel, R.; Bittner, F.; Magalon, A.; Walburger, A. A sulfurtransferase is essential for activity of formate dehydrogenases in Escherichia coli. J. Biol. Chem. 2012, 287, 4671–4678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnoux, P.; Ruppelt, C.; Oudouhou, F.; Lavergne, J.; Siponen, M.I.; Toci, R.; Mendel, R.R.; Bittner, F.; Pignol, D.; Magalon, A.; et al. Sulphur shuttling across a chaperone during molybdenum cofactor maturation. Nat. Commun. 2015, 6, 6148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwanhold, N.; Iobbi-Nivol, C.; Lehmann, A.; Leimkühler, S. Same but different: Comparison of two system-specific molecular chaperones for the maturation of formate dehydrogenases. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed]
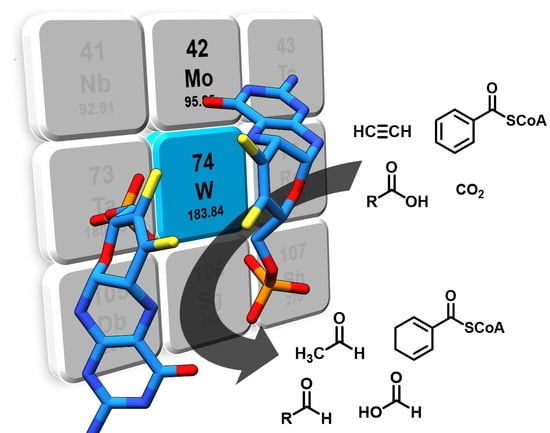
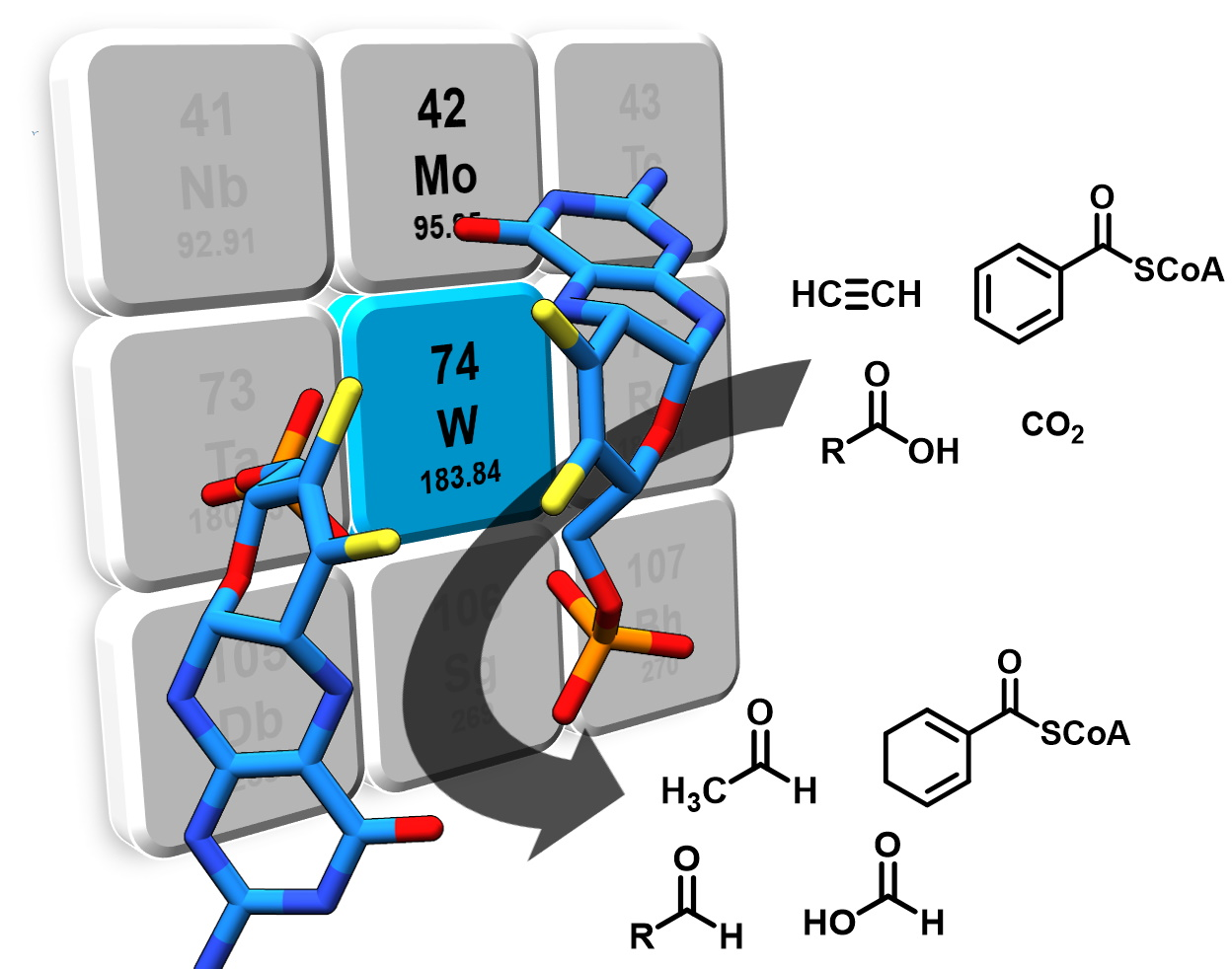
Figure 1.
Tungsten cofactor (Wco) found in members of the dimethyl sulfoxide reductase (DMSOR) and aldehyde oxidoreductase (AOR) enzyme families. The former contains either Mo-bis-MPT guanine dinucleotide (MGD) or W-bis-MGD, the latter in most cases use W-bis-metallopterin (MPT) as the active site cofactor.
Figure 1.
Tungsten cofactor (Wco) found in members of the dimethyl sulfoxide reductase (DMSOR) and aldehyde oxidoreductase (AOR) enzyme families. The former contains either Mo-bis-MPT guanine dinucleotide (MGD) or W-bis-MGD, the latter in most cases use W-bis-metallopterin (MPT) as the active site cofactor.

Figure 2.
Reactions catalyzed by obligately and facultatively tungsten-dependent enzymes. Abbreviations: AOR: aldehyde oxidoreductase; BCR: class II benzoyl-CoA reductase; AH: acetylene hydratase; FDH: formate dehydrogenase; FWD: tungsten-containing formylmethanofuran dehydrogenase; TSR: thiosulfate reductase; DMSOR: dimethyl sulfoxide reductase; NAR: membrane-bound nitrate reductase; NAP: periplasmic nitrate reductase.
Figure 2.
Reactions catalyzed by obligately and facultatively tungsten-dependent enzymes. Abbreviations: AOR: aldehyde oxidoreductase; BCR: class II benzoyl-CoA reductase; AH: acetylene hydratase; FDH: formate dehydrogenase; FWD: tungsten-containing formylmethanofuran dehydrogenase; TSR: thiosulfate reductase; DMSOR: dimethyl sulfoxide reductase; NAR: membrane-bound nitrate reductase; NAP: periplasmic nitrate reductase.

Figure 3.
Phylogenetic trees of the large subunits of selected enzymes of the AOR family (A) and the DMSOR family (B). Clades or subclades containing tungsten enzymes are labeled in light-orange (bacteria), dark-orange (archaea) or violet (enzymes working with either tungsten or molybdenum, for better visualization additionally marked with a violet dot), and those containing molybdenum enzymes are labeled in light- or dark-blue (bacterial and archaeal enzymes, respectively). Enzyme clades of unknown metal content are not colored. Members of the DMSOR subfamilies 1–3 are highlighted by the outer circle as indicated. Species epithets for unaffiliated AORs: Pa: Pyrobaculum aerophilum; Dg: Desulfovibrio gigas; Cf: Clostridium formicoaceticum. Abbreviations: AOR, FOR, GAPOR, WOR5: archaeal aldehyde oxidoreductases of various specificities (for details see text); BamB: active site subunit of class II benzoyl-CoA reductases; WOR4, YdhV, AOR1: archaeal or bacterial Mo- or W-containing enzymes of unknown function; GOR: glyceraldehyde-3-phosphate oxidoreductases from thermophilic bacteria; FDH: formate dehydrogenases; NAP/NAS: periplasmic and assimilatory nitrate reductases; FMD/FWD: molybdenum- and tungsten-dependent formylmethanofuran dehydrogenases; AH-like: acetylene hydratases and similar hypothetical enzymes; TSR/PSR: thiosulfate/polysulfide reductases; PAD: phenylacetyl-CoA dehydrogenases; ARR, ARS: arsenate reductases/arsenite oxidases; DMSOR: dimethyl sulfoxide reductases; TMAOR: trimethylaminoxide reductases; NAR/anammox: chemolithotrophic nitrite oxidoreductases and putative nitrate reductases from anammox bacteria; NAR-6: nitrate reductases and similar clades; CLR/SER: chlorate/selenate reductases and dimethyl sulfide dehydrogenases; EBDH: ethylbenzene dehydrogenases and related enzymes.
Figure 3.
Phylogenetic trees of the large subunits of selected enzymes of the AOR family (A) and the DMSOR family (B). Clades or subclades containing tungsten enzymes are labeled in light-orange (bacteria), dark-orange (archaea) or violet (enzymes working with either tungsten or molybdenum, for better visualization additionally marked with a violet dot), and those containing molybdenum enzymes are labeled in light- or dark-blue (bacterial and archaeal enzymes, respectively). Enzyme clades of unknown metal content are not colored. Members of the DMSOR subfamilies 1–3 are highlighted by the outer circle as indicated. Species epithets for unaffiliated AORs: Pa: Pyrobaculum aerophilum; Dg: Desulfovibrio gigas; Cf: Clostridium formicoaceticum. Abbreviations: AOR, FOR, GAPOR, WOR5: archaeal aldehyde oxidoreductases of various specificities (for details see text); BamB: active site subunit of class II benzoyl-CoA reductases; WOR4, YdhV, AOR1: archaeal or bacterial Mo- or W-containing enzymes of unknown function; GOR: glyceraldehyde-3-phosphate oxidoreductases from thermophilic bacteria; FDH: formate dehydrogenases; NAP/NAS: periplasmic and assimilatory nitrate reductases; FMD/FWD: molybdenum- and tungsten-dependent formylmethanofuran dehydrogenases; AH-like: acetylene hydratases and similar hypothetical enzymes; TSR/PSR: thiosulfate/polysulfide reductases; PAD: phenylacetyl-CoA dehydrogenases; ARR, ARS: arsenate reductases/arsenite oxidases; DMSOR: dimethyl sulfoxide reductases; TMAOR: trimethylaminoxide reductases; NAR/anammox: chemolithotrophic nitrite oxidoreductases and putative nitrate reductases from anammox bacteria; NAR-6: nitrate reductases and similar clades; CLR/SER: chlorate/selenate reductases and dimethyl sulfide dehydrogenases; EBDH: ethylbenzene dehydrogenases and related enzymes.

Figure 4.
Structure of formaldehyde:ferredoxin oxidoreductase from P. furiosus, in complex with glutarate (pdb 1B4N) at 2.4 Å solution. Left panel: overall structure; right panel W-bis-MPT and [4Fe-4S] cofactor. The tungsten atom is depicted in blue, Mg2+ is shown in light green, and Ca2+ in dark green.
Figure 4.
Structure of formaldehyde:ferredoxin oxidoreductase from P. furiosus, in complex with glutarate (pdb 1B4N) at 2.4 Å solution. Left panel: overall structure; right panel W-bis-MPT and [4Fe-4S] cofactor. The tungsten atom is depicted in blue, Mg2+ is shown in light green, and Ca2+ in dark green.

Figure 5.
Scheme of the class II BCR-complex from G. metallireducens. The endergonic electron transfer from reduced ferredoxin to benzoyl-CoA may be driven by an exergonic reduction of NAD+ and/or menaquinone (MQ).
Figure 5.
Scheme of the class II BCR-complex from G. metallireducens. The endergonic electron transfer from reduced ferredoxin to benzoyl-CoA may be driven by an exergonic reduction of NAD+ and/or menaquinone (MQ).

Figure 6.
Structure of the BamBC components of class II benzoyl-CoA reductase from G. metallireducens (pdb 4Z3W-Z). Left panel: overall structure; the catalytic BamB subunit depicted in red contains the W-bis-MPT cofactor, BamC (light green) contains 3 [4Fe-4S] clusters. Right panel: W-bis-MPT, [4Fe-4S] cluster and the substrate benzoyl-CoA. Tungsten (blue) is coordinated by five thiols (four from the dithiolenes and one from Cys322), and by an inorganic ligand (magenta). A Mg2+ bridging the phosphate groups of the two MPTs is shown in green.
Figure 6.
Structure of the BamBC components of class II benzoyl-CoA reductase from G. metallireducens (pdb 4Z3W-Z). Left panel: overall structure; the catalytic BamB subunit depicted in red contains the W-bis-MPT cofactor, BamC (light green) contains 3 [4Fe-4S] clusters. Right panel: W-bis-MPT, [4Fe-4S] cluster and the substrate benzoyl-CoA. Tungsten (blue) is coordinated by five thiols (four from the dithiolenes and one from Cys322), and by an inorganic ligand (magenta). A Mg2+ bridging the phosphate groups of the two MPTs is shown in green.
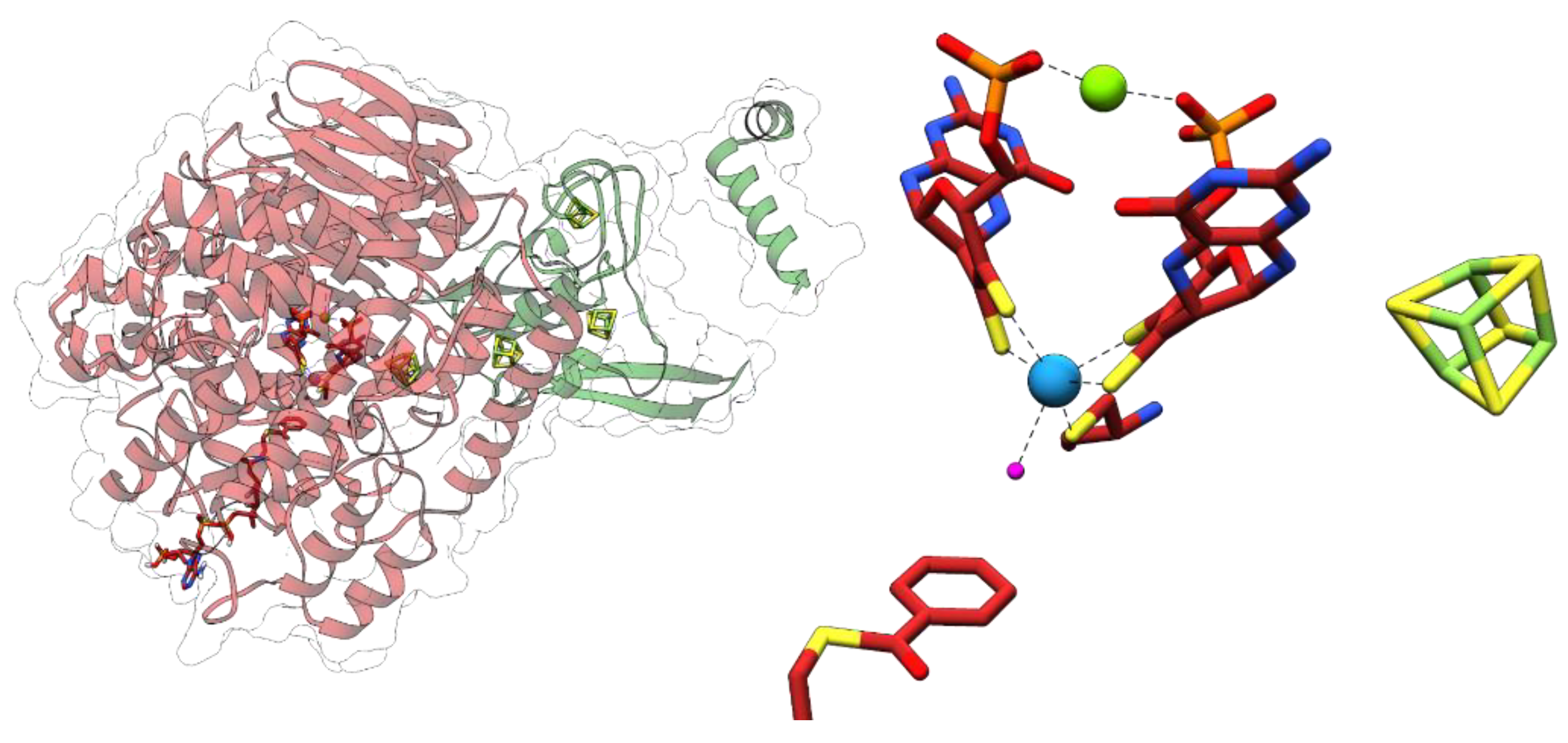
Figure 7.
Structure of acetylene hydratase from P. acetylenicus (pdb 2E7Z) at 1.26 Å resolution. Left panel: overall structure. Right panel: W-bis-MGD and [4Fe-4S] cofactors. Tungsten is coordinated by five thiols (four from the dithiolenes and one from Cys141), and by a water molecule. The tungsten atom is depicted in blue.
Figure 7.
Structure of acetylene hydratase from P. acetylenicus (pdb 2E7Z) at 1.26 Å resolution. Left panel: overall structure. Right panel: W-bis-MGD and [4Fe-4S] cofactors. Tungsten is coordinated by five thiols (four from the dithiolenes and one from Cys141), and by a water molecule. The tungsten atom is depicted in blue.

Figure 8.
Moco biosynthesis in E. coli. In prokaryotes, the steps from GTP to MPT-adenosine monophosphate (AMP) are catalyzed by similar enzymes for Moco and Wco synthesis. YdhV from E. coli is the only characterized molybdenum-containing AOR family member [126].
Figure 8.
Moco biosynthesis in E. coli. In prokaryotes, the steps from GTP to MPT-adenosine monophosphate (AMP) are catalyzed by similar enzymes for Moco and Wco synthesis. YdhV from E. coli is the only characterized molybdenum-containing AOR family member [126].
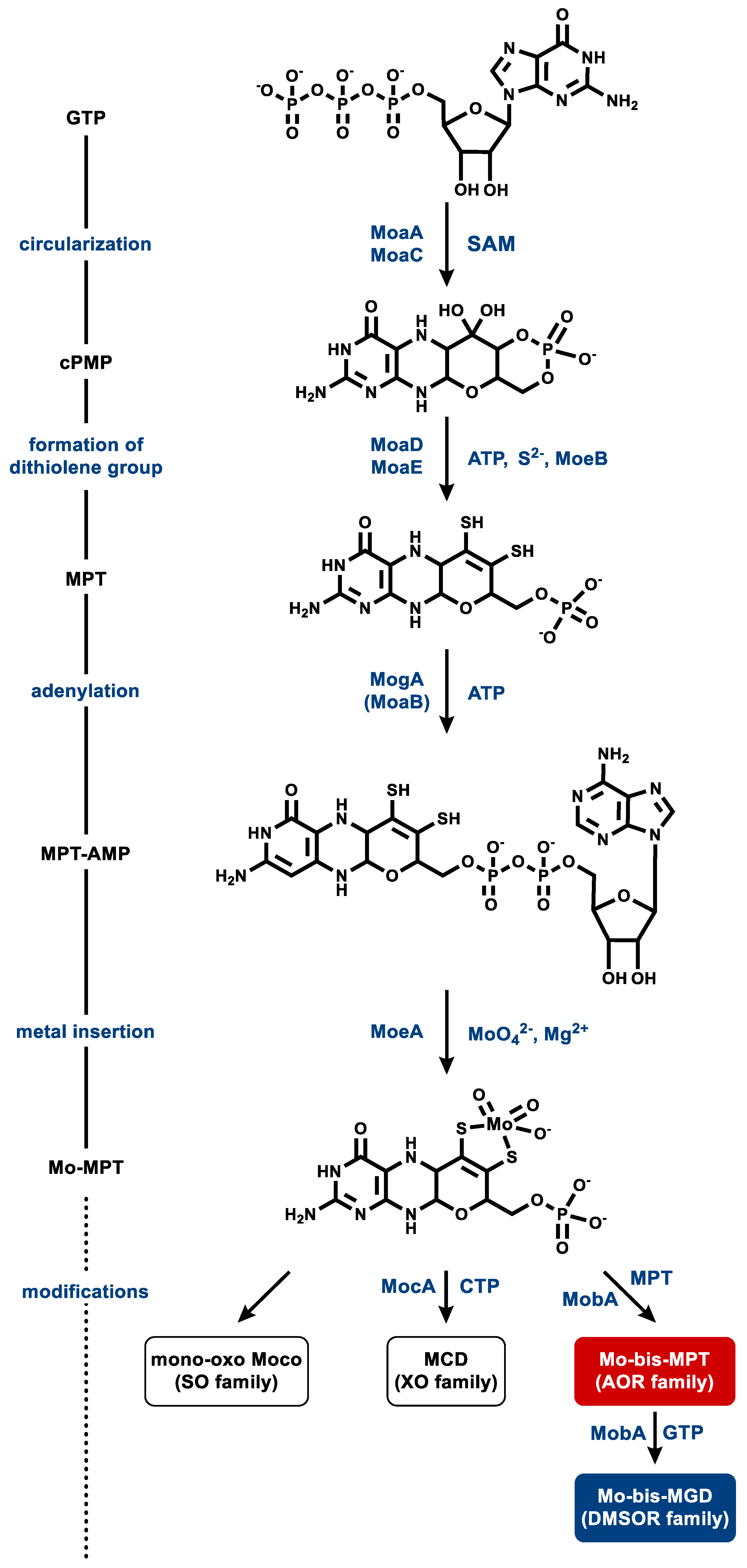
Figure 9.
Genomic location of moeA paralogues in bacteria that produce Wco- and Moco-containing enzymes simultaneously.
Figure 9.
Genomic location of moeA paralogues in bacteria that produce Wco- and Moco-containing enzymes simultaneously.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Seelmann, C.S.; Willistein, M.; Heider, J.; Boll, M. Tungstoenzymes: Occurrence, Catalytic Diversity and Cofactor Synthesis. Inorganics 2020, 8, 44. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8080044
AMA Style
Seelmann CS, Willistein M, Heider J, Boll M. Tungstoenzymes: Occurrence, Catalytic Diversity and Cofactor Synthesis. Inorganics. 2020; 8(8):44. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8080044
Chicago/Turabian StyleSeelmann, Carola S., Max Willistein, Johann Heider, and Matthias Boll. 2020. "Tungstoenzymes: Occurrence, Catalytic Diversity and Cofactor Synthesis" Inorganics 8, no. 8: 44. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics8080044
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.