
The Carcinogenic Properties of Overlooked yet Prevalent Polycyclic Aromatic Hydrocarbons in Human Lung Epithelial Cells

 ,
,  , and
, and 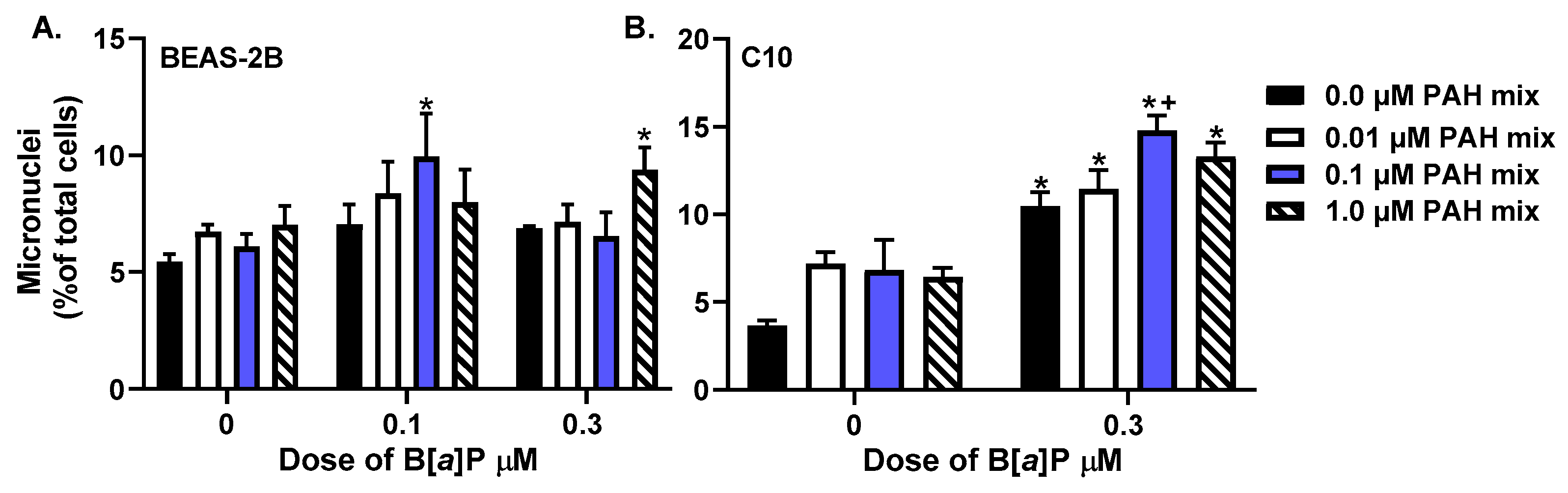{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. Micronucleus Assay
2.4. Cell Cycle Analysis
2.5. Scalpel-Loaded Dye Transfer Assay (SL/DT)
2.6. Experiments for DNA Adducts and Immunoblots
2.7. DNA Extraction and Quantitation
2.8. Analysis of Anti-BPDE-DNA Adducts
2.9. CYP1B1 Immunoblots
2.10. Statistical Analysis
3. Results
3.1. Micronuclei Formation in Response to PAHs
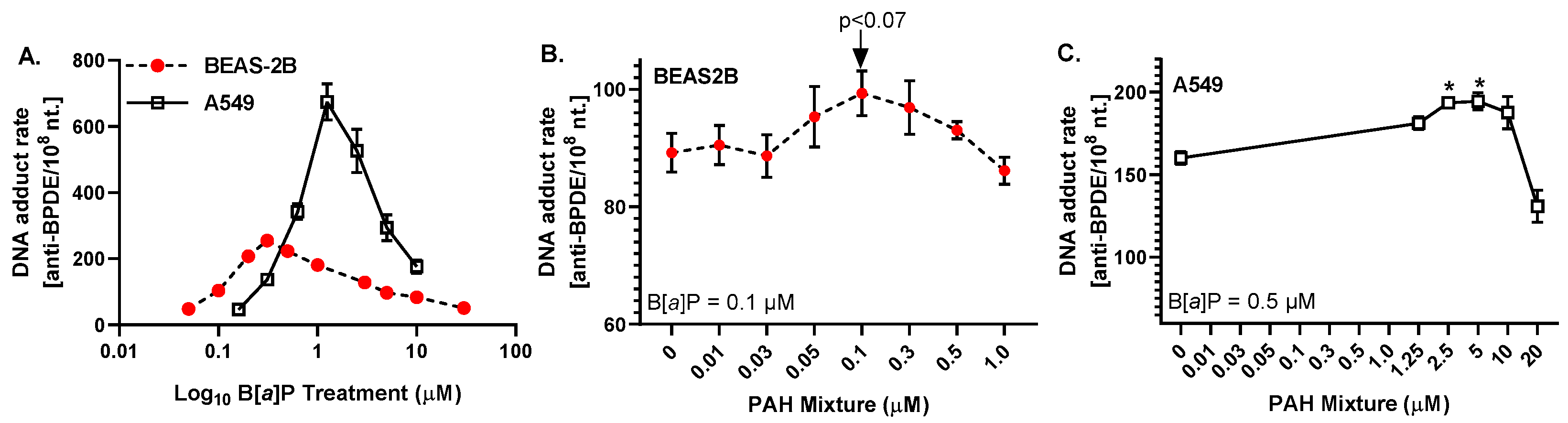
3.2. Anti-BPDE-DNA Adduct Formation in Response to PAHs
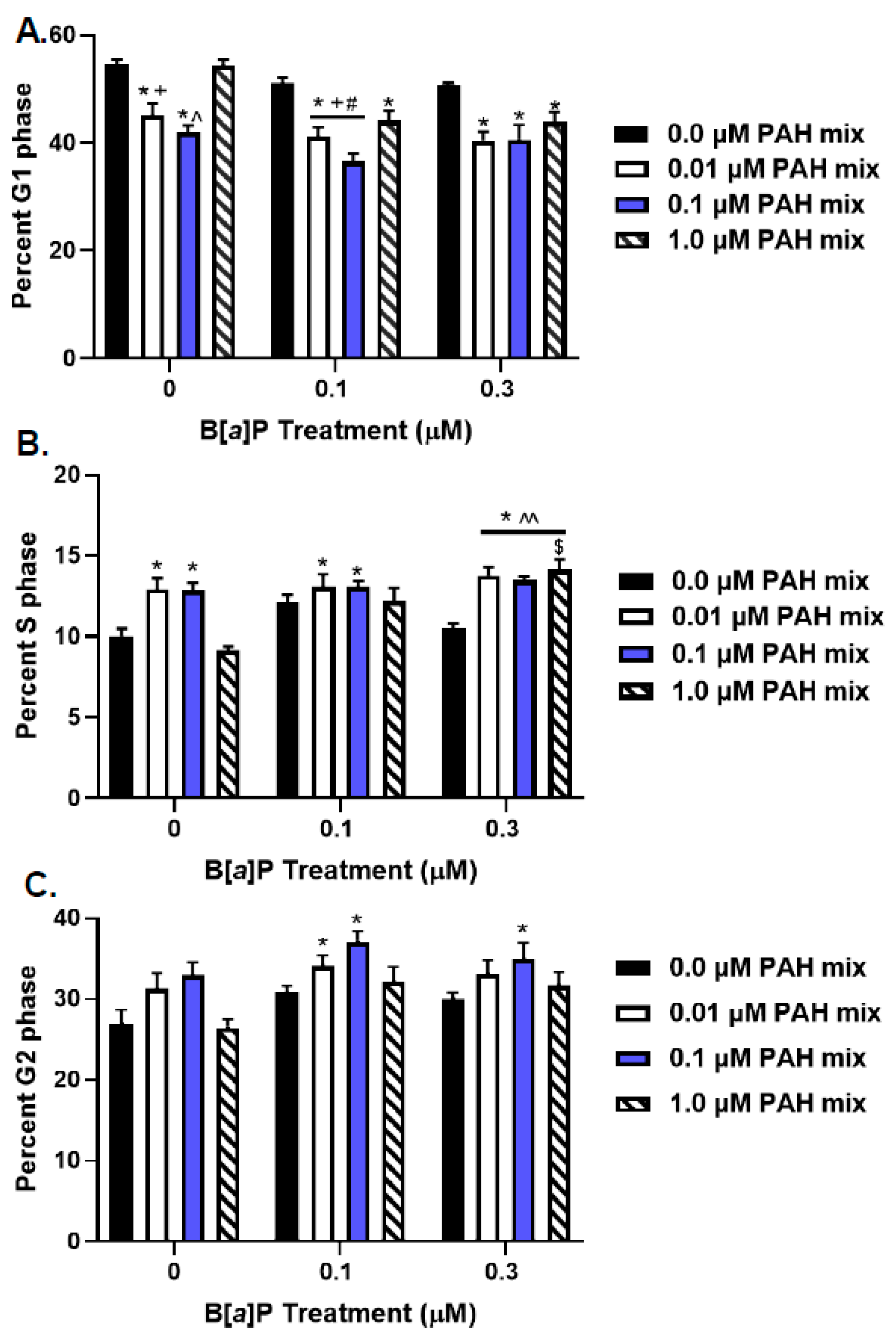
3.3. Cell Cycle Analysis of PAH-Treated Lung Epithelial Cells
3.4. GJIC in Response to PAHs
3.5. Cytochrome p450 1B1 Protein Expression in Response to LMW PAHs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Agency for Toxic Substances and Disease Registry. Toxicology Profile for Polyaromatic Hydrocarbons; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- International Agency for Research on Cancer (IARC). Diesel and Gasoline Engine Exhausts and Some Nitroarenes; IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer (IARC): Lyon, France, 2013; Volume 105. [Google Scholar]
- International Agency for Research on Cancer (IARC). Personal Habits and Indoor Combustions; IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer (IARC): Lyon, France, 2012; Volume 100E. [Google Scholar]
- Hong, W.J.; Jia, H.; Ma, W.L.; Sinha, R.K.; Moon, H.B.; Nakata, H.; Minh, N.H.; Chi, K.H.; Li, W.L.; Kannan, K.; et al. Distribution, Fate, Inhalation Exposure and Lung Cancer Risk of Atmospheric Polycyclic Aromatic Hydrocarbons in Some Asian Countries. Environ. Sci. Technol. 2016, 50, 7163–7174. [Google Scholar] [CrossRef]
- Lee, H.L.; Hsieh, D.P.; Li, L.A. Polycyclic aromatic hydrocarbons in cigarette sidestream smoke particulates from a Taiwanese brand and their carcinogenic relevance. Chemosphere 2010, 82, 477–482. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, R.; Lv, J. Spatial and Seasonal Variations of Polycyclic Aromatic Hydrocarbons (PAHs) in Ambient Particulate Matter (PM10, PM2.5) in Three Mega-Cities in China and Identification of Major Contributing Source Types. Bull. Environ. Contam. Toxicol. 2016, 96, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.L.; Campbell, L.; Donatuto, J.; Heidt, M.; Kile, M.; Massey Simonich, S.L. Impact of local and regional sources of PAHs on tribal reservation air quality in the U.S. Pacific Northwest. Sci. Total Environ. 2020, 710, 136412. [Google Scholar] [CrossRef]
- Hoppe-Jones, C.; Griffin, S.C.; Gulotta, J.J.; Wallentine, D.D.; Moore, P.K.; Beitel, S.C.; Flahr, L.M.; Zhai, J.; Zhou, J.J.; Littau, S.R.; et al. Evaluation of fireground exposures using urinary PAH metabolites. J. Expo. Sci. Environ. Epidemiol. 2021, 31, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.R.; Yamamoto, M.; Shaari, H.; Hayashi, R.; Seki, O.; Mohd Tahir, N.; Fadzil, M.F.; Sulaiman, A. The significance of pyrogenic polycyclic aromatic hydrocarbons in Borneo peat core for the reconstruction of fire history. PLoS ONE 2021, 16, e0256853. [Google Scholar] [CrossRef]
- Leroy-Cancellieri, V.; Cancellieri, D.; Leoni, E. Characterization of PAHs trapped in the soot from the combustion of various Mediterranean species. Atmosphere 2021, 12, 965. [Google Scholar] [CrossRef]
- Moir, D.; Rickert, W.S.; Levasseur, G.; Larose, Y.; Maertens, R.; White, P.; Desjardins, S. A comparison of mainstream and sidestream marijuana and tobacco cigarette smoke produced under two machine smoking conditions. Chem. Res. Toxicol. 2008, 21, 494–502. [Google Scholar] [CrossRef]
- Achutan, C.; West, C.; Mueller, C.; Bernert, J.T.; Bernard, B. Environmental tobacco smoke exposure among casino dealers. J. Occup. Environ. Med. 2011, 53, 346–351. [Google Scholar] [CrossRef]
- Beatty, A.L.; Haight, T.J.; Redberg, R.F. Associations between respiratory illnesses and secondhand smoke exposure in flight attendants: A cross-sectional analysis of the Flight Attendant Medical Research Institute Survey. Environ. Health 2011, 10, 81. [Google Scholar] [CrossRef] [Green Version]
- Pilkington, P.A.; Gray, S.; Gilmore, A.B. Health impacts of exposure to second hand smoke (SHS) amongst a highly exposed workforce: Survey of London casino workers. BMC Public Health 2007, 7, 257. [Google Scholar] [CrossRef] [PubMed]
- Crawford, C.B.; Quinn, B. The interactions of microplastics and chemical pollutants. In Microplastic Pollutants; Crawford, C.B., Quinn, B., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 131–157. [Google Scholar]
- International Agency for Research on Cancer (IARC). Chemical Agents and Related Occupations; IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer (IARC): Lyon, France, 2012; Volume 100F. [Google Scholar]
- International Agency for Research on Cancer (IARC). Some Non-Heterocyclic Polycyclic Aromatic Hydrocarbons and Some Related Exposures; IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer (IARC): Lyon, France, 2010; Volume 92. [Google Scholar]
- Bojes, H.K.; Pope, P.G. Characterization of EPA’s 16 priority pollutant polycyclic aromatic hydrocarbons (PAHs) in tank bottom solids and associated contaminated soils at oil exploration and production sites in Texas. Regul. Toxicol. Pharmacol. 2007, 47, 288–295. [Google Scholar] [CrossRef]
- United States Environmental Protection Agency, Polycyclic aromatic hydrocarbons, 15 listings. Rep. Carcinog. 2002, 10, 201–204.
- Bauer, A.K.; Velmurugan, K.; Plöttner, S.; Siegrist, K.J.; Romo, D.; Welge, P.; Brüning, T.; Xiong, K.N.; Käfferlein, H.U. Environmentally prevalent polycyclic aromatic hydrocarbons can elicit co-carcinogenic properties in an in vitro murine lung epithelial cell model. Arch. Toxicol. 2018, 92, 1311–1322. [Google Scholar] [CrossRef] [Green Version]
- Osgood, R.S.; Upham, B.L.; Bushel, P.R.; Velmurugan, K.; Xiong, K.N.; Bauer, A.K. Secondhand Smoke-Prevalent Polycyclic Aromatic Hydrocarbon Binary Mixture-Induced Specific Mitogenic and Pro-inflammatory Cell Signaling Events in Lung Epithelial Cells. Toxicol. Sci. 2017, 157, 156–171. [Google Scholar] [CrossRef] [Green Version]
- Osgood, R.S.; Upham, B.L.; Hill, T., 3rd; Helms, K.L.; Velmurugan, K.; Babica, P.; Bauer, A.K. Polycyclic aromatic hydrocarbon-induced signaling events relevant to inflammation and tumorigenesis in lung cells are dependent on molecular structure. PLoS ONE 2013, 8, e65150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegrist, K.J.; Romo, D.; Upham, B.L.; Armstrong, M.; Quinn, K.; Vanderlinden, L.; Osgood, R.S.; Velmurugan, K.; Elie, M.; Manke, J.; et al. Early Mechanistic Events Induced by Low Molecular Weight Polycyclic Aromatic Hydrocarbons in Mouse Lung Epithelial Cells: A Role for Eicosanoid Signaling. Toxicol. Sci. 2019, 169, 180–193. [Google Scholar] [CrossRef]
- Schick, S.F.; Farraro, K.F.; Perrino, C.; Sleiman, M.; van de Vossenberg, G.; Trinh, M.P.; Hammond, S.K.; Jenkins, B.M.; Balmes, J. Thirdhand cigarette smoke in an experimental chamber: Evidence of surface deposition of nicotine, nitrosamines and polycyclic aromatic hydrocarbons and de novo formation of NNK. Tob. Control 2014, 23, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, L.; Zheng, X.; Zhang, S.; Song, S.; Li, J.; Hao, J. Characterization and source apportionment of particulate PAHs in the roadside environment in Beijing. Sci. Total Environ. 2014, 470–471, 76–83. [Google Scholar] [CrossRef]
- Guillen, M.D.; Palencia, G.; Sopelana, P.; Ibargoitia, M.L. Occurrence of polycyclic aromatic hydrocarbons in artisanal Palmero cheese smoked with two types of vegetable matter. J. Dairy Sci. 2007, 90, 2717–2725. [Google Scholar] [CrossRef]
- Marczynski, B.; Pesch, B.; Wilhelm, M.; Rossbach, B.; Preuss, R.; Hahn, J.U.; Rabstein, S.; Raulf-Heimsoth, M.; Seidel, A.; Rihs, H.P.; et al. Occupational exposure to polycyclic aromatic hydrocarbons and DNA damage by industry: A nationwide study in Germany. Arch. Toxicol. 2009, 83, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Pesch, B.; Kappler, M.; Straif, K.; Marczynski, B.; Preuss, R.; Rossbach, B.; Rihs, H.P.; Weiss, T.; Rabstein, S.; Pierl, C.; et al. Dose-response modeling of occupational exposure to polycyclic aromatic hydrocarbons with biomarkers of exposure and effect. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1863–1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talaska, G.; Thoroman, J.; Schuman, B.; Kafferlein, H.U. Biomarkers of polycyclic aromatic hydrocarbon exposure in European coke oven workers. Toxicol. Lett. 2014, 231, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Vondracek, J.; Svihalkova-Sindlerova, L.; Pencikova, K.; Marvanova, S.; Krcmar, P.; Ciganek, M.; Neca, J.; Trosko, J.E.; Upham, B.; Kozubik, A.; et al. Concentrations of methylated naphthalenes, anthracenes, and phenanthrenes occurring in Czech river sediments and their effects on toxic events associated with carcinogenesis in rat liver cell lines. Environ. Toxicol. Chem. 2007, 26, 2308–2316. [Google Scholar] [CrossRef] [PubMed]
- Fustinoni, S.; Campo, L.; Cirla, P.E.; Martinotti, I.; Buratti, M.; Longhi, O.; Foa, V.; Bertazzi, P. Dermal exposure to polycyclic aromatic hydrocarbons in asphalt workers. Occup. Environ. Med. 2010, 67, 456–463. [Google Scholar] [CrossRef]
- Avanzo, J.L.; Mesnil, M.; Hernandez-Blazquez, F.J.; da Silva, T.C.; Fukumasu, H.; Mori, C.M.; Yamasaki, H.; Dagli, M.L. Altered expression of connexins in urethane-induced mouse lung adenomas. Life Sci. 2006, 79, 2202–2208. [Google Scholar] [CrossRef] [PubMed]
- Avanzo, J.L.; Mesnil, M.; Hernandez-Blazquez, F.J.; Mackowiak, I.I.; Mori, C.M.; da Silva, T.C.; Oloris, S.C.; Garate, A.P.; Massironi, S.M.; Yamasaki, H.; et al. Increased susceptibility to urethane-induced lung tumors in mice with decreased expression of connexin43. Carcinogenesis 2004, 25, 1973–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, A.K.; Cho, H.Y.; Miller-Degraff, L.; Walker, C.; Helms, K.; Fostel, J.; Yamamoto, M.; Kleeberger, S.R. Targeted deletion of Nrf2 reduces urethane-induced lung tumor development in mice. PLoS ONE 2011, 6, e26590. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, H.; Krutovskikh, V.; Mesnil, M.; Tanaka, T.; Zaidan, D.M.; Omori, Y. Role of connexin (gap junction) genes in cell growth control and carcinogenesis. C. R. Acad. Sci. III 1999, 322, 151–159. [Google Scholar] [CrossRef]
- Rosenkranz, M.; Rosenkranz, H.S.; Klopman, G. Intercellular communication, tumor promotion and non-genotoxic carcinogenesis: Relationships based upon structural considerations. Mutat. Res. 1997, 381, 171–188. [Google Scholar] [CrossRef]
- Nahta, R.; Al-Mulla, F.; Al-Temaimi, R.; Amedei, A.; Andrade-Vieira, R.; Bay, S.N.; Brown, D.G.; Calaf, G.M.; Castellino, R.C.; Cohen-Solal, K.A.; et al. Mechanisms of environmental chemicals that enable the cancer hallmark of evasion of growth suppression. Carcinogenesis 2015, 36 (Suppl. 1), S2–S18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegrist, K.J.; Reynolds, S.H.; Kashon, M.L.; Lowry, D.T.; Dong, C.; Hubbs, A.F.; Young, S.H.; Salisbury, J.L.; Porter, D.W.; Benkovic, S.A.; et al. Genotoxicity of multi-walled carbon nanotubes at occupationally relevant doses. Part. Fibre Toxicol. 2014, 11, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegrist, K.J.; Reynolds, S.H.; Porter, D.W.; Mercer, R.R.; Bauer, A.K.; Lowry, D.; Cena, L.; Stueckle, T.A.; Kashon, M.L.; Wiley, J.; et al. Mitsui-7, heat-treated, and nitrogen-doped multi-walled carbon nanotubes elicit genotoxicity in human lung epithelial cells. Part. Fibre Toxicol. 2019, 16, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shetty, S.K.; Bhandary, Y.P.; Marudamuthu, A.S.; Abernathy, D.; Velusamy, T.; Starcher, B.; Shetty, S. Regulation of airway and alveolar epithelial cell apoptosis by p53-Induced plasminogen activator inhibitor-1 during cigarette smoke exposure injury. Am. J. Respir. Cell Mol. Biol. 2012, 47, 474–483. [Google Scholar] [CrossRef]
- Feng, F.; Wu, Y.; Zhang, S.; Liu, Y.; Qin, L.; Wu, Y.; Yan, Z.; Wu, W. Macrophages Facilitate Coal Tar Pitch Extract-Induced Tumorigenic Transformation of Human Bronchial Epithelial Cells Mediated by NF-kappaB. PLoS ONE 2012, 7, e51690. [Google Scholar] [CrossRef] [Green Version]
- Stueckle, T.A.; Lu, Y.; Davis, M.E.; Wang, L.; Jiang, B.H.; Holaskova, I.; Schafer, R.; Barnett, J.B.; Rojanasakul, Y. Chronic occupational exposure to arsenic induces carcinogenic gene signaling networks and neoplastic transformation in human lung epithelial cells. Toxicol. Appl. Pharmacol. 2012, 261, 204–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malkinson, A.M.; Dwyer-Nield, L.D.; Rice, P.L.; Dinsdale, D. Mouse lung epithelial cell lines--tools for the study of differentiation and the neoplastic phenotype. Toxicology 1997, 123, 53–100. [Google Scholar] [CrossRef]
- Giard, D.J.; Aaronson, S.A.; Todaro, G.J.; Arnstein, P.; Kersey, J.H.; Dosik, H.; Parks, W.P. In vitro cultivation of human tumors: Establishment of cell lines derived from a series of solid tumors. J. Natl. Cancer Inst. 1973, 51, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zhang, M.; Barnes, P.F. Chemokine production by a human alveolar epithelial cell line in response to Mycobacterium tuberculosis. Infect. Immun. 1998, 66, 1121–1126. [Google Scholar] [CrossRef] [Green Version]
- Tajima, H.; Tajiki-Nishino, R.; Watanabe, Y.; Kurata, K.; Fukuyama, T. Activation of aryl hydrocarbon receptor by benzo[a]pyrene increases interleukin 33 expression and eosinophil infiltration in a mouse model of allergic airway inflammation. J. Appl. Toxicol. 2020, 40, 1545–1553. [Google Scholar] [CrossRef]
- Plöttner, S.; Bastian, L.A.; Käfferlein, H.U.; Brüning, T. Effects of benzo[a]pyrene, aromatic amines, and a combination of both on CYP1A1 activities in RT-4 human bladder papilloma cells. J. Toxicol. Environ. Health Part A 2016, 79, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Tai, M.H.; Upham, B.L.; Olson, L.K.; Tsao, M.S.; Reed, D.N., Jr.; Trosko, J.E. Cigarette smoke components inhibited intercellular communication and differentiation in human pancreatic ductal epithelial cells. Int. J. Cancer. 2007, 120, 1855–1862. [Google Scholar] [CrossRef] [PubMed]
- Upham, B.L. Role of integrative signaling through gap junctions in toxicology. Curr. Protoc. Toxicol. 2011, 47, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Fraser, K.; Kodali, V.; Yanamala, N.; Birch, M.E.; Cena, L.; Casuccio, G.; Bunker, K.; Lersch, T.L.; Evans, D.E.; Stefaniak, A.; et al. Physicochemical characterization and genotoxicity of the broad class of carbon nanotubes and nanofibers used or produced in U.S. facilities. Part. Fibre Toxicol. 2020, 17, 62. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Kim, M.J.; Lee, S.Y.; Oh, S.M.; Chung, K.H. Genotoxic effects of silver nanoparticles stimulated by oxidative stress in human normal bronchial epithelial (BEAS-2B) cells. Mutat. Res. 2011, 726, 129–135. [Google Scholar] [CrossRef]
- Brozman, O.; Novak, J.; Bauer, A.K.; Babica, P. Airborne PAHs inhibit gap junctional intercellular communication and activate MAPKs in human bronchial epithelial cell line. Environ. Toxicol. Pharmacol. 2020, 79, 103422. [Google Scholar] [CrossRef]
- Ding, Y.S.; Yan, X.J.; Jain, R.B.; Lopp, E.; Tavakoli, A.; Polzin, G.M.; Stanfill, S.B.; Ashley, D.L.; Watson, C.H. Determination of 14 polycyclic aromatic hydrocarbons in mainstream smoke from U.S. brand and non-U.S. brand cigarettes. Environ. Sci. Technol. 2006, 40, 1133–1138. [Google Scholar] [CrossRef]
- La Voie, E.J.; Coleman, D.T.; Rice, J.E.; Geddie, N.G.; Hoffmann, D. Tumor-initiating activity, mutagenicity, and metabolism of methylated anthracenes. Carcinogenesis 1985, 6, 1483–1488. [Google Scholar] [CrossRef]
- Lodovici, M.; Akpan, V.; Evangelisti, C.; Dolara, P. Sidestream tobacco smoke as the main predictor of exposure to polycyclic aromatic hydrocarbons. J. Appl. Toxicol. 2004, 24, 277–281. [Google Scholar] [CrossRef]
- Severson, R.F.; Snook, M.E.; Higman, H.C.; Chortyk, O.T.; Akin, F.J. Isolation, identification, and quantification of polynuclear aromatic hydrocarbons in tobacco smoke. In Carcinogenesis—A Comprehensive Survey. Vol. 1. Polynuclear Aromatic Hydrocarbons: Chemistry, Metabolism, and Carcinogenesis; Freudenthal, R.I., Jones, P.W., Eds.; Raven Press: New York, NY, USA, 1976; pp. 253–270. [Google Scholar]
- White, P.A. The genotoxicity of priority polycyclic aromatic hydrocarbons in complex mixtures. Mutat. Res. 2002, 515, 85–98. [Google Scholar] [CrossRef]
- Fang, M.D.; Lee, C.L.; Yu, C.S. Distribution and source recognition of polycyclic aromatic hydrocarbons in the sediments of Hsin-ta Harbour and adjacent coastal areas, Taiwan. Mar. Pollut. Bull. 2003, 46, 941–953. [Google Scholar] [CrossRef]
- Zakaria, M.P.; Takada, H.; Tsutsumi, S.; Ohno, K.; Yamada, J.; Kouno, E.; Kumata, H. Distribution of polycyclic aromatic hydrocarbons (PAHs) in rivers and estuaries in Malaysia: A widespread input of petrogenic PAHs. Environ. Sci. Technol. 2002, 36, 1907–1918. [Google Scholar] [CrossRef] [PubMed]
- Navarro, K.M.; Cisneros, R.; Schweizer, D.; Chowdhary, P.; Noth, E.M.; Balmes, J.R.; Hammond, S.K. Incident command post exposure to polycyclic aromatic hydrocarbons and particulate matter during a wildfire. J. Occup. Environ. Hyg. 2019, 16, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, J.; Marques, J.E.; Mansilha, C.; Flores, D. Wildfires effects on organic matter of soils from Caramulo Mountain (Portugal): Environmental implications. Environ. Sci. Pollut. Res. Int. 2021, 28, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.S.; Soshilov, A.A.; He, G.; DeGroot, D.E.; Zhao, B. Exactly the same but different: Promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci. 2011, 124, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, G.I.; Melvin, W.T.; Greenlee, W.F.; Burke, M.D. Regulation, function, and tissue-specific expression of cytochrome P450 CYP1B1. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 297–316. [Google Scholar] [CrossRef]
- Shimada, T.; Guengerich, F.P. Inhibition of human cytochrome P450 1A1-, 1A2-, and 1B1-mediated activation of procarcinogens to genotoxic metabolites by polycyclic aromatic hydrocarbons. Chem. Res. Toxicol. 2006, 19, 288–294. [Google Scholar] [CrossRef]
- Shimada, T. Inhibition of Carcinogen-Activating Cytochrome P450 Enzymes by Xenobiotic Chemicals in Relation to Antimutagenicity and Anticarcinogenicity. Toxicol. Res. 2017, 33, 79–96. [Google Scholar] [CrossRef]
- Shimada, T.; Murajama, N.; Tanaka, K.; Takenaka, S.; Imai, Y.; Hopkins, N.E.; Foroozesh, M.K.; Alworth, W.L.; Yamazaki, H.; Guengerich, F.P.; et al. Interaction of polycyclic aromatic hydrocarbons with human cytochrome P450 1B1 in inhibiting catalytic activity. Chem. Res. Toxicol. 2008, 21, 2313–2323. [Google Scholar]
- Guo, W.; Liu, X.; Lee, S.; Park, N.H. High O6-methylguanine methyl transferase activity is frequently found in human oral cancer cells with p53 inactivation. Int. J. Oncol. 1999, 15, 817–821. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scanlon, S.E.; Scanlon, C.D.; Hegan, D.C.; Sulkowski, P.L.; Glazer, P.M. Nickel induces transcriptional down-regulation of DNA repair pathways in tumorigenic and non-tumorigenic lung cells. Carcinogenesis 2017, 38, 627–637. [Google Scholar]
- Upham, B.L.; Blaha, L.; Babica, P.; Park, J.S.; Sovadinova, I.; Pudrith, C.; Rummel, A.M.; Weis, L.M.; Sai, K.; Tithof, P.K.; et al. Tumor promoting properties of a cigarette smoke prevalent polycyclic aromatic hydrocarbon as indicated by the inhibition of gap junctional intercellular communication via phosphatidylcholine-specific phospholipase C. Cancer Sci. 2008, 99, 696–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upham, B.L.; Sovadinova, I.; Babica, P. Gap Junctional Intercellular Communication: A Functional Biomarker to Assess Adverse Effects of Toxicants and Toxins, and Health Benefits of Natural Products. J. Vis. Exp. 2016, 118, e54281. [Google Scholar] [CrossRef]
- Decordier, I.; Dillen, L.; Cundari, E.; Kirsch-Volders, M. Elimination of micronucleated cells by apoptosis after treatment with inhibitors of microtubules. Mutagenesis 2002, 17, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Balmes, J.R. The Changing Nature of Wildfires: Impacts on the Health of the Public. Clin. Chest Med. 2020, 41, 771–776. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer (IARC). Outdoor Air Pollution; IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer (IARC): Lyon, France, 2016; Volume 109. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauer, A.K.; Siegrist, K.J.; Wolff, M.; Nield, L.; Brüning, T.; Upham, B.L.; Käfferlein, H.U.; Plöttner, S. The Carcinogenic Properties of Overlooked yet Prevalent Polycyclic Aromatic Hydrocarbons in Human Lung Epithelial Cells. Toxics 2022, 10, 28. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics10010028
Bauer AK, Siegrist KJ, Wolff M, Nield L, Brüning T, Upham BL, Käfferlein HU, Plöttner S. The Carcinogenic Properties of Overlooked yet Prevalent Polycyclic Aromatic Hydrocarbons in Human Lung Epithelial Cells. Toxics. 2022; 10(1):28. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics10010028
Chicago/Turabian StyleBauer, Alison K., Katelyn J. Siegrist, Melanie Wolff, Lindsey Nield, Thomas Brüning, Brad L. Upham, Heiko U. Käfferlein, and Sabine Plöttner. 2022. "The Carcinogenic Properties of Overlooked yet Prevalent Polycyclic Aromatic Hydrocarbons in Human Lung Epithelial Cells" Toxics 10, no. 1: 28. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics10010028