Production of a Recombinant Single-Domain Antibody for Gluten Detection in Foods Using the Pichia pastoris Expression System

 , , ,
, , ,  , and
, and
Abstract
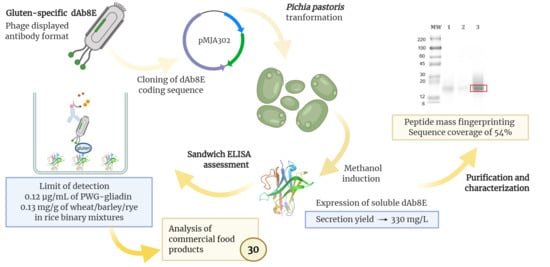
:
1. Introduction
2. Materials and Methods
2.1. Materials and Chemicals
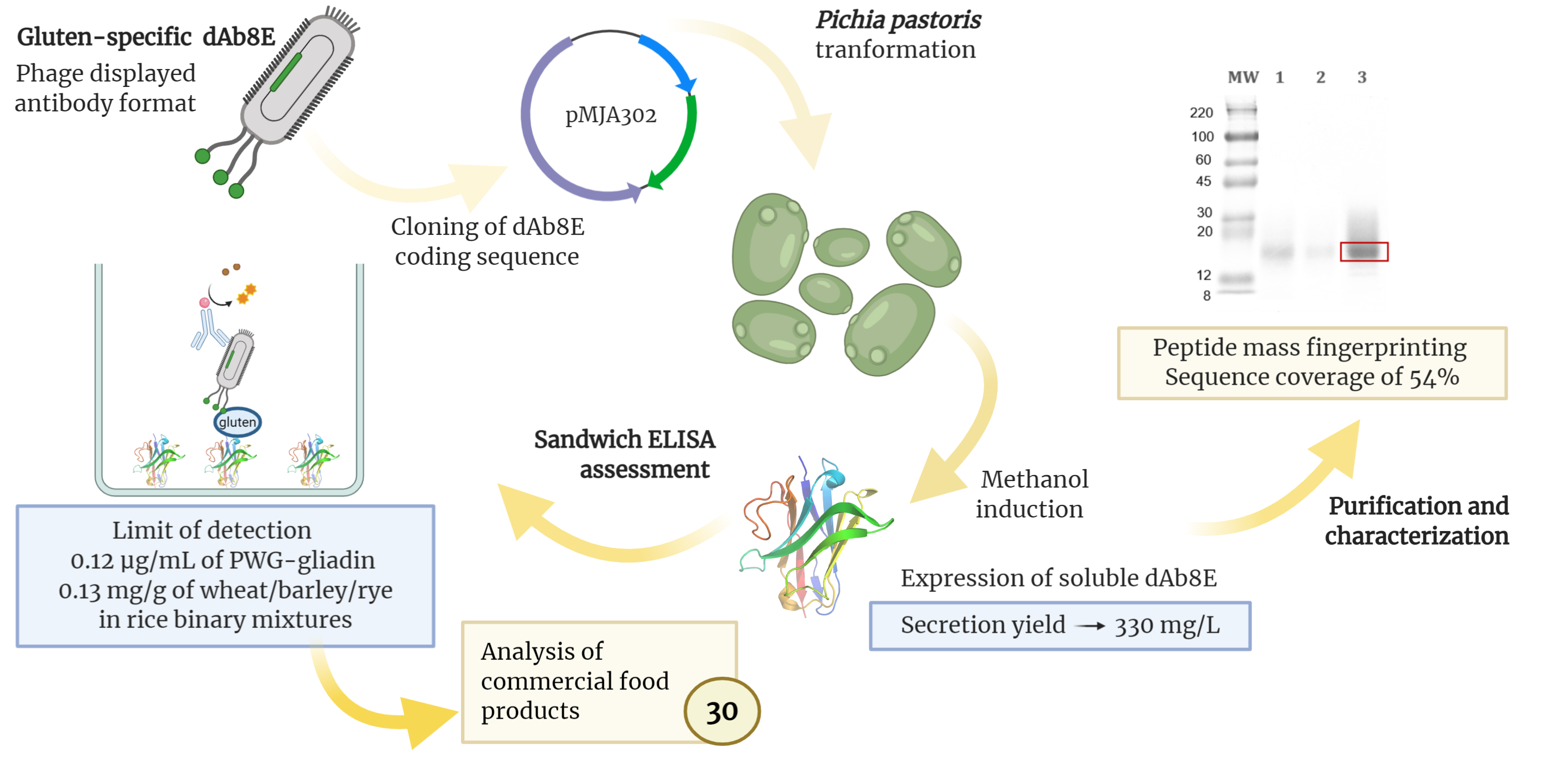
2.2. Vector Construction
2.3. Transformation of Escherichia coli
2.4. Transformation and Induction of Pichia pastoris
2.5. Characterization of the Antibody Fragment dAb8E
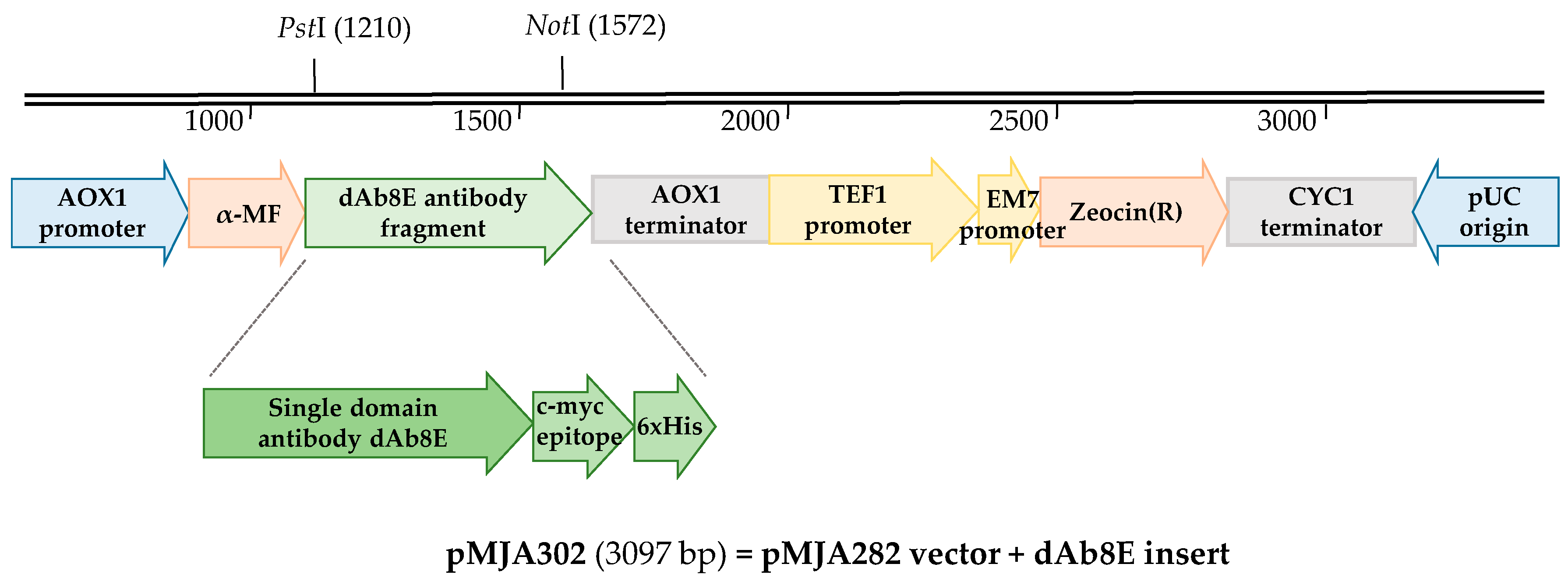
2.5.1. Analysis of the Expression in the Supernatant of Pichia pastoris
2.5.2. Purification of the Recombinant Protein dAb8E
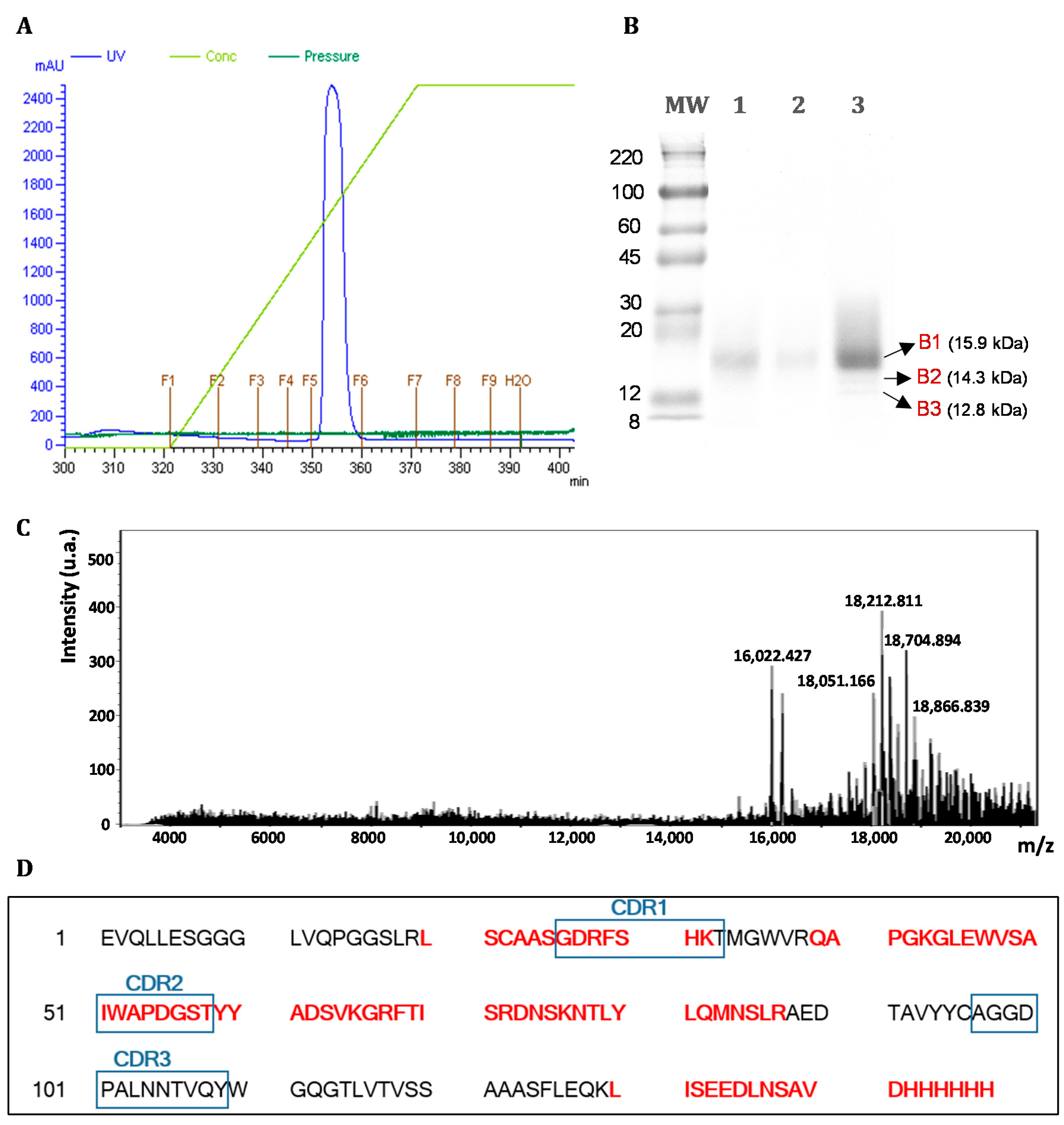
2.5.3. Protein Identification by Matrix-Assisted Laser Desorption/Ionisation-Time of Flight Mass Spectrometry
2.6. Assessment of ELISA Methods Based on the Purified dAb8E for the Detection of Gluten in Foodstuffs
2.6.1. Preparation of Protein Extracts and Reference Materials
2.6.2. Indirect Enzyme-Linked Immunosorbent Assay (ELISA)
2.6.3. Sandwich Enzyme-Linked Immunosorbent Assay (ELISA) with Phage-dAb8E as Detection Antibody
2.6.4. Assay Validation
3. Results and Discussion
3.1. Vector Construction
3.2. Expression and Purification of Soluble Antibody Fragments dAb8E
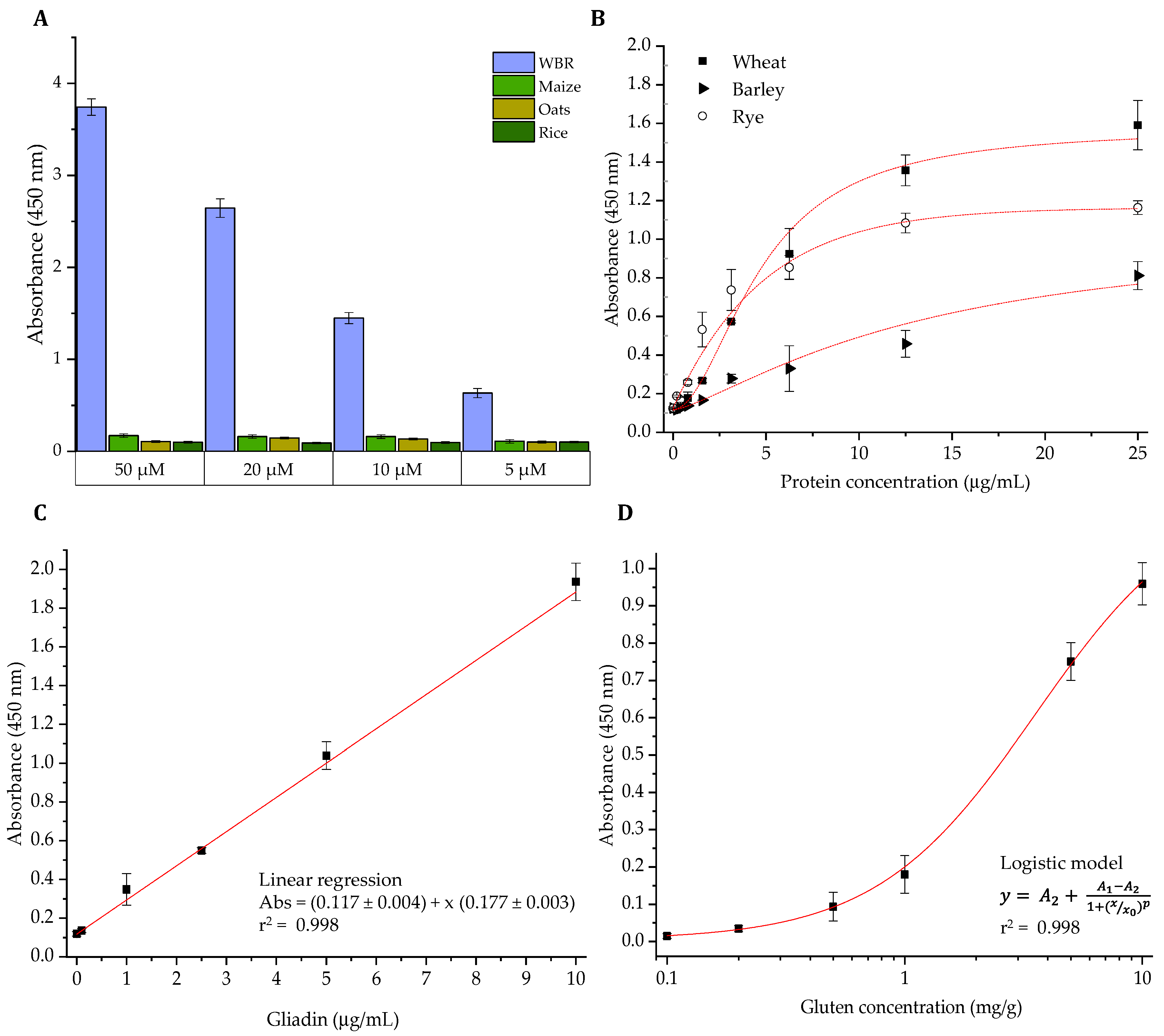
3.3. Development of an ELISA Methodology for the Analysis of Gluten in Foods
3.4. Comparative Quantification of Gluten in Commercial Food Products
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Procedure for Obtaining Escherichia coli Competent Cells
Appendix A.2. Procedure for Obtaining Pichia pastoris Electrocompletent Cells
References
- Savage, J.; Johns, C.B. Food allergy: Epidemiology and natural history. Immunol. Allergy Clin. North. Am. 2015, 35, 45–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapone, A.; Bai, J.C.; Ciacci, C.; Dolinsek, J.; Green, P.H.R.; Hadjivassiliou, M.; Kaukinen, K.; Rostami, K.; Sanders, D.S.; Schumann, M.; et al. Spectrum of gluten-related disorders: Consensus on new nomenclature and classification. BMC Med. 2012, 10, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elli, L.; Branchi, F.; Tomba, C.; Villalta, D.; Norsa, L.; Ferretti, F.; Roncoroni, L.; Bardella, M.T. Diagnosis of gluten related disorders: Celiac disease, wheat allergy and non-celiac gluten sensitivity. World J. Gastroenterol. 2015, 21, 7110–7119. [Google Scholar] [CrossRef] [PubMed]
- Valenti, S.; Corica, D.; Ricciardi, L.; Romano, C. Gluten-related disorders: Certainties, questions and doubts. Ann. Med. 2017, 49, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Al-Bawardy, B.; Codipilly, D.C.; Rubio-Tapia, A.; Bruining, D.H.; Hansel, S.L.; Murray, J.A. Celiac disease: A clinical review. Abdom. Radiol. 2017, 42, 351–360. [Google Scholar] [CrossRef]
- Melini, F.; Melini, V. Immunological methods in gluten risk analysis: A snapshot. Safety 2018, 4, 56. [Google Scholar] [CrossRef] [Green Version]
- Prado, M.; Ortea, I.; Vial, S.; Rivas, J.; Calo-Mata, P.; Barros-Velázquez, J. Advanced DNA- and protein-based methods for the detection and investigation of food allergens. Crit. Rev. Food Sci. Nutr. 2016, 56, 2511–2542. [Google Scholar] [CrossRef]
- Thompson, T.; Méndez, E. Commercial Assays to Assess Gluten Content of Gluten-Free Foods: Why They Are Not Created Equal. J. Am. Diet. Assoc. 2008, 108, 1682–1687. [Google Scholar] [CrossRef]
- European Commision Commission Implementing Regulation (EU). No. 828/2014 of 30 July 2014 on the requirements for the provision of information to consumers on the absence or reduced presence of gluten in food. Off. J. Eur. Union 2014, L228, 5–8. [Google Scholar]
- Bruins-Slot, I.D.; Bremer, M.G.E.G.; van der Fels-Klerx, I.; Hamer, R.J. Evaluating the performance of gluten ELISA test kits: The numbers do not tell the tale. Cereal Chem. 2015, 92, 513–521. [Google Scholar] [CrossRef]
- Rzychon, M.; Brohée, M.; Cordeiro, F.; Haraszi, R.; Ulberth, F.; O’Connor, G. The feasibility of harmonizing gluten ELISA measurements. Food Chem. 2017, 234, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sapienza, G.; Rossotti, M.A.; Tabares-da Rosa, S. Single-domain antibodies as versatile affinity reagents for analytical and diagnostic applications. Front. Immunol. 2017, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Henry, K.A.; Mackenzie, C.R. Antigen recognition by single-domain antibodies: Structural latitudes and constraints. MAbs 2018, 10, 815–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghassabeh, H.-G.; Devoogdt, N.; De Pauw, P.; Vincke, C.; Muyldermans, S. Nanobodies and their potential applications. Nanomedicine 2013, 8, 1013–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Li, Y.; Wang, H.; Samad, A.; Zhou, J.; Zhu, L.; Zhang, Y.; He, J.; Fan, X.; Jin, T. Screening of nanobody specific for peanut major allergen Ara h 3 by phage display. J. Agric. Food Chem. 2019, 67, 11219–11229. [Google Scholar] [CrossRef]
- Madrid, R.; García-García, A.; González, I.; Martín, R.; García, T. Phage displayed domain antibodies (dAb) for detection of allergenic pistachio proteins in foods. Foods 2020, 9, 1230. [Google Scholar] [CrossRef]
- Doña, V.; Urrutia, M.; Bayardo, M.; Alzogaray, V.; Goldbaum, F.A.; Chirdo, F.G. Single domain antibodies are specially suited for quantitative determination of gliadins under denaturing conditions. J. Agric. Food Chem. 2010, 58, 918–926. [Google Scholar] [CrossRef]
- García-García, A.; Madrid, R.; González, I.; García, T.; Martín, R. A novel approach to produce phage single domain antibody fragments for the detection of gluten in foods. Food Chem. 2020, 321, 10. [Google Scholar] [CrossRef]
- Liu, Z.H.; Huang, D.; Fu, X.J.; Cheng, P.; Du, E.Q. Comparison of three commonly used fusion tags for the expression of nanobodies in the cytoplasm of Escherichia coli. Biotechnol. Biotechnol. Equip. 2018, 32, 462–469. [Google Scholar] [CrossRef] [Green Version]
- Damasceno, L.M.; Huang, C.J.; Batt, C.A. Protein secretion in Pichia pastoris and advances in protein production. Appl. Microbiol. Biotechnol. 2012, 93, 31–39. [Google Scholar] [CrossRef]
- Jeong, K.J.; Jang, S.H.; Velmurugan, N. Recombinant antibodies: Engineering and production in yeast and bacterial hosts. Biotechnol. J. 2011, 6, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Emberson, L.M.; Trivett, A.J.; Blower, P.J.; Nicholls, P.J. Expression of an anti-CD33 single-chain antibody by Pichia pastoris. J. Immunol. Methods 2005, 305, 135–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarei, N.; Vaziri, B.; Shokrgozar, M.A.; Mahdian, R.; Fazel, R.; Khalaj, V. High efficient expression of a functional humanized single-chain variable fragment (scFv) antibody against CD22 in Pichia pastoris. Appl. Microbiol. Biotechnol. 2014, 98, 10023–10039. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhou, Y.; Yu, J.; Liu, W.; Li, F.; Xian, M.; Nian, R.; Song, H.; Feng, D. An efficient constitutive expression system for Anti-CEACAM5 nanobody production in the yeast Pichia pastoris. Protein Expr. Purif. 2019, 155, 43–47. [Google Scholar] [CrossRef] [PubMed]
- de Marco, A. Recombinant antibody production evolves into multiple options aimed at yielding reagents suitable for application-specific needs. Microb. Cell Fact. 2015, 14, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourasadi, S.; Mousavi Gargari, S.L.; Rajabibazl, M.; Nazarian, S. Efficient production of nanobodies against urease activity of Helicobacter pylori in Pichia pastoris. Turk. J. Med. Sci. 2017, 47, 695–701. [Google Scholar] [CrossRef] [PubMed]
- García-García, A.; Madrid, R.; Sohrabi, H.; de la Cruz, S.; García, T.; Martín, R.; González, I. A sensitive and specific real-time PCR targeting DNA from wheat, barley and rye to track gluten contamination in marketed foods. LWT Food Sci. Technol. 2019, 114, 9. [Google Scholar] [CrossRef]
- Cregg, J.M.; Russell, K.A. Pichia Protocols; Higgins, D.R., Cregg, J.M., Eds.; Humana Press: Totowa, NJ, USA, 1998; Volume 103. [Google Scholar]
- de la Cruz, S.; Alcocer, M.; Madrid, R.; García, A.; Martín, R.; González, I.; García, T. Production of in vivo biotinylated scFv specific to almond (Prunus dulcis) proteins by recombinant Pichia pastoris. J. Biotechnol. 2016, 227, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Sechi, S.; Chait, B.T. Modification of cysteine residues by alkylation. A tool in peptide mapping and protein identification. Anal. Chem. 1998, 70, 5150–5158. [Google Scholar] [CrossRef]
- de la Cruz, S.; López-Calleja, I.M.; Alcocer, M.; González, I.; Martín, R.; García, T. Selection of recombinant antibodies by phage display technology and application for detection of allergenic Brazil Nut (Bertholletia excelsa) in processed foods. J. Agric. Food Chem. 2013, 61, 10310–10319. [Google Scholar] [CrossRef]
- Magnusson, B.; Örnemark, U. Eurachem Guide: The Fitness for Purpose of Analytical Methods. A Laboratory Guide to Method Validation and Related Topics, 2nd ed.; 2014; ISBN 9789187461590. Available online: http://www.eurachem.org (accessed on 9 December 2020).
- Lin-Cereghino, G.P.; Stark, C.M.; Kim, D.; Chang, J.; Shaheen, N.; Poerwanto, H.; Agari, K.; Moua, P.; Low, L.K.; Tran, N.; et al. The effect of α-mating factor secretion signal mutations on recombinant protein expression in Pichia pastoris. Gene 2013, 519, 311–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bretthauer, R.K.; Castellino, F.J. Glycosylation of Pichia pastoris -derived proteins. Biotechnol. Appl. Biochem. 1999, 30, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Wagner-Rousset, E.; Bussat, M.-C.; Lokteff, M.; Klinguer-Hamour, C.; Haeuw, J.-F.; Goetsch, L.; Wurch, T.; Dorsselaer, A.; Corvaia, N. Trends in Glycosylation, Glycoanalysis and Glycoengineering of Therapeutic Antibodies and Fc-Fusion Proteins. Curr. Pharm. Biotechnol. 2008, 9, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Rahbarizadeh, F.; Rasaee, M.J.; Forouzandeh, M.; Allameh, A.A. Over expression of anti-MUC1 single-domain antibody fragments in the yeast Pichia pastoris. Mol. Immunol. 2006, 43, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Weidner, M.; Taupp, M.; Hallam, S.J. Expression of recombinant proteins in the methylotrophic yeast Pichia pastoris. J. Vis. Exp. 2010, 36, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Karkut, T.; Chamankhah, M.; Alting-Mees, M.; Hemmingsen, S.M.; Hegedus, D. Optimal conditions for the expression of a single-chain antibody (scFv) gene in Pichia pastoris. Protein Expr. Purif. 2003, 28, 321–330. [Google Scholar] [CrossRef]
- Doña, V.V.; Fossati, C.A.; Chirdo, F.G. Interference of denaturing and reducing agents on the antigen/antibody interaction. Impact on the performance of quantitative immunoassays in gliadin analysis. Eur. Food Res. Technol. 2008, 226, 591–602. [Google Scholar] [CrossRef]
- Oldfield, P.R. Understanding the matrix effect in immunoassays. Bioanalysis 2014, 6, 1425–1427. [Google Scholar] [CrossRef]
- Martín-Fernández, B.; Costa, J.; Oliveira, M.B.P.P.; López-Ruiz, B.; Mafra, I. Screening new gene markers for gluten detection in foods. Food Control. 2015, 56, 57–63. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sandwich dAb8E | Sandwich mAbR5 | |
|---|---|---|
| (A) Category of products that declared wheat, barley, rye and/or gluten in their labelling | ||
| Breakfast cereal 1 | >100 | >100 |
| Breakfast cereal 2 | >100 | >100 |
| Chocolate bar | >100 | >100 |
| Bread 1 | >100 | >100 |
| Bulgur | >100 | >100 |
| Soup | >100 | >100 |
| (B) Category of products that declared may contain traces of wheat, barley, rye and/or gluten | ||
| Rice cake | <LOD | 13.1 ± 0.2 * |
| Pre-cooked meal | <LOD | <LOD |
| Snack 1 | >100 | >100 |
| Seeds | <LOD | 13.5 ± 0.4 |
| Breakfast cereal 3 | 27 ± 0.3 | 26.4 ± 0.3 |
| (C) Category of products that did not declare wheat, barley, rye nor gluten in their labelling | ||
| Breakfast cereal 4 | >100 | >100 |
| Breakfast cereal 5 | 23.9 ± 0.2 | 28.2 ± 0.7 |
| Breakfast cereal 6 | 28.6 ± 0.1 | 77.5 ± 0.5 |
| Oats beverage 1 | <LOD | 15.6 ± 0.1 |
| Oats beverage 2 | <LOD | <LOD |
| Oats bran | <LOD | 14.3 ± 0.3 |
| (D) Category of products certified as gluten free | ||
| Breakfast cereal 7 | <LOD | <LOD |
| Nut bar | 26.5 ± 0.3 | 35.2 ± 0.2 |
| Bread 2 | <LOD | <LOD |
| Bread 3 | <LOD | <LOD |
| Biscuit 1 | <LOD | <LOD |
| Biscuit 2 | <LOD | <LOD |
| Snack 2 | <LOD | <LOD |
| Pasta 1 | <LOD | <LOD |
| Pasta 2 | <LOD | <LOD |
| Pasta 3 | <LOD | <LOD |
| Pasta 4 | <LOD | <LOD |
| Flour | <LOD | <LOD |
| Bakery | <LOD | <LOD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-García, A.; Madrid, R.; Garcia-Calvo, E.; Mendoza-Chamizo, B.; García, T.; Martin, R. Production of a Recombinant Single-Domain Antibody for Gluten Detection in Foods Using the Pichia pastoris Expression System. Foods 2020, 9, 1838. https://0-doi-org.brum.beds.ac.uk/10.3390/foods9121838
García-García A, Madrid R, Garcia-Calvo E, Mendoza-Chamizo B, García T, Martin R. Production of a Recombinant Single-Domain Antibody for Gluten Detection in Foods Using the Pichia pastoris Expression System. Foods. 2020; 9(12):1838. https://0-doi-org.brum.beds.ac.uk/10.3390/foods9121838
Chicago/Turabian StyleGarcía-García, Aina, Raquel Madrid, Eduardo Garcia-Calvo, Belén Mendoza-Chamizo, Teresa García, and Rosario Martin. 2020. "Production of a Recombinant Single-Domain Antibody for Gluten Detection in Foods Using the Pichia pastoris Expression System" Foods 9, no. 12: 1838. https://0-doi-org.brum.beds.ac.uk/10.3390/foods9121838