
Current View in Platinum Drug Mechanisms of Peripheral Neurotoxicity

Abstract
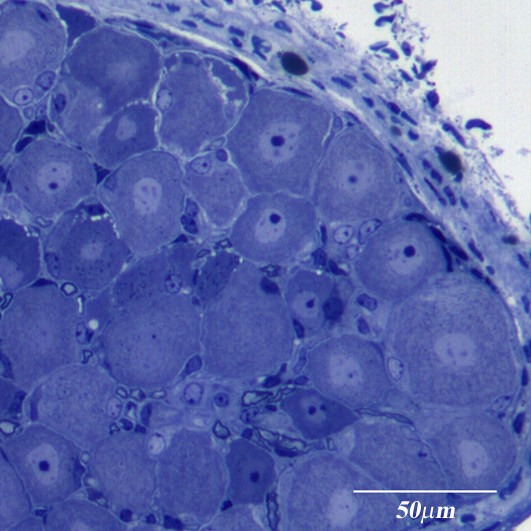
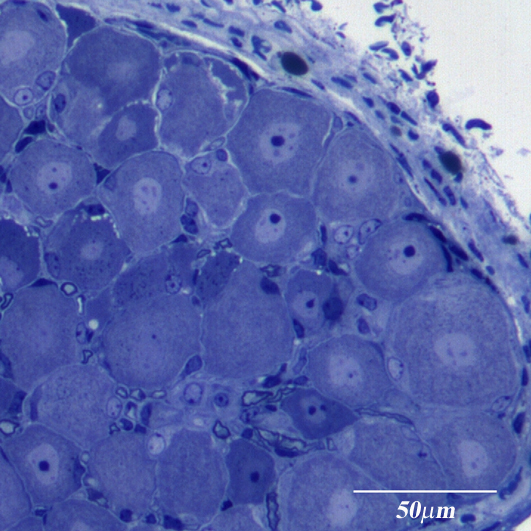
:
{kind=link}
1. Introduction
2. Platinum Drug General Toxicity
3. Platinum Drug Neurotoxicity
4. Main mechanisms of Platinum Drug Neurotoxicity
4.1. Nuclear DNA Damage
4.2. Mitochondrial DNA Damage and Oxidative Stress
4.3. Ion Channels and Role of Calcium Signaling
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Windebank, A.J.; Grisold, W. Chemotherapy-induced neuropathy. J. Peripher. Nerv. Syst. 2008, 13, 27–46. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Wani, W.A.; Saleem, K.; Haque, A. Platinum compounds: A hope for future cancer chemotherapy. Anti-Cancer Agents Med. Chem. 2013, 13, 296–306. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- McWhinney, S.R.; Goldberg, R.M.; McLeod, H.L. Platinum neurotoxicity pharmacogenetics. Mol. Cancer Ther. 2009, 8, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Miltenburg, N.C.; Boogerd, W. Chemotherapy-induced neuropathy: A comprehensive survey. Cancer Treat. Rev. 2014, 40, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Xiao, J.; Yang, Y.; Cao, B. Oxaliplatin-based doublets versus cisplatin or carboplatin-based doublets in the first-line treatment of advanced nonsmall cell lung cancer. Medicine 2015, 94, e1072. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Kyritsis, A.P.; Makatsoris, T.; Kalofonos, H.P. Chemotherapy-induced peripheral neuropathy in adults: A comprehensive update of the literature. Cancer Manag. Res. 2014, 6, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Grisold, W.; Cavaletti, G.; Windebank, A.J. Peripheral neuropathies from chemotherapeutics and targeted agents: Diagnosis, treatment, and prevention. Neuro-Oncology 2012, 14, iv45–iv54. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Bruna, J.; Marmiroli, P.; Cavaletti, G. Chemotherapy-induced peripheral neurotoxicity (CIPN): An update. Crit. Rev. Oncol./Hematol. 2012, 82, 51–77. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Cavaletti, G.; Briani, C.; Velasco, R.; Bruna, J.; Campagnolo, M.; Alberti, P.; Bergamo, F.; Cortinovis, D.; Cazzaniga, M.; et al. Clinical pattern and association of oxaliplatin acute neurotoxicity: A prospective study in 170 patients with colorectal cancer. Cancer 2013, 119, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Glendenning, J.L.; Barbachano, Y.; Norman, A.R.; Dearnaley, D.P.; Horwich, A.; Huddart, R.A. Long-term neurologic and peripheral vascular toxicity after chemotherapy treatment of testicular cancer. Cancer 2010, 116, 2322–2331. [Google Scholar] [CrossRef] [PubMed]
- Strumberg, D.; Brugge, S.; Korn, M.W.; Koeppen, S.; Ranft, J.; Scheiber, G.; Reiners, C.; Möckel, C.; Seeber, S.; Scheulen, M.E. Evaluation of long-term toxicity in patients after cisplatin-based chemotherapy for non-seminomatous testicular cancer. Ann. Oncol. 2002, 13, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Park, S.B.; Lin, C.S.; Krishnan, A.V.; Goldstein, D.; Friedlander, M.L.; Kiernan, M.C. Long-term neuropathy after oxaliplatin treatment: Challenging the dictum of reversibility. Oncologist 2011, 16, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Briani, C.; Argyriou, A.A.; Izquierdo, C.; Velasco, R.; Campagnolo, M.; Alberti, P.; Frigeni, B.; Cacciavillani, M.; Bergamo, F.; Cortinovis, D.; et al. Long-term course of oxaliplatin-induced polyneuropathy: A prospective 2-year follow-up study. J. Peripher. Nerv. Syst. 2014, 19, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Beijers, A.J.; Mols, F.; Vreugdenhil, G. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Support Care Cancer 2014, 22, 1999–2007. [Google Scholar] [CrossRef] [PubMed]
- Pietrangeli, A.; Leandri, M.; Terzoli, E.; Jandolo, B.; Garufi, C. Persistence of high-dose oxaliplatin-induced neuropathy at long-term follow-up. Eur. Neurol. 2006, 56, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, E.E.; Huitema, A.D.; Boogerd, W.; Beijnen, J.H.; Schellens, J.H. Persistent neuropathy after treatment with cisplatin and oxaliplatin. Acta Oncol. 2008, 48, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Kostova, I. Platinum complexes as anticancer agents. Recent Pat. Anticancer Drug Discov. 2006, 1, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Mezencev, R. Interactions of cisplatin with non-DNA targets and their influence on anticancer activity and drug toxicity: The complex world of the platinum complex. Curr. Cancer Drug Targets 2015, 14, 794–816. [Google Scholar] [CrossRef] [PubMed]
- Rabik, C.A.; Dolan, E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Ciarimboli, G. Membrane transporters as mediators of Cisplatin effects and side effects. Scientifica (Cairo) 2012, 2012, 473829. [Google Scholar] [CrossRef] [PubMed]
- Zlatanova, J.; Yaneva, J.; Leuba, S.H. Proteins that specifically recognize cisplatin-damaged DNA: A clue to anticancer activity of cisplatin. FASEB J. 1998, 12, 791–799. [Google Scholar] [PubMed]
- Kartalou, M.; Essigmann, J.M. Recognition of cisplatin adducts by cellular proteins. Mutat. Res. 2001, 478, 1–21. [Google Scholar] [CrossRef]
- Cepeda, V.; Fuertes, M.A.; Castilla, J.; Alonso, C.; Quevedo, C.; Pérez, J.M. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med. Chem. 2007, 7, 3–18. [Google Scholar] [CrossRef] [PubMed]
- SiddiK, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed]
- Frey, U.; Ranford, J.D.; Sadler, P.J. Ring-opening reactions of the anticancer drug carboplatin: NMR characterization of cis-[Pt(NH3)2(CBDCA-O)(5'-GMP-N7)] in solution. Inorg. Chem. 1993, 32, 1333–1340. [Google Scholar] [CrossRef]
- Kasparkova, J.; Vojtiskova, M.; Natile, G.; Brabec, V. Unique properties of DNA interstrand cross-links of antitumor oxaliplatin and the effect of chirality of the carrier ligand. Chemistry 2008, 14, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Alcindor, T.; Beauger, N. Oxaliplatin: A review in the era of molecularly targeted therapy. Curr. Oncol. 2011, 18, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Ta, L.E.; Espeset, L.; Podratz, J.; Windebank, A.J. Neurotoxicity of oxaliplatin and cisplatin for dorsal root ganglion neurons correlates with platinum-DNA binding. Neurotoxicology 2006, 27, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L.R. New platinum antitumor complexes. Crit. Rev. Oncol. Hematol. 1993, 15, 191–219. [Google Scholar] [CrossRef]
- Albers, J.W.; Chaudhry, V.; Cavaletti, G.; Donehower, R.C. Interventions for preventing neuropathy caused by cisplatin and related compounds. Cochrane Database Syst. Rev. 2011, 16. [Google Scholar] [CrossRef] [Green Version]
- Carozzi, V.A.; Canta, A.; Chiorazzi, A. Chemotherapy-induced peripheral neuropathy: What do we know about mechanisms? Neurosci. Lett. 2015, 596, 90–107. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Raber, J.; Eriksson, L.A. Hydrolysis process of the second generation platinum-based anticancer drug cis-amminedichlorocyclohex-ylamineplatinum(II). J. Phys. Chem B. 2005, 109, 12195–12205. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.M.; Chiganças, V.; Galhardo Rda, S.; Carvalho, H.; Menck, C.F. The eukaryotic nucleotide excision repair pathway. Biochimic 2003, 85, 1083–1099. [Google Scholar] [CrossRef]
- Dzagnidze, A.; Katsarava, Z.; Makhalova, J.; Liedert, B.; Yoon, M.S.; Kaube, H.; Limmroth, V.; Thomale, J. Repair capacity for platinum-DNA adducts determines the severity of cisplatin-induced peripheral neuropathy. J. Neurosci. 2007, 27, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Nouspikel, T.; Hanawalt, P.C. DNA repair in terminally differentiated cells. DNA Repair (Amst.). 2002, 1, 59–75. [Google Scholar] [CrossRef]
- Gill, J.S.; Windebank, A.J. Cisplatin-induced apoptosis in rat dorsal root ganglion neurons is associated with attempted entry into the cell cycle. J. Clin. Investig. 1998, 101, 2842–2850. [Google Scholar] [CrossRef] [PubMed]
- Alaedini, A.; Xiang, Z.; Kim, H.; Sung, Y.J.; Latov, N. Up-regulation of apoptosis and regeneration genes in the dorsal root ganglia during cisplatin treatment. Exp. Neurol. 2008, 210, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hara, R.; Singh, G.; Sancar, A.; Lippard, S.J. Nucleotide excision repair from site-specifically platinum-modified nucleosomes. Biochemistry 2003, 42, 6747–6753. [Google Scholar] [CrossRef] [PubMed]
- Arnould, S.; Hennebelle, I.; Canal, P.; Bugat, R.; Guichard, S. Cellular determinants of oxaliplatin sensitivity in colon cancer cell lines. Eur. J. Cancer 2003, 39, 112–119. [Google Scholar] [CrossRef]
- Köberle, B.; Grimaldi, K.A.; Sunters, A.; Hartley, J.A.; Kelland, L.R.; Masters, J.R. DNA repair capacity and cisplatin sensitivity of human testis tumour cells. Int. J. Cancer 1997, 70, 551–555. [Google Scholar] [CrossRef]
- Chaney, S.G.; Vaisman, A. Specificity of platinum-DNA adduct repair. J. Inorg. Biochem. 1999, 77, 71–81. [Google Scholar] [CrossRef]
- Park, S.; Lippard, S.J. Binding interaction of HMGB4 with cisplatin-modified DNA. Biochemistry 2012, 51, 6728–6737. [Google Scholar] [CrossRef] [PubMed]
- Zamble, D.B.; Lippard, S.J. Cisplatin and DNA repair in cancer chemotherapy. Trends Biochem. Sci. 1995, 20, 435–439. [Google Scholar] [CrossRef]
- Gonzalez, V.M.; Fuertes, M.A.; Alonso, C.; Perez, J.M. Is cisplatin-induced cell death always produced by apoptosis? Mol. Pharmacol. 2001, 59, 657–663. [Google Scholar] [PubMed]
- Jung, Y.; Mikata, Y.; Lippard, S.J. Kinetic studies of the TATA-binding protein interaction with cisplatin-modified DNA. J. Biol. Chem. 2001, 276, 43589–43596. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.M.; Jamieson, E.R.; Lippard, S.J. Enhanced binding of the TATA-binding protein to TATA boxes containing flanking cisplatin 1,2-cross-links. Biochemistry 2000, 39, 8259–8265. [Google Scholar] [CrossRef] [PubMed]
- Podratz, J.L.; Knight, A.M.; Ta, L.E.; Staff, N.P.; Gass, J.M.; Genelin, K.; Schlattau, A.; Lathroum, L.; Windebank, A.J. Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol. Dis. 2011, 41, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Guo, C.; Vasko, M.R.; Kelley, M.R. Implications of apurinic/apyrimidinic endonuclease in reactive oxygen signaling response after cisplatin treatment of dorsal root ganglion neurons. Cancer Res. 2008, 68, 6425–6434. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.H.; Bennett, G.J. Effects of mitochondrial poisons on the neuropathic pain produced by the chemotherapeutic agents, paclitaxel and oxaliplatin. Pain 2012, 153, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.J.; Doyle, T.; Salvemini, D. Mitotoxicity in distal symmetrical sensory peripheral neuropathies. Nat. Rev. Neurol. 2014, 10, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Cavaletti, G.; Antonacopoulou, A.; Genazzani, A.A.; Briani, C.; Bruna, J.; Terrazzino, S.; Velasco, R.; Alberti, P.; Campagnolo, M.; et al. Voltage-gated sodium channel polymorphisms play a pivotal role in the development of oxaliplatin-induced peripheral neurotoxicity: Results from a prospective multicenter study. Cancer 2013, 119, 3570–3577. [Google Scholar] [CrossRef] [PubMed]
- Areti, A.; Yerra, V.G.; Naidu, V.; Kumar, A. Oxidative stress and nerve damage: role in chemotherapy induced peripheral neuropathy. Redox Biol. 2014, 18, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Tanaka, T.; Takahama, U. Cisplatin generates superoxide anion by interaction with DNA in a cell-free system. Biochem. Biophys. Res. Commun. 1994, 203, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Ozben, T. Oxidative stress and apoptosis: Impact on cancer therapy. J. Pharm. Sci. 2007, 96, 2181–2196. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E.; Prasad, A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol. Cancer Ther. 2011, 10, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Zanardelli, M.; Micheli, L.; Cinci, L.; Failli, P.; Ghelardini, C.; di Cesare Mannelli, L. Oxaliplatin neurotoxicity involves peroxisome alterations. PPARγ agonism as preventive pharmacological approach. PLoS ONE 2014, 9, e102758. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Fahimi, H.D. Peroxisomes and oxidative stress. Biochim. Biophys. Acta 2006, 1763, 1755–1666. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.F.; Juan, S.H.; Chen, J.J.; Chao, Y.C.; Chen, H.H.; Lian, W.S.; Lu, C.Y.; Chang, C.I.; Chiu, T.H.; Lin, H. Pravastatin attenuates carboplatin-induced cardiotoxicity via inhibition of oxidative stress associated apoptosis. Apoptosis 2008, 13, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Yüce, A.; Ateşşahin, A.; Ceribaşi, A.O.; Aksakal, M. Ellagic acid prevents cisplatin-induced oxidative stress in liver and heart tissue of rats. Basic Clin. Pharmacol. Toxicol. 2007, 101, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Işeri, S.; Ercan, F.; Gedik, N.; Yüksel, M.; Alican, I. Simvastatin attenuates cisplatin-induced kidney and liver damage in rats. Toxicology 2007, 230, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Yoshida, M.; Nishijima, H.; Yokosuka, M.; Iigo, M.; Ohtani-Kaneko, R.; Shimada, A.; Hasegawa, T.; Akama, Y.; Hirata, K. Melatonin, a pineal secretory product with antioxidant properties, protects against cisplatin-induced nephrotoxicity in rats. J. Pineal Res. 2001, 30, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Carozzi, V.A.; Marmiroli, P.; Cavaletti, G. The role of oxidative stress and anti-oxidant treatment in platinum-induced peripheral neurotoxicity. Curr. Cancer Drug Targets. 2010, 10, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Mukhopadhyay, P.; Rajesh, M.; Patel, V.; Mukhopadhyay, B.; Gao, B.; Haskó, G.; Pacher, P. Cannabidiol attenuates cisplatin-induced nephrotoxicity by decreasing oxidative/nitrosative stress, inflammation, and cell death. J. Pharmacol Exp. Ther. 2009, 328, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Melli, G.; Taiana, M.; Camozzi, F.; Triolo, D.; Podini, P.; Quattrini, A.; Taroni, F.; Lauria, G. Alpha-lipoic acid prevents mitochondrial damage and neurotoxicity in experimental chemotherapy neuropathy. Exp. Neurol. 2008, 214, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Custódio, J.B.; Cardoso, C.M.; Santos, M.S.; Almeida, L.M.; Vicente, J.A.; Fernandes, M.A. Cisplatin impairs rat liver mitochondrial functions by inducing changes on membrane ion permeability: Prevention by thiol group protecting agents. Toxicology 2009, 259, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Saad, S.Y.; Najjar, T.A.; Alashari, M. Role of non-selective adenosine receptor blockade and phosphodiesterase inhibition in cisplatin-induced nephrogonadal toxicity in rats. Clin. Exp. Pharmacol. Physiol. 2004, 31, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Mollman, J.E. Cisplatin neurotoxicity. N. Engl. J. Med. 1990, 322, 126–127. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare Mannelli, L.; Zanardelli, M.; Failli, P.; Ghelardini, C. Oxaliplatin-induced oxidative stress in nervous system-derived cellular models: Could it correlate with in vivo neuropathy? Free Radic. Biol. Med. 2013, 61, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Xiao, W.H.; Bennett, G.J. Functional deficits in peripheral nerve mitochondria in rats with paclitaxel- and oxaliplatin-evoked painful peripheral neuropathy. Exp. Neurol. 2011, 232, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Nassini, R.; Gees, M.; Harrison, S.; de Siena, G.; Materazzi, S.; Moretto, N.; Failli, P.; Preti, D.; Marchetti, N.; Cavazzini, A.; et al. Oxaliplatin elicits mechanical and cold allodynia in rodents via TRPA1 receptor stimulation. Pain 2011, 152, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.J.; Ji, R.R. Targeting astrocyte signaling for chronic pain. Neurotherapeutics 2010, 7, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Salvemini, D.; Neumann, W. Targeting peroxynitrite driven nitroxidative stress with synzymes: A novel therapeutic approach in chronic pain management. Life Sci. 2010, 86, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Karashima, Y.; Talavera, K.; Everaerts, W.; Janssens, A.; Kwan, K.Y.; Vennekens, R.; Nilius, B.; Voets, T. TRPA1 acts as a cold sensor in vitro and in vivo. Proc. Natl Acad. Sci. USA 2009, 106, 1273–1278. [Google Scholar] [CrossRef] [PubMed]
- Joseph, E.K.; Chen, X.; Bogen, O.; Levine, J.D. Oxaliplatin acts on IB4-positive nociceptors to induce an oxidative stress-dependent acute painful peripheral neuropathy. J. Pain 2008, 9, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare Mannelli, L.; Zanardelli, M.; Failli, P.; Ghelardini, C. Oxaliplatin-induced neuropathy: Oxidative stress as pathological mechanism. Protective effect of silibinin. J. Pain 2012, 13, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Coriat, R.; Alexandre, J.; Nicco, C.; Quinquis, L.; Benoit, E.; Chéreau, C.; Lemaréchal, H.; Mir, O.; Borderie, D.; Tréluyer, J.M.; et al. Treatment of oxaliplatin-induced peripheral neuropathy by intravenous mangafodipir. J. Clin. Investig. 2014, 124, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Husain, K.; Whitworth, C.; Hazelrigg, S.; Rybak, L. Carboplatin-induced oxidative injury in rat inferior colliculus. Int. J. Toxicol. 2003, 22, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Park, S.B.; Lin, C.S-Y.; Krishnan, A.V.; Goldstein, D.; Friedlander, M.L.; Kiernan, M.C. Dose Effects of oxaliplatin on persistent and transient Na+ conductances and the development of neurotoxicity. PLoS ONE 2011, 6, e18469. [Google Scholar] [CrossRef] [PubMed]
- Grolleau, F.; Gamelin, L.; Boisdron-Celle, M.; Lapied, B.; Pelhate, M.; Gamelin, E. A possible explanation for a neurotoxic effect of the anticancer agent oxaliplatin on neuronal voltage-gated sodium channels. J. Neurophysiol. 2001, 85, 2293–2297. [Google Scholar] [PubMed]
- Wu, S.; Chen, B.; Wu, Y.; Peng, H.; Chen, L. The mechanism of the actions of oxaliplatin on ion currents and action potentials in differentiated NG108-15 neuronal cells. Neurotoxicology 2009, 30, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Sittl, R.; Lampert, A.; Huth, T.; Schuy, E.T.; Link, A.S.; Fleckenstein, J.; Alzheimer, C.; Grafe, P.; Carr, R.W. Anticancer drug oxaliplatin induces acute cooling-aggravated neuropathy via sodium channel subtype NaV1. 6-resurgent and persistent current. Proc. Natl Acad. Sci. USA 2012, 109, 6704–6709. [Google Scholar] [CrossRef] [PubMed]
- Deuis, J.R.; Zimmermann, K.; Romanovsky, A.A.; Possani, L.D.; Cabot, P.J.; Lewis, R.J.; Vetter, I. An animal model of oxaliplatin-induced cold allodynia reveals a crucial role for NaV1.6 in peripheral pain pathways. Pain 2013, 154, 1749–1757. [Google Scholar] [CrossRef] [PubMed]
- Avan, A.; Postma, T.J.; Ceresa, C.; Avan, A.; Cavaletti, G.; Giovannetti, E.; Peters, G.J. Platinum-induced neurotoxicity and preventive strategies: Past, present and future. Oncologist 2015, 20, 411–432. [Google Scholar] [CrossRef] [PubMed]
- Benoit, E.; Brienza, S.; Dubois, J.M. Oxaliplatin, an anticancer agent that affects both Na+ and K+ channels in frog peripheral myelinated axons. Gen. Physiol. Biophys. 2006, 25, 263–276. [Google Scholar] [PubMed]
- Kagiava, A.; Tsingotjidou, A.; Emmanouilides, C.; Theophilidis, G. The effects of oxaliplatin, an anticancer drug, on potassium channels of the peripheral myelinated nerve fibres of the adult rat. Neurotoxicology 2008, 29, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Descoeur, J.; Pereira, V.; Pizzoccaro, A.; Francois, A.; Ling, B.; Maffre, V.; Couette, B.; Busserolles, J.; Courteix, C.; Noel, J.; et al. Oxaliplatin-induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors. EMBO Mol. Med. 2011, 3, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Benbow, J.H.; Mann, T.; Keeler, C.; Fan, C.; Hodsdon, M.E.; Lolis, E.; DeGray, B.; Ehrlich, B.E. Inhibition of paclitaxel-induced decreases in calcium signaling. J. Biol. Chem. 2012, 287, 37907–37916. [Google Scholar] [CrossRef] [PubMed]
- Schulze, C.; McGowan, M.; Jordt, S.E.; Ehrlich, B.E. Prolonged oxaliplatin exposure alters intracellular calcium signaling: A new mechanism to explain oxaliplatin-associated peripheral neuropathy. Clin. Colorectal Cancer 2011, 10, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Kawashiri, T.; Egashira, N.; Kurobe, K.; Tsutsumi, K.; Yamashita, Y.; Ushio, S.; Yano, T.; Oishi, R. L type Ca2+ channel blockers prevent oxaliplatin-induced cold hyperalgesia and TRPM8 overexpression in rats. Mol. Pain 2012, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Tatsushima, Y.; Egashira, N.; Narishige, Y.; Fukui, S.; Kawashiri, T.; Yamauchi, Y.; Oishi, R. Calcium channel blockers reduce oxaliplatin-induced acute neuropathy: A retrospective study of 69 male patients receiving modified FOLFOX6 therapy. Biomed. Pharmacother. 2013, 67, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, K.; Okada, N.; Miyazaki, T.; Sano, M.; Ishida, H. Effect of calcium and magnesium on neurotoxicity and blood platinum concentrations in patients receiving mFOLFOX6 therapy: A prospective randomized study. Int. J. Clin. Oncol. 2010, 15, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Han, C.H.; Khwaounjoo, P.; Kilfoyle, D.H.; Hill, A.; McKeage, M.J. Phase I drug-interaction study of effects of calcium and magnesium infusions on oxaliplatin pharmacokinetics and acute neurotoxicity in colorectal cancer patients. BMC Cancer 2013, 13, 495. [Google Scholar] [CrossRef] [PubMed]
- Loprinzi, C.L.; Qin, R.; Dakhil, S.R.; Fehrenbacher, L.; Flynn, K.A.; Atherton, P.; Seisler, D.; Qamar, R.; Lewis, G.C.; Grothey, A. Phase III randomized, placebo-controlled, double-blind study of intravenous calcium and magnesium to prevent oxaliplatin-induced sensory neurotoxicity (N08CB/Alliance). J. Clin. Oncol. 2014, 32, 997–1005. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiorazzi, A.; Semperboni, S.; Marmiroli, P. Current View in Platinum Drug Mechanisms of Peripheral Neurotoxicity. Toxics 2015, 3, 304-321. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics3030304
Chiorazzi A, Semperboni S, Marmiroli P. Current View in Platinum Drug Mechanisms of Peripheral Neurotoxicity. Toxics. 2015; 3(3):304-321. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics3030304
Chicago/Turabian StyleChiorazzi, Alessia, Sara Semperboni, and Paola Marmiroli. 2015. "Current View in Platinum Drug Mechanisms of Peripheral Neurotoxicity" Toxics 3, no. 3: 304-321. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics3030304