
Nickel’s Role in Pancreatic Ductal Adenocarcinoma: Potential Involvement of microRNAs

 ,
,  ,
,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Sampling
2.2. Human Sample Analysis
2.3. Pancreatic Cell Cultures and Nickel Treatment
2.4. Chemicals and Antibodies
2.5. LC50 Assays to Determine Nickel Toxicity after 48 h of Exposure
2.6. Changes in Protein Expression
2.7. Caspase 3/7 Kinetic Assays
2.8. RNA Extraction and Analysis of miR Expression
2.9. Statistical Analysis
3. Results
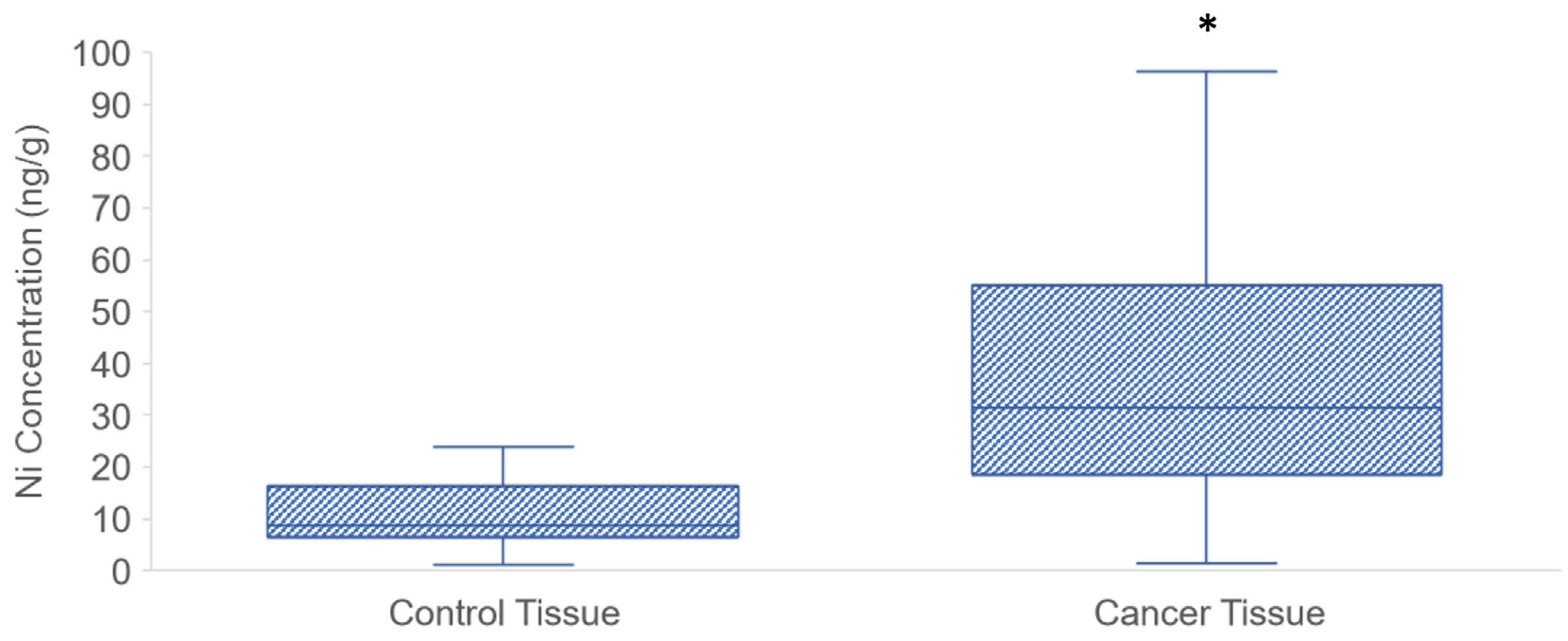
3.1. Ni Concentration in PDAC Tissue Samples
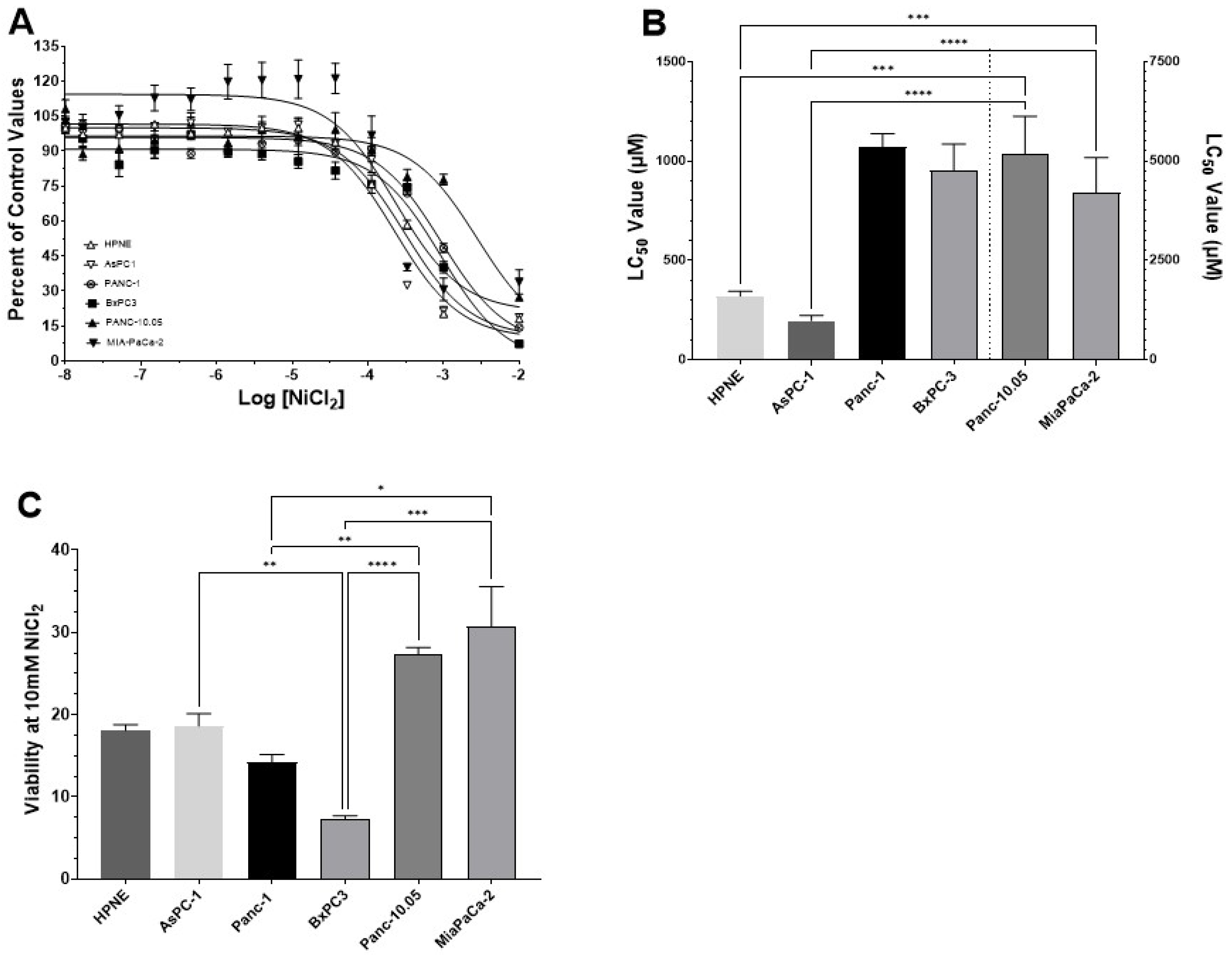
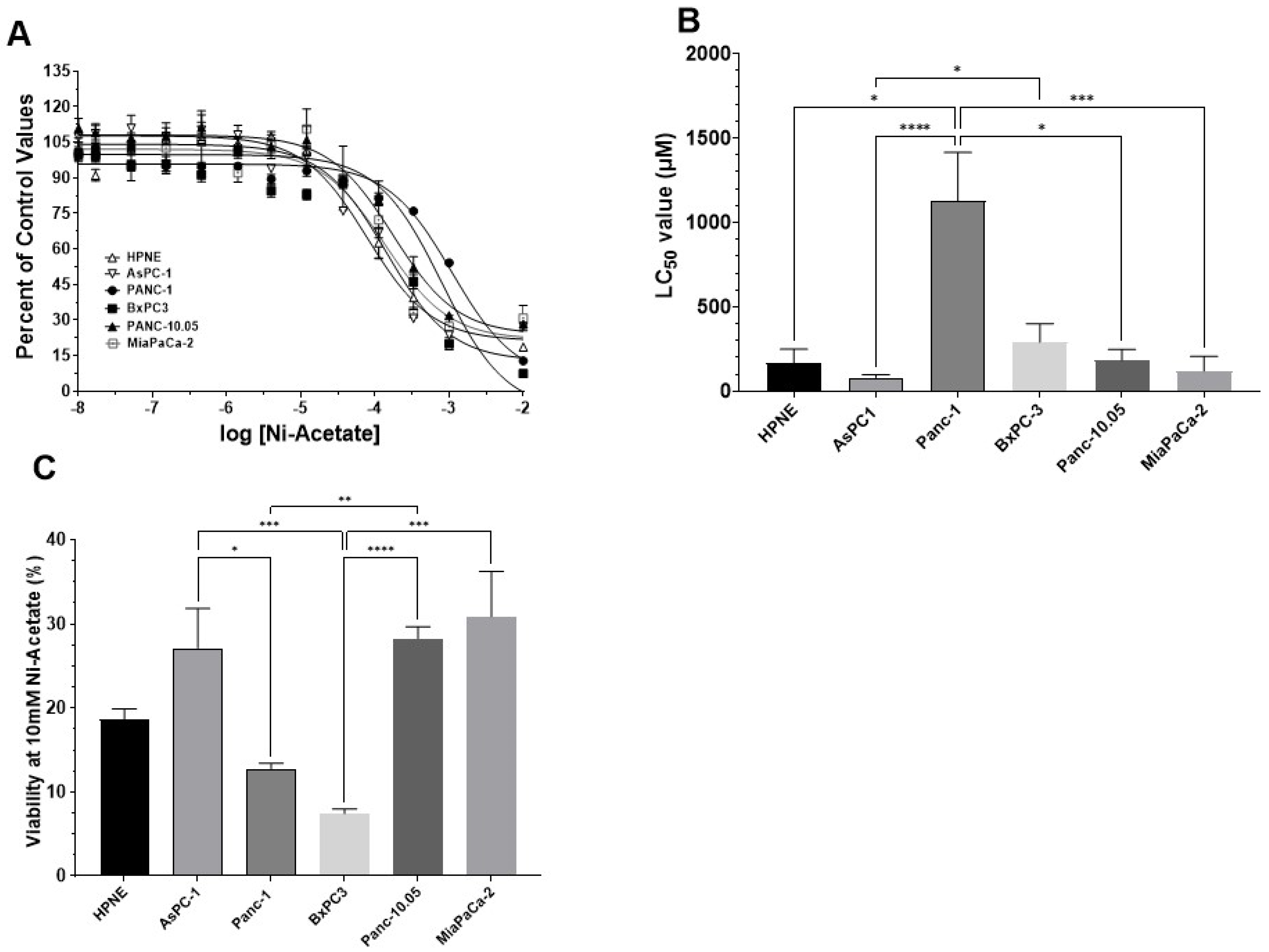
3.2. Determination of LC50 Values for NiCl2 and Ni-Acetate in PDAC Cell Lines
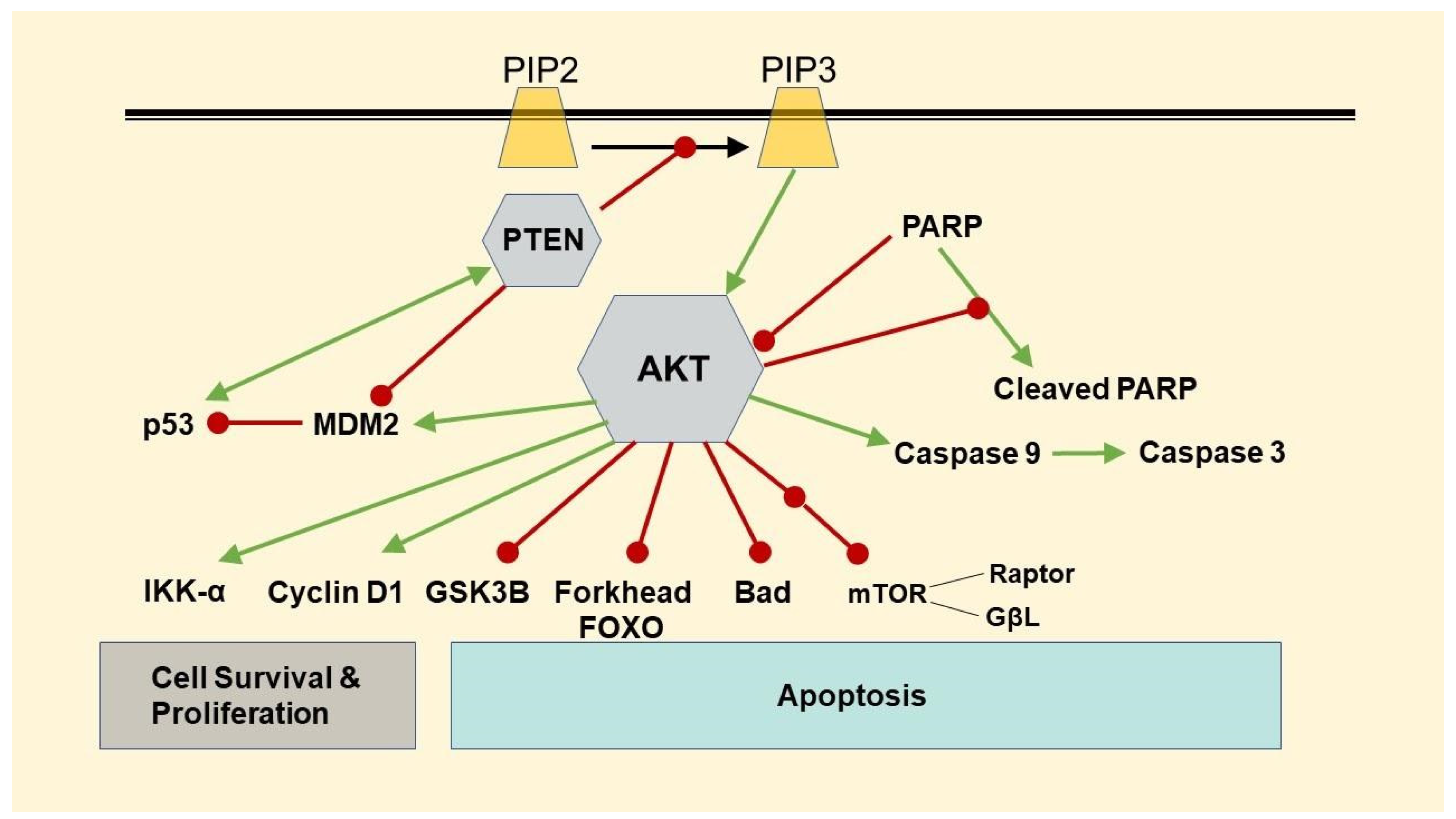
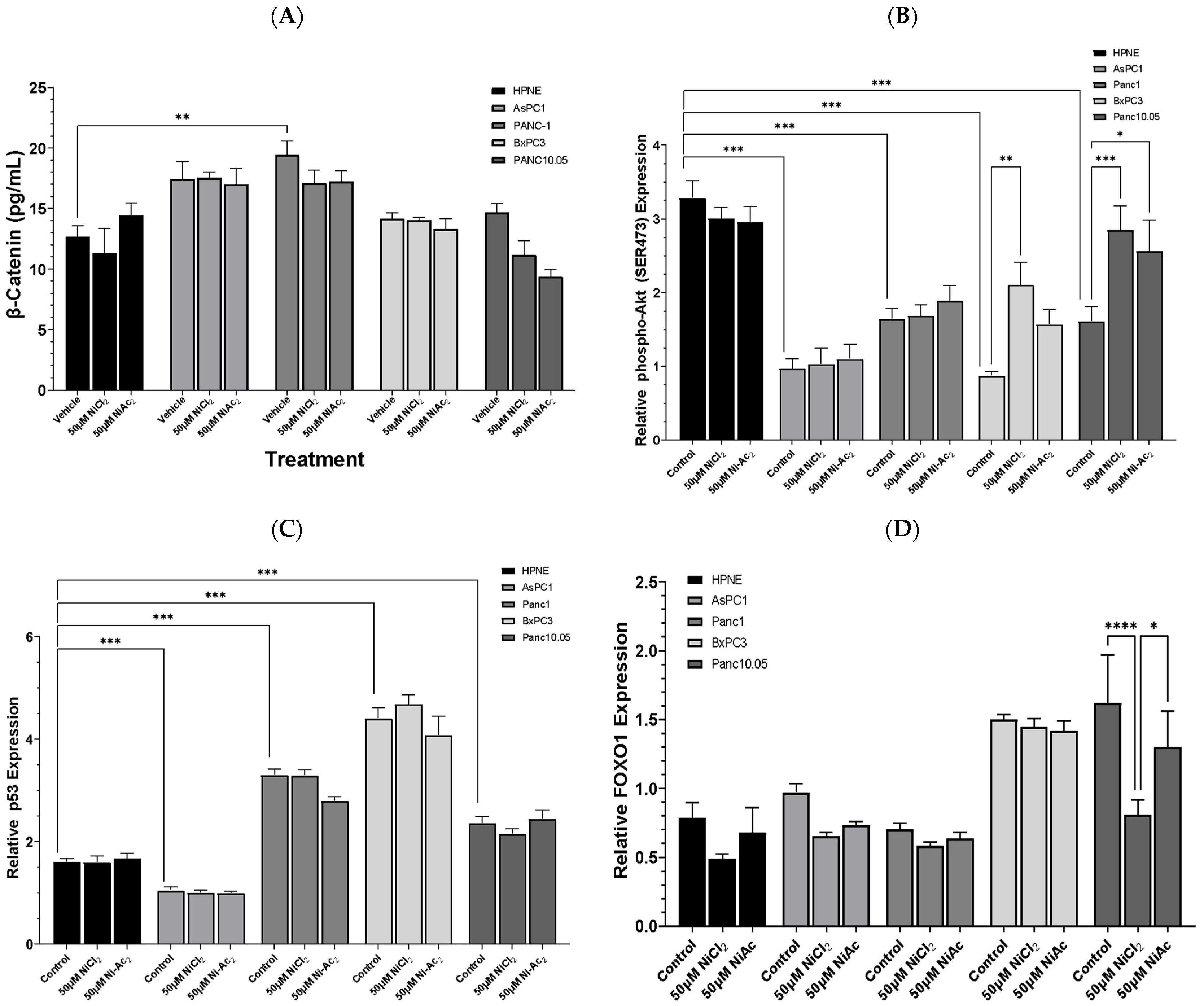
3.3. Changes in Protein Expressions of β-Catenin, Phospho-AKT, p53, and FOXO-1
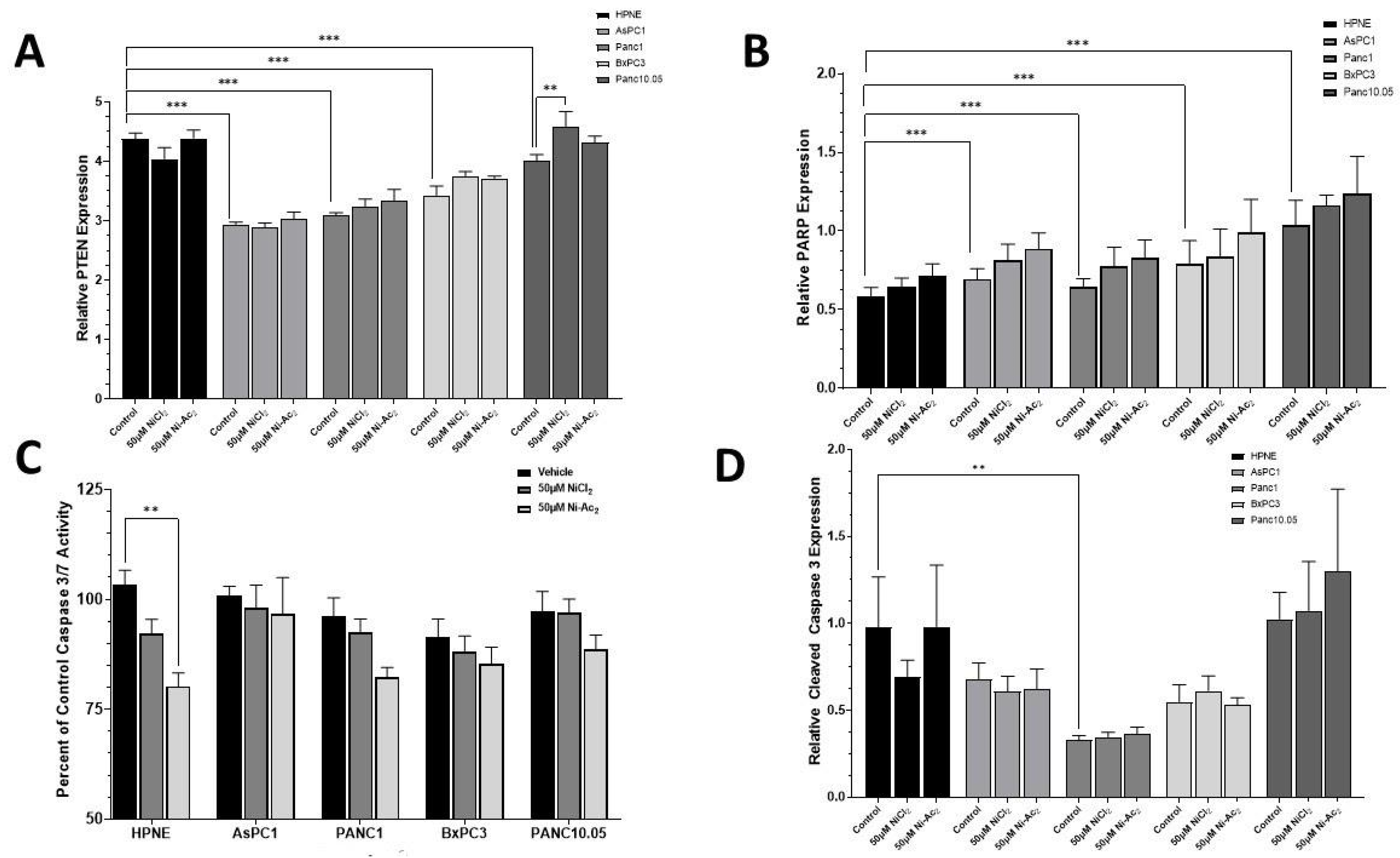
3.4. Changes in Protein Expressions of PTEN and PARP, and Caspase 3/7 Activity
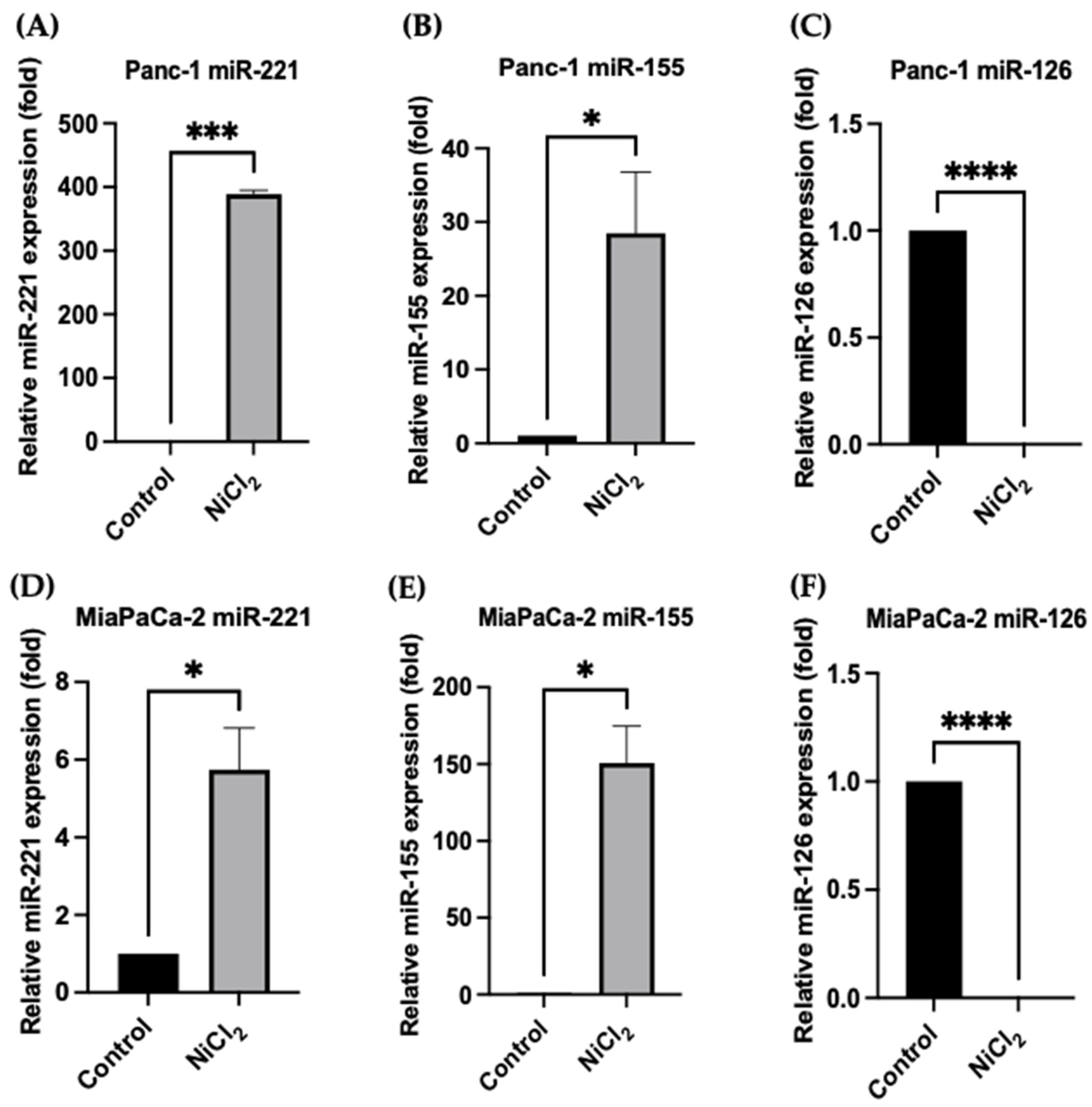
3.5. Changes in miR Expression Levels in Response to Nickel Chloride Treatments in PDAC Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM9 | A disintegrin and metalloproteinase 9 |
| AKT | Ak strain transforming |
| Bad | Bcl-2-associated death promoter |
| CD | Cadmium |
| CDKNs | Cyclin-dependent kinases |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| DMSO | Dimethylsulfoxide |
| EGFR | Epidermal growth factor receptor |
| ERK | Extracellular receptor kinase |
| FBS | Foetal bovine serum |
| FOXO-1 | Forkhead box protein O1 |
| GSK3β | Glycogen Synthase Kinase 3 Beta |
| HER2 | Human epidermal growth factor receptor 2 |
| HIF-1 | Hypoxia-inducible-factor-1 |
| HPNE | Human pancreatic nestin-expressing |
| HRP | Horseradish peroxidase |
| IARC | International Agency for Research on Cancer |
| IKK-ɑ | IkappaB kinase |
| K-RAS | Kirsten rat sarcoma viral homolog |
| LC50 | Lethal concentration 50%: concentration that is lethal to half the population of cells |
| MDM2 | Mouse double minute 2 homolog or E3 ubiquitin-protein ligase |
| miRs/miRNAs | microRNAs |
| mRNAs | Messenger RNAs |
| mTOR | mechanistic target of rapamycin |
| Ni | Nickel |
| Ni-acetate | Nickel acetate |
| NiCl2 | Nickel chloride |
| PanIN | Pancreatic intraepithelial neoplasia |
| PARP | Poly (ADP-ribose) polymerase |
| PBS | Phosphate-buffered saline |
| PDAC | Pancreatic ductal adenocarcinoma |
| phospho-AKT | Phosphorylated protein kinase B (PKB) |
| PI3K | Phosphatidylinositol 3-kinase |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PIP3 | Phosphatidylinositol 3,4,5-trisphosphate |
| PTEN | Phosphatase and tensin homolog |
| RhoA | Ras homolog family member A |
| ROCK | Rho-associated kinase |
| SD | Standard deviation |
| tp53 | Tumour protein 53 |
| UTRs | Untranslated regions |
References
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Gourgou-Bourgade, S.; Bascoul-Mollevi, C.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Boige, V.; et al. Impact of FOLFIRINOX compared with gemcitabine on quality of life in patients with metastatic pancreatic cancer: Results from the PRODIGE 4/ACCORD 11 randomized trial. J. Clin. Oncol. 2013, 31, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobuzio-Donahue, C.A.; Velculescu, V.E.; Wolfgang, C.L.; Hruban, R.H. Genetic basis of pancreas cancer development and progression: Insights from whole-exome and whole-genome sequencing. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 4257–4265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortoglou, M.; Wallace, D.; Buha Djordjevic, A.; Djordjevic, V.; Arisan, E.D.; Uysal-Onganer, P. MicroRNA-Regulated Signaling Pathways: Potential Biomarkers for Pancreatic Ductal Adenocarcinoma. Stresses 2021, 1, 30–47. [Google Scholar] [CrossRef]
- Mortoglou, M.; Tabina, Z.K.; Arisan, E.D.; Kocher, H.M.; Uysal-Onganer, P. Non-coding RNAs in pancreatic ductal adenocarcinoma: New approaches for better diagnosis and therapy. Transl. Oncol. 2021, 14, 101090. [Google Scholar] [CrossRef]
- Barone, E.; Corrado, A.; Gemignani, F.; Landi, S. Environmental risk factors for pancreatic cancer: An update. Arch. Toxicol. 2016, 90, 2617–2642. [Google Scholar] [CrossRef]
- Buha, A.; Wallace, D.; Matovic, V.; Schweitzer, A.; Oluic, B.; Micic, D.; Djordjevic, V. Cadmium Exposure as a Putative Risk Factor for the Development of Pancreatic Cancer: Three Different Lines of Evidence. BioMed Res. Int. 2017, 2017, 1981837. [Google Scholar] [CrossRef]
- Djordjevic, V.; Wallace, D.; Schweitzer, A.; Boricic, N.; Knezevic, D.; Matic, S.; Grubor, N.; Kerkez, M.; Radenkovic, D.; Bulat, Z.; et al. Environmental cadmium exposure and pancreatic cancer: Evidence from case control, animal and in vitro studies. Environ. Int. 2019, 128, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.R.; Buha Djordjevic, A. Heavy metal and pesticide exposure: A mixture of potential toxicity and carcinogenicity. Curr. Opin. Toxicol. 2020, 19, 72–79. [Google Scholar] [CrossRef]
- IARC. Nickel and Nickel Compounds. 2018. Available online: https://monographs.iarc.who.int/wp-content/uploads/2018/06/mono100C-10.pdf (accessed on 12 December 2021).
- Oller, A.R.; Costa, M.; Oberdorster, G. Carcinogenicity assessment of selected nickel compounds. Toxicol. Appl. Pharmacol. 1997, 143, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Chen, Q.Y.; Li, A.H.; Costa, M. The Role of Non-Coding RNAs Involved in Nickel-Induced Lung Carcinogenic Mechanisms. Inorganics 2019, 7, 81. [Google Scholar] [CrossRef] [Green Version]
- Cameron, K.S.; Buchner, V.; Tchounwou, P.B. Exploring the molecular mechanisms of nickel-induced genotoxicity and carcinogenicity: A literature review. Rev. Environ. Health 2011, 26, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Tomás, Á.; Pumarega, J.; Alguacil, J.; Amaral, A.F.S.; Malats, N.; Pallarès, N.; Gasull, M.; Porta, M. Concentrations of trace elements and KRAS mutations in pancreatic ductal adenocarcinoma. Environ. Mol. Mutagen. 2019, 60, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.; Buha-Djordjevic, A.; Benton, A. Toxicity of Organic and Inorganic Nickel in Pancreatic Cell Cultures: Comparison to Cadmium. Arh. farm. 2020, 70, 344–359. [Google Scholar] [CrossRef]
- Yamamoto, S.; Tomita, Y.; Hoshida, Y.; Morooka, T.; Nagano, H.; Dono, K.; Umeshita, K.; Sakon, M.; Ishikawa, O.; Ohigashi, H.; et al. Prognostic Significance of Activated Akt Expression in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2004, 10, 2846–2850. [Google Scholar] [CrossRef] [Green Version]
- Massihnia, D.; Avan, A.; Funel, N.; Maftouh, M.; Van Krieken, A.; Granchi, C.; Raktoe, R.; Boggi, U.; Aicher, B.; Minutolo, F.; et al. Phospho-Akt overexpression is prognostic and can be used to tailor the synergistic interaction of Akt inhibitors with gemcitabine in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 9. [Google Scholar] [CrossRef]
- Soares, H.P.; Ni, Y.; Kisfalvi, K.; Sinnett-Smith, J.; Rozengurt, E. Different Patterns of Akt and ERK Feedback Activation in Response to Rapamycin, Active-Site mTOR Inhibitors and Metformin in Pancreatic Cancer Cells. PLoS ONE 2013, 8, e57289. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, S.; Hosseini, M.; Shahidsales, S.; Maftouh, M.; Ferns, G.A.; Ghayour-Mobarhan, M.; Hassanian, S.M.; Avan, A. Targeting the Akt/PI3K Signaling Pathway as a Potential Therapeutic Strategy for the Treatment of Pancreatic Cancer. Curr. Med. Chem. 2017, 24, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.L.; Dubois, C.L.; Schaeffer, D.F.; Samani, A.; Taghizadeh, F.; Cowan, R.W.; Rhim, A.D.; Stiles, B.L.; Valasek, M.; Sander, M. Loss of Pten and Activation of Kras Synergistically Induce Formation of Intraductal Papillary Mucinous Neoplasia From Pancreatic Ductal Cells in Mice. Gastroenterology 2018, 154, 1509–1523. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Elpek, K.G.; Vinjamoori, A.; Zimmerman, S.M.; Chu, G.C.; Yan, H.; Fletcher-Sananikone, E.; Zhang, H.; Liu, Y.; Wang, W.; et al. PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-κB-cytokine network. Cancer Discov. 2011, 1, 158–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P., IV; Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor β signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [Green Version]
- Jahedi, H.; Fahud, A.L.; Lim, C.L. Role of p53 family isoforms in enhancing aggressiveness and chemoresistance in pancreatic cancer (Review). World Acad. Sci. J. 2019, 1, 236–246. [Google Scholar] [CrossRef]
- Moffat, G.T.; O’Reilly, E.M. The role of PARP inhibitors in germline BRCA-associated pancreatic ductal adenocarcinoma. Clin. Adv. Hematol. Oncol. 2020, 18, 168–179. [Google Scholar]
- Chi, J.; Chung, S.Y.; Parakrama, R.; Fayyaz, F.; Jose, J.; Saif, M.W. The role of PARP inhibitors in BRCA mutated pancreatic cancer. Therap. Adv. Gastroenterol. 2021, 14, 1–11. [Google Scholar] [CrossRef]
- Gallyas, F.; Sumegi, B.; Szabo, C. Role of akt activation in PARP inhibitor resistance in cancer. Cancers 2020, 12, 532. [Google Scholar] [CrossRef] [Green Version]
- Essers, M.A.G.; De Vries-Smits, L.M.M.; Barker, N.; Polderman, P.E.; Burgering, B.M.T.; Korswagen, K.C. Functional Interaction Between ß-Catenin and FOXO in Oxidative Stress Signaling. Science 2005, 308, 1181–1184. [Google Scholar] [CrossRef]
- Song, W.; Wang, L.; Wang, L.; Li, Q. Interplay of miR-21 and FoxO1 modulates growth of pancreatic ductal adenocarcinoma. Tumor Biol. 2015, 36, 4741–4745. [Google Scholar] [CrossRef]
- Al-Zoughbi, W.; Schauer, S.; Pichler, M.; Hoefler, G. Early Loss of Forkhead Transcription Factor, O Subgroup, Member 1 Protein in the Development of Pancreatic Ductal Adenocarcinoma. Pathobiology 2018, 85, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Dong, X.; Wang, L.; Xue, Y.; Jia, X.; Song, W.; Li, Q. MiR-27a-regulated FOXO1 promotes pancreatic ductal adenocarcinoma cell progression by enhancing Wnt/β-catenin signaling activity. Am. J. Transl. Res. 2019, 11, 3069–3080. [Google Scholar] [PubMed]
- Song, W.; Li, Q.; Wang, L.; Huang, W.; Wang, L. FoxO1-negative cells are cancer stem-like cells in pancreatic ductal adenocarcinoma. Sci. Rep. 2015, 5, 10081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, J.; Wang, F.; Liu, C.; Dong, X.; Xue, Y.; Jia, X.; Song, W.; Li, Q. FOXO1-regulated lncRNA LINC01197 inhibits pancreatic adenocarcinoma cell proliferation by restraining Wnt/β-catenin signaling. J. Exp. Clin. Cancer Res. 2019, 38, 179. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Amirkhah, R.; Schmitz, U.; Linnebacher, M.; Wolkenhauer, O.; Farazmand, A. MicroRNA-mRNA interactions in colorectal cancer and their role in tumour progression. Genes Chromosomes Cancer 2015, 54, 129–141. [Google Scholar] [CrossRef]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef]
- Meltzer, P.S. Cancer genomics: Small RNAs with big impacts. Nature 2005, 435, 745–746. [Google Scholar] [CrossRef]
- Słotwiński, R.; Lech, G.; Słotwińska, S.M. MicroRNAs in pancreatic cancer diagnosis and therapy. Cent.-Eur. J. Immunol. 2018, 43, 314–324. [Google Scholar] [CrossRef] [Green Version]
- Uysal-Onganer, P.; D’Alessio, S.; Mortoglou, M.; Kraev, I.; Lange, S. Peptidylarginine Deiminase Inhibitor Application, Using Cl-Amidine, PAD2, PAD3 and PAD4 Isozyme-Specific Inhibitors in Pancreatic Cancer Cells, Reveals Roles for PAD2 and PAD3 in Cancer Invasion and Modulation of Extracellular Vesicle Signatures. Int. J. Mol. Sci. 2021, 22, L1396. [Google Scholar] [CrossRef]
- Uysal-Onganer, P.; MacLatchy, A.; Mahmoud, R.; Kraev, I.; Thompson, P.R.; Inal, J.M.; Lange, S. Peptidylarginine Deiminase Isozyme-Specific PAD2, PAD3 and PAD4 Inhibitors Differentially Modulate Extracellular Vesicle Signatures and Cell Invasion in Two Glioblastoma Multiforme Cell Lines. Int. J. Mol. Sci. 2020, 21, 1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dart, D.A.; Arisan, D.E.; Owen, S.; Hao, C.; Jiang, W.G.; Uysal-Onganer, P. Wnt-11 Expression Promotes Invasiveness and Correlates with Survival in Human Pancreatic Ductal Adeno Carcinoma. Genes 2019, 10, 921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arisan, E.D.; Rencuzogullari, O.; Cieza-Borrella, C.; Miralles Arenas, F.; Dwek, M.; Lange, S.; Uysal-Onganer, P. MiR-21 Is Required for the Epithelial–Mesenchymal Transition in MDA-MB-231 Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 1557. [Google Scholar] [CrossRef]
- Lanning, B.; Webber, J.; Uysal-Onganer, P.; Jiang, W.G.; Clayton, A.; Dart, D.A. Prostate Cancer Cell Extracellular Vesicles Increase Mineralisation of Bone Osteoblast Precursor Cells in an In Vitro Model. Biology 2021, 10, 318. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, A.; Hong, S.M.; Hruban, R.H.; Goggins, M. MicroRNA alterations of pancreatic intraepithelial neoplasias. Clin. Cancer Res. 2012, 18, 981–992. [Google Scholar] [CrossRef] [Green Version]
- Bloomston, M.; Frankel, W.L.; Petrocca, F.; Volinia, S.; Alder, H.; Hagan, J.P.; Liu, C.G.; Bhatt, D.; Taccioli, C.; Croce, C.M. MicroRNA Expression Patterns to Differentiate Pancreatic Adenocarcinoma from Normal Pancreas and Chronic Pancreatitis. JAMA 2007, 297, 1901–1908. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Liu, J.; Wang, L.; Wu, H.; Zhou, C.; Zhu, H.; Xu, N.; Xie, Y. Association of Serum MicroRNA Expression in Hepatocellular Carcinomas Treated with Transarterial Chemoembolization and Patient Survival. PLoS ONE 2014, 9, e109347. [Google Scholar] [CrossRef]
- Tao, K.; Yang, J.; Guo, Z.; Hu, Y.; Sheng, H.; Gao, H.; Yu, H. Prognostic value of mir-221-3p, mir-342-3p and mir-491-5p expression in colon cancer. Am. J. Transl. Res. 2014, 6, 391–401. [Google Scholar]
- Yau, T.O.; Wu, C.W.; Dong, Y.; Tang, C.M.; Ng, S.S.M.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. microRNA-221 and microRNA-18a identification in stool as potential biomarkers for the non-invasive diagnosis of colorectal carcinoma. Br. J. Cancer 2014, 111, 1765–1771. [Google Scholar] [CrossRef]
- Zheng, Q.; Peskoe, S.B.; Ribas, J.; Rafiqi, F.; Kudrolli, T.; Meeker, A.K.; de Marzo, A.M.; Platz, E.A.; Lupold, S.E. Investigation of mir-21, mir-141, and mir-221 expression levels in prostate adenocarcinoma for associated risk of recurrence after radical prosta-tectomy. Prostate 2014, 74, 1655–1662. [Google Scholar] [CrossRef] [Green Version]
- Papaconstantinou, I.G.; Manta, A.; Gazouli, M.; Lyberopoulou, A.; Lykoulis, P.M.; Polymeneas, G.; Voros, D. Expression of microRNAs in patients with pancreatic cancer and its prognostic significance. Pancreas 2013, 42, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 2140–2141. [Google Scholar] [CrossRef] [PubMed]
- Greither, T.; Grochola, L.F.; Udelnow, A.; Lautenschläger, C.; Wurl, P.; Taubert, H. Elevated expression of microRNAs 155, 203, 210 and 222 in pancreatic tumours is associated with poorer survival. Int. J. Cancer 2010, 126, 73–80. [Google Scholar] [CrossRef]
- Feng, R.; Chen, X.; Yu, Y.; Su, L.; Yu, B.; Li, J.; Cai, Q.; Yan, M.; Liu, B.; Zhu, Z. miR-126 functions as a tumour suppressor in human gastric cancer. Cancer Lett. 2010, 298, 50–63. [Google Scholar] [CrossRef]
- Sempere, L.F.; Preis, M.; Yezefski, T.; Ouyang, H.; Suriawinata, A.A.; Silahtaroglu, A.; Conejo-Garcia, J.R.; Kauppinen, S.; Wells, W.; Korc, M. Fluorescence-based codetection with protein markers reveals distinct cellular compartments for altered MicroRNA expression in solid tumours. Clin. Cancer Res. 2010, 16, 4246–4255. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Huang, Z.; Xu, L.; Zhu, M.; Zhang, L.; Zhang, H.; Wang, X.; Li, H.; Zhu, W.; Shu, Y.; et al. A panel of 13-miRNA signature as a potential biomarker for predicting survival in pancreatic cancer. Oncotarget 2016, 7, 69616–69624. [Google Scholar] [CrossRef] [Green Version]
- Jiao, L.R.; Frampton, A.E.; Jacob, J.; Pellegrino, L.; Krell, J.; Giamas, G.; Tsim, N.; Vlavianos, P.; Cohen, P.; Ahmad, R.; et al. MicroRNAs targeting oncogenes are down-regulated in pancreatic malignant transfor-mation from benign tumours. PLoS ONE 2012, 7, e32068. [Google Scholar] [CrossRef] [Green Version]
- Garajová, I.; le Large, T.Y.; Frampton, A.E.; Rolfo, C.; Voortman, J.; Giovannetti, E. Molecular Mechanisms Underlying the Role of MicroRNAs in the Chemoresistance of Pancreatic Cancer. BioMed Res. Int. 2014, 2014, 1–17. [Google Scholar] [CrossRef]
- Mortoglou, M.; Buha Djordjevic, A.; Djordjevic, V.; Collins, H.; York, L.; Mani, K.; Valle, E.; Wallace, D.; Uysal-Onganger, P. Role of microRNAs in response to cadmium chloride in pancreatic ductal adenocarcinoma. Arch. Toxicol. 2021, 96, 467–485. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Komoto, M.; Nakata, B.; Amano, R.; Yamada, N.; Yashiro, M.; Ohira, M.; Wakasa, K.; Hirakawa, K. HER2 overexpression correlates with survival after curative resection of pancreatic cancer. Cancer Sci. 2009, 100, 1243–1247. [Google Scholar] [CrossRef]
- Su, A.; He, S.; Tian, B.; Hu, W.; Zhang, Z. MicroRNA-221 Mediates the Effects of PDGF-BB on Migration, Proliferation, and the Epithelial-Mesenchymal Transition in Pancreatic Cancer Cells. PLoS ONE 2013, 8, e71309. [Google Scholar] [CrossRef] [Green Version]
- Yasaei, H.; Gilham, E.; Pickles, J.C.; Roberts, T.P.; O’Donovan, M.; Newbold, R.F. Carcinogen-specific mutational and epigenetic alterations in INK4A, INK4B and p53 tumour-suppressor genes drive induced senescence bypass in normal diploid mammalian cells. Oncogene 2013, 32, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Salnikow, K.; Davidson, T.; Zhang, Q.; Chen, L.C.; Su, W.; Costa, M. The involvement of hypoxia-inducible transcription factor-1-dependent pathway in nickel carcinogenesis. Cancer Res. 2003, 63, 3524–3530. [Google Scholar]
- Rezuke, W.N.; Knight, J.A.; Sunderman, F.W. Reference Values for Nickel Concentrations in Human Tissues and Bile. Am. J. Ind. Med. 1987, 11, 419–426. [Google Scholar] [CrossRef]
- Amaral, A.F.S.; Porta, M.; Silverman, D.T.; Milne, R.L.; Kogevinas, M.; Rothman, N.; Cantor, K.P.; Jackson, B.P.; Pumarega, J.A.; López, T.; et al. Pancreatic cancer risk and levels of trace elements. Gut 2012, 61, 1583–1588. [Google Scholar] [CrossRef] [Green Version]
- Salcedo-Bellido, I.; Gutiérrez-González, E.; García-Esquinas, E.; Fernández de Larrea-Baz, N.; Navas-Acien, A.; Téllez-Plaza, M.; Pastor-Barriuso, R.; Lope, V.; Gómez-Ariza, J.L.; García-Barrera, T.; et al. Toxic metals in toenails as biomarkers of exposure: A review. Environ. Res. 2021, 197, 111028. [Google Scholar] [CrossRef]
- Vinceti, M.; Venturelli, M.; Sighinolfi, C.; Trerotoli, P.; Bonvicini, F.; Ferrari, A.; Bianchi, G.; Serio, G.; Bergomi, M.; Vivoli, G. Case-control study of toenail cadmium and prostate cancer risk in Italy. Sci. Total Environ. 2007, 373, 77–81. [Google Scholar] [CrossRef]
- Anđelković, M.; Buha Đorđević, A.; Antonijević Miljaković, E.; Javorac, D.; Čolaković, N.; Oprić, S.; Petročević, S.; Granić, M.; Kotur-Stevuljević, J.; Antonijević, B.; et al. Cadmium tissue level in women diagnosed with breast cancer—A case control study. Environ. Res. 2021, 199, 111300. [Google Scholar] [CrossRef]
- Pamphletti, R.; Satgunaseelan, L.; Jew, S.K.; Doble, P.A.; Bishop, D.P. Elemental bioimaging shows mercury and other toxic metals in normal breast tissue and in breast cancers. PLoS ONE 2020, 15, e0228226. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Gawad, M.; Elsobky, E.; Abdel-Hameed, M.; Abdel-Rahim, M.; Harraz, A.; Shokeir, A.A.; Ali-El-Dein, B. Quantitative and qualitative evaluation of toxic metals and trace elements in the tissues of renal cell carcinoma compared with the adjacent non-cancerous and control kidney tissues. Environ. Sci. Pollut. Res. Int. 2020, 27, 30460–30467. [Google Scholar] [CrossRef]
- Rahil-Khazen, R.; Bolann, B.J.; Myking, A.; Ulvik, R.J. Multi-element analysis of trace element levels in human autopsy tissues by using inductively coupled atomic emission spectrometry technique (ICP-AES). J. Trace Elem. Med. Biol. 2002, 16, 15–25. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, N.; Zhang, Y.; Wu, Q.; Chen, R.; Gao, J.; Chang, P.; Liu, Q.; Fan, T.; Li, J.; et al. Element contents in organs and tissues od chinese adult men. Chin. Med. Sci. J. 2007, 22, 71–82. [Google Scholar] [PubMed]
- Lo, A.-C.; Soliman, A.S.; El-Ghawalby, N.; Abdel-Wahab, M.; Fathy, O.; Khaled, H.M.; Omar, S.; Hamilton, S.R.; Greenson, J.K.; Abbruzzese, J.L. Lifestyle, occupational, and reproductive factors in relation to pancreatic cancer risk. Pancreas 2007, 35, 120–129. [Google Scholar] [CrossRef]
- Fryzek, J.P.; Garabrant, D.H.; Harlow, S.D.; Severson, R.K.; Gillespie, B.W.; Schenk, M.; Schottenfeld, D. A case–control study of self-reported exposures to pesticides and pancreas cancer in southeastern Michigan. Int. J. Cancer 1997, 72, 62–67. [Google Scholar] [CrossRef]
- Luckett, B.G.; Su, L.J.; Rood, J.C.; Fontham, E.T. Cadmium exposure and pancreatic cancer in south Louisiana. J. Environ. Public Health 2012, 2012, 180–186. [Google Scholar] [CrossRef]
- Antwi, S.O.; Eckert, E.C.; Sabaque, S.V.; Leof, E.R.; Hawthorne, K.M.; Bamlet, W.R.; Chaffee, K.G.; Oberg, A.L.; Petersen, G.M. Exposure to environmental chemicals and heavy metals, and risk of pancreatic cancer. Cancer Causes Control. 2015, 26, 1583–1591. [Google Scholar] [CrossRef] [Green Version]
- Ojajarvi, A.; Partanen, T.; Ahlbom, A.; Hakulinen, T.; Kauppinen, T.; Weiderpass, E.; Wesseling, C. Estimating the relative risk of pancreatic cancer associated with exposure agents in job title data in a hierarchical Bayesian meta-analysis. Scand. J. Work Environ. Health 2007, 33, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Alguacil, J.; Kauppinen, T.; Porta, M.; Partanen, T.; Malats, N.; Kogevinas, M.; Benavides, F.G.; Obiols, J.; Bernal, F.; Rifà, J.; et al. Risk of pancreatic cancer and occupational exposures in Spain. PANKRAS II Study Group. Ann. Occup. Hyg. 2000, 44, 391–403. [Google Scholar] [CrossRef]
- Andreotti, G.; Silverman, D.T. Occupational risk factors and pancreatic cancer: A review of recent findings. Mol. Carcinog. 2012, 51, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Santibañez, M.; Vioque, J.; Alguacil, J.; de la Hera, M.G.; Moreno-Osset, E.; Carrato, A.; Porta, M.; Kauppinen, T. Occupational exposures and risk of pancreatic cancer. Eur. J. Epidemiol. 2010, 25, 721–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojajarvi, I.A.; Partanen, T.J.; Ahlbom, A.; Boffetta, P.; Hakulinen, T.; Jourenkova, N.; Kauppinen, T.P.; Kogevinas, M.; Porta, M.; Vainio, H.U.; et al. Occupational exposures and pancreatic cancer: A meta-analysis. Occup. Environ. Med. 2000, 57, 316–324. [Google Scholar] [CrossRef] [Green Version]
- Arita, A.; Costa, M. Epigenetics in metal carcinogenesis: Nickel, arsenic, chromium and cadmium. Metallomics 2009, 1, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jornot, L.; Petersen, H.; Junod, A.F. Hydrogen peroxide-induced DNA damage is independent of nuclear calcium but dependent on redox-active ions. Biochem. J. 1998, 335, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Bucher, J.R.; Tien, M.; Aust, S.D. The requirement for ferric in the initiation of lipid peroxidation by chelated ferrous iron. Biochem. Biophys. Res. Commum. 1983, 11, 777–784. [Google Scholar] [CrossRef]
- Valko, M.; Morris, H.; Cronin, M.T.D. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Wang, X.; Zhao, L.; Chen, K.; Deng, J. Nickel chloride (NiCl2) in hepatic toxicity: Apoptosis, G2/M cell cycle arrest and inflammatory response. Aging 2016, 8, 3009–3027. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Deng, J.; Liu, H.; Jian, Z.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, X.; Wang, X.; et al. Nickel carcinogenesis mechanism: Cell cycle dysregulation. Environm. Sci. Pollut. Res. 2021, 28, 4893–4901. [Google Scholar] [CrossRef]
- Ota, H.; Shionome, T.; Suguro, H.; Saito, S.; Ueki, Y.A.; Asano, M. Nickel chloride administration prevents the growth of oral squamous cell carcinoma. Oncotarget 2018, 9, 24107–24121. [Google Scholar] [CrossRef] [Green Version]
- Eckers, A.; Reimann, K.; Klotz, L.-O. Nickel and copper ion-induced stress signalign in human hepatoma cells: Analysis of phosphoinositide 3′-kinase/Akt signaling. Biometals 2009, 22, 307–316. [Google Scholar] [CrossRef]
- Wang, J.; Chen, S.; Wang, M.; Ko, J.; Wu, C.; Chen, C.; Lin, H.; Chang, Y. Nickel-induced VEGF expression via regulation of Akt, ERK1/2, NFκB, and AMPK pathways in H460 cells. Environ. Toxicol. 2019, 34, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Pan, J.-J.; Chang, Q.-S.; Wang, X.; Son, Y.-O.; Liu, J.; Zhang, Z.; Bi, Y.-Y. Activation of Akt/GSK3β and Akt/Bcl-2 signaling pathways in nickel-transformed BEAS-2B cells. Int. J. Oncol. 2011, 39, 1285–1294. [Google Scholar] [CrossRef] [Green Version]
- Terpilowska, S.; Siwicka-Gieroba, D.; Siwicki, A.K. Cell viability in normal fibroblasts and liver cancer cells after treatment with iron (III), nickel (II), and their mixture. J. Vet. Res. 2018, 62, 535–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.C.; Yang, C.Y.; Hung, D.Z.; Su, C.C.; Chen, K.L.; Yen, C.C.; Ho, T.J.; Su, Y.C.; Huang, C.F.; Chen, C.H.; et al. Nickel(II) induced JNK activation-regulated mitochondria-dependent apoptotic pathway leading to cultured rat pancreatic β-cell death. Toxicology 2011, 289, 103–111. [Google Scholar] [CrossRef]
- Long, P.M.; Tighe, S.W.; Driscoll, H.E.; Moffett, J.R.; Namboodiri, A.M.A.; Viapiano, M.S.; Lawler, S.E.; Jaworski, D.M. Acetate supplementation induces growth arrest of NG2/PDGFRα-positive oligodendroglioma-derived tumor-initiating cells. PLoS ONE 2013, 8, e80714. [Google Scholar] [CrossRef] [Green Version]
- Marques, C.; Oliveira, C.S.F.; Alves, S.; Chaves, S.R.; Coutinho, O.P.; Côrte-Real, M.; Preto, A. Acetate-induced apoptosis in colorectal carcinoma cells involves lysosomal membrane permeabilization and cathepsin D release. Cell Death Dis. 2013, 4, e507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Bi, L.; Chi, Y.; Aoki, K.; Misumi, J. Effect of sodium acetate on cell proliferation and induction of proinflammatory cytokines: A preliminary evaluation. Food Chem. Toxicol. 2005, 43, 1773–1780. [Google Scholar] [CrossRef]
- Guo, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Wang, X.; Zhao, L.; Wu, B.; Chen, K.; Deng, J. Nickel chloride-induced apoptosis via mitochondria-and Fas-mediated caspase-dependent pathways in broiler chickens. Oncotarget 2016, 7, 79747–79760. [Google Scholar] [CrossRef] [Green Version]
- Daoud, A.Z.; Mulholland, E.J.; Cole, G.; McCarthy, H.O. MicroRNAs in Pancreatic Cancer: Biomarkers, prognostic, anD therapeutic modulators. BMC Cancer 2019, 19, 1130. [Google Scholar] [CrossRef] [Green Version]
- Humphries, B.; Wang, Z.; Yang, C. The role of microRNAs in metal carcinogen-induced cell malignant transformation and tumorigenesis. Food Chem Toxicol. 2016, 98, 58–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Shamy, M.; Costa, M. Nickel and epigenetic gene silencing. Genes 2013, 4, 583–595. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Lu, Y.; Xu, S.; Mao, L.; Zhang, L.; Duan, W.; Liu, C.; Pi, H.; Zhang, Y.; Zhong, M.; et al. MiRNA-210 modulates a nickel-induced cellular energy metabolism shift by repressing the iron-sulfur cluster assembly proteins ISCU1/2 in Neuro-2a cells. Cell Death Dis. 2014, 5, e1090. [Google Scholar] [CrossRef] [PubMed]
- Kulshreshtha, R.; Ferracin, M.; Wojcik, S.E.; Garzon, R.; Alder, H.; Agosto-Perez, F.J.; Davuluri, R.; Liu, C.G.; Croce, C.M.; Negrini, M.; et al. A microRNA signature of hypoxia. Mol. Cell. Biol. 2007, 27, 1859–1867. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Liu, L.Z.; Jiang, B.H. Dysregulation of microRNAs in metal-induced angiogenesis and carcinogenesis. Semin. Cancer Biol. 2021, 76, 279–286. [Google Scholar] [CrossRef]
- Li, J.; Davidson, G.; Huang, Y.; Jiang, B.H.; Shi, X.; Costa, M.; Huang, C. Nickel compounds act through phosphatidylinositol-3-kinase/Akt-dependent, p70(S6k)-independent pathway to induce hypoxia inducible factor transactivation and Cap43 expression in mouse epidermal Cl41 cells. Cancer Res. 2004, 64, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Baradaran, B.; Shahbazi, R.; Khordadmehr, M. Dysregulation of key microRNAs in pancreatic cancer development. Biomedicine & Pharmacotherapy 2019, 109, 1008–1015. [Google Scholar]
- Uddin, M.H.; Al-Hallak, M.N.; Philip, P.A.; Mohammad, R.M.; Viola, N.; Wagner, K.-U.; Azmi, A.S. Exosomal microRNA in Pancreatic Cancer Diagnosis, Prognosis, and Treatment: From Bench to Bedside. Cancers 2021, 13, 2777. [Google Scholar] [CrossRef]
- Almas, I.; Afzal, S.; Idrees, M.; Ashraf, M.U.; Amin, I.; Shahid, M.; Zahid, K.; Zahid, S. Role of circulatory microRNAs in the pathogenesis of hepatitis C virus. Virusdisease 2017, 28, 360–367. [Google Scholar] [CrossRef]
- Han, Z.-B.; Chen, H.-Y.; Fan, J.-W.; Wu, J.-Y.; Tang, H.-M.; Peng, Z.-H. Up-regulation of microRNA-155 promotes cancer cell invasion and predicts poor survival of hepatocellular carcinoma following liver transplantation. J. Cancer Res. Clin. Oncol. 2012, 138, 153–161. [Google Scholar] [CrossRef]
- Sasi, W.; Jiang, W.G.; Sharma, A.; Mokbel, K. Higher expression levels of SOCS 1,3,4,7 are associated with earlier tumour stage and better clinical outcome in human breast cancer. BMC Cancer 2010, 10, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Li, H.; Yu, J.-P.; Wang, S.E.; Ren, X.-B. Role of SOCS1 in tumor progression and therapeutic application. Int. J. Cancer 2012, 130, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Guo, Y.Q.; Li, X.Y.; Deng, M.Z.; Shen, Z.H.; Bo, C.B.; Dai, Y.F.; Huang, M.Y.; Yang, Z.Y.; Quan, Y.S.; et al. MicroRNA-126 inhibits colon cancer cell proliferation and invasion by targeting the chemokine (C-X-C motif) receptor 4 and Ras homolog gene family, member A, signaling pathway. Oncotarget 2016, 7, 60230–60244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.; Xiong, S.; Chen, Q.; Zhu, S.; Zhou, X. Novel role of microRNA-126 in digestive system cancers: From bench to bedside (Review). Oncol. Lett. 2019, 17, 31–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metals (µM ± S.D.) | ||
|---|---|---|
| Cell Type | NiCl2 | Ni-Acetate |
| HPNE | 319.8 ± 24.1 | 167.3 ± 28.7 |
| AsPC-1 | 196.3 ± 26.9 | 81.3 ± 6.1 |
| Panc-1 | 1068 ± 69.3 # | 1128 ± 101.3 †,* |
| BxPC-3 | 953.5 ± 132.5 * | 290.6 ± 38.8 # |
| Panc-10.05 | >5000 † | 183.5 ± 22.8 |
| MiaPaCa-2 | 4209 ± 875 † | 119.3 ± 33.1 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mortoglou, M.; Manić, L.; Buha Djordjevic, A.; Bulat, Z.; Đorđević, V.; Manis, K.; Valle, E.; York, L.; Wallace, D.; Uysal-Onganer, P. Nickel’s Role in Pancreatic Ductal Adenocarcinoma: Potential Involvement of microRNAs. Toxics 2022, 10, 148. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics10030148
Mortoglou M, Manić L, Buha Djordjevic A, Bulat Z, Đorđević V, Manis K, Valle E, York L, Wallace D, Uysal-Onganer P. Nickel’s Role in Pancreatic Ductal Adenocarcinoma: Potential Involvement of microRNAs. Toxics. 2022; 10(3):148. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics10030148
Chicago/Turabian StyleMortoglou, Maria, Luka Manić, Aleksandra Buha Djordjevic, Zorica Bulat, Vladimir Đorđević, Katherine Manis, Elizabeth Valle, Lauren York, David Wallace, and Pinar Uysal-Onganer. 2022. "Nickel’s Role in Pancreatic Ductal Adenocarcinoma: Potential Involvement of microRNAs" Toxics 10, no. 3: 148. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics10030148