
The Photochemistry of Fe2(S2C3H6)(CO)6(µ-CO) and Its Oxidized Form, Two Simple [FeFe]-Hydrogenase CO-Inhibited Models. A DFT and TDDFT Investigation

 and
and
Abstract
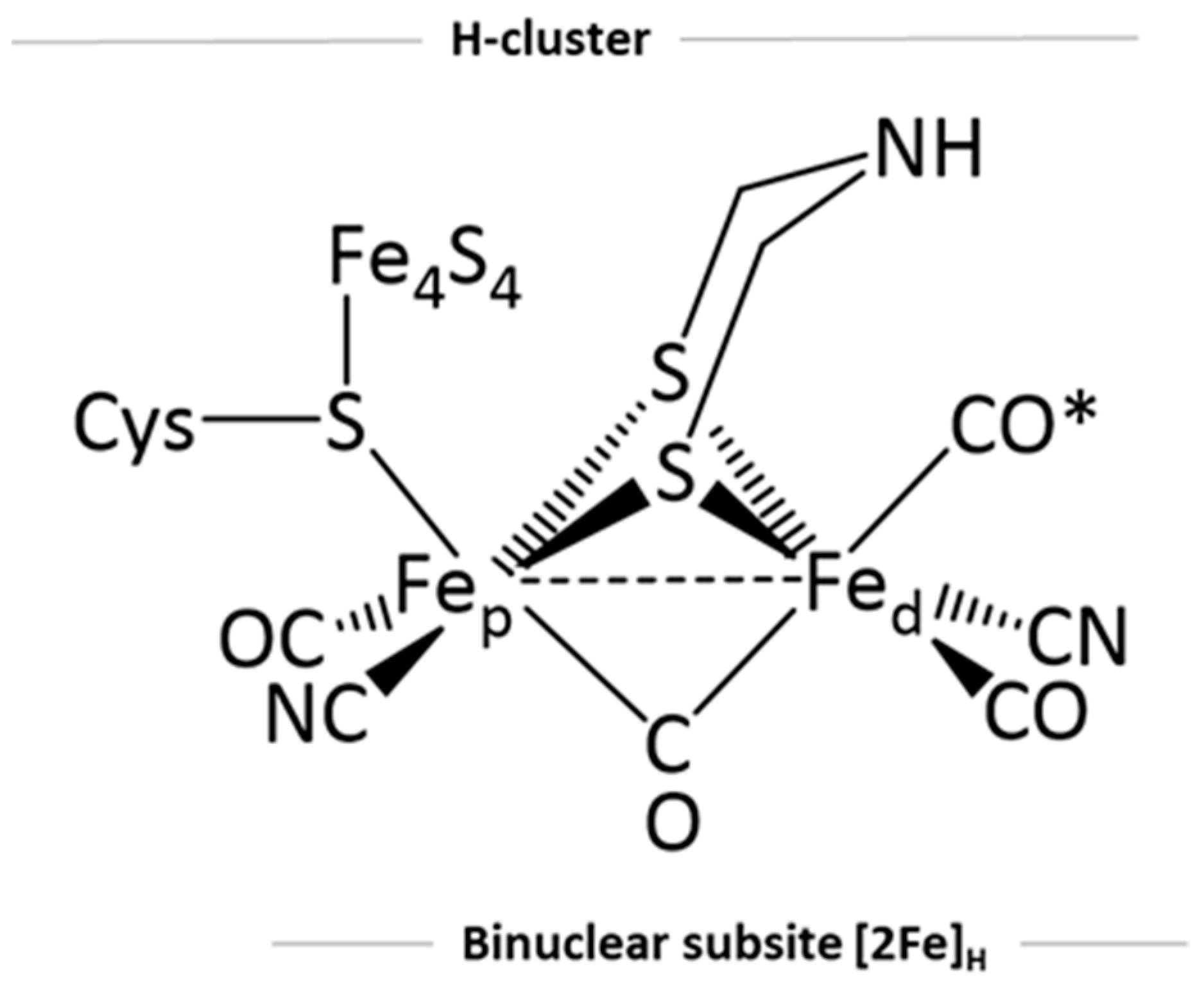
:1. Introduction
2. Results and Discussion
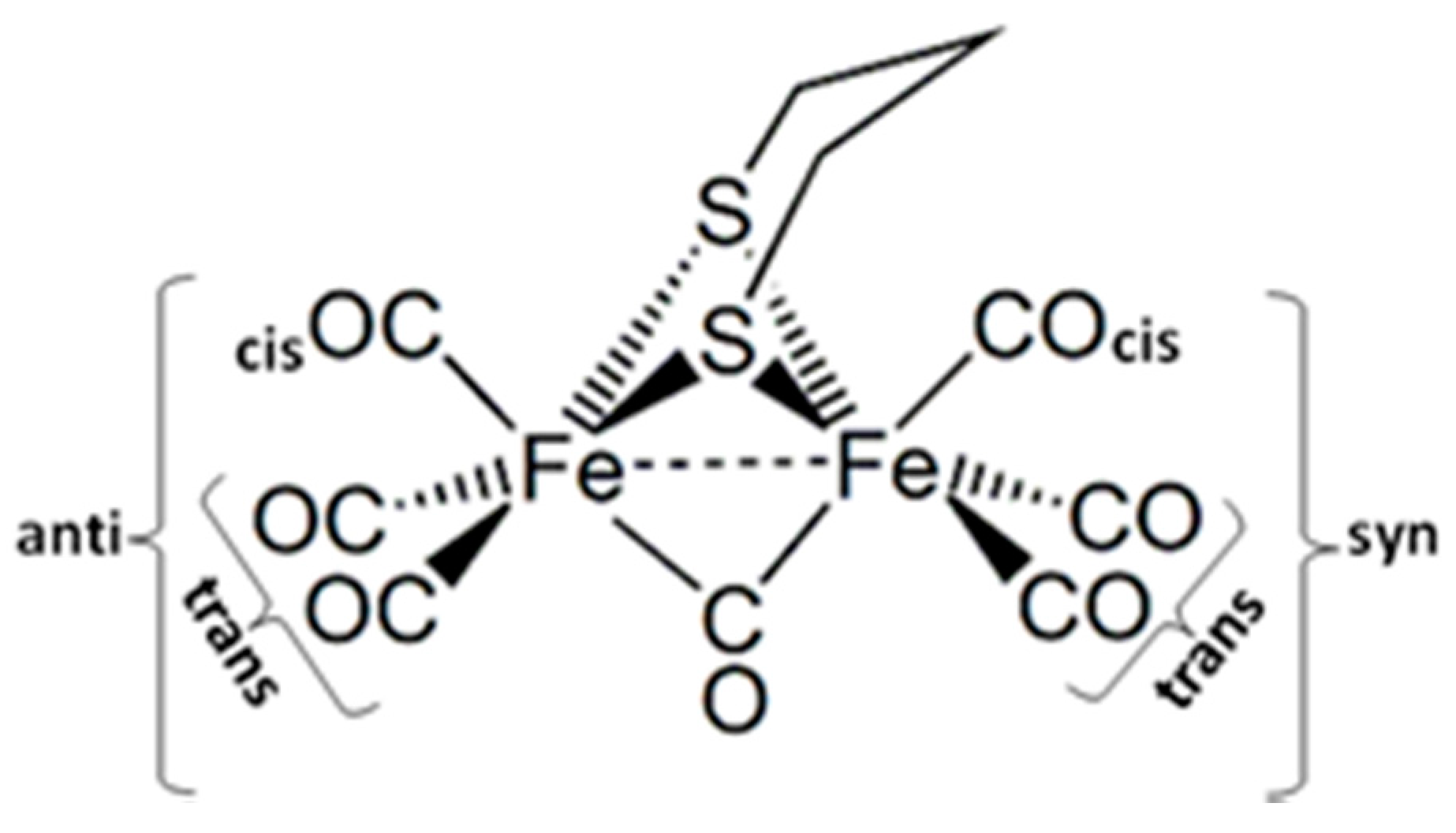
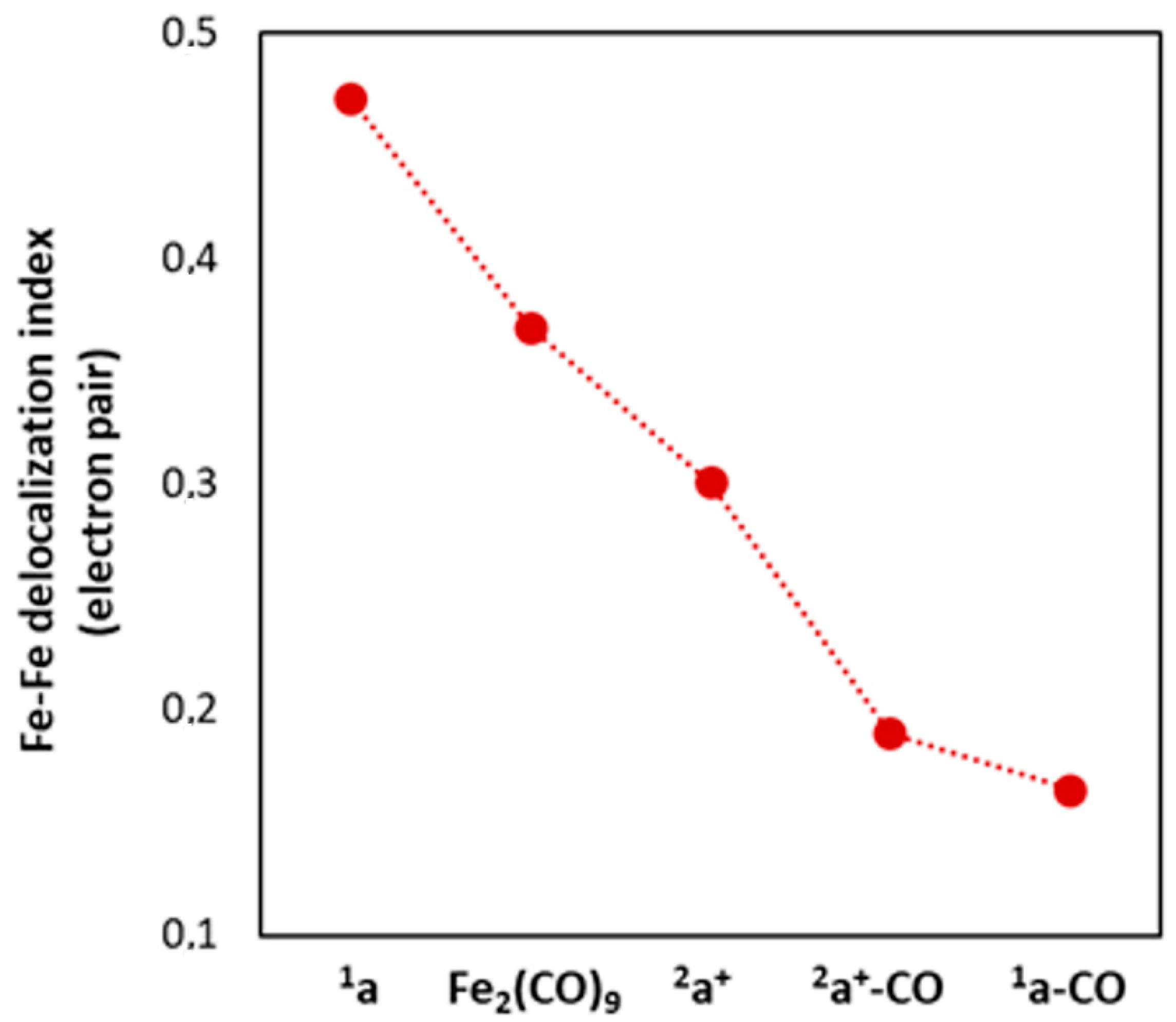
2.1. Ground States
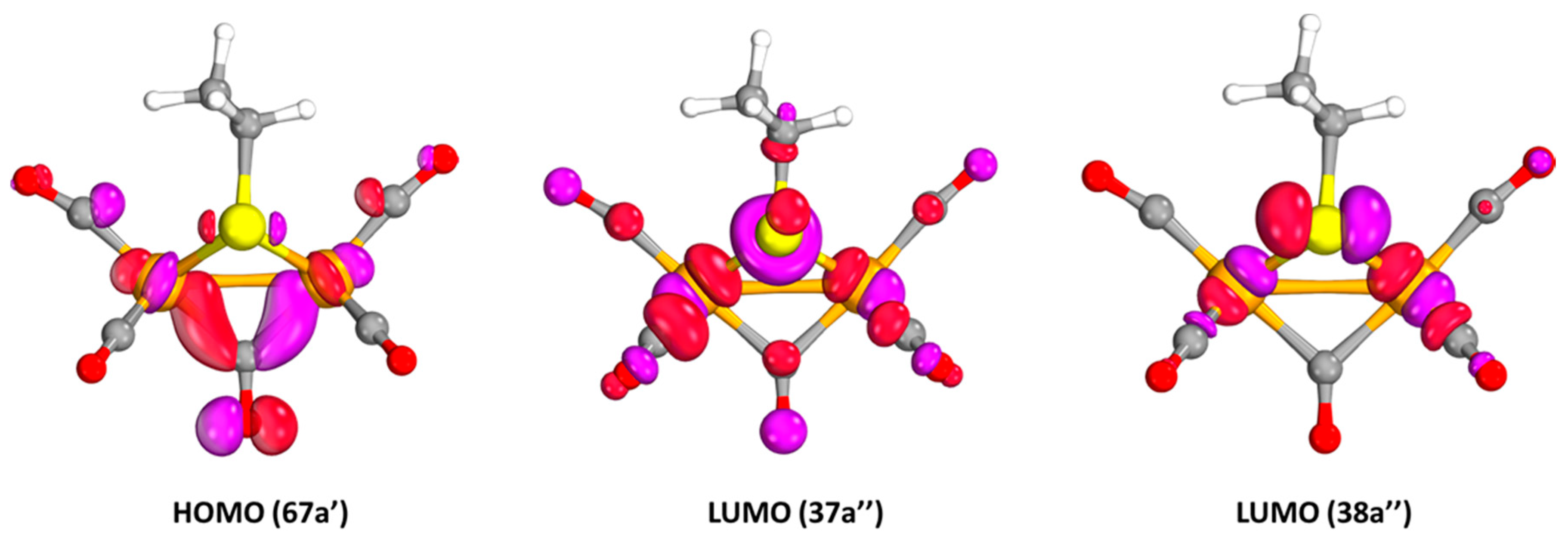
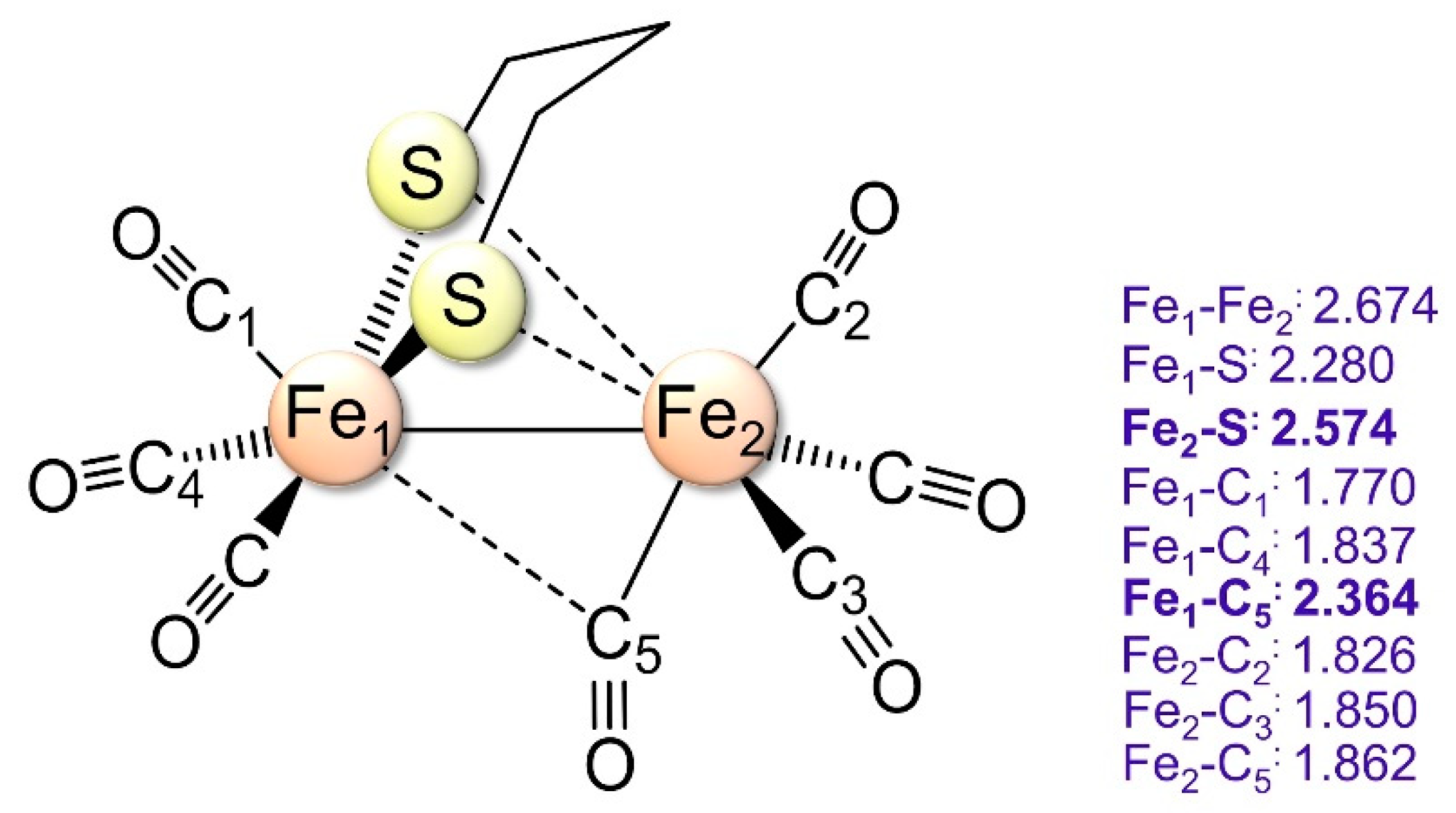
2.1.1. 1a–CO and 2a+–CO Ground State Properties
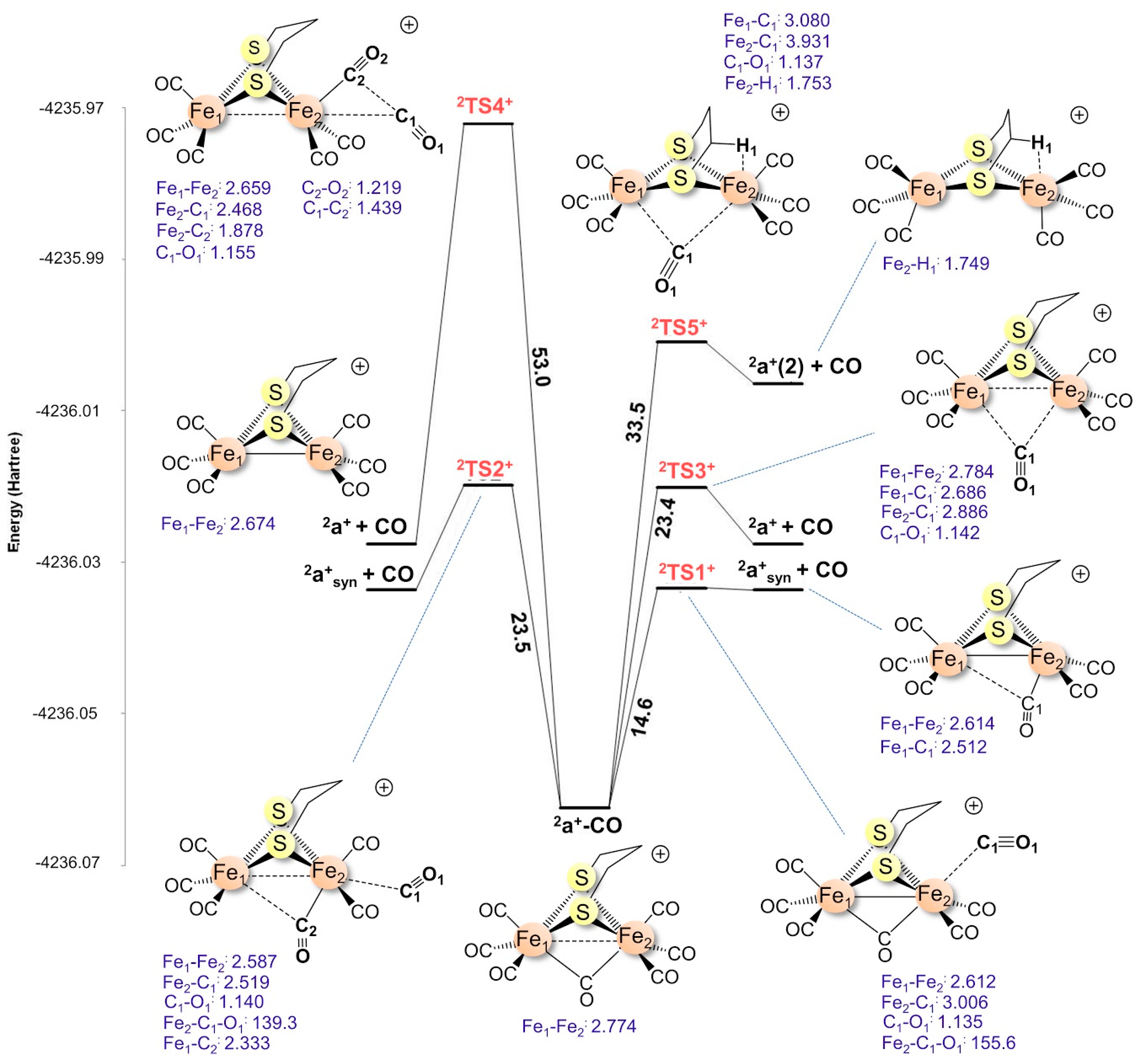
2.1.2. 1a–CO CO Dissociation Transition States
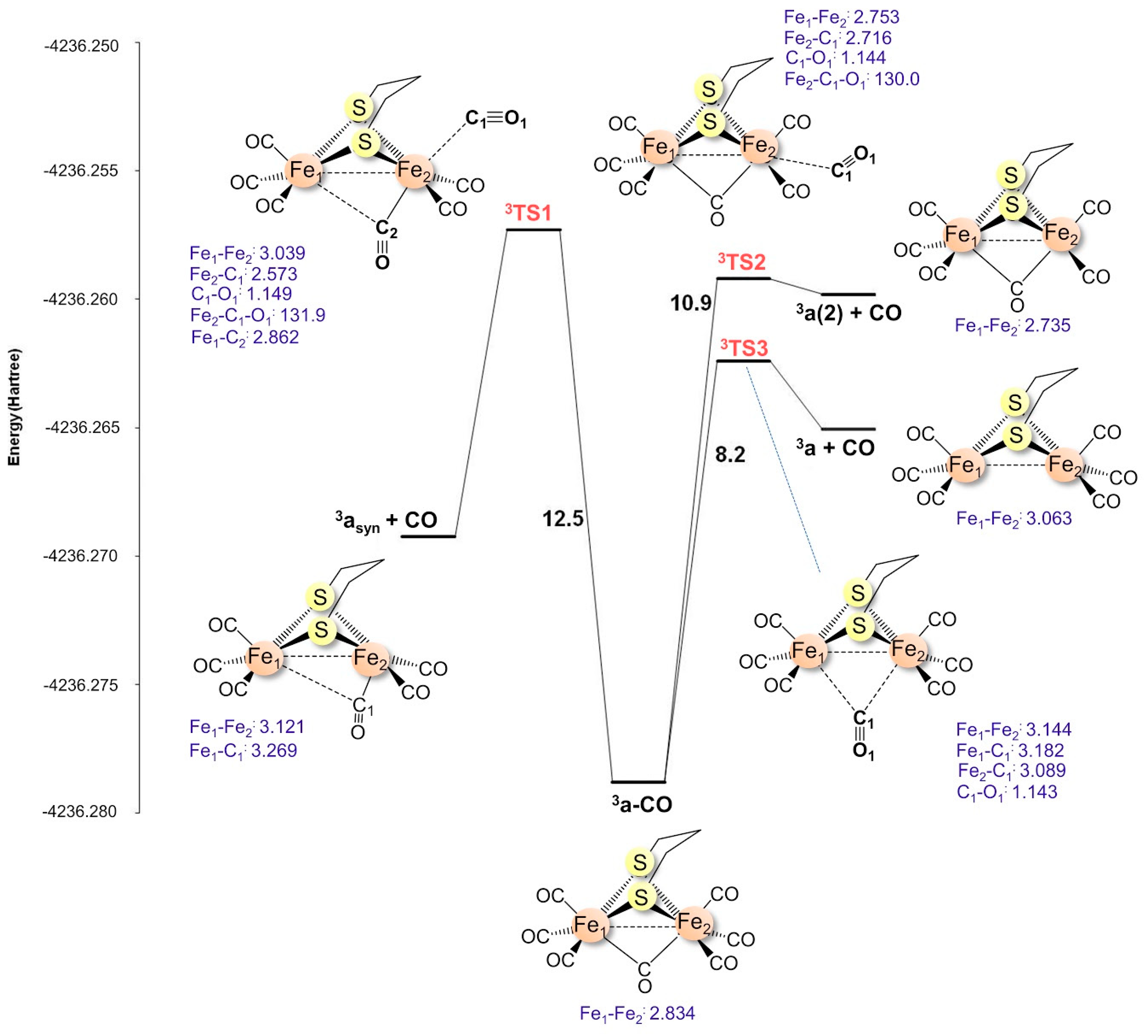
2.1.3. Transition States for 2a+–CO Dissociation
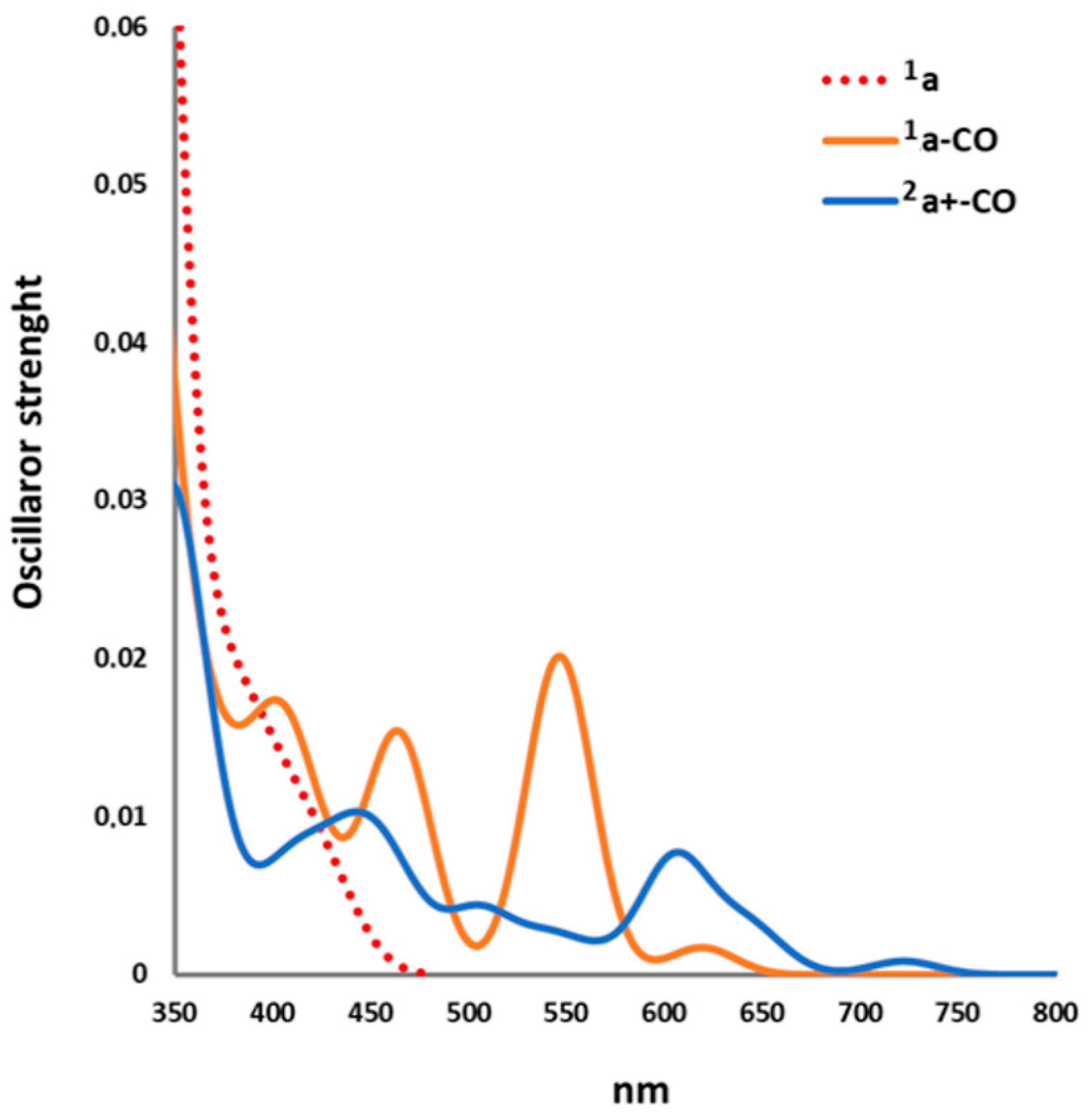
2.2. Excited States
2.2.1. Electronic Transitions
2.2.2. 3a–CO Lowest Triplet State
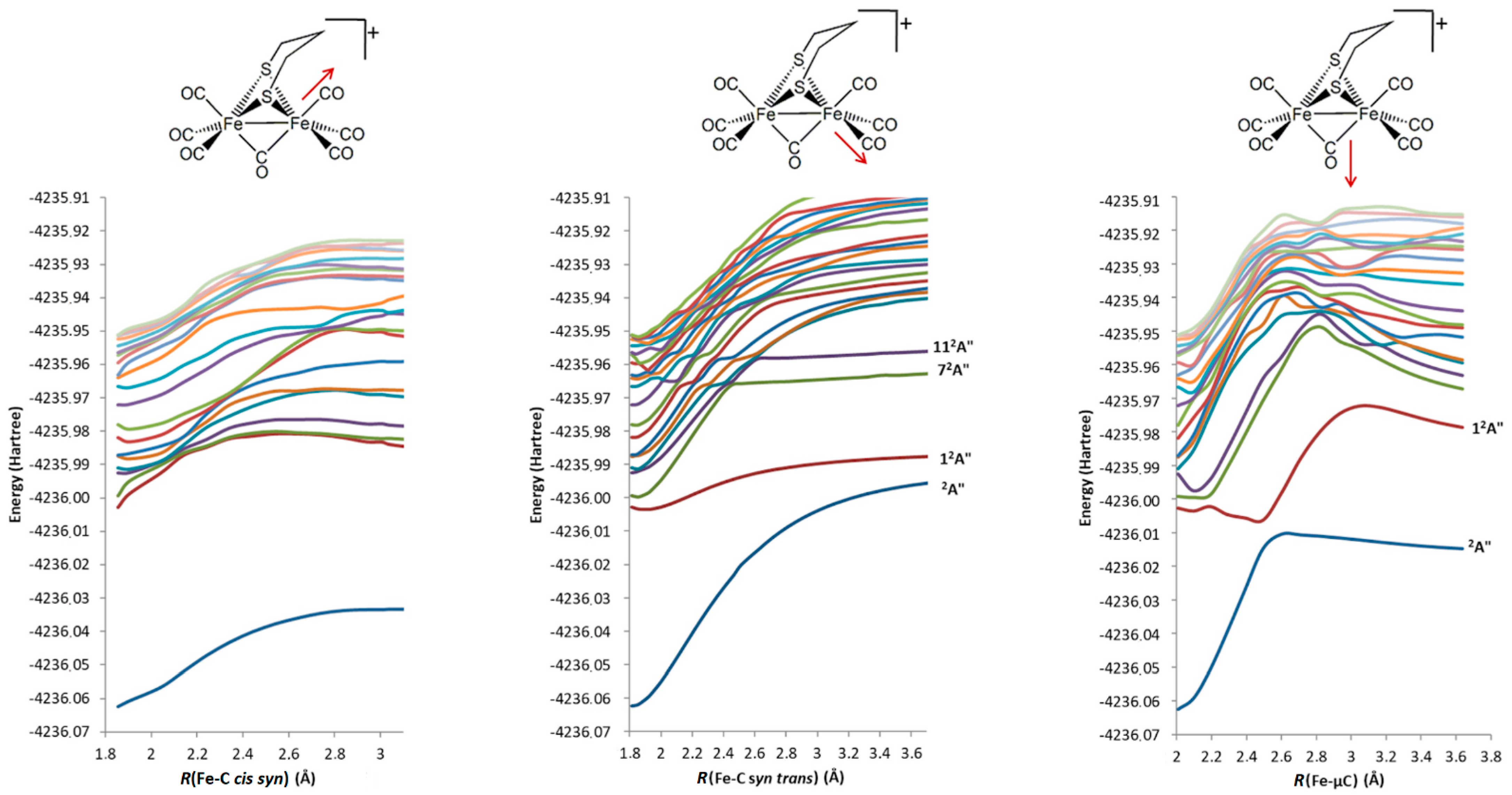
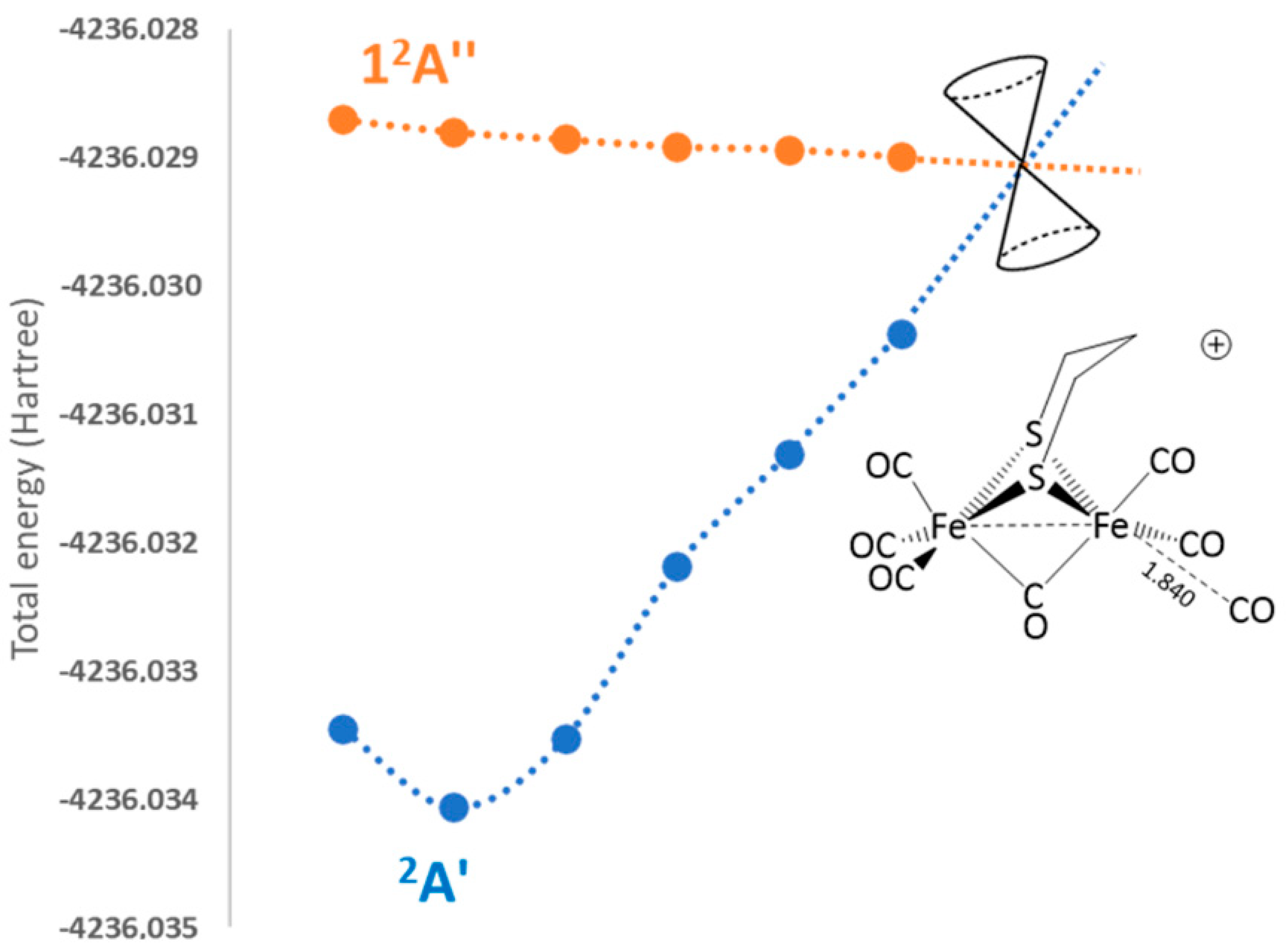
2.2.3. 2a+–CO 12A″ Excited-State PES Exploration
2.2.4. 2a+–CO Excited States PES Scanning
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef]
- Kaur-Ghumaan, S.; Stein, M. [NiFe] hydrogenases: How close do structural and functional mimics approach the active site? Dalton Trans. 2014, 43, 9392–9405. [Google Scholar] [CrossRef] [Green Version]
- Kleinhaus, J.T.; Wittkamp, F.; Yadav, S.; Siegmund, D.; Apfel, U.-P. [FeFe]-Hydrogenases: Maturation and reactivity of enzymatic systems and overview of biomimetic models. Chem. Soc. Rev. 2020. [Google Scholar] [CrossRef]
- Tard, C.; Pickett, C.J. Structural and Functional Analogues of the Active Sites of the [Fe]-, [NiFe]-, and [FeFe]-Hydrogenases†. Chem. Rev. 2009, 109, 2245–2274. [Google Scholar] [CrossRef]
- Frey, M. Hydrogenases: Hydrogen-activating enzymes. Chembiochem 2002, 3, 153–160. [Google Scholar] [CrossRef]
- Reihlen, H.; Gruhl, A.; v. Hessling, G. Über den photochemischen und oxydativen Abbau von Carbonylen. Justus Liebig’s Ann. Chem. 1929, 472, 268–287. [Google Scholar] [CrossRef]
- Peters, J.W.; Lanzilotta, W.N.; Lemon, B.J.; Seefeldt, L.C. X-ray crystal structure of the Fe-only hydrogenase (CpI) from Clostridium pasteurianum to 1.8 angstrom resolution. Science 1998, 282, 1853–1858. [Google Scholar] [CrossRef]
- Arrigoni, F.; Bertini, L.; Breglia, R.; Greco, C.; De Gioia, L.; Zampella, G. Catalytic H2 evolution/oxidation in [FeFe]-hydrogenase biomimetics: Account from DFT on the interplay of related issues and proposed solutions. New J. Chem. 2020, 44, 17596–17615. [Google Scholar] [CrossRef]
- Camara, J.M.; Rauchfuss, T.B. Combining acid-base, redox and substrate binding functionalities to give a complete model for the [FeFe]-hydrogenase. Nat. Chem. 2011, 4, 26–30. [Google Scholar] [CrossRef] [Green Version]
- Greco, C. H2 binding and splitting on a new-generation [FeFe]-hydrogenase model featuring a redox-active decamethylferrocenyl phosphine ligand: A theoretical investigation. Inorg. Chem. 2013, 52, 1901–1908. [Google Scholar] [CrossRef]
- Fan, H.J.; Hall, M.B. A capable bridging ligand for Fe-only hydrogenase: Density functional calculations of a low-energy route for heterolytic cleavage and formation of dihydrogen. J. Am. Chem. Soc. 2001, 123, 3828–3829. [Google Scholar] [CrossRef] [PubMed]
- Lacey, A.L.D.; De Lacey, A.L.; Stadler, C.; Cavazza, C.; Claude Hatchikian, E.; Fernandez, V.M. FTIR Characterization of the Active Site of the Fe-hydrogenase fromDesulfovibrio desulfuricans. J. Am. Chem. Soc. 2000, 122, 11232–11233. [Google Scholar] [CrossRef]
- Lemon, B.J.; Peters, J.W. Binding of exogenously added carbon monoxide at the active site of the fe-only hydrogenase (CpI) from clostridium pasteurianum. Biochemistry 1999, 38, 12969–12973. [Google Scholar] [CrossRef]
- Greco, C.; Bruschi, M.; Heimdal, J.; Fantucci, P.; De Gioia, L.; Ryde, U. Structural insights into the active-ready form of [FeFe]-hydrogenase and mechanistic details of its inhibition by carbon monoxide. Inorg. Chem. 2007, 46, 7256–7258. [Google Scholar] [CrossRef] [PubMed]
- Bertsch, J.; Müller, V. Bioenergetic constraints for conversion of syngas to biofuels in acetogenic bacteria. Biotechnol. Biofuels 2015, 8, 210. [Google Scholar] [CrossRef] [Green Version]
- Zilberman, S.; Stiefel, E.I.; Cohen, M.H.; Car, R. Resolving the CO/CN ligand arrangement in CO-inactivated [FeFe] hydrogenase by first principles density functional theory calculations. Inorg. Chem. 2006, 45, 5715–5717. [Google Scholar] [CrossRef]
- Duan, J.; Mebs, S.; Laun, K.; Wittkamp, F.; Heberle, J.; Happe, T.; Hofmann, E.; Apfel, U.-P.; Winkler, M.; Senger, M.; et al. Geometry of the Catalytic Active Site in [FeFe]-Hydrogenase Is Determined by Hydrogen Bonding and Proton Transfer. ACS Catal. 2019, 9, 9140–9149. [Google Scholar] [CrossRef]
- Razavet, M.; Borg, S.J.; George, S.J.; Best, S.P.; Fairhurst, S.A.; Pickett, C.J. Transient FTIR spectroelectrochemical and stopped-flow detection of a mixed valence Fe(i)–Fe(ii) bridging carbonyl intermediate with structural elements and spectroscopic characteristics of the di-iron sub-site of all-iron hydrogenase. Chem. Comm. 2002, 700–701. [Google Scholar] [CrossRef]
- Greco, C.; Bruschi, M.; Fantucci, P.; De Gioia, L. Relation between coordination geometry and stereoelectronic properties in DFT models of the CO-inhibited [FeFe]-hydrogenase cofactor. J. Organomet. Chem. 2009, 694, 2846–2853. [Google Scholar] [CrossRef]
- Baffert, C.; Bertini, L.; Lautier, T.; Greco, C.; Sybirna, K.; Ezanno, P.; Etienne, E.; Soucaille, P.; Bertrand, P.; Bottin, H.; et al. CO disrupts the reduced H-cluster of FeFe hydrogenase. A combined DFT and protein film voltammetry study. J. Am. Chem. Soc. 2011, 133, 2096–2099. [Google Scholar] [CrossRef]
- Bitterwolf, T.E. Organometallic photochemistry at the end of its first century. J. Organomet. Chem. 2004, 689, 3939–3952. [Google Scholar] [CrossRef]
- Chen, Z.; Lemon, B.J.; Huang, S.; Swartz, D.J.; Peters, J.W.; Bagley, K.A. Infrared Studies of the CO-Inhibited Form of the Fe-Only Hydrogenase from Clostridium pasteurianum I: Examination of Its Light Sensitivity at Cryogenic Temperatures†. Biochemistry 2002, 41, 2036–2043. [Google Scholar] [CrossRef]
- Albracht, S.P.J.; Roseboom, W.; Claude Hatchikian, E. The active site of the [FeFe]-hydrogenase from Desulfovibrio desulfuricans. I. Light sensitivity and magnetic hyperfine interactions as observed by electron paramagnetic resonance. J. Biol. Inorg. Chem. 2006, 11, 88–101. [Google Scholar] [CrossRef]
- Roseboom, W.; De Lacey, A.L.; Fernandez, V.M.; Claude Hatchikian, E.; Albracht, S.P.J. The active site of the [FeFe]-hydrogenase from Desulfovibrio desulfuricans. II. Redox properties, light sensitivity and CO-ligand exchange as observed by infrared spectroscopy. J. Biol. Inorg. Chem. 2006, 11, 102–118. [Google Scholar] [CrossRef]
- Darensbourg, M.Y.; Lyon, E.J.; Zhao, X.; Georgakaki, I.P. The organometallic active site of [Fe]hydrogenase: Models and entatic states. Proc. Natl. Acad. Sci. USA 2003, 100, 3683–3688. [Google Scholar] [CrossRef] [Green Version]
- Gloaguen, F.; Rauchfuss, T.B. Small molecule mimics of hydrogenases: Hydrides and redox. Chem. Soc. Rev. 2009, 38, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Greco, C.; Zampella, G.; Bertini, L.; Bruschi, M.; Fantucci, P.; De Gioia, L. Insights into the mechanism of electrocatalytic hydrogen evolution mediated by Fe2(S2C3H6)(CO)6: The simplest functional model of the Fe-hydrogenase active site. Inorg. Chem. 2007, 46, 108–116. [Google Scholar] [CrossRef]
- Bertini, L.; Greco, C.; De Gioia, L.; Fantucci, P. DFT/TDDFT Exploration of the Potential Energy Surfaces of the Ground State and Excited States of Fe2(S2C3H6)(CO)6: A Simple Functional Model of the [FeFe] Hydrogenase Active Site. J. Phys. Chem. A. 2009, 113, 5657–5670. [Google Scholar] [CrossRef]
- Tyler, D.R. 19-Electron organometallic adducts. Acc. Chem. Res. 1991, 24, 325–331. [Google Scholar] [CrossRef]
- Astruc, D. Nineteen-electron complexes and their role in organometallic mechanisms. Chem. Rev. 1988, 88, 1189–1216. [Google Scholar] [CrossRef]
- Thauer, R.K.; Käufer, B.; Zähringer, M.; Jungermann, K. The reaction of the iron-sulfur protein hydrogenase with carbon monoxide. Eur. J. Biochem. 1974, 42, 447–452. [Google Scholar] [CrossRef]
- Bertini, L.; Alberto, M.E.; Arrigoni, F.; Vertemara, J.; Fantucci, P.; Bruschi, M.; Zampella, G.; De Gioia, L. On the photochemistry of Fe2(edt)(CO)4(PMe3)2, a [FeFe]-hydrogenase model: A DFT/TDDFT investigation. Int. J. Quantum Chem. 2018, 118, e25537. [Google Scholar] [CrossRef]
- Bertini, L.; Greco, C.; Fantucci, P.; De Gioia, L. TDDFT modeling of the CO-photolysis of Fe2(S2C3H6)(CO)6, a model of the [FeFe]-hydrogenase catalytic site. Int. J. Quantum Chem. 2014, 114, 851–861. [Google Scholar] [CrossRef]
- Fletcher, S.C.; Poliakoff, M.; Turner, J.J. Structure and reactions of octacarbonyldiiron: An IR spectroscopic study using carbon-13 monoxide, photolysis with plane-polarized light, and matrix isolation. Inorg. Chem. 1986, 25, 3597–3604. [Google Scholar] [CrossRef]
- Bertini, L.; Bruschi, M.; De Gioia, L.; Fantucci, P. Structure and energetics of Fe2(CO)8 singlet and triplet electronic states. J. Phys. Chem. A 2007, 111, 12152–12162. [Google Scholar] [CrossRef] [PubMed]
- Sensi, M.; Baffert, C.; Greco, C.; Caserta, G.; Gauquelin, C.; Saujet, L.; Fontecave, M.; Roy, S.; Artero, V.; Soucaille, P.; et al. Reactivity of the Excited States of the H-Cluster of FeFe Hydrogenases. J. Am. Chem. Soc. 2016, 138, 13612–13618. [Google Scholar] [CrossRef] [Green Version]
- Sensi, M.; Baffert, C.; Fradale, L.; Gauquelin, C.; Soucaille, P.; Meynial-Salles, I.; Bottin, H.; de Gioia, L.; Bruschi, M.; Fourmond, V.; et al. Photoinhibition of FeFe Hydrogenase. ACS Catal. 2017, 7, 7378–7387. [Google Scholar] [CrossRef]
- Bertini, L.; Greco, C.; Bruschi, M.; Fantucci, P.; De Gioia, L. CO Affinity and Bonding Properties of [FeFe] Hydrogenase Active Site Models. A DFT Study. Organometallics 2010, 29, 2013–2025. [Google Scholar] [CrossRef]
- Arrigoni, F.; Mohamed Bouh, S.; De Gioia, L.; Elleouet, C.; Pétillon, F.Y.; Schollhammer, P.; Zampella, G. Influence of the Dithiolate Bridge on the Oxidative Processes of Diiron Models Related to the Active Site of [FeFe] Hydrogenases. Chem. Eur. J. 2017, 23, 4364–4372. [Google Scholar] [CrossRef]
- Arrigoni, F.; Bouh, S.M.; Elleouet, C.; Pétillon, F.Y.; Schollhammer, P.; De Gioia, L.; Zampella, G. Electrochemical and Theoretical Investigations of the Oxidatively Induced Reactivity of the Complex [Fe2(CO)4(κ2-dmpe)(μ-adtBn)] Related to the Active Site of [FeFe] Hydrogenases. Chem. Eur. J. 2018, 24, 15036–15051. [Google Scholar] [CrossRef]
- Chouffai, D.; Zampella, G.; Capon, J.-F.; De Gioia, L.; Le Goff, A.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J. Electrochemical and Theoretical Studies of the Impact of the Chelating Ligand on the Reactivity of [Fe2(CO)4(κ2-LL)(μ-pdt)] Complexes with Different Substrates (LL = IMe-CH2-IMe, dppe; IMe = 1-Methylimidazol-2-ylidene). Organometallics 2012, 31, 1082–1091. [Google Scholar] [CrossRef]
- Macchi, P. Chemical bonding in transition metal carbonyl clusters: Complementary analysis of theoretical and experimental electron densities. Coord. Chem. Rev. 2003, 238–239, 383–412. [Google Scholar] [CrossRef]
- Gatti, C.; Lasi, D. Source function description of metal-metal bonding in d-block organometallic compounds. Faraday Discuss. 2007, 135, 55–78. [Google Scholar] [CrossRef] [PubMed]
- Bertini, L.; Greco, C.; De Gioia, L.; Fantucci, P. Time-dependent density functional theory study of Fe2(CO)9 low-lying electronic excited states. J. Phys. Chem. A 2006, 110, 12900–12907. [Google Scholar] [CrossRef]
- Bertini, L.; Fantucci, P.; De Gioia, L. On the Photochemistry of the Low-Lying Excited State of Fe2(CO)6S2. A DFT and QTAIM Investigation. Organometallics 2011, 30, 487–498. [Google Scholar] [CrossRef]
- Bertini, L.; Fantucci, P.; De Gioia, L.; Zampella, G. Excited state properties of diiron dithiolate hydrides: Implications in the unsensitized photocatalysis of H2 evolution. Inorg. Chem. 2013, 52, 9826–9841. [Google Scholar] [CrossRef]
- Balasubramani, S.G.; Chen, G.P.; Coriani, S.; Diedenhofen, M.; Frank, M.S.; Franzke, Y.J.; Furche, F.; Grotjahn, R.; Harding, M.E.; Hättig, C.; et al. TURBOMOLE: Modular program suite for ab initio quantum-chemical and condensed-matter simulations. J. Chem. Phys. 2020, 152, 184107. [Google Scholar] [CrossRef] [PubMed]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A Gen. Phys. 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B Condens. Matter 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J.Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Optimized accurate auxiliary basis sets for RI-MP2 and RI-CC2 calculations for the atoms Rb to Rn. Theor. Chem. Acc. 1997, 97, 119–124. [Google Scholar] [CrossRef]
- Goy, R.; Bertini, L.; Rudolph, T.; Lin, S.; Schulz, M.; Zampella, G.; Dietzek, B.; Schacher, F.H.; De Gioia, L.; Sakai, K.; et al. Photocatalytic Hydrogen Evolution Driven by [FeFe] Hydrogenase Models Tethered to Fluorene and Silafluorene Sensitizers. Chem. Eur. J. 2017, 23, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, F.; Rizza, F.; Vertemara, J.; Breglia, R.; Greco, C.; Bertini, L.; Zampella, G.; De Gioia, L. Rational Design of Fe2 (μ-PR2)(L)6 Coordination Compounds Featuring Tailored Potential Inversion. Chemphyschem 2020, 21, 2279–2292. [Google Scholar] [CrossRef]
- Greco, C.; Fantucci, P.; De Gioia, L.; Suarez-Bertoa, R.; Bruschi, M.; Talarmin, J.; Schollhammer, P. Electrocatalytic dihydrogen evolution mechanism of [Fe2(CO)4(κ2-Ph2PCH2CH2PPh2)(μ-S(CH2)3S)] and related models of the [FeFe]-hydrogenases active site: A DFT investigation. Dalton Trans. 2010, 39, 7320. [Google Scholar] [CrossRef] [PubMed]
- Sicolo, S.; Bruschi, M.; Bertini, L.; Zampella, G.; Filippi, G.; Arrigoni, F.; De Gioia, L.; Greco, C. Towards biomimetic models of the reduced [FeFe]-hydrogenase that preserve the key structural features of the enzyme active site; a DFT investigation. Int. J. Hydrog. Energy 2014, 39, 18565–18573. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CO | i | ΔG‡ | |
|---|---|---|---|
| 1a–CO | syn cis | 145.5i | 9.7 |
| syn trans | 102.3i | 36.7 | |
| μ-CO | 77.2i | 41.5 | |
| 2a+–CO | syn cis | 76.4i | 14.6 |
| syn trans | 124.8i | 53.0 | |
| μ-CO | 40.6i | 33.5 | |
| 3a–CO | syn cis | 111.4i | 11.5 |
| syn trans | 99.6i | 12.5 | |
| μ-CO | 54.1i | 8.2 |
| 1a–CO | nm | f | 1e | 2a+–CO | nm | f | 1e |
|---|---|---|---|---|---|---|---|
| 11A″ | 767.8 | 7·10−8 | 67a′ → 37a″ | 12A″ | 764.4 | 1·10−5 | 67(α)a′ → 37(α)a″ |
| 21A″ | 619.1 | 2·10−3 | 67a′ → 38a″ | 22A″ | 722.1 | 9·10−4 | 67(α)a′ → 38(α)a″ |
| 11A′ | 546.4 | 0.02 | 67a′ → 68a′ | 12A′ | 652.4 | 1·10−3 | 66(β)a′ → 67(β)a′ (65%) 65(β)a′ → 67(β)a′ (19%) 67(α)a′ → 68(α)a′ (12%) |
| 21A′ | 464.1 | 1·10−2 | 67a′ → 69a′ | 22A′ | 637.7 | 2·10−3 | 65(β)a′ → 67(β)a′ (46%) 66(β)a′ → 67(β)a′ (33%) 67(α)a′ → 68(α)a′(17%) |
| 31A″ | 429.9 | 6·10−6 | 67a′ → 39a″ | 32A″ | 608.6 | 1·10−4 | 36(β)a″→ 67(β)a′ |
| 31A′ | 422.1 | 1·10−3 | 67a′ → 70a′ | 32A′ | 604.9 | 7·10−3 | 67(α)a′ → 68(α)a′ (52%) 65(β)a′ → 67(β)a′ (33%) 63(β)a′ → 67(β)a′ (11%) |
| 41A″ | 407.5 | 1·10−3 | 67a′ → 40a″ | 42A″ | 566.2 | 9·10−3 | 35(β)a″ → 67(β)a′ |
| 51A″ | 405.6 | 3·10−4 | 67a′ → 41a″ (63%) 67a′ → 40a″ (34%) | 42A′ | 540.1 | 2·10−3 | 64(β)a′ → 67(β)a′ |
| 41A′ | 403.4 | 3·10−3 | 67a′ → 71a′(69%) 67a′ → 72a′(27%) | 52A′ | 504.8 | 4·10−3 | 63(β)a′ → 67(β)a′ (63%) 67(α)a′ → 70(α)a′ (17%) 67(α)a′ → 68(α)a′ (14%) |
| 61A″ | 400.3 | 3·10−4 | 66a′ → 37a″ (62%) 67a′ → 41a″ (33%) | 52A″ | 475.6 | 2·10−7 | 34(β)a″ → 67(β)a′(81%) 67(α)a′ → 37(α)a″(16%) |
| 62A′ | 463.4 | 4·10−3 | 67(α)a′ → 69(α)a′ | ||||
| 62A″ | 458.9 | 2·10−5 | 67(α)a′ → 39(α)a″ (82%) 34(β)a″ → 67(β)a′ (16%) | ||||
| 72A′ | 442.5 | 7·10−3 | 67(α)a′ → 70(α)a′ (78%) 63(β)a′ → 67(β)a′ (15%) | ||||
| 72A″ | 433.0 | 5·10−7 | 66(α)a′ → 38(α)a″ (42%) 66(β)a′ → 38(β)a″ (27%) 66(β)a′ → 37(β)a″ (14%) 65(β)a′ → 37(β)a″ (5%) | ||||
| 82A″ | 430.3 | 3·10−7 | 65(α)a′ → 37(α)a″ (34%) 65(β)a′ → 37(β)a″ (21%) 65(β)a′ → 38(β)a″ (13%) 66(β)a′ → 38(β)a″ (11%) | ||||
| 82A′ | 412.8 | 2·10−3 | 62(β)a′ → 67(β)a′ (31%) 36(α)a″ → 37(α)a″ (28%) 36(β)a″ → 37(β)a″ (18%) 36(α)a″ → 38(α)a″ (8%) | ||||
| 92A″ | 414.5 | 1·10−3 | 67(α)a′ → 40(α)a″ | ||||
| 102A″ | 410.2 | 2·10−3 | 66(α)a′ → 37(α)a″ (33%) 66(β)a′ → 37(β)a″ (21%) 67(α)a′ → 41(α)″ (9%) 66(β)a′ → 38(β)a″ (9%) | ||||
| 112A″ | 409.9 | 8·10−3 | 67(α)a′ → 41(α)a″ (87%) 66(β)a′ → 37(β)a″ (4%) | ||||
| 92A′ | 408.3 | 7·10−4 | 36(α)a″ → 38(α)a″ (33%) 36(β)a″ → 38(β)a″ (31%) 36(α)a″ → 37(α)a″ (15%) 66(β)a′ → 68(β)a′ (5%) | ||||
| 102A′ | 405.1 | 1·10−3 | 62(β)a′ → 67(β)a′ (44%) 36(β)a″ → 37(β)a″ (27%) 36(α)a″ → 37(α)a″ (9%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arrigoni, F.; Zampella, G.; De Gioia, L.; Greco, C.; Bertini, L. The Photochemistry of Fe2(S2C3H6)(CO)6(µ-CO) and Its Oxidized Form, Two Simple [FeFe]-Hydrogenase CO-Inhibited Models. A DFT and TDDFT Investigation. Inorganics 2021, 9, 16. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9020016
Arrigoni F, Zampella G, De Gioia L, Greco C, Bertini L. The Photochemistry of Fe2(S2C3H6)(CO)6(µ-CO) and Its Oxidized Form, Two Simple [FeFe]-Hydrogenase CO-Inhibited Models. A DFT and TDDFT Investigation. Inorganics. 2021; 9(2):16. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9020016
Chicago/Turabian StyleArrigoni, Federica, Giuseppe Zampella, Luca De Gioia, Claudio Greco, and Luca Bertini. 2021. "The Photochemistry of Fe2(S2C3H6)(CO)6(µ-CO) and Its Oxidized Form, Two Simple [FeFe]-Hydrogenase CO-Inhibited Models. A DFT and TDDFT Investigation" Inorganics 9, no. 2: 16. https://0-doi-org.brum.beds.ac.uk/10.3390/inorganics9020016