Phylogenetic Integration Reveals the Zebrafish Core Microbiome and Its Sensitivity to Environmental Exposures

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Study Inclusion Criteria
2.2. S rRNA Gene Sequence Quality Control and Amplicon Sequence Variant Clustering
2.3. Phylogenetic Analysis
2.4. Ecophylogenetic Discovery of Core Clades
2.5. Linear Regression
3. Results
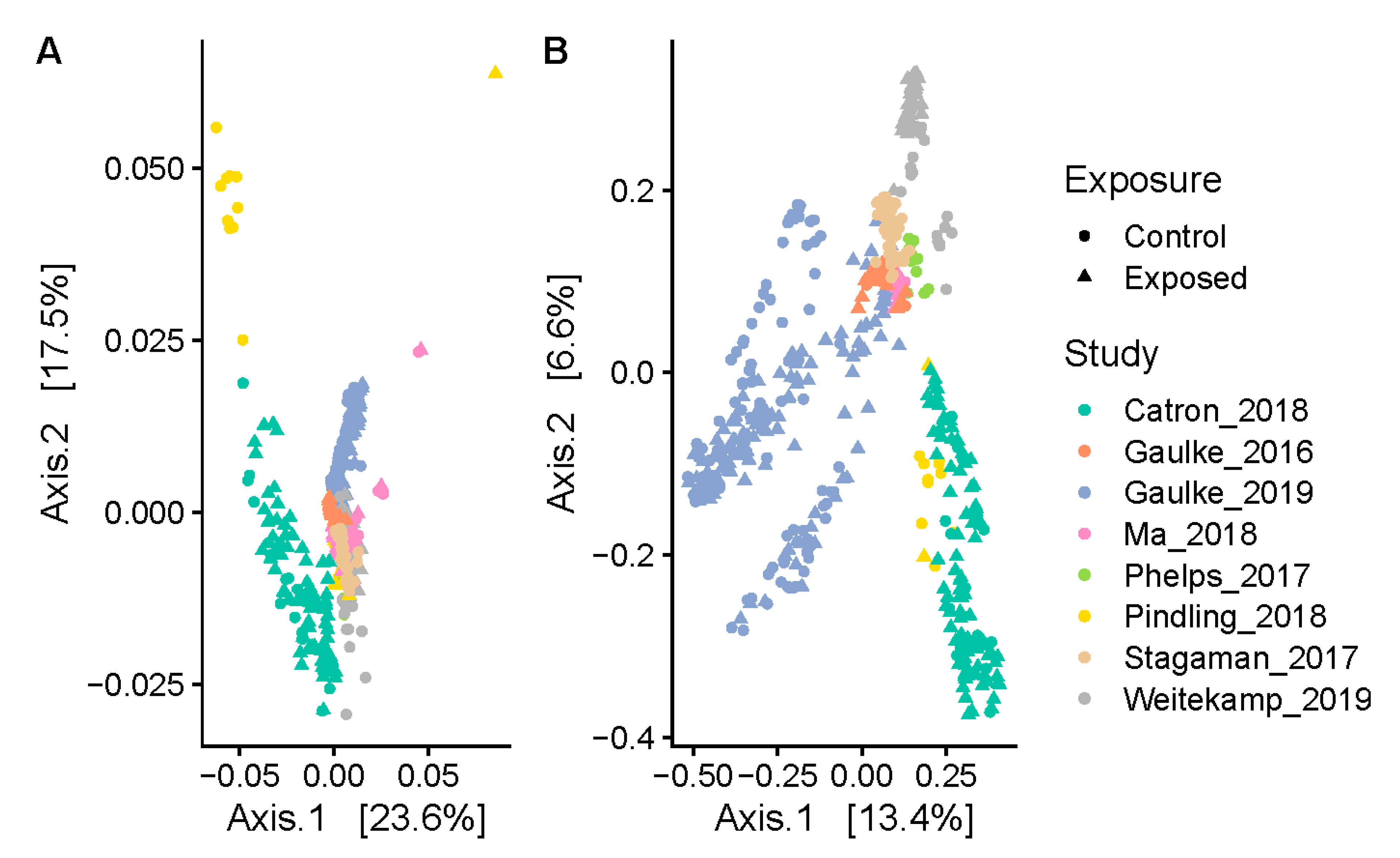
3.1. Study and Age Effects Drive the Phylogenetic Composition of the Zebrafish Gut Microbiome
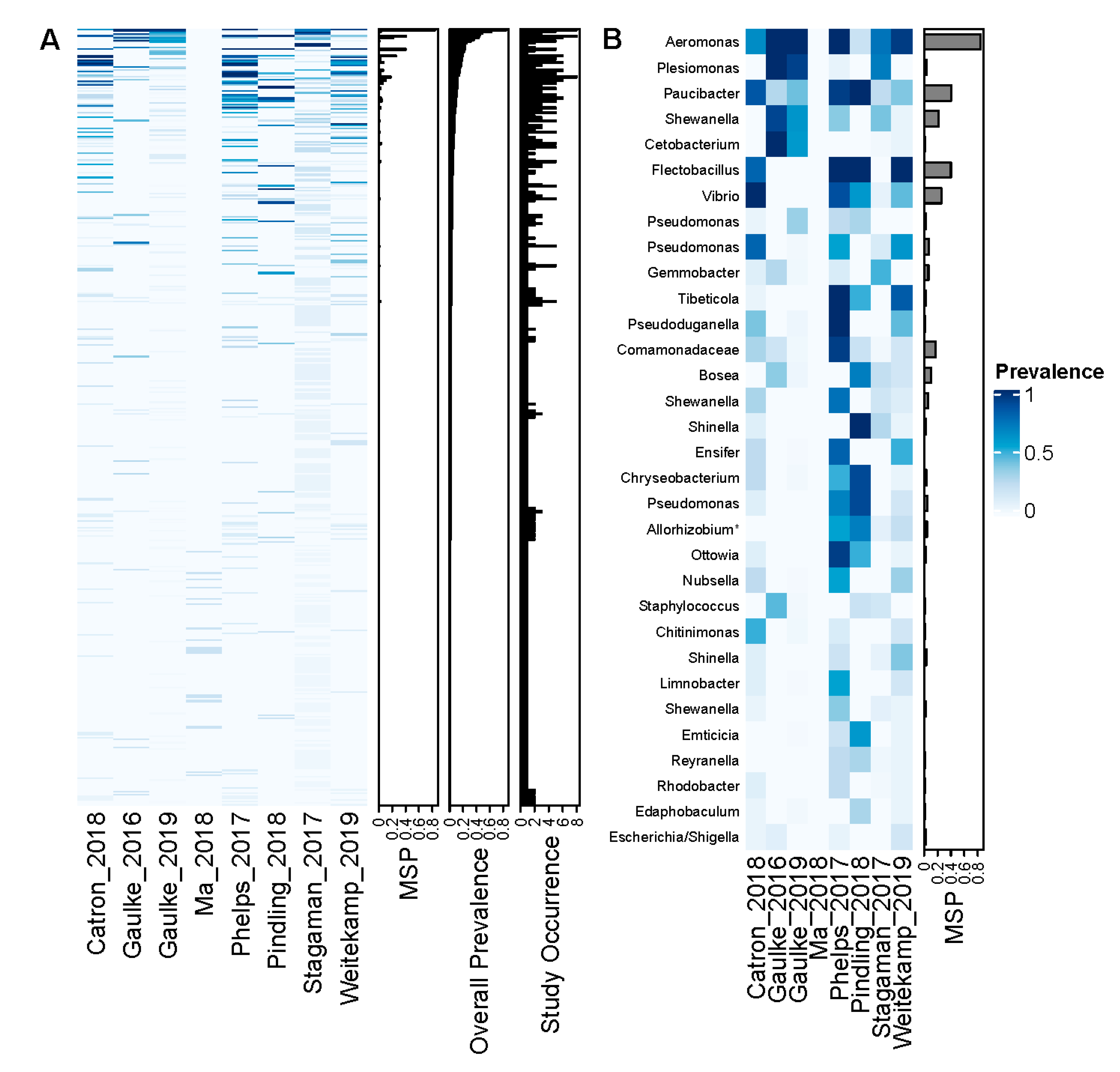
3.2. A Small Set of ASVs Are Common to Zebrafish across Studies
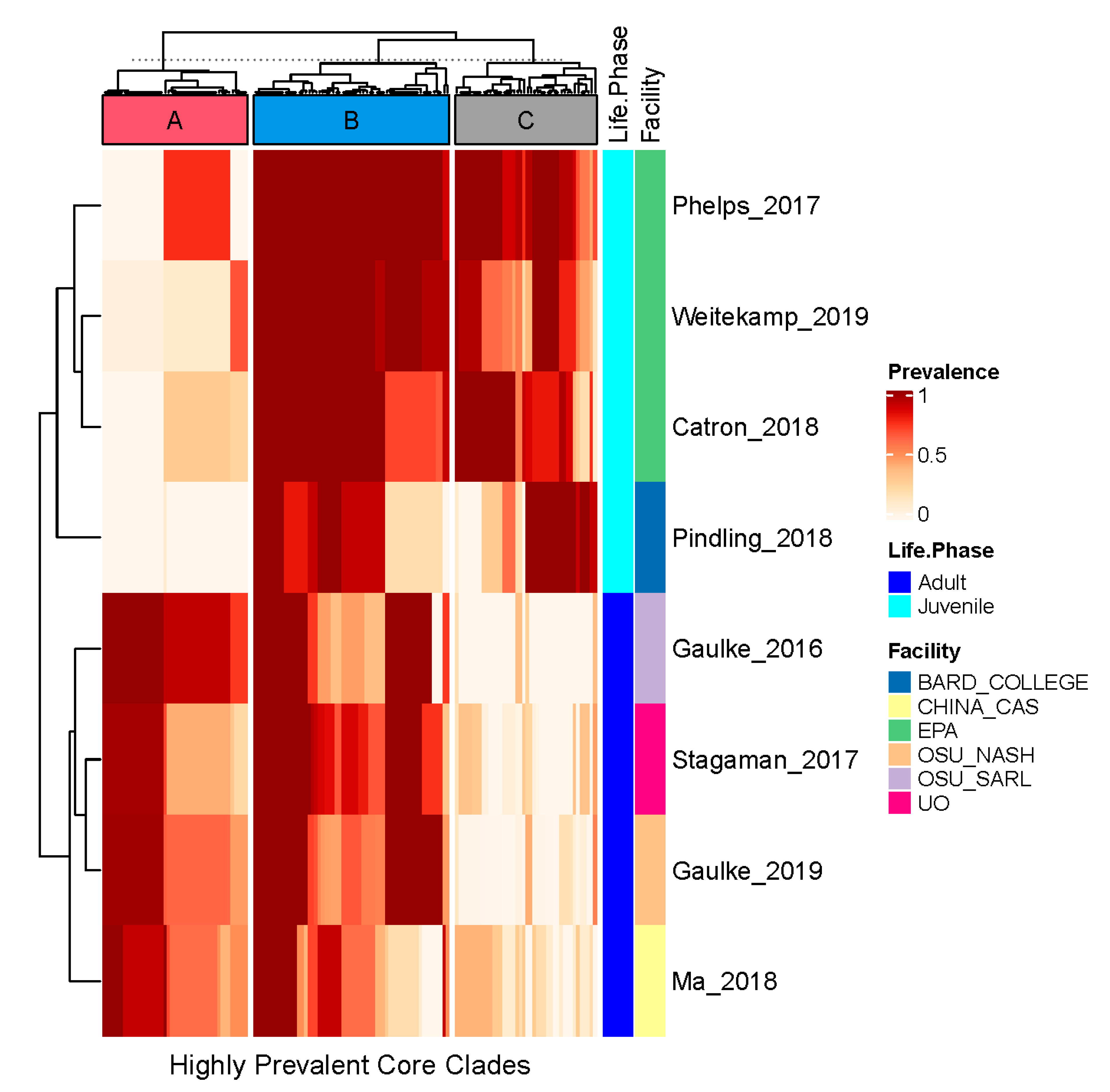
3.3. A Diverse Set of Clades Comprise the Core Zebrafish Gut Microbiome
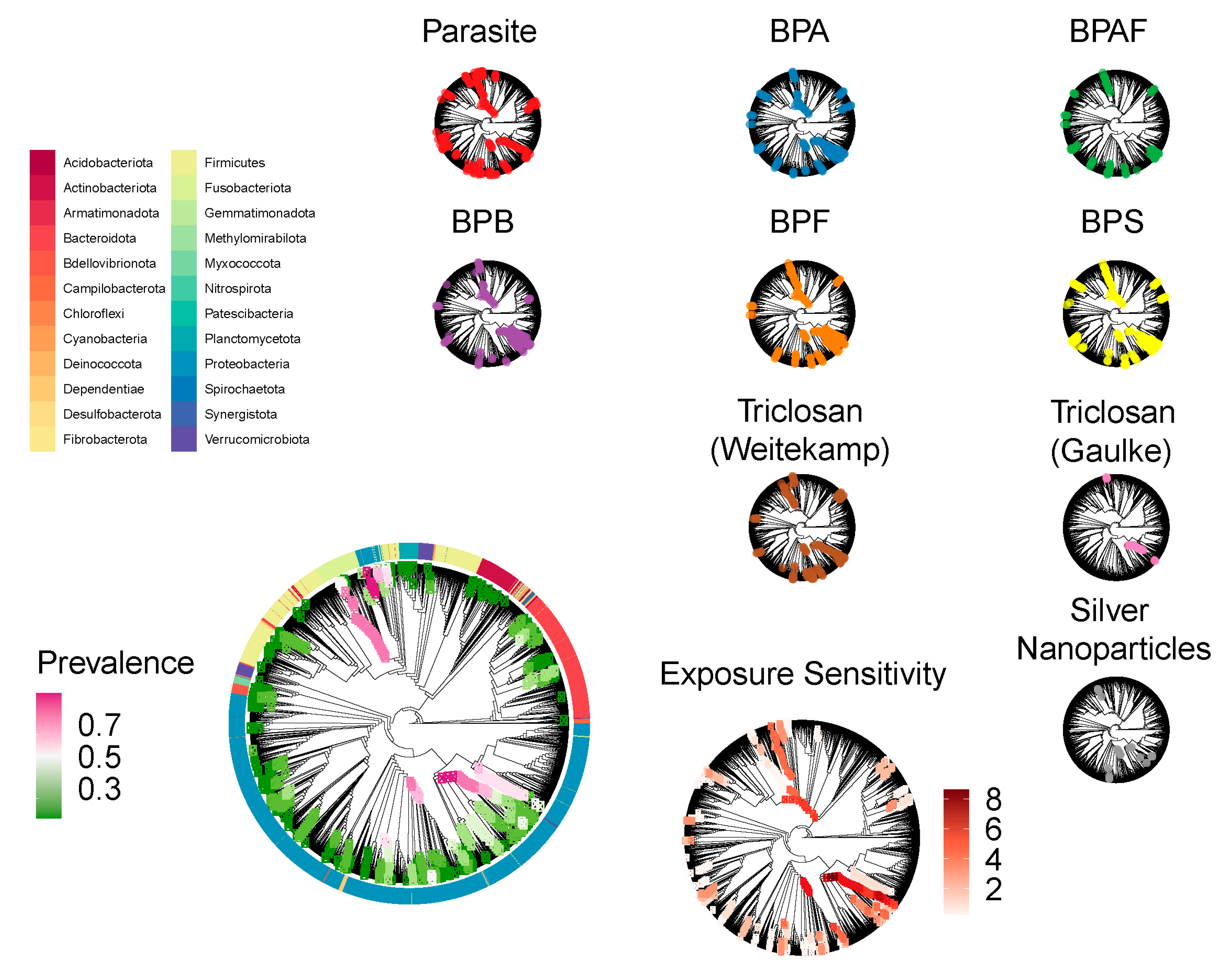
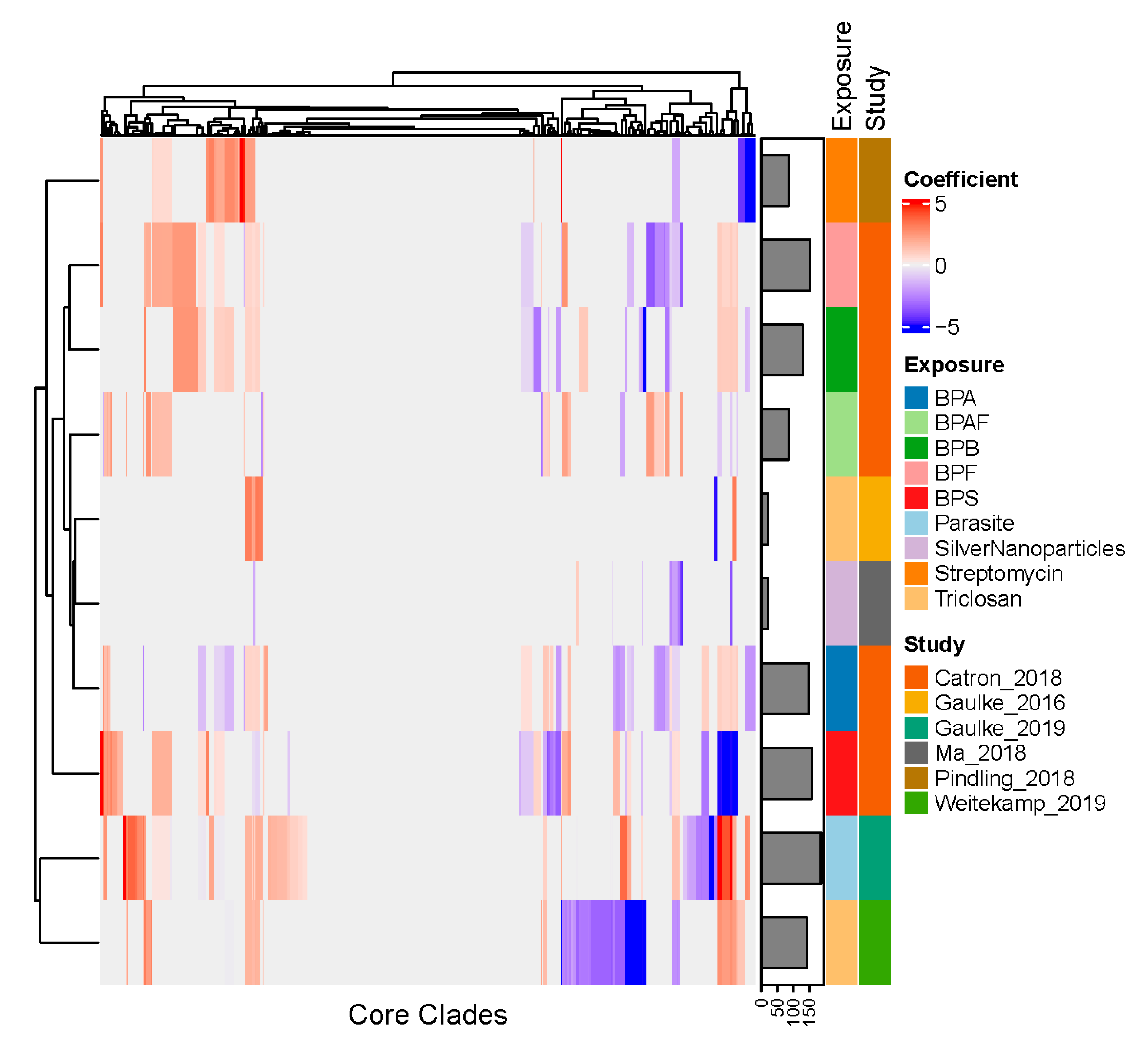
3.4. Core Clades Differentially Respond to Environmental Exposures
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stagaman, K.; Sharpton, T.J.; Guillemin, K. Zebrafish Microbiome Studies Make Waves. Lab. Anim. 2020, 49, 201–207. [Google Scholar] [CrossRef]
- Gaulke, C.A.; Barton, C.L.; Proffitt, S.; Tanguay, R.L.; Sharpton, T.J. Triclosan Exposure Is Associated with Rapid Restructuring of the Microbiome in Adult Zebrafish. PLoS ONE 2016, 11, e0154632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pindling, S.; Azulai, D.; Zheng, B.; Dahan, D.; Perron, G.G. Dysbiosis and Early Mortality in Zebrafish Larvae Exposed to Subclinical Concentrations of Streptomycin. FEMS Microbiol. Lett. 2018, 365. [Google Scholar] [CrossRef]
- Weitekamp, C.A.; Phelps, D.; Swank, A.; McCord, J.; Sobus, J.R.; Catron, T.; Keely, S.; Brinkman, N.; Zurlinden, T.; Wheaton, E.; et al. Triclosan-Selected Host-Associated Microbiota Perform Xenobiotic Biotransformations in Larval Zebrafish. Toxicol. Sci. 2019, 172, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Song, L.; Lei, Y.; Jia, P.; Lu, C.; Wu, J.; Xi, C.; Strauss, P.R.; Pei, D.-S. Sex Dependent Effects of Silver Nanoparticles on the Zebrafish Gut Microbiota. Environ. Sci. Nano 2018, 5, 740–751. [Google Scholar] [CrossRef]
- Catron, T.R.; Keely, S.P.; Brinkman, N.E.; Zurlinden, T.J.; Wood, C.E.; Wright, J.R.; Phelps, D.; Wheaton, E.; Kvasnicka, A.; Gaballah, S.; et al. Host Developmental Toxicity of BPA and BPA Alternatives Is Inversely Related to Microbiota Disruption in Zebrafish. Toxicol. Sci. 2019, 167, 468–483. [Google Scholar] [CrossRef]
- Gaulke, C.A.; Martins, M.L.; Watral, V.G.; Humphreys, I.R.; Spagnoli, S.T.; Kent, M.L.; Sharpton, T.J. A Longitudinal Assessment of Host-Microbe-Parasite Interactions Resolves the Zebrafish Gut Microbiome’s Link to Pseudocapillaria tomentosa Infection and Pathology. Microbiome 2019, 7, 10. [Google Scholar] [CrossRef]
- Rawls, J.F.; Samuel, B.S.; Gordon, J.I. Gnotobiotic Zebrafish Reveal Evolutionarily Conserved Responses to the Gut Microbiota. Proc. Natl. Acad. Sci. USA 2004, 101, 4596–4601. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.H.; Franzosa, E.A.; Huttenhower, C.; Guillemin, K. A Conserved Bacterial Protein Induces Pancreatic Beta Cell Expansion during Zebrafish Development. eLife 2016, 5, e20145. [Google Scholar] [CrossRef]
- Phelps, D.; Brinkman, N.E.; Keely, S.P.; Anneken, E.M.; Catron, T.R.; Betancourt, D.; Wood, C.E.; Espenschied, S.T.; Rawls, J.F.; Tal, T. Microbial Colonization Is Required for Normal Neurobehavioral Development in Zebrafish. Sci. Rep. 2017, 7, 11244. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, B.W.; Koch, B.E.V.; Spaink, H.P.; Peijnenburg, W.J.G.M.; Vijver, M.G. Colonizing Microbiota Protect Zebrafish Larvae against Silver Nanoparticle Toxicity. Nanotoxicology 2020, 14, 725–739. [Google Scholar] [CrossRef] [PubMed]
- Parker, K.D.; Albeke, S.E.; Gigley, J.P.; Goldstein, A.M.; Ward, N.L. Microbiome Composition in Both Wild-Type and Disease Model Mice Is Heavily Influenced by Mouse Facility. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armour, C.R.; Nayfach, S.; Pollard, K.S.; Sharpton, T.J. A Metagenomic Meta-Analysis Reveals Functional Signatures of Health and Disease in the Human Gut Microbiome. mSystems 2019, 4, e00332-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shade, A.; Handelsman, J. Beyond the Venn Diagram: The Hunt for a Core Microbiome. Environ. Microbiol. 2012, 14, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Risely, A. Applying the Core Microbiome to Understand Host–Microbe Systems. J. Anim. Ecol. 2020, 89, 1549–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Gordon, J.I. The Core Gut Microbiome, Energy Balance and Obesity. J. Physiol. 2009, 587, 4153–4158. [Google Scholar] [CrossRef]
- Roeselers, G.; Mittge, E.K.; Stephens, W.Z.; Parichy, D.M.; Cavanaugh, C.M.; Guillemin, K.; Rawls, J.F. Evidence for a Core Gut Microbiota in the Zebrafish. ISME J. 2011, 5, 1595–1608. [Google Scholar] [CrossRef] [Green Version]
- Stephens, W.Z.; Burns, A.R.; Stagaman, K.; Wong, S.; Rawls, J.F.; Guillemin, K.; Bohannan, B.J.M. The composition of the zebrafish intestinal microbial community varies across development. ISME J. 2016, 10, 644–654. [Google Scholar] [CrossRef] [Green Version]
- Stagaman, K.; Burns, A.R.; Guillemin, K.; Bohannan, B.J. The Role of Adaptive Immunity as an Ecological Filter on the Gut Microbiota in Zebrafish. ISME J. 2017, 11, 1630–1639. [Google Scholar] [CrossRef]
- Gaulke, C.A.; Arnold, H.K.; Humphreys, I.R.; Kembel, S.W.; O’Dwuer, J.P.; Sharpton, T.J. Ecophylogenetics Clarifies the Evolutionary Association Between Mammals and Their Gut Microbiota. mBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. 2019. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 5 December 2020).
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Dwyer, J.P.; Kembel, S.W.; Sharpton, T.J. Backbones of Evolutionary History Test Biodiversity Theory for Microbes. Proc. Natl. Acad. Sci. USA 2015, 112, 8356–8361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Schliep, K. Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Revell, L.J. Phytools: An R Package for Phylogenetic Comparative Biology (and Other Things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [Green Version]
- Venables, W.N.; Ripley, B.D. Modern Applied Statistics with S, 4th ed.; Springer: New York, NY, USA, 2002. [Google Scholar]
- Rolig, A.S.; Sweeney, E.G.; Kaye, L.E.; DeSantis, M.D.; Perkins, A.; Banse, A.V.; Hamilton, M.K.; Guillemin, K. A Bacterial Immunomodulatory Protein with Lipocalin-like Domains Facilitates Host–Bacteria Mutualism in Larval Zebrafish. eLife 2018, 7, e37172. [Google Scholar] [CrossRef]
- Rolig, A.S.; Parthasarathy, R.; Burns, A.R.; Bohannan, B.J.M.; Guillemin, K. Individual Members of the Microbiota Disproportionately Modulate Host Innate Immune Responses. Cell Host Microbe 2015, 18, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stressmann, F.A.; Bernal-Bayard, J.; Perez-Pascual, D.; Audrain, B.; Rendueles, O.; Briolat, V.; Bruchmann, S.; Volant, S.; Ghozlane, A.; Häussler, S.; et al. Mining Zebrafish Microbiota Reveals Key Community-Level Resistance against Fish Pathogen Infection. ISME J. 2020. Available online: https://pubmed.ncbi.nlm.nih.gov/33077888/ (accessed on 1 December 2020). [CrossRef] [PubMed]
- Wiles, T.J.; Jemielita, M.; Baker, R.P.; Schlomann, B.H.; Logan, S.L.; Ganz, J.; Melancon, E.; Eisen, J.S.; Guillemin, K.; Parthasarathy, R. Host Gut Motility Promotes Competitive Exclusion within a Model Intestinal Microbiota. PLoS Biol. 2016, 14, e1002517. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lead Author | Year | Facility ¥ | Number of Individuals | Age (dpf) | Exposure Conditions | SRA Bioproject Identifier | Citation |
|---|---|---|---|---|---|---|---|
| Catron | 2018 | EPA | 118 | 10 | Bisphenol A or related metabolites € | NA * | [6] |
| Gaulke | 2016 | SARL | 45 | 270 | Triclosan | SRP071910 | [2] |
| Gaulke | 2019 | OSU ZPV | 210 | 127–213 | P. tomentosa | SRA708553 | [7] |
| Ma | 2018 | CAS | 132 | 120 | Silver nanoparticles | PRJNA348716 | [5] |
| Phelps | 2017 | EPA | 32 | 10 | N/A | NA * | [10] |
| Pindling | 2018 | Bard College | 10 | 96 | Subclinical streptomycin | SRP139123 | [3] |
| Stagaman | 2017 | Huestis | 198 | 9, 75 | N/A | SRA527217 | [19] |
| Weitekamp | 2019 | EPA | 53 | 6, 10 | Triclosan | NA * | [4] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharpton, T.J.; Stagaman, K.; Sieler, M.J., Jr.; Arnold, H.K.; Davis, E.W., II. Phylogenetic Integration Reveals the Zebrafish Core Microbiome and Its Sensitivity to Environmental Exposures. Toxics 2021, 9, 10. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics9010010
Sharpton TJ, Stagaman K, Sieler MJ Jr., Arnold HK, Davis EW II. Phylogenetic Integration Reveals the Zebrafish Core Microbiome and Its Sensitivity to Environmental Exposures. Toxics. 2021; 9(1):10. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics9010010
Chicago/Turabian StyleSharpton, Thomas J., Keaton Stagaman, Michael J. Sieler, Jr., Holly K. Arnold, and Edward W. Davis, II. 2021. "Phylogenetic Integration Reveals the Zebrafish Core Microbiome and Its Sensitivity to Environmental Exposures" Toxics 9, no. 1: 10. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics9010010