Mechanistic Implications of Biomass-Derived Particulate Matter for Immunity and Immune Disorders


1
Laboratory of Mucosal Exposome and Biomodulation, Department of Integrative Biomedical Sciences, Pusan National University, Yangsan 50612, Korea
2
Department of Internal Medicine, College of Medicine and Research Institute for Convergence of Biomedical Science and Technology, Pusan National University Yangsan Hospital, Yangsan 50612, Korea
3
Department of Pharmacology, Inje University College of Medicine, Busan 47392, Korea
4
Biomedical Research Institute, Pusan National University, Yangsan 50612, Korea
*
Author to whom correspondence should be addressed.
Toxics 2021, 9(2), 18; https://0-doi-org.brum.beds.ac.uk/10.3390/toxics9020018
Submission received: 9 November 2020
/
Revised: 4 January 2021
/
Accepted: 15 January 2021
/
Published: 20 January 2021
(This article belongs to the Special Issue Molecular Basis of Air-Pollution-Induced Disease Risk)
Abstract
:Particulate matter (PM) is a major and the most harmful component of urban air pollution, which may adversely affect human health. PM exposure has been associated with several human diseases, notably respiratory and cardiovascular diseases. In particular, recent evidence suggests that exposure to biomass-derived PM associates with airway inflammation and can aggravate asthma and other allergic diseases. Defective or excess responsiveness in the immune system regulates distinct pathologies, such as infections, hypersensitivity, and malignancies. Therefore, PM-induced modulation of the immune system is crucial for understanding how it causes these diseases and highlighting key molecular mechanisms that can mitigate the underlying pathologies. Emerging evidence has revealed that immune responses to biomass-derived PM exposure are closely associated with the risk of diverse hypersensitivity disorders, including asthma, allergic rhinitis, atopic dermatitis, and allergen sensitization. Moreover, immunological alteration by PM accounts for increased susceptibility to infectious diseases, such as tuberculosis and coronavirus disease-2019 (COVID-19). Evidence-based understanding of the immunological effects of PM and the molecular machinery would provide novel insights into clinical interventions or prevention against acute and chronic environmental disorders induced by biomass-derived PM.
1. Introduction
Particulate matter (PM) is a term used to describe a mixture of pollutants, containing a complex mixture of smoke, dust, and other solid particles, as well as liquid droplets, present in the air [1]. PM vary in size, shape, and chemical composition. PM is not just carbon black, but is also a carrier of some detrimental factors, including heavy metals, allergens, microbial components, and other organic toxicants [2,3]. The World Health Organization (WHO) and the International Agency for Research on Cancer (IARC) have shown that airborne PM can contain Group 1 carcinogens [4,5,6]. Following the inhalation of PM into the human airway, the airway epithelial layer is the first barrier to systemic exposure. In addition to airway distress, including dyspnea, cough, and aggravation of asthma, exposure to PM has been closely associated with the risk of lung cancer [7,8]. Additionally, PM is responsible for chronic respiratory diseases, such as chronic obstructive pulmonary disease (COPD) [9,10,11]. PM can penetrate different tissues and cause severe oxidative damage wherever deposited [12,13]. Furthermore, circulatory exposure to PM through the respiratory tract is directly linked to an elevated risk of arrhythmia, myocardial infarction, premature delivery, low birth weight, and even premature death in cardiovascular or respiratory comorbidities [14,15,16,17]. In particular, the immune system is a sensitive tissue to airway PM and allergen exposure and is involved in diverse tissue disorders. Based on the hypothesis that immune responses are closely linked to airway and systemic distress during PM exposure.
It has been well known that biomass fuel combustion, as well as wildland and agricultural fires, exhale large quantities of harmful air pollutants to both ambient and indoor air, which is associated with increased incidences of respiratory disease-related morbidity and mortality [18,19]. Exposure to PM derived from woodsmoke causes various health effects, such as systemic and airway inflammation, and increases the incidence of asthma and allergic diseases [20,21,22]. Furthermore, exposure to wood smoke particles (WSP) is associated with various types of lung distress, including acute respiratory infection (ARI) and COPD [23,24,25,26].
Many previous studies addressed the health effects of different types of PM exposure [27,28,29,30,31]. Although investigations attempted to assess the effect of PM on the immune systems [32,33], mechanistic pathways involved in immune alteration-related pathologic events have not been fully established. In this review, we systemically discussed the immune alteration-related events and associated the disease outcomes with cellular and molecular evidence in humans and animal models in response to PM, including biomass-derived pollutants. The comprehensive review would provide insights into strategies of prevention and novel intervention with environmental immune diseases.
2. Source and Characteristics of PM
PM is a mixture of solid and liquid particles suspended in the air, varying in size, number, surface area, chemical composition, and sources. Among the various methods of PM classification, the aerodynamic diameter is one appropriate criterion to define transportability in the atmosphere and inhaling ability through the respiratory tract. PM is categorized as PM10, PM2.5, and PM0.1, according to mean particulate size fractions. Based on “aerodynamic equivalent diameter” (AED), PM is categorized into three classes, including coarse particles or PM10 (ranging from 2.5 to 10 μm); fine particles or PM2.5 (smaller than 2.5 μm); ultrafine particles or PM0.1 (UFP, smaller than 0.1 μm) [34,35]. Regardless of the PM composition, differently sized PMs are expected to have different particle velocities for settling in the respiratory tract [36]. Generally, PM contains various chemical constituents, including inorganic ions (e.g., sulfates, nitrates, ammonium, sodium, potassium, calcium, magnesium, and chloride), heavy metals (including cadmium, copper, nickel, vanadium, and zinc), black carbon, and organic compounds (e.g., polycyclic aromatic hydrocarbons, and polyaromatic hydrocarbons (PAHs)) [28,37,38]. The main sources of coarse particles (PM10) include abraded soil, dust from road and construction sites, and oil combustion products with bio-aerosols, such as fungi, bacteria, pollen, and endotoxins. Particles that are derived from direct emissions owing to combustion processes of gasoline, oil, diesel, wood burning, and coal burning are primary sources of fine (PM2.5) and ultrafine particles (PM0.1) [38,39]. The woodsmoke poses the pollutants like carbon monoxide (CO), nitrogen oxides (NOx), methane, chlorinated dioxin, volatile organic compounds (VOC), PAH, and PM [18,40,41]. The general properties and primary sources of fine (PM2.5) and coarse (PM10) particles are described in Table 1.
Particle Size-Dependent Translocation and Toxicity in the Airway Mucosa
PM2.5 contains high levels of transition metals associated with increased potential of oxidative stress in the tissue. Moreover, PAHs, richly distributed in UFP (PM0.1), can generate oxidative stress, whereas their levels are low in both coarse (PM10) and fine (PM2.5) particles [33]. Generally, PM10 settles in the upper respiratory tract and is substantially associated with several diseases, including cardiovascular diseases [42,43], cancers [7,44,45], tuberculosis [46,47], and asthma [48,49]. Furthermore, PM2.5 induces vascular inflammation, leading to a potentially increased risk of atherosclerotic cardiovascular complications [50]. In addition to airway disorders, another susceptible target of circulating PM is the cardiovascular system as the incidence of ischemic heart disease and cardiac injury increases with PM exposure. A study investigating fine particles and cardiac injury revealed that PM2.5 exposure led to a substantial increase in interleukin 4 (IL-4) and IL-13 levels and a decrease in interferon γ (IFN-γ) in the myocardium of rats [51]. Taken together, the particulate size is the deterministic factor of translocation and actions in the airway mucosa and other tissues.
3. Mucosal Exposure and Innate Immune Responses to PM
Innate Immune Regulation and Signaling in the Lung Barrier
When biomass burning-derived ultrafine and fine particles enter into the airway through inhalation from ambient air, they can reach the alveolar tissue. Compared to most deposited particles, ultrafine particles can cross the airway-blood barrier and enter the pulmonary and systemic circulation [52,53,54]. Although most inhaled PM0.2 and PM2.0 particles are deposited in the acinar region of the lung, some of them can be released into circulation with time and can be found in the kidney and liver [55]. The well-known mechanism of nanoparticles passing through the air-blood barrier in the acinar region is the process of endocytosis or diffusion [56,57,58]. However, the delivery of PM0.2 and PM2.0 particles to blood circulation and extrapulmonary organs might be mediated by particle-internalizing macrophages [55]. Following the inhalation of pollutants, such as PM into the human airway, airway epithelial cells are the first defense of the respiratory system. When barrier epithelial cells are exposed to PM (especially combustion-derived PMs), oxidative stress and inflammation are elevated in the lung [59], thus disrupting epithelial barrier integrity. Moreover, smooth muscles are thickened, and the epithelial-mesenchymal transition is induced [60]. Through the epithelial-mesenchymal transition, lung epithelial cells display reduced levels of E-cadherin, representing the loss of characteristic features of epithelial polarity and intercellular junctions. In addition to barrier disruption, the innate immune responses to PM in human airway epithelial cells and alveolar macrophages are affected by changes in pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) and nucleotide-binding domain and leucine-rich repeat (NLR) containing inflammasomes [61,62,63,64]. Household air pollution from biomass fuel combustion triggers dose-dependent production of proinflammatory cytokines and alters phagocytosis activity in human alveolar macrophages [65]. TLRs are representative PRRs that play pivotal roles in innate immune responses to infection in epithelial cells and phagocytes. The mechanisms involved in PM-induced innate immune responses are mainly through the production of proinflammatory cytokines and chemokines via activation of the TLR signaling pathway [1,66,67]. There is ample evidence demonstrating that PM modulates cytokine and chemokine levels, although the effects vary depending on the airway exposure regime [61,68]. TLRs can modulate myeloid differentiation primary response protein 88 (MyD88), resulting in the activation of the nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (IκBα) and nuclear factor kappa B (NF-κB) inhibitory protein, which activates the expression of proinflammatory genes [69,70,71,72,73]. Moreover, airway epithelial cells exposed to PM10 can activate the NLRP3 inflammasome, subsequently facilitating the release of a mature form of IL-1β and IL-18, whose intracellular expression was induced by NF-κB signaling [64]. Furthermore, PM2.5 can activate TLR4 and the NLRP3 inflammasome in cooperation with mitogen-activated protein kinase (MAPK) pathways in monocytes [61]. IL-1β can recruit immune cells, including neutrophils and lymphocytes [74,75]. Additionally, PM induced granulocyte/macrophage colony-stimulating factor (GM-CSF) and CC chemokine ligand (CCL)-20 production. Notably, GM-CSF can recruit mature dendritic cells (DCs) [76,77,78]. The expression of GM-CSF, leukemia inhibitory factor (LIF), IL-1α, and IL-8 was reportedly increased via PRR action induced by PM10 exposure [78,79]. Therefore, ambient PM-activated PRR signaling has a pivotal role in early proinflammatory cell activation and recruitment for further immune responses in the human lung, as well as in circulation. Further studies are required to clarify specific components of the PM mixture that activate PRR signaling during early innate immunity and proinflammatory responses (Figure 1).
4. Adaptive Immune Response to PM Exposure
4.1. Influence of PM Exposure on T Cell Population
Herein, we focused on the relationship between adaptive immunity and exposure to PM by reviewing recent studies with mutual interests. Currently, most studies have revealed that T lymphocyte-mediated adaptive immune responses are associated with the pathophysiology following PM exposure. CD4+T cells, also known as T helper cells (Th cells), are key components of the adaptive immune system. The Th cells can be divided into three types, such as Th type-1 (Th1), Th type-2 (Th2), and Th type-17 (Th17) cells, displaying unique cytokine secretion phenotypes with distinct functional characteristics [80]. After the differentiation of Th cells, Th1 and Th2 cells play distinct roles in immune responses. Th1 cells are triggered by the polarizing cytokine IL-12 and secrete IFN-γ and IL-2, facilitating the cell-mediated response, typically against intracellular bacteria and protozoa [81]. Th2 cells are triggered by the polarizing cytokines IL-4 and IL-2, and are involved in humoral immune responses via their effector cytokines, such as IL-4, IL-5, IL-9, IL-10, IL-13, and IL-25 [80,82]. Several studies have investigated the influence of PM on Th1/Th2 responses, suggesting diverse answers to this question. First, some studies revealed that the Th1 profile can be affected by PM [28,83,84]. Levels of IL-12 and IFN-γ, the Th1 cytokines, were increased in a dose-dependent manner following PM exposure [84,85]. Additionally, the production of numerous cytokines, including IL-6, IL-1β, and GM-CSF, except IL-10, increased in human alveolar macrophages (AMs) exposed to PM10 [67,86,87], which accounts for alterations in the T cell cytokine-mediated cellular immunity in response to PM in the airway mucosa.
PM can also affect Th2 cytokine profiles. For example, diesel extract particles (DEP) favor Th2-type responses, whereas black carbon particles upregulate both Th1 and Th2 cytokine production [88]. Moreover, fine particles induced lung inflammation through the production of IL-13 and IL-25 in alveolar macrophages [89]. IL-13 is structurally and functionally similar to IL-4 and plays an important role in the defense against parasite infections and allergic diseases. Furthermore, IL-13 promotes fibrosis in the chronic inflammation state and stimulates mucus production by pulmonary epithelial cells [90,91,92]. In addition, IL-13 affects B cells to induce isotype switching, resulting in a switch from B cells to immunoglobulin E (IgE) [93]. IL-25 is structurally similar to IL-17 and is produced by several cell types, including T cells, mast cells, and macrophages. IL-25 induces the production of other Th2-type cytokines, such as IL-4, IL-5, and IL-13 [94,95]. When PM and specific allergens are inhaled simultaneously, T cell-mediated inflammation increases IL-4, IL-5, IL-13, GM-CSF, chemokine (C-C motif) ligand 5 (CCL5), monocyte-chemotactic protein-3 (MCP-3), and macrophage inflammatory protein-1 (MIP-1) [88]. Moreover, the upregulation of transcription factor GATA Binding Protein 3 (GATA3; Th2 maker) and the downregulation of T-bet (Th2 maker) have been identified in the nasal mucosa of PM2.5-exposed rats [96]. An increase in Th2-related cytokines and a decrease in Th1-related cytokines disrupt the balance between Th1 and Th2 cells, with skewing toward Th2, which is closely related to immunotoxicity in the cardiovascular system. Therefore, depending on the exposure regimes and host factors, differential regulation of helper T cell population is markedly involved in PM-induced immune regulation.
4.1.1. AMs as Pivotal Mediators of T Cell Skewing
As stated above, PM2.5 disrupts the balance of Th1/Th2 cells. Numerous studies have revealed that this imbalance is induced at the transcriptional level, as well as by various cytokines [97,98]. AMs are known as the principal cells that process airborne particles in the lung and act as APCs to activate the adaptive immune response, although not as efficiently as DCs [99]. Hence, we postulate that antigen presentation stimulates immature T lymphocytes in the respiratory tract to become either mature helper T cells or mature cytotoxic T cells. Moreover, major histocompatibility complex (MHC) II expression by macrophages is an important part of presenting antigens to stimulate T cells. Expression level of MHC II increases when AMs phagocytize PM containing endotoxins, such as lipopolysaccharide [100]. Reportedly, PM promotes B7 and cluster of differentiation 40 (CD40) expression in DCs and monocytes [101,102,103]. Therefore, naïve T cells can differentiate into Th1 or Th2 cells. Some studies have reported that PM induces an imbalance of Th1/Th2 via TLR-MyD88 signaling [62,63]. Additionally, studies have reported that the TLR4-MyD88 dependent pathway plays an important role in recognizing the PM2.5 and Th2 differentiation process [62,63]. PM affects transcription factors, such as NF-κB, which play a pivotal role in adaptive immune responses. Activation of NF-κB promotes T cell activation, proliferation, and survival [104]. Several studies have reported that the activation of NF-κB correlates with the size and composition of PM [105,106,107]. On fulfilling specific conditions, NF-κB signaling can be upregulated, and MHC II and costimulatory molecules can be sequentially activated.
4.1.2. Adjuvant-Like Actions of PM in T Cell Regulation
Reportedly, the effect of PM as an adjuvant has been assessed on the adaptive response [108]. In this context, an adjuvant is a substance that boosts the immune response to antigens on treatment with antigens. Therefore, PM2.5 was administered only during the sensitization period with house dust mite (HDM) as the allergen. The results showed that PM acted as an adjuvant by promoting inflammation and allergic sensitization, whereas other groups failed to induce important inflammatory responses. Another notable result was that the PAH content in PM promoted Th17-responses and led to the activation of the aryl hydrocarbon receptor (AhR). PM exposure stimulated the expression of Th17 cytokines, including IL-17 and IL-22, as well as that of retinoic acid-related orphan receptor gamma t (RORγt), the Th17 lineage determining transcription factor. Following allergen sensitization, the stimulated expression of AhR in DCs promoted T lymphocyte activation and differentiation into Th17 cells. Furthermore, PM induced allergic sensitization via Th2 associated inflammation [108]. Th17 subsets are involved in the recruitment of neutrophils and induction of inflammation in response to PM exposure.
4.2. PM-Induced Suppression of T Cell Immune Responses
Some reports have demonstrated that PM exposure suppressed T cell-mediated cellular immune responses [59,60,78], which poses a risk of respiratory infection. DC is the APC capable of initiating the primary T cell response. In the lung, local DCs react with the PM that penetrates the respiratory tract barrier, bridging innate immunity with adaptive immunity by antigen presentation. Although PM enhanced GM-CSF-induced DC maturation, it attenuated IL-5, IL-13, and IFN-γ secretion, thus lowering the frequencies of alloantigen-specific T helper type 1 effector cells [78]. This indicates that PM exposure may impair the T helper 1 response and increase susceptibility to pulmonary pathogens [78]. In response to PM exposure, DCs tend to mature into distinct cell phenotypes, which promote immune suppression rather than immune stimulation [109]. These tolerogenic, immunosuppressive DCs significantly decreased T helper type 2 and significantly increased regulatory T cells.
Furthermore, accumulated evidence has revealed that PM can deteriorate the host immune system by activating regulatory T cells and IL-10. In vivo experiments in neonatal mice showed that environmentally persistent free radicals significantly increased the levels of pulmonary regulatory T cells and immunosuppressive cytokine IL-10 following influenza infection [110,111]. Consequently, the protective T cell function was suppressed, and the disease severity of influenza was enhanced, indicating that PM exposure may interfere with human immune suppression via regulatory T cells and immunosuppressive cytokines [110,111]. In mice, compared with filtered air (FA) exposed/HDM-challenged mice, UFP-exposed offspring demonstrated lower white blood cell counts in the bronchoalveolar lavage fluid, as well as less pronounced peribronchiolar inflammation. Furthermore, mice prenatally exposed to UFP significantly elevates circulating IL-10 levels, resulting in the upregulation of regulatory T cells and reduced levels of inflammatory cytokines, IL-13 and IL-17, suggesting suppression of Th2/Th17 responses [112]. Collectively, these findings verify the lowered immune response against allergens and pathogens in PM-exposed subjects.
4.3. Controversy Regarding the Effect of PM on Adaptive Immunity
PM has been well known to affect public health and is considered an economic burden, especially in developing countries. Several scientific studies have confirmed that the pathophysiology of this process is associated with adaptive immunity [33,62,63,96]. Previous studies have suggested that PM stimulates adaptive immunity; conversely, some findings report that PM inhibits the adaptive immune response [109,110,111]. These two opposing results may seem problematic, but the research we introduced is not mutually exclusive. As experiments are performed in distinct settings in different laboratories, PM can either stimulate or suppress immune responses by T lymphocytes. Further studies are necessary to determine factors that determine the balance between stimulation and suppression in PM-associated adaptive immunity. The composition or size of the PM, age of the patient or laboratory animals, and degree of exposure are possible factors that should be closely monitored.
5. Effects of PM on Host Resistance to Infection
5.1. Roles of Airway Macrophages in Response to Infection and PM Exposure
For microbes to establish infection in the lower respiratory tract, they must first avoid the innate immune system, composed of the epithelial cell barrier, antimicrobial molecules in mucus, AMs, neutrophils, natural killer cells, and DCs. AMs detect pathogen-associated molecular patterns via TLRs. Through TLR activation, a pathogen-triggered immune reaction cascade is stimulated [32,33]. This cascade includes cytokine release, AMs, and reactive oxygen species (ROS) originating from neutrophils, phagocytosis, and intracellular killing. An excessive immune response could destroy barrier defense and increase the risk of developing other infections, such as tuberculosis [113].
Macrophages act as the first line of defense in recognizing tissue injury in response to PM or microorganisms. Macrophages are mainly involved in the process of phagocytosis, and eradication of bacteria, as well as other harmful organisms [11]. In general, monocytes can be differentiated into either activated macrophage M1 or M2 with distinct phenotypes and functionality corresponding to the Th1/Th2 paradigm [114]. Many Th1-derived proinflammatory cytokines are involved in activating M1 macrophages, which can mediate host defense against the various infections of bacteria, viruses, and protozoa. The main function of M2 macrophages is an anti-inflammatory response by stimulating adaptive Th2 immunity, as well as the regulation of angiogenesis, tissue remodeling, and wound healing [114]. An imbalance of M1/M2 polarization is associated with the development of various immunological disorders [115,116]. Therefore, it is pivotal to maintain the balance between M1 and M2 macrophages for the host homeostasis. Exposure to PM2.5 induces M1 macrophage-dependent inflammation by increasing secretions of IFN-γ, and IL-17 and IL-21 in CD4+T cells, indicating activation or enrichment of Th1 and Th17 responses, respectively [117]. Mechanistically, exposure to PM2.5 particles promotes M1 polarization through TLR4-activated JNK and p38 pathways [118]. In contrast, chronic exposure to PM2.5 reprograms cellular polarization toward M2 macrophage-involved adverse outcomes, including sustained inflammation and emphysematous lesions. Moreover, chronic exposure to PM2.5 downregulates the expression of histone deacetylase 2, which is crucial for deleterious airway remodeling, facilitating COPD [119]. Depending on the exposure regimes, differentially activated macrophages play crucial roles in disease development and progression via specific molecular pathways (Figure 2).
5.2. PM Exposure in Bacterial Infection
During bacterial infection, it is recognized by their PRRs like TLRs activates macrophages to produce M1-like proinflammatory cytokines, such as TNF-α, IL-1, IL-6, IL-12, and NO that can eliminate the invading bacterial pathogens [120]. Some bacterial pathogens prevent M1-like polarization, in that case driving the polarization toward an M2 phenotype to lessen the inflammatory response. Mycobacterium (M.) tuberculosis infection induces M1 polarization, which secretes the proinflammatory mediators, such as TNF-α, IL-6, IL-12, CCL5, and chemokine C-C motif ligand (CXCL18) [121]. However, during the formation and development of tuberculous granulomas, the active state of macrophages underwent M1-like to M2-like transition [122].
A potential link between long-term exposure to suspended PM and increased rates of pulmonary tuberculosis (TB) has been reported [47,123,124]. To address the effects of PM on the natural course of tuberculosis infection, researchers reported that PM impaired human peripheral blood mononuclear cell (PBMC) immune reactions toward M. tuberculosis, via the downregulation of TLR-dependent cytokines [47]. More specifically, DEP as a part of urban ambient PM2.5 interfered with human anti-mycobacterial immunity [28,46]. Following the exposure of PBMCs to DEP, researchers observed the suppression of expression of M. tuberculosis-induced TLRs 3, 4, 7, and 10, as well as NF-κB-induced expression of chemokines and cytokines, including IFN-γ, TNF-α, IL-1β, and IL-6, which play an important role in the control of M. tuberculosis. In vitro and in vivo M. tuberculosis infections promoted epithelial cells in the airway, provoking anti-mycobacterial reactions, including the release of cytokines, chemokines, and antimicrobial peptides. A549 cells, a type II alveolar epithelial cell line, generated IL-8 and MCP-1, which attract neutrophils, T cells, basophils, and monocytes, in the early stages of M. tuberculosis infection [46]. Furthermore, A549 cells produce human β-defensin 2 (BD-2) and 3 under the influence of mycobacterial lipids and different M. tuberculosis strains. Human BD-2 and 3 are tiny cationic peptides that are affiliated with a family of antimicrobial peptides. As they possess antimicrobial characteristics and modulate the immune response, human BD-2 and 3 play key roles in the early control of M. tuberculosis infection by reducing the pulmonary bacterial load [46].
On investigating PM2.5 and T cell functions in human PBMCs, it has been observed that urban PM may negatively affect the mechanisms of M. tuberculosis-specific human T cell functions [47,125]. Exposure to PM2.5 reduced the capacity of PBMCs to restrict the growth of M. tuberculosis and downregulated the expression of the early activation marker, CD69, during infection on CD3+T cells. Furthermore, these findings suggest that PM2.5 exposure decreased the production of IFN-γ in CD3+, TNF-α in CD3+, and CD14+ PBMC, which were infected by M. tuberculosis, as well as the M. tuberculosis-induced expression of T-box transcription factor (TBX21; T-bet) [47,125]. Notably, in contrast to these results, PM2.5 exposure increased the expression levels of anti-inflammatory cytokines, such as IL-10 in CD3+ and CD14+ PBMCs. These data revealed that PM2.5 damaged crucial anti-mycobacterial T cell immune functions [125]. Another study investigated whether in vitro exposure to urban air pollution PM alters human immune responses to M. tuberculosis in human PBMCs obtained from IFN-γ release assay (IGRA+) and IGRA− healthy study subjects. This study was performed in Iztapalapa, a highly populated tuberculosis-endemic municipality of Mexico City with elevated outdoor air pollution levels, and researchers collected monthly PM2.5 and PM10 from rainy, cold-dry, and warm-dry seasons to evaluate whether seasonal changes affect the human immune response to M. tuberculosis. This result corroborated previous research findings, demonstrating that, except for PM2.5 from the cold-dry season, pre-exposure to all seasonal PM reduced M. tuberculosis phagocytosis by PBMC. Additionally, M. tuberculosis-induced IFN-γ production was suppressed in PM2.5 and PM10 pre-exposed PBMC from IGRA+ subjects. Hence, both pre-exposure to PM10 and PM2.5 resulted in a greater loss of M. tuberculosis growth control [47] As there exists a close mechanistic association between PM exposure and response to M. tuberculosis, the epidemiological evidence in populations with high tuberculosis prevalence should be explored, including the Korean peninsula, who also present a high risk of PM exposure.
5.3. PM Exposure in Viral Infection
A previous study has demonstrated that high doses of PM10 suppressed respiratory tract inflammatory responses to the respiratory syncytial virus (RSV), a frequent cause of viral pneumonia in infants and the elderly [126]. Therefore, the hosts displayed lower viral immunity after simultaneous exposure to RSV and PM, compared with the defense against a single infection by RSV. Coronavirus disease 2019 (COVID-19), a novel disease, has emerged as a major public health concern. The primary symptoms of COVID-19 include respiratory tract symptoms, such as fever, cough, fatigue, pneumonia, and acute respiratory distress syndrome, remaining asymptomatic in some cases [127]. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) appears to be associated with PM, which can increase the severity of COVID-19 outcomes [128]. In particular, the spread of COVID-19 has been observed in northern Italian cities, where daily PM concentrations are higher than the annual average [129]. Like other viruses, SARS-CoV-2 could spread rapidly through the aerosol by utilizing the PM as a carrier. Notably, SARS-CoV-2 RNA was detected using three viral markers of PM in the area, thus suggesting a potent simultaneous exposure to SARS-CoV-2 and hazardous PM chemicals. Moreover, populations that live in areas with high levels of PM would be susceptible to acute or chronic inflammatory states owing to the dysregulated immune function induced by toxic chemicals released by PM. PM-insulted immune system would be inefficient in defending against viral infection [130]. In addition to the impaired antiviral responses, PM-induced production of free radicals can contribute to lung cell toxicity during SARS-CoV-2 infection [131,132]. Moreover, PM-activated inflammatory cells and cytokines can exacerbate pulmonary inflammation and tissue injuries in patients with COVID-19 [131]. Long-term and short-term exposures to high levels of pollutants correlated with an increase in COVID-19 incidence worldwide [131]. However, additional issues need to be addressed, including the particle size to which the virus binds, and more importantly, the duration for which the virus remains active in different age groups, as elderly subjects were found to be increasingly susceptible to COVID-19 worldwide.
6. PM-Linked Hypersensitivity
Exposure to air pollution increases the risk of asthma, allergic rhinitis, atopic dermatitis (AD), and allergen sensitization. The effects of air pollution exposure can be displayed differently depending on the timing of exposure, individual sensitivities, and simultaneous exposure to allergens.
6.1. Effect on Acute and Chronic Hypersensitivity Diseases
Previous studies have suggested that PM can increase hyperresponsiveness in the respiratory system. Diseases representing respiratory allergies include allergic rhinitis and asthma. For instance, urban levels of air pollution can induce pulmonary hyperresponsiveness in rats; however, the effect was reversed under the nonpolluted condition [133]. Moreover, PM can enhance immunological responses and inflammatory reactions in the nasal mucosa, but not the development of allergic rhinitis. Short-term exposure to PM (PM10 and PM2.5) increased the exacerbation of asthma, as well as the number of emergency cases [134]. Although there is a lack of evidence illustrating that PM can cause respiratory allergic diseases, such as asthma or allergic rhinitis, it is well accepted that PM is associated with exacerbations of asthma or allergic rhinitis [134]. It needs to be clarified whether short-term, peak exposure, or long-term exposure to PM is greatly associated with asthma exacerbation. Furthermore, PM exposure can induce allergic inflammation with T helper cell 2 (Th2) and Th17 differentiation, which can exacerbate allergic asthma [135]. Additionally, it can increase the risk of hospitalization in COPD. COPD is not a disease that is induced by hyperresponsivity—but following hyperactivity in the airways of COPD patients, disease exacerbation can occur because hyperactivity induces airway inflammation, thus narrowing the airway. Hence, COPD can be exacerbated by PM-associated hypersensitivity.
6.2. PM Enhances Allergen Sensitization
PM can enhance the immune response to specific allergens and increase IgE production. In a previous study, exposure to DEP increased IgE production in the nasal mucosa [136]. DEP exposure can only induce the production of IgE, but not of IgA, IgM, IgG, and albumin. In the lavage fluid, IgE-secreting cells were reportedly increased, but the number of IgA-secreting cells was not elevated. Additionally, epsilon mRNA production was altered. These results suggest that the local production of IgE is altered based on both quantitative and qualitative aspects, confirming that DEP can be an allergen and induces allergen specific IgE production. Moreover, ragweed specific IgE production was more highly induced upon a challenge of DEP and ragweed allergens when compared to that with ragweed allergens alone [137]. DEP and aeroallergens demonstrated synergistic effects and promoted allergen-induced allergic respiratory disease. Therefore, it can enhance allergen specific IgE production. Notably, DEP can elicit symptoms, act on mast cells, and enhance allergic responses, presenting more severe symptoms following high-dose, short-term exposure. These findings demonstrate that DEP can further augment allergic responses to other allergens.
Air pollutants, such as PM, can enhance allergen sensitization by carrying aeroallergens into the airway. Following the introduction of aeroallergens into the airway, airway epithelial cells promote oxidative stress and inflammation, increasing the antigenicity of allergens [138,139,140,141]. DEP alone did not induce the release of histamine and symptoms; the DEP and allergen + placebo groups demonstrated extremely low histamine levels. However, the histamine level increased by approximately three-fold in the DEP and allergen groups when compared to that in the allergen alone group. This implies that DEP did not induce an allergic response alone, but can increase the allergic response to the allergen, increasing the severity. Therefore, according to this study, PM does not act as an allergen alone, but augments allergic responses to other allergens, and hence, is harmful and can exacerbate other allergic diseases.
6.3. Effect of Prenatal and Early-Life Exposure on Hypersensitivity Diseases
It is well known that early-life exposure to fine PM in the air is associated with adult infant respiratory diseases, such as pneumonia and childhood asthma, with additional evidence suggesting that UFPs may affect childhood respiratory disease. PM can exacerbate respiratory allergic diseases, such as asthma and allergic rhinitis. In clinical evaluations, exposure to PM10 in the first year of life increased the incidence of asthma at 12 years of age [142]. Moreover, subjects born at locations with high PM2.5 exposure tend to display an increased incidence of asthma at seven years of age [143]. However, extensive investigations are warranted, as several studies have observed no correlation between the prevalence of allergic rhinitis or asthma incidence and PM exposure [144,145].
Several epidemiological studies have reported that living in places with high levels of air pollutants from birth increases the risk of allergic disease [91,108,144,146]. Additionally, if the mother resided adjacent to the main road, the offspring presented an increased risk of allergen sensitization and asthma. Studies have shown that exposure to air pollution during growth, even at an extremely young age, can affect lung function in adults. An 8-year follow-up of 1,759 students aged between 10 and 18 years in 12 schools in California, USA, revealed that those highly exposed to air pollution showed poorer lung functions than those who were not highly exposed, with 4.9 times higher risk [147]. Furthermore, a large cross-sectional study investigating children aged 6–14 years, in 10 cities in South Korea, observed that exposure to car-related air pollution increased the risk of asthma, rhinitis, allergen sensitization, and lowered lung functions in children living adjacent to major roads. In addition to subjective symptoms, effects on objective indicators of allergic diseases were recorded [146]. Moreover, short-term exposure to ambient temperature, relative humidity, diurnal temperature range (DTR), rainfall, and air pollutants, such as PM, were significantly associated with AD symptoms in infants and young children living in a temperate region [148]. The symptoms, including itching, sleep disturbance, erythema, dry skin, oozing, and edema, were significantly higher in subjects exposed to PM10 than in the control.
Asthma is characterized by variable airway obstruction, hyperresponsivity, and airway inflammation. In addition, asthma is a representative disease associated with hyperresponsiveness. However, the contribution of PM to asthma development remains inconsistent. Many birth cohort studies have been conducted. For example, in a cohort study in Asia, children with increased exposure to PM during gestational weeks 6–22 or the postnatal period demonstrated a higher incidence of asthma [149]. In this study, PM2.5 exposure in both the prenatal and postnatal periods induced later development of asthma. However, another study suggests that there is no significant correlation between the incidence of asthma or wheezing and exposure to PM10 and nitrogen dioxide (NO2) [150], with the prevalence of asthma determined as 15.2–23.3%. The prevalence was the lowest at age three and the highest at age five. The mean exposure to NO2 decreased from the first year of life to the eleventh year of life; the mean exposure to PM10 reportedly decreased.
6.4. Molecular Etiologies of PM-Linked Hypersensitivity
6.4.1. PM-Derived Oxidative Stress
PM causes oxidative damages in the pulmonary system [151]. The main adverse outcomes from exposure to PM are associated with oxidative stress from the disruption of oxidant redox-cycling reaction in the biological system. Although the pulmonary tissue contains the antioxidant defense systems [152], the generation of reactive oxygen species (ROS) plays pivotal roles in the pathologic process during PM exposure. PM can pose ROS in itself [50], or endogenous ROS can be generated through catalytic activity in vivo via cellular redox reactions stimulated by redox-active components of PM [151]. ROS are highly reactive molecules, including superoxide radical (O2¯), hydrogen peroxide (H2O2), and hydroxyl radical ∙OH [50]. Among ROS radicals, ∙OH is one of the most dangerous molecules that can react quickly with DNA and induce damage [153]. PM can catalyze ·OH generation mainly from transition metals and quinones [151], which are expected to affect the upper airways (75%) and lower parts in the lung (42–46%), respectively. PM generates hydroxyl radicals mostly sourced from biomass burning, incomplete combustion, and mineral dust [151,154,155]. Moreover, PM-derived redox-active components and oxidation products of the lipid membrane can serve as ligands for the aryl hydrocarbon receptor (AhR) via modulation of the xenobiotic responsive element (XRE) [135]. AhR-XRE signaling mediates the expression of ROS-producing metabolic enzyme systems, including cytochrome p450 monooxygenase (CYP), proinflammatory cytokines, and NADPH oxidase in the airways [156,157,158,159]. CYP and NADPH oxidase are pivotal players in endogenous production of ROS during PM exposure (Figure 3). Mechanistically, in addition to DNA damage and cytotoxicity, ROS and inflammatory insult can cause mitochondrial injury, which is the crucial step of pulmonary and cardiovascular distress during exposure to air pollutants [113,160].
Following exposure to air pollution, such as fine dust, the immune response to oxidative damage is altered in airway epithelial cells, with inhibited phagocytic action inducing various inflammatory reactions to increase inflammatory mediators, and reduced inflammatory damage in the airway [12,13,33]. In response to PM-induced oxidative stress, antioxidant defense systems are also triggered by NF-E2-related factor 2 (Nrf2), a master transcription factor via modulation of the antioxidant responsive element (ARE) [161]. The Nrf2-ARE-associated antioxidant system fails to activate protection against the oxidative and proinflammatory burst, leading to lung and extrapulmonary tissue damages during PM-induced hypersensitivity (Figure 3). In a cohort study, air pollution levels and lung function were evaluated in children in areas presenting severe air pollution in South Africa, revealing that decreased lung function was associated with mutations in the glutathione S-transferase P gene (GSTP1), which is involved in the metabolism of the antioxidant glutathione following air pollutant exposure [162]. When the gene is mutated, the effective antioxidant reaction against oxidative damage induced by air pollution is downregulated. ROS induces direct damage to proteins and lipids, impairs skin barrier function, and exacerbates atopic dermatitis (AD). In animal studies, AD exacerbation, resulting from oxidative skin damage, has been reported following air pollution exposure and the Th2 immune response [148].
6.4.2. Inflammatory Insults
Increased expression of inflammatory mediators in the airway following exposure to air pollution causes an imbalance in immune responses and increases the likelihood of developing allergic diseases [113,163,164]. In 2003, Lambert et al. showed that exposure to fine dust activated the Th2 immune response after RSV infection, instead of before the usual viral defense mechanisms [165]. The concentrations of inflammatory cells in the bronchoalveolar fluid and Th2 cytokines, such as IL-13, were compared between RSV infected mice exposed to fine dust and RSV infected mice without fine dust exposure. Notably, a lower Th1 immune response was observed in animals presenting RSV infection and fine dust exposure. Exposure to fine dust resulted in an imbalance in the Th1/Th2 immune response after RSV infection, increased airway inflammation, and presented characteristic phenomena of asthma, such as airway sensitivity. It was confirmed that allergen specific Th2/Th17 cells increased in the lung and induced allergen reminiscence [166]. In the Cincinnati Childhood Allergy and Air Pollution Study, children were reportedly sensitized to inhaled allergens. When exposed to high levels of air pollution early in life, sensitization is more pronounced, and the prevalence of asthma is increased. However, there is no evidence supporting clinical outcomes in children. In particular, simultaneous exposure to allergens and air pollution during the early postnatal period amplifies allergic recall and promotes the development of asthma.
In addition to PM, various other matters are known to promote and exacerbate asthma; however, the pathogenesis of asthma induced by these compounds remains unclear. Airway macrophages could react quickly to the inhaled PM as an initial barrier. In patients with severe asthma, airway macrophages are unable to perform their functions, including the phagocytosis of inhaled PM. Th2 cells are essential for IgE isotype switching in B lymphocytes, which may stimulate allergic inflammatory responses, as well as airway hyperresponsiveness [167,168]. Furthermore, PM exposure increases the level of proinflammatory cytokines and induces macrophage polarization, especially inflammatory M1 polarization through the ROS pathway [169]. In certain situations, PM can induce M2 polarization. PM exposure has been associated with chronic inflammatory and autoimmune diseases. Typically, macrophages are cells related to the modulation of chronic inflammation, along with various infectious and autoimmune diseases. Depending on the type of stimulus, macrophages can be polarized into distinct functional phenotypes: M1-type proinflammatory/microbicidal cells, M2-type anti-inflammatory/suppressor cells, or a mixed state of activation [170]. Reportedly, owing to innate immunity, PM can be related to several diseases, including asthma and tuberculosis. Nevertheless, information regarding the pathophysiological aspects of these diseases and their association with innate immunity remains scarce. Various factors could influence the incidence, severity, and pathogenesis of these diseases, including the period of exposure, exposure duration, particle size and components of PM, ethnicity, age, and gender [36].
6.4.3. Crosstalk with Allergens
Air pollution increases the permeability of airway epithelial cells and increases sensitization by increasing the exposure of immune cells or antigenicity of external allergens [171,172]. Although allergic rhinitis and AD have not been extensively associated with PM exposure, the inflammatory responses through the nose and skin can be expected to promote sensitization. Air pollutants play an important auxiliary role in allergen sensitization, including DEP and PAH, which are components of fine dust and automobile exhaust. DEP, a major component of vehicle exhaust, increased the allergenicity, inducing allergen sensitization through increased Th2 cytokines and mast cell activation [166]. In the US, a 5-year follow-up was performed in children between the ages of 0–5 years living adjacent to the main road. Reportedly, the group exposed to air pollution demonstrated higher serum total IgE levels than those not exposed to air pollution. It can be interpreted that during the process of allergic development, allergens with air pollution-related substances are carried into the airway and cause sensitization. Another study has reported that the concentration of Th2 cytokines in rhinorrhea increased by more than 16-fold following simultaneous exposure to pollen and DEP when compared with exposure to pollen allergens alone [166], suggesting PM-induced crosstalk with allergen responses.
6.4.4. Epigenetics and miRNA Regulation during PM-Linked Hypersensitivity
Environmental exposure during pregnancy or early childbirth is closely linked to the development of allergic diseases, and in recent years, the mechanism has been gradually revealed in the development of welfare genetics. Welfare genetic changes are those in which the gene itself does not change owing to environmental exposure, but changes are observed in gene expression and activity. Changes in gene expression due to environmental exposure can occur over life, but it is postulated that effects are further pronounced in babies and infants. Studies have investigated fetal exposure to harmful environments, reporting that changes in the asthma risk may be substantiated by, for example, exposure to car exhaust and passive smoking in the womb, along with the risk of developing allergic diseases [173]. Exposure to a harmful environment during pregnancy acts on regulatory T cells and induces DNA methylation of allergy-related substances, such as FoxP3, contributing to the development of allergic diseases [174].
In genetically predisposed individuals, allergic diseases are susceptible to interaction with environmental factors. In particular, the inflammatory regulatory response in allergic diseases is presumed to involve gene regulation of the relevant immune system component; however, it has been recently noted that miRNAs affect the expression of these genes. miRNAs are extremely small noncoding RNAs, only 21–25 bases in size, that regulate gene expression by binding to mainly specific mRNAs, which inhibit or promote protein synthesis of target genes [175]. Reportedly, miRNAs regulate several genes in cells, thereby regulating immune cell growth, differentiation, and proliferation in allergic diseases. To date, several miRNAs associated with AD have been revealed, with dysregulation, such as increased or decreased miRNA expression in patients with AD when compared with healthy, atopic skin. Furthermore, studies have shown that the number of low regulatory T cells in cord blood was associated with increased miRNA-223 expression, affecting atopic pathogenesis [176].
7. Conclusions
In this review, we systemically discussed the immune alteration-associated disease outcomes with cellular and molecular evidence in response to PM, including biomass-derived pollutants. Epidemiological and experimental studies have demonstrated that all particles are not equally toxic, but cause different adverse health effects. Generally, the particulate size is the deterministic factor of translocation and actions in the airway mucosa and other tissues. Although most fine particles deposits in the air-blood barrier tissues, UFPs, including some components of WSP, can easily migrate to different regions of the body, leading to local and systemic immunological responses. Although the migration of most fine particulates to the circulation is limited, PM-derived or endogenously produced ROS and inflammatory cytokines can mediate acute and chronic distress, including infection and hypersensitivity during PM exposure.
Despite controversies about whether PM can either stimulate or inhibit adaptive immune responses, PM-induced cellular, and molecular alterations can contribute to outcomes of human diseases, including infection, hypersensitivity, and chronic diseases depending on chemical composition or size of the PM and exposure regimes (frequency, duration, and age). Exposure to PM, including WSP, alters the macrophage activity through the pattern-recognition signaling, leading to the production of proinflammatory cytokines and activation of innate immune cells in the airway and circulation. Depending on the exposure regimes, differentially activated macrophages play crucial roles in disease development and progression via specific molecular pathways. In particular, exposure to PM2.5 induces M1 macrophage-dependent inflammation by increasing the secretions of Th1 and Th17 cytokines through PRR-activated signaling, while chronic exposure to PM2.5 can reprogram cellular polarization toward M2 macrophage-involved sustained inflammation, fibrosis, and deleterious airway remodeling. In terms of host responses to infection, PRR-activated macrophages produce M1-like proinflammatory cytokines, destroying the invading pathogens. However, some bacterial pathogens tend to prevent M1-like polarization, facilitating M2-like transition during the chronic inflammation and fibrosis in association with polarization toward Th2-lined immune response to PM exposure.
However, there are still a few questions that need to be addressed for a more comprehensive understanding. First, the mechanistic implications of the immune response and mode of action of different PM mixtures have not been fully elucidated with comparative investigations for diverse exposure regimes. In addition to individual chemical-specific toxicity, integrative effects of PM mixture on human immunity-related disease processes need to be further investigated. In particular, clinical evaluation of simultaneous exposure to the emerging microbes and air pollutants are increasing in interest. Epidemic investigation in the population under the biomass-associated air pollution would provide crucial insights to predict unexpected risks in an emerging microbe-abundant society. Second, target cellular molecules linked with immune dysregulation mediated by PM actions remain elusive. The identification of target molecules and their mechanistic network would help identify new strategies to inhibit the adversary health effects of biomass-derived PM, including WSP. The present review provides insights into novel immunotherapeutic interventions against PM-induced acute and chronic environmental disorders. Moreover, it is warranted to address climate change- or deforesting-associated PM exposure and health for the sustainable planet.
Author Contributions
Conceptualization, Y.M.; writing—original draft preparation, Y.M., S.-J.L.; writing—review and editing, Y.M., A.N.; verification and visualization, S.B.P.; supervision, Y.M.; project administration, Y.M.; funding acquisition, Y.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a 2-Year Research Grant of Pusan National University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mishra, R.; Krishnamoorthy, P.; Gangamma, S.; Raut, A.A.; Kumar, H. Particulate matter (PM10) enhances RNA virus infection through modulation of innate immune responses. Environ. Pollut. 2020, 266, 115148. [Google Scholar] [CrossRef]
- Namork, E.; Johansen, B.V.; Løvik, M. Detection of allergens adsorbed to ambient air particles collected in four European cities. Toxicol. Lett. 2006, 165, 71–78. [Google Scholar] [CrossRef]
- Vincent, R.; Goegan, P.; Johnson, G.; Brook, J.R.; Kumarathasan, P.; Bouthillier, L.; Burnett, R.T. Regulation of Promoter-CAT Stress Genes in HepG2 Cells by Suspensions of Particles from Ambient Air. Fundam. Appl. Toxicol. 1997, 39, 18–32. [Google Scholar] [CrossRef]
- Pope, C.A., 3rd; Burnett, R.T.; Thun, M.J.; Calle, E.E.; Krewski, D.; Ito, K.; Thurston, G.D. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. J. Am. Med. Assoc. 2002, 287, 1132–1141. [Google Scholar] [CrossRef] [Green Version]
- Loomis, D.A.N.A.; Huang, W.; Chen, G. The International Agency for Research on Cancer (IARC) evaluation of the carcinogenicity of outdoor air pollution: Focus on China. Chin. J. Cancer 2014, 33, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Loomis, D.; Grosse, Y.; Lauby-Secretan, B.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Baan, R.; Mattock, H.; Straif, K. The carcinogenicity of outdoor air pollution. Lancet Oncol. 2013, 14, 1262–1263. [Google Scholar] [CrossRef]
- Hamra, G.B.; Guha, N.; Cohen, A.; Laden, F.; Raaschou-Nielsen, O.; Samet, J.M.; Vineis, P.; Forastiere, F.; Saldiva, P.; Yorifuji, T.; et al. Outdoor Particulate Matter Exposure and Lung Cancer: A Systematic Review and Meta-Analysis. Environ. Health Perspect. 2014, 122, 906–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhang, L.-W.; Huang, J.-J.; Song, F.-J.; Zhang, L.-P.; Qian, Z.-M.; Trevathan, E.; Mao, H.-J.; Han, B.; Vaughn, M.; et al. Long-term exposure to urban air pollution and lung cancer mortality: A 12-year cohort study in Northern China. Sci. Total. Environ. 2016, 571, 855–861. [Google Scholar] [CrossRef]
- Wen, C.P.; Gao, W. PM 2.5: An important cause for chronic obstructive pulmonary disease? Lancet Planet. Health 2018, 2, e105–e106. [Google Scholar] [CrossRef]
- Khafaie, M.A.; Salvi, S.S.; Yajnik, C.S.; Ojha, A.; Khafaie, B.; Gore, S.D. Air pollution and respiratory health among diabetic and non-diabetic subjects in Pune, India—Results from the Wellcome Trust Genetic Study. Environ. Sci. Pollut. Res. 2017, 24, 15538–15546. [Google Scholar] [CrossRef]
- Ling, S.H.; van Eeden, S.F. Particulate matter air pollution exposure: Role in the development and exacerbation of chronic obstructive pulmonary disease. Int. J. Chronic Obstr. Pulm. Dis. 2009, 4, 233–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.M.; Kim, H.R.; Park, Y.J.; Lee, S.Y.; Chung, K.H. Organic extracts of urban air pollution particulate matter (PM2.5)-induced genotoxicity and oxidative stress in human lung bronchial epithelial cells (BEAS-2B cells). Mutat. Res. Toxicol. Environ. Mutagen. 2011, 723, 142–151. [Google Scholar] [CrossRef]
- Harrison, C.M.; Pompilius, M.; Pinkerton, K.E.; Ballinger, S.W. Mitochondrial Oxidative Stress Significantly Influences Atherogenic Risk and Cytokine-Induced Oxidant Production. Environ. Health Perspect. 2011, 119, 676–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinmayr, G.; Pedersen, M.; Stafoggia, M.; Andersen, Z.J.; Galassi, C.; Munkenast, J.; Jaensch, A.; Oftedal, B.; Krog, N.H.; Aamodt, G.; et al. Particulate matter air pollution components and incidence of cancers of the stomach and the upper aerodigestive tract in the European Study of Cohorts of Air Pollution Effects (ESCAPE). Environ. Int. 2018, 120, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Crinnion, W. Particulate Matter Is a Surprisingly Common Contributor to Disease. Integr. Med. 2017, 16, 8–12. [Google Scholar]
- Zhang, P.; Dong, G.; Sun, B.; Zhang, L.; Chen, X.; Ma, N.; Yu, F.; Guo, H.; Huang, H.; Lee, Y.L.; et al. Long-Term Exposure to Ambient Air Pollution and Mortality Due to Cardiovascular Disease and Cerebrovascular Disease in Shenyang, China. PLoS ONE 2011, 6, e20827. [Google Scholar] [CrossRef] [PubMed]
- Katanoda, K.; Sobue, T.; Satoh, H.; Tajima, K.; Suzuki, T.; Nakatsuka, H.; Takezaki, T.; Nakayama, T.; Nitta, H.; Tanabe, K.; et al. An Association Between Long-Term Exposure to Ambient Air Pollution and Mortality from Lung Cancer and Respiratory Diseases in Japan. J. Epidemiology 2011, 21, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Riddervold, I.S.; Bønløkke, J.H.; Olin, A.-C.; Grønborg, T.K.; Schlünssen, V.; Skogstrand, K.; Hougaard, D.M.; Massling, A.; Sigsgaard, T. Effects of wood smoke particles from wood-burning stoves on the respiratory health of atopic humans. Part. Fibre Toxicol. 2012, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- Jamriska, M.; Thomas, S.; Morawska, L.; Clark, B.A. Relation between indoor and outdoor exposure to fine particles near a busy arterial road. Indoor Air 1999, 9, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Erlandsson, L.; Lindgren, R.; Nääv, Åsa; Krais, A.M.; Strandberg, B.; Lundh, T.; Boman, C.; Isaxon, C.; Hansson, S.R.; Malmqvist, E. Exposure to wood smoke particles leads to inflammation, disrupted proliferation and damage to cellular structures in a human first trimester trophoblast cell line. Environ. Pollut. 2020, 264, 114790. [Google Scholar] [CrossRef]
- Rokoff, L.B.; Koutrakis, P.; Garshick, E.; Karagas, M.R.; Oken, E.; Gold, D.R.; Fleisch, A.F. Wood Stove Pollution in the Developed World: A Case to Raise Awareness Among Pediatricians. Curr. Probl. Pediatr. Adolesc. Health Care 2017, 47, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Ghio, A.J.; Soukup, J.M.; Case, M.; Dailey, L.; Richards, J.; Berntsen, J.; Devlin, R.B.; Stone, S.; Rappold, A. Exposure to wood smoke particles produces inflammation in healthy volunteers. Occup. Environ. Med. 2011, 69, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Orozco-Levi, M.; Garcia-Aymerich, J.; Villar, J.; Ramírez-Sarmiento, A.; Antó, J.M.; Gea, J. Wood smoke exposure and risk of chronic obstructive pulmonary disease. Eur. Respir. J. 2006, 27, 542–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajpandey, M. Domestic smoke pollution and chronic bronchitis in a rural community of the Hill Region of Nepal. Thorax 1984, 39, 337–339. [Google Scholar] [CrossRef] [Green Version]
- Ezzati, M.; Kammen, D.M. Quantifying the effects of exposure to indoor air pollution from biomass combustion on acute respiratory infections in developing countries. Environ. Health Perspect. 2001, 109, 481–488. [Google Scholar] [CrossRef]
- Smith, K.R.; Samet, J.M.; Romieu, I.; Bruce, N. Indoor air pollution in developing countries and acute lower respiratory infections in children. Thorax 2000, 55, 518–532. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.E. Airborne Particulate Matter: Human Exposure and Health Effects. J. Occup. Environ. Med. 2018, 60, 392–423. [Google Scholar] [CrossRef]
- Wei, T.; Tang, M. Biological effects of airborne fine particulate matter (PM 2.5) exposure on pulmonary immune system. Environ. Toxicol. Pharmacol. 2018, 60, 195–201. [Google Scholar] [CrossRef]
- Mukherjee, A.; Agrawal, M. A Global Perspective of Fine Particulate Matter Pollution and Its Health Effects. Rev. Environ. Contam. Toxicol. 2018, 244, 5–51. [Google Scholar] [CrossRef]
- Alfaro-Moreno, E.; Nawrot, T.S.; Nemmar, A.; Nemery, B. Particulate matter in the environment: Pulmonary and cardiovascular effects. Curr. Opin. Pulm. Med. 2007, 13, 98–106. [Google Scholar] [CrossRef]
- Gu, X.-Y.; Chu, X.; Zeng, X.-L.; Bao, H.-R.; Liu, X.-J. Effects of PM2.5 exposure on the Notch signaling pathway and immune imbalance in chronic obstructive pulmonary disease. Environ. Pollut. 2017, 226, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Glencross, D.A.; Ho, T.-R.; Camiña, N.; Hawrylowicz, C.M.; Pfeffer, P.E. Air pollution and its effects on the immune system. Free Radic. Biol. Med. 2020, 151, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Miyata, R.; Van Eeden, S.F. The innate and adaptive immune response induced by alveolar macrophages exposed to ambient particulate matter. Toxicol. Appl. Pharmacol. 2011, 257, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Deng, L.; Miao, Y.; Guo, X.; Li, Y. Particle deposition in the human lung: Health implications of particulate matter from different sources. Environ. Res. 2019, 169, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Idani, E.; Geravandi, S.; Akhzari, M.; Goudarzi, G.; Alavi, N.; Yari, A.R.; Mehrpour, M.; Khavasi, M.; Bahmaei, J.; Bostan, H.; et al. Characteristics, sources, and health risks of atmospheric PM10-bound heavy metals in a populated middle eastern city. Toxin Rev. 2020, 39, 266–274. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, J.; Yang, X.; Zhang, Y.; Chen, Z. The Role and Potential Pathogenic Mechanism of Particulate Matter in Childhood Asthma: A Review and Perspective. J. Immunol. Res. 2020, 2020, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.; Daher, N.; Kam, W.; Shafer, M.M.; Ning, Z.; Schauer, J.J.; Sioutas, C. Spatial and temporal variation of chemical composition and mass closure of ambient coarse particulate matter (PM10–2.5) in the Los Angeles area. Atmos. Environ. 2011, 45, 2651–2662. [Google Scholar] [CrossRef]
- Kim, K.-H.; Kabir, E.; Kabir, S. A review on the human health impact of airborne particulate matter. Environ. Int. 2015, 74, 136–143. [Google Scholar] [CrossRef]
- Bernstein, J.A.; Alexis, N.; Barnes, C.; Bernstein, I.L.; Nel, A.; Peden, D.; Diaz-Sanchez, D.; Tarlo, S.M.; Williams, P.B. Health effects of air pollution. J. Allergy Clin. Immunol. 2004, 114, 1116–1123. [Google Scholar] [CrossRef]
- Naeher, L.P.; Brauer, M.; Lipsett, M.; Zelikoff, J.T.; Simpson, C.D.; Koenig, J.Q.; Smith, K.R. Woodsmoke Health Effects: A Review. Inhal. Toxicol. 2007, 19, 67–106. [Google Scholar] [CrossRef]
- Larson, T.V.; Koenig, J.Q. Wood Smoke: Emissions and Noncancer Respiratory Effects. Annu. Rev. Public Health 1994, 15, 133–156. [Google Scholar] [CrossRef] [PubMed]
- Dastoorpoor, M.; Sekhavatpour, Z.; Masoumi, K.; Mohammadi, M.J.; Aghababaeian, H.; Khanjani, N.; Hashemzadeh, B.; Vahedian, M. Air pollution and hospital admissions for cardiovascular diseases in Ahvaz, Iran. Sci. Total. Environ. 2019, 652, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Aghababaeian, H.; Dastoorpoor, M.; Ghasemi, A.; Kiarsi, M.; Khanjani, N.; Ahvazi, L.A. Cardiovascular and respiratory emergency dispatch due to short-term exposure to ambient PM10 in Dezful, Iran. J. Cardiovasc. Thorac. Res. 2019, 11, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.-H.; Kao, S.-W.; Tantoh, D.M.; Ko, P.-C.; Lan, S.-J.; Liaw, Y.-P. Association between fine particulate matter and oral cancer among Taiwanese men. J. Investig. Med. 2018, 67, 34–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consonni, D.; Carugno, M.; De Matteis, S.; Nordio, F.; Randi, G.; Bazzano, M.; Caporaso, N.E.; Tucker, M.A.; Bertazzi, P.A.; Pesatori, A.C.; et al. Outdoor particulate matter (PM10) exposure and lung cancer risk in the EAGLE study. PLoS ONE 2018, 13, e0203539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivas-Santiago, C.E.; Sarkar, S.; Cantarella, P.; Osornio-Vargas, A.; Quintana-Belmares, R.; Meng, Q.; Kirn, T.J.; Ohman Strickland, P.; Chow, J.C.; Watson, J.G.; et al. Air Pollution Particulate Matter Alters Antimycobacterial Respiratory Epithelium Innate Immunity. Infect. Immun. 2015, 83, 2507–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Rivas-Santiago, C.; Ibironke, O.A.; Carranza, C.; Meng, Q.; Osornio-Vargas, A.R.; Zhang, J.; Torres, M.; Chow, J.C.; Watson, J.G.; et al. Season and size of urban particulate matter differentially affect cytotoxicity and human immune responses to Mycobacterium tuberculosis. PLoS ONE 2019, 14, e0219122. [Google Scholar] [CrossRef] [Green Version]
- Saygin, M.; Gonca, T.; Ozturk, O.; Has, M.; Caliskan, S.; Has, Z.G. To Investigate the Effects of Air Pollution (PM10 and SO2) on the Respiratory Diseases Asthma and Chronic Obstructive Pulmonary Disease. Turk. Thorac. J. 2017, 18, 33–39. [Google Scholar] [CrossRef]
- Tecer, L.H.; Alagha, O.; Karaca, F.; Tuncel, G.; Eldes, N. Particulate Matter (PM2.5, PM10-2.5, and PM10) and Children’s Hospital Admissions for Asthma and Respiratory Diseases: A Bidirectional Case-Crossover Study. J. Toxicol. Environ. Health Part A 2008, 71, 512–520. [Google Scholar] [CrossRef]
- Dellinger, B.; Pryor, W.A.; Cueto, R.; Squadrito, G.L.; Hegde, V.; Deutsch, W.A. Role of Free Radicals in the Toxicity of Airborne Fine Particulate Matter. Chem. Res. Toxicol. 2001, 14, 1371–1377. [Google Scholar] [CrossRef]
- Zhao, J.; Xie, Y.; Qian, C.; Li, L.; Jiang, R.; Kan, H.; Chen, R.; Song, W. Imbalance of Th1 and Th2 cells in cardiac injury induced by ambient fine particles. Toxicol. Lett. 2012, 208, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Nemmar, A.; Hoet, P.H.M.; Vanquickenborne, B.; Dinsdale, D.; Thomeer, M.; Hoylaerts, M.F.; Vanbilloen, H.; Mortelmans, L.; Nemery, B. Passage of Inhaled Particles into the Blood Circulation in Humans. Circulation 2002, 105, 411–414. [Google Scholar] [CrossRef] [Green Version]
- Nemmar, A.; Vanbilloen, H.; Hoylaerts, M.F.; Hoet, P.H.M.; Verbruggen, A.; Nemery, B. Passage of Intratracheally Instilled Ultrafine Particles from the Lung into the Systemic Circulation in Hamster. Am. J. Respir. Crit. Care Med. 2001, 164, 1665–1668. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, S.; Karg, E.; Roth, C.; Schulz, H.; Ziesenis, A.; Heinzmann, U.; Schramel, P.; Heyder, J. Pulmonary and systemic distribution of inhaled ultrafine silver particles in rats. Environ. Health Perspect. 2001, 109, 547–551. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Li, Y.; Li, G.; Zhang, Y.; Li, J.; Haosheng, C. Fluorescent reconstitution on deposition of PM2.5 in lung and extrapulmonary organs. Proc. Natl. Acad. Sci. USA 2019, 116 (Suppl. 4), 2488–2493. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.; Hwang, S.; Jin, H.; Kim, D.; Minai-Tehrani, A.; Yoon, H.; Choi, M.; Yoon, T.; Han, D.; Kang, Y.; et al. Body Distribution of Inhaled Fluorescent Magnetic Nanoparticles in the Mice. J. Occup. Health 2008, 50, 1–6. [Google Scholar] [CrossRef]
- Sarlo, K.; Blackburn, K.L.; Clark, E.D.; Grothaus, J.; Chaney, J.; Neu, S.; Flood, J.; Abbott, D.; Bohne, C.; Casey, K.; et al. Tissue distribution of 20nm, 100nm and 1000nm fluorescent polystyrene latex nanospheres following acute systemic or acute and repeat airway exposure in the rat. Toxicology 2009, 263, 117–126. [Google Scholar] [CrossRef]
- Pillay, V.; Murugan, K.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; Du Toit, L.C. Parameters and characteristics governing cellular internalization and trans-barrier trafficking of nanostructures. Int. J. Nanomed. 2015, 10, 2191–2206. [Google Scholar] [CrossRef] [Green Version]
- Balakrishna, S.; Saravia, J.; Thevenot, P.; Ahlert, T.; Lomnicki, S.M.; Dellinger, B.; Cormier, S. Environmentally persistent free radicals induce airway hyperresponsiveness in neonatal rat lungs. Part. Fibre Toxicol. 2011, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Thevenot, P.T.; Saravia, J.; Jin, N.; Giaimo, J.D.; Chustz, R.E.; Mahne, S.; Kelley, M.A.; Hebert, V.Y.; Dellinger, B.; Dugas, T.R.; et al. Radical-Containing Ultrafine Particulate Matter Initiates Epithelial-to-Mesenchymal Transitions in Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2013, 48, 188–197. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Huang, K.; Liu, J.; Wu, S.; Shen, D.; Dai, P.; Li, C. Fine particulate matter from pig house induced immune response by activating TLR4/MAPK/NF-κB pathway and NLRP3 inflammasome in alveolar macrophages. Chemosphere 2019, 236, 124373. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Ichinose, T.; Song, Y.; Yoshida, Y.; Bekki, K.; Arashidani, K.; Yoshida, S.; Nishikawa, M.; Takano, H.; Shibamoto, T.; et al. Desert dust induces TLR signaling to trigger Th2-dominant lung allergic inflammation via a MyD88-dependent signaling pathway. Toxicol. Appl. Pharmacol. 2016, 296, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Shoenfelt, J.; Mitkus, R.J.; Zeisler, R.; Spatz, R.O.; Powell, J.; Fenton, M.J.; Squibb, K.A.; Medvedev, A.E. Involvement of TLR2 and TLR4 in inflammatory immune responses induced by fine and coarse ambient air particulate matter. J. Leukoc. Biol. 2009, 86, 303–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, J.A.; Hirota, S.A.; Warner, S.M.; Stefanowicz, D.; Shaheen, F.; Beck, P.L.; Macdonald, J.A.; Hackett, T.-L.; Sin, D.D.; Van Eeden, S.; et al. The airway epithelium nucleotide-binding domain and leucine-rich repeat protein 3 inflammasome is activated by urban particulate matter. J. Allergy Clin. Immunol. 2012, 129, 1116–1125.e6. [Google Scholar] [CrossRef]
- Rylance, J.; Fullerton, D.G.; Scriven, J.; Aljurayyan, A.N.; Mzinza, D.; Barrett, S.; Wright, A.K.A.; Wootton, D.G.; Glennie, S.J.; Baple, K.; et al. Household Air Pollution Causes Dose-Dependent Inflammation and Altered Phagocytosis in Human Macrophages. Am. J. Respir. Cell Mol. Biol. 2015, 52, 584–593. [Google Scholar] [CrossRef] [Green Version]
- Bauer, R.N.; Diaz-Sanchez, D.; Jaspers, I. Effects of air pollutants on innate immunity: The role of Toll-like receptors and nucleotide-binding oligomerization domain–like receptors. J. Allergy Clin. Immunol. 2012, 129, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Becker, S.; Fenton, M.J.; Soukup, J.M. Involvement of Microbial Components and Toll-like Receptors 2 And 4 in Cytokine Responses to Air Pollution Particles. Am. J. Respir. Cell Mol. Biol. 2002, 27, 611–618. [Google Scholar] [CrossRef]
- Hirota, J.A.; Marchant, D.J.; Singhera, G.K.; Moheimani, F.; Dorscheid, D.R.; Carlsten, C.; Sin, D.; Knight, D. Urban particulate matter increases human airway epithelial cell IL-1β secretion following scratch wounding and H1N1 influenza A exposurein vitro. Exp. Lung Res. 2015, 41, 353–362. [Google Scholar] [CrossRef]
- Adachi, O.; Kawai, T.; Takeda, K.; Matsumoto, M.; Tsutsui, H.; Sakagami, M.; Nakanishi, K.; Akira, S. Targeted Disruption of the MyD88 Gene Results in Loss of IL-1- and IL-18-Mediated Function. Immunology 1998, 9, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R.; Preston-Hurlburt, P.; Kopp, E.; Stadlen, A.; Chen, C.; Ghosh, S.; Janeway, C.A., Jr. MyD88 Is an Adaptor Protein in the hToll/IL-1 Receptor Family Signaling Pathways. Mol. Cell 1998, 2, 253–258. [Google Scholar] [CrossRef]
- Mukaida, N.; Mahe, Y.; Matsushima, K. Cooperative interaction of nuclear factor-kappa B- and cis-regulatory enhancer binding protein-like factor binding elements in activating the interleukin-8 gene by pro-inflammatory cytokines. J. Biol. Chem. 1990, 265, 21128–21133. [Google Scholar] [CrossRef]
- Yang, Z.; Kong, B.; Mosser, D.M.; Zhang, X. TLRs, macrophages, and NK cells: Our understandings of their functions in uterus and ovary. Int. Immunopharmacol. 2011, 11, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.-Y.; Yoon, S.R.; Kim, T.-D.; Choi, I.; Jung, H. Toll-Like Receptors in Natural Killer Cells and Their Application for Immunotherapy. J. Immunol. Res. 2020, 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.-U.; Kamada, N.; Muñoz-Planillo, R.; Kim, Y.-G.; Kim, D.; Koizumi, Y.; Hasegawa, M.; Himpsl, S.D.; Browne, H.P.; Lawley, T.D.; et al. Distinct Commensals Induce Interleukin-1β via NLRP3 Inflammasome in Inflammatory Monocytes to Promote Intestinal Inflammation in Response to Injury. Immunology 2015, 42, 744–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Reibman, J.; Hsu, Y.; Chen, L.-C.; Bleck, B.; Gordon, T. Airway Epithelial Cells Release MIP-3α/CCL20 in Response to Cytokines and Ambient Particulate Matter. Am. J. Respir. Cell Mol. Biol. 2003, 28, 648–654. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, C.H.; Roberts, A.I.; Das, J.; Xu, G.; Ren, G.; Zhang, Y.; Zhang, L.; Yuan, Z.R.; Tan, H.S.W.; et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: What we do and don’t know. Cell Res. 2006, 16, 126–133. [Google Scholar] [CrossRef]
- Matthews, N.C.; Faith, A.; Pfeffer, P.E.; Lu, H.; Kelly, F.; Hawrylowicz, C.M.; Lee, T.H. Urban Particulate Matter Suppresses Priming of Th1 Cells by GM-CSF-Activated Human Dendritic Cells. Am. J. Respir. Cell Mol. Biol. 2013, 50, 281–291. [Google Scholar] [CrossRef]
- Fujii, T.; Hayashi, S.; Hogg, J.C.; Vincent, R.; Van Eeden, S.F. Particulate Matter Induces Cytokine Expression in Human Bronchial Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2001, 25, 265–271. [Google Scholar] [CrossRef]
- Kaiko, G.E.; Horvat, J.C.; Beagley, K.W.; Hansbro, P.M. Immunological decision-making: How does the immune system decide to mount a helper T-cell response? Immunology 2008, 123, 326–338. [Google Scholar] [CrossRef]
- Trinchieri, G.; Pflanz, S.; Kastelein, R. The IL-12 Family of Heterodimeric Cytokines. Immunology 2003, 19, 641–644. [Google Scholar] [CrossRef] [Green Version]
- Kidd, P. Th1/Th2 balance: The hypothesis, its limitations, and implications for health and disease. Altern. Med. Rev. J. Clin. Ther. 2003, 8, 223–246. [Google Scholar]
- Zhao, H.; Ma, J.K.; Barger, M.W.; Mercer, R.R.; Millecchia, L.; Schwegler-Berry, D.; Castranova, V.; Ma, J.Y. Reactive Oxygen Species- and Nitric Oxide-Mediated Lung Inflammation and Mitochondrial Dysfunction in Wild-Type and iNOS-Deficient Mice Exposed to Diesel Exhaust Particles. J. Toxicol. Environ. Health Part A 2009, 72, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Park, E.-J.; Roh, J.; Kim, Y.; Park, K.; Kim, D.-S.; Yu, S.-D. PM 2.5 collected in a residential area induced Th1-type inflammatory responses with oxidative stress in mice. Environ. Res. 2011, 111, 348–355. [Google Scholar] [CrossRef]
- Huang, K.-L.; Liu, S.-Y.; Chou, C.C.K.; Lee, Y.-H.; Cheng, T.-J. The effect of size-segregated ambient particulate matter on Th1/Th2-like immune responses in mice. PLoS ONE 2017, 12, e0173158. [Google Scholar] [CrossRef] [Green Version]
- Van Eeden, S.F.; Tan, W.C.; Suwa, T.; Mukae, H.; Terashima, T.; Fujii, T.; Qui, D.; Vincent, R.; Hogg, J.C. Cytokines Involved in the Systemic Inflammatory Response Induced by Exposure to Particulate Matter Air Pollutants (PM10). Am. J. Respir. Crit. Care Med. 2001, 164, 826–830. [Google Scholar] [CrossRef]
- Bach, N.; Bølling, A.K.; Brinchmann, B.C.; Totlandsdal, A.I.; Skuland, T.; Holme, J.; Låg, M.; Schwarze, P.E.; Øvrevik, J. Cytokine responses induced by diesel exhaust particles are suppressed by PAR-2 silencing and antioxidant treatment, and driven by polar and non-polar soluble constituents. Toxicol. Lett. 2015, 238, 72–82. [Google Scholar] [CrossRef]
- Hamilton, R.F.; Holian, A.; Morandi, M.T. A comparison of asbestos and urban particulate matter in the in vitro modification of human alveolar macrophage anti-gen-presenting cell function. Exp. Lung Res. 2004, 30, 147–162. [Google Scholar] [CrossRef]
- Kang, C.-M.; Jang, A.-S.; Ahn, M.-H.; Shin, J.-A.; Kim, J.-H.; Choi, Y.-S.; Rhim, T.-Y.; Park, C.-S. Interleukin-25 and Interleukin-13 Production by Alveolar Macrophages in Response to Particles. Am. J. Respir. Cell Mol. Biol. 2005, 33, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Homer, R.J.; Wang, Z.; Chen, Q.; Geba, G.P.; Wang, J.; Zhang, Y.; Elias, J.A. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J. Clin. Investig. 1999, 103, 779–788. [Google Scholar] [CrossRef] [Green Version]
- Gour, N.; Wills-Karp, M. IL-4 and IL-13 signaling in allergic airway disease. Cytokine 2015, 75, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.S.; Wynn, T. Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal Immunol. 2009, 2, 103–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.. IL-13 effector functions. Annu. Rev. Immunol. 2003, 21, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Fort, M.M.; Cheung, J.; Yen, D.; Li, J.; Zurawski, S.M.; Lo, S.; Menon, S.; Clifford, T.; Hunte, B.; Lesley, R.; et al. IL-25 Induces IL-4, IL-5, and IL-13 and Th2-Associated Pathologies In Vivo. Immunity 2001, 15, 985–995. [Google Scholar] [CrossRef] [Green Version]
- Corrigan, C.J.; Wang, W.; Meng, Q.; Fang, C.; Eid, G.; Caballero, M.R.; Lv, Z.; An, Y.; Wang, Y.-H.; Liu, Y.-J.; et al. Allergen-induced expression of IL-25 and IL-25 receptor in atopic asthmatic airways and late-phase cutaneous responses. J. Allergy Clin. Immunol. 2011, 128, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.-Q.; Dong, W.-Y.; Xu, J.; Hong, Z.-C.; Zhao, R.-W.; Deng, C.; Zhuang, G.; Zhang, R. T-Helper Type 1-T-Helper Type 2 Shift and Nasal Remodeling after Fine Particulate Matter Exposure in a Rat Model of Allergic Rhinitis. Am. J. Rhinol. Allergy 2017, 31, 148–155. [Google Scholar] [CrossRef]
- Zhang, X.; Zhong, W.; Meng, Q.; Lin, Q.; Fang, C.; Huang, X.; Li, C.; Huang, Y.; Tan, J. Ambient PM2.5 exposure exacerbates severity of allergic asthma in previously sensitized mice. J. Asthma 2015, 52, 785–794. [Google Scholar]
- He, M.; Ichinose, T.; Kobayashi, M.; Arashidani, K.; Yoshida, S.; Nishikawa, M.; Takano, H.; Sun, G.; Shibamoto, T. Differences in allergic inflammatory responses between urban PM2.5 and fine particle derived from desert-dust in murine lungs. Toxicol. Appl. Pharmacol. 2016, 297, 41–55. [Google Scholar] [CrossRef]
- Galli, S.J.; Borregaard, N.; Wynn, T. Phenotypic and functional plasticity of cells of innate immunity: Macrophages, mast cells and neutrophils. Nat. Immunol. 2011, 12, 1035–1044. [Google Scholar] [CrossRef] [Green Version]
- Alexis, N.E.; Lay, J.C.; Zeman, K.; Bennett, W.E.; Peden, D.B.; Soukup, J.M.; Devlin, R.B.; Becker, S. Biological material on inhaled coarse fraction particulate matter activates airway phagocytes in vivo in healthy volunteers. J. Allergy Clin. Immunol. 2006, 117, 1396–1403. [Google Scholar] [CrossRef]
- Williams, M.A.; Rangasamy, T.; Bauer, S.M.; Killedar, S.; Karp, M.; Kensler, T.W.; Yamamoto, M.; Breysse, P.; Biswal, S.; Georas, S.N. Disruption of the transcription factor Nrf2 promotes pro-oxidative dendritic cells that stimulate Th2-like immunoresponsiveness upon activation by ambient particulate matter. J. Immunol. 2008, 181, 4545–4559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, J.M.S.S.; Soukup, J. Coarse(PM 2.5–10), Fine(PM 2.5), and Ultrafine Air Pollution Particles Induce/Increase Immune Costimulatory Receptors on Human Blood-Derived Monocytes but not on Alveolar Macrophages. J. Toxicol. Environ. Health Part A 2003, 66, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Porter, M.; Karp, M.; Killedar, S.; Bauer, S.M.; Guo, J.; Williams, D.; Breysse, P.; Georas, S.N.; Williams, M.A. Diesel-Enriched Particulate Matter Functionally Activates Human Dendritic Cells. Am. J. Respir. Cell Mol. Biol. 2007, 37, 706–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerondakis, S.; Siebenlist, U. Roles of the NF- B Pathway in Lymphocyte Development and Function. Cold Spring Harb. Perspect. Biol. 2009, 2, a000182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Ning, Z.; Majumdar, R.; Cui, J.; Takabe, W.; Jen, N.; Sioutas, C.; Hsiai, T.K. Ultrafine particles from diesel vehicle emissions at different driving cycles induce differential vascular pro-inflammatory responses: Implication of chemical components and NF-κB signaling. Part. Fibre Toxicol. 2010, 7, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Nadra, I.; Boccaccini, A.R.; Philippidis, P.; Whelan, L.C.; McCarthy, G.M.; Haskard, D.O.; Landis, R. Effect of particle size on hydroxyapatite crystal-induced tumor necrosis factor alpha secretion by macrophages. Atherosclerosis 2008, 196, 98–105. [Google Scholar] [CrossRef]
- Ding, M.; Kisin, E.; Zhao, J.; Bowman, L.; Lu, Y.; Jiang, B.; Leonard, S.; Vallyathan, V.; Castranova, V.; Murray, A. Size-dependent effects of tungsten carbide–cobalt particles on oxygen radical production and activation of cell signaling pathways in murine epidermal cells. Toxicol. Appl. Pharmacol. 2009, 241, 260–268. [Google Scholar] [CrossRef]
- Castañeda, A.R.; Vogel, C.F.A.; Bein, K.J.; Hughes, H.; Smiley-Jewell, S.; Pinkerton, K.E. Ambient particulate matter enhances the pulmonary allergic immune response to house dust mite in a BALB/c mouse model by augmenting Th2- and Th17-immune responses. Physiol. Rep. 2018, 6, e13827. [Google Scholar] [CrossRef]
- Jiang, A.; Bloom, O.; Ono, S.; Cui, W.; Unternaehrer, J.; Jiang, S.; Whitney, J.A.; Connolly, J.; Banchereau, J.; Mellman, I. Disruption of E-Cadherin-Mediated Adhesion Induces a Functionally Distinct Pathway of Dendritic Cell Maturation. Immunology 2007, 27, 610–624. [Google Scholar] [CrossRef] [Green Version]
- Saravia, J.; You, D.; Thevenot, P.; Lee, G.I.; Shrestha, B.; Lomnicki, S.; Cormier, S. Early-life exposure to combustion-derived particulate matter causes pulmonary immunosuppression. Mucosal Immunol. 2014, 7, 694–704. [Google Scholar] [CrossRef]
- Jaligama, S.; Saravia, J.; You, D.; Yadav, N.; Lee, G.I.; Shrestha, B.; Cormier, S. Regulatory T cells and IL10 suppress pulmonary host defense during early-life exposure to radical containing combustion derived ultrafine particulate matter. Respir. Res. 2017, 18, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rychlik, K.A.; Secrest, J.R.; Lau, C.; Pulczinski, J.; Zamora, M.L.; Leal, J.; Langley, R.; Myatt, L.G.; Raju, M.; Chang, R.C.-A.; et al. In utero ultrafine particulate matter exposure causes offspring pulmonary immunosuppression. Proc. Natl. Acad. Sci. USA 2019, 116, 3443–3448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.G.; Kinney, P.L.; Chillrud, S.N.; Jack, D. A Systematic Review of Innate Immunomodulatory Effects of Household Air Pollution Secondary to the Burning of Biomass Fuels. Ann. Glob. Health 2015, 81, 368–374. [Google Scholar] [CrossRef]
- Zhu, L.; Zhao, Q.; Yang, T.; Ding, W.; Zhao, Y. Cellular Metabolism and Macrophage Functional Polarization. Int. Rev. Immunol. 2014, 34, 82–100. [Google Scholar] [CrossRef]
- Shen, Y.; Song, J.; Wang, Y.; Chen, Z.; Zhang, L.; Yu, J.; Zhu, D.; Zhong, M. M2 macrophages promote pulmonary endothelial cells regeneration in sepsis-induced acute lung injury. Ann. Transl. Med. 2019, 7, 142. [Google Scholar] [CrossRef]
- Vlahos, R.; Bozinovski, S. Role of alveolar macrophages in chronic obstructive pulmonary disease. Front. Immunol. 2014, 5, 435. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.-Y.; Huang, D.; Zhang, H.-J.; Wang, S.; Chen, X.-F. Exposure to particulate matter 2.5 (PM2.5) induced macrophage-dependent inflammation, characterized by increased Th1/Th17 cytokine secretion and cytotoxicity. Int. Immunopharmacol. 2017, 50, 139–145. [Google Scholar] [CrossRef]
- Shi, Q.; Zhao, L.; Xu, C.; Zhang, L.; Zhao, H. High Molecular Weight Hyaluronan Suppresses Macrophage M1 Polarization and Enhances IL-10 Production in PM2.5-Induced Lung Inflammation. Molecules 2019, 24, 1766. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Zhao, Y.; Wang, Q.; Chen, H.; Zhou, X. Fine particulate matter exposure promotes M2 macrophage polarization through inhibiting histone deacetylase 2 in the pathogenesis of chronic obstructive pulmonary disease. Ann. Transl. Med. 2020, 8, 1303. [Google Scholar] [CrossRef]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [Green Version]
- Chandra, V.; Mahajan, S.; Saini, A.; Dkhar, H.K.; Nanduri, R.; Raj, E.B.; Kumar, A.; Gupta, P. Human IL10 Gene Repression by Rev-erbα Ameliorates Mycobacterium tuberculosis Clearance. J. Biol. Chem. 2013, 288, 10692–10702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Luo, Q.; Guo, Y.; Chen, J.; Xiong, G.; Peng, Y.; Ye, J.; Li, J. Mycobacterium tuberculosis-Induced Polarization of Human Macrophage Orchestrates the Formation and Development of Tuberculous Granulomas In Vitro. PLoS ONE 2015, 10, e0129744. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.S.; Schoenbach, V.J.; Richardson, D.B.; Gammon, M.D. Particulate air pollution and susceptibility to the development of pulmonary tuberculosis disease in North Carolina: An ecological study. Int. J. Environ. Health Res. 2014, 24, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Cui, L.; Hou, L.; Yu, C.; Tao, N.; Liu, J.; Li, Y.; Zhou, C.; Yang, G.; Li, H. Ambient Air Pollution Exposures and Newly Diagnosed Pulmonary Tuberculosis in Jinan, China: A Time Series Study. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Ibironke, O.; Carranza, C.; Sarkar, S.; Torres, M.; Choi, H.T.; Nwoko, J.; Black, K.; Quintana-Belmares, R.O.; Osornio-Vargas, A.R.; Strickland, P.O.; et al. Urban Air Pollution Particulates Suppress Human T-Cell Responses to Mycobacterium Tuberculosis. Int. J. Environ. Res. Public Health 2019, 16, 4112. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.M.S.S. Exposure to urban air particulates alters the macrophage-mediated inflammatory response to respiratory viral infection. J. Toxicol. Environ. Health Part A 1999, 57, 445–457. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Tanwar, V.; Adelstein, J.M.; Wold, L.E. Double Trouble: Combined Cardiovascular Effects of Particulate Matter Exposure and COVID-19. Cardiovasc. Res. 2020, 117, 85–95. [Google Scholar] [CrossRef]
- Setti, L.; Passarini, F.; De Gennaro, G.; Barbieri, P.; Perrone, M.G.; Borelli, M.; Palmisani, J.; Di Gilio, A.; Torboli, V.; Fontana, F.; et al. SARS-Cov-2RNA found on particulate matter of Bergamo in Northern Italy: First evidence. Environ. Res. 2020, 188, 109754. [Google Scholar] [CrossRef]
- Kaan, P.M.; Hegele, R.G. Interaction between respiratory syncytial virus and particulate matter in guinea pig alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 2003, 28, 697–704. [Google Scholar] [CrossRef]
- Comunian, S.; Dongo, D.; Milani, C.; Palestini, P. Air Pollution and COVID-19: The Role of Particulate Matter in the Spread and Increase of COVID-19′s Morbidity and Mortality. Int. J. Environ. Res. Public Health 2020, 17, 4487. [Google Scholar] [CrossRef] [PubMed]
- Mantecca, P.; Sancini, G.; Moschini, E.; Farina, F.; Gualtieri, M.; Rohr, A.; Miserocchi, G.; Palestini, P.; Camatini, M. Lung toxicity induced by intratracheal instillation of size-fractionated tire particles. Toxicol. Lett. 2009, 189, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Pereira, P.; Saldiva, P.; Sakae, R.; Böhm, G.; Martins, M. Urban Levels of Air Pollution Increase Lung Responsiveness in Rats. Environ. Res. 1995, 69, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.-Y.; Ding, H.; Jiang, L.-N.; Chen, S.-W.; Zheng, J.-P.; Qiu, M.; Zhou, Y.-X.; Chen, Q.; Guan, W.-J. Association between Air Pollutants and Asthma Emergency Room Visits and Hospital Admissions in Time Series Studies: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0138146. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Thevenot, P.; Saravia, J.; Ahlert, T.; Cormier, S.A. Radical-Containing Particles Activate Dendritic Cells and Enhance Th17 Inflammation in a Mouse Model of Asthma. Am. J. Respir. Cell Mol. Biol. 2011, 45, 977–983. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Sanchez, D.; Dotson, A.R.; Takenaka, H.; Saxon, A. Diesel exhaust particles induce local IgE production in vivo and alter the pattern of IgE messenger RNA isoforms. J. Clin. Investig. 1994, 94, 1417–1425. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Sanchez, D.; Tsien, A.; Fleming, J.; Saxon, A. Combined diesel exhaust particulate and ragweed allergen challenge markedly enhances human in vivo nasal ragweed-specific IgE and skews cytokine production to a T helper cell 2-type pattern. J. Immunol. 1997, 158, 2406–2413. [Google Scholar]
- Diaz-Sanchez, D.; Penichet-Garcia, M.; Saxon, A. Diesel exhaust particles directly induce activated mast cells to degranulate and increase histamine levels and symptom severity. J. Allergy Clin. Immunol. 2000, 106, 1140–1146. [Google Scholar] [CrossRef]
- Castañeda, A.R.; Bein, K.; Smiley-Jewell, S.; Pinkerton, K.E. Fine particulate matter (PM2.5) enhances allergic sensitization in BALB/cmice. J. Toxicol. Environ. Health Part A 2017, 80, 197–207. [Google Scholar] [CrossRef]
- Yang, S.-I. Particulate matter and childhood allergic diseases. Korean J. Pediatr. 2018, 62, 22–29. [Google Scholar] [CrossRef]
- Joubert, A.I.; Geppert, M.; Johnson, L.; Mills-Goodlet, R.; Michelini, S.; Korotchenko, E.; Duschl, A.; Weiss, R.; Horejs-Höck, J.; Himly, M. Mechanisms of Particles in Sensitization, Effector Function and Therapy of Allergic Disease. Front. Immunol. 2020, 11, 1334. [Google Scholar] [CrossRef] [PubMed]
- Gruzieva, O.; Bergström, A.; Hulchiy, O.; Kull, I.; Lind, T.; Melén, E.; Moskalenko, V.; Pershagen, G.; Bellander, T. Exposure to Air Pollution from Traffic and Childhood Asthma Until 12 Years of Age. Epidemiology 2013, 24, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Carlsten, C.; DyBuncio, A.; Becker, A.; Chan-Yeung, M.; Brauer, M. Traffic-related air pollution and incident asthma in a high-risk birth cohort. Occup. Environ. Med. 2010, 68, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, E.; Standl, M.; Cyrys, J.; Berdel, D.; von Berg, A.; Bauer, C.P.; Krämer, U.; Sugiri, D.; Lehmann, I.; Koletzko, S.; et al. A longitudinal analysis of associations between traffic-related air pollution with asthma, allergies and sensitization in the GINIplus and LISAplus birth cohorts. PeerJ 2013, 1, e193. [Google Scholar] [CrossRef] [PubMed]
- Gehring, U.; Wijga, A.H.; Hoek, G.; Bellander, T.; Berdel, D.; Brueske, I.; Fuertes, E.; Gruzieva, O.; Heinrich, J.; Hoffmann, B.; et al. Exposure to air pollution and development of asthma and rhinoconjunctivitis throughout childhood and adolescence: A population-based birth cohort study. Lancet Respir. Med. 2015, 3, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Jung, D.Y.; Leem, J.H.; Kim, H.C.; Kim, J.H.; Hwang, S.S.; Lee, J.Y.; Kim, B.J.; Hong, Y.C.; Hong, S.J.; Kwon, H.J. Effect of traffic-related air pollution on allergic disease: Results of the children’s health and environmental research. Allergy Asthma Immunol. Res. 2015, 7, 359–366. [Google Scholar] [CrossRef] [Green Version]
- Gauderman, W.J.; Avol, E.; Gilliland, F.; Vora, H.; Thomas, D.; Berhane, K.; McConnell, R.; Kuenzli, N.; Lurmann, F.; Rappaport, E.; et al. The effect of air pollution on lung development from 10 to 18 years of age. N. Engl. J. Med. 2004, 351, 1057–1067. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-M.; Kim, J.; Han, Y.; Jeon, B.-H.; Cheong, H.-K.; Ahn, K. Short-term effects of weather and air pollution on atopic dermatitis symptoms in children: A panel study in Korea. PLoS ONE 2017, 12, e0175229. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.-R.; Chen, W.-T.; Tang, Y.-H.; Hwang, B.-F. Fine particulate matter exposure during pregnancy and infancy and incident asthma. J. Allergy Clin. Immunol. 2019, 143, 2254–2262.e5. [Google Scholar] [CrossRef]
- Mölter, A.; Agius, R.; De Vocht, F.; Lindley, S.; Gerrard, W.; Custovic, A.; Simpson, A. Effects of long-term exposure to PM10 and NO2 on asthma and wheeze in a prospective birth cohort. J. Epidemiol. Community Health 2014, 68, 21–28. [Google Scholar] [CrossRef]
- Wu, N.; Lu, B.; Chen, J.; Li, X. Size distributions of particle-generated hydroxyl radical (·OH) in surrogate lung fluid (SLF) solution and their potential sources. Environ. Pollut. 2021, 268, 115582. [Google Scholar] [CrossRef]
- Pardo, M.; Xu, F.; Shemesh, M.; Qiu, X.; Barak, Y.; Zhu, T.; Rudich, Y. Nrf2 protects against diverse PM2.5 components-induced mitochondrial oxidative damage in lung cells. Sci. Total. Environ. 2019, 669, 303–313. [Google Scholar] [CrossRef]
- Risom, L.; Møller, P.; Loft, S. Oxidative stress-induced DNA damage by particulate air pollution. Mutat. Res. Mol. Mech. Mutagen. 2005, 592, 119–137. [Google Scholar] [CrossRef]
- Vidrio, E.; Jung, H.; Anastasio, C. Generation of hydroxyl radicals from dissolved transition metals in surrogate lung fluid solutions. Atmos. Environ. 2008, 42, 4369–4379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, A.K.; Sioutas, C.; Miguel, A.H.; Kumagai, Y.; Schmitz, D.A.; Singh, M.; Eiguren-Fernandez, A.; Froines, J.R. Redox activity of airborne particulate matter at different sites in the Los Angeles Basin. Environ. Res. 2005, 99, 40–47. [Google Scholar] [CrossRef]
- Harmon, A.C.; Hebert, V.Y.; Cormier, S.; Subramanian, B.; Reed, J.R.; Backes, W.L.; Dugas, T.R. Particulate matter containing environmentally persistent free radicals induces AhR-dependent cytokine and reactive oxygen species production in human bronchial epithelial cells. PLoS ONE 2018, 13, e0205412. [Google Scholar] [CrossRef] [Green Version]
- Pardo, M.; Shafer, M.M.; Rudich, A.; Schauer, J.J.; Rudich, Y. Single Exposure to near Roadway Particulate Matter Leads to Confined Inflammatory and Defense Responses: Possible Role of Metals. Environ. Sci. Technol. 2015, 49, 8777–8785. [Google Scholar] [CrossRef] [PubMed]
- Kopf, P.; Walker, M.K. 2,3,7,8-Tetrachlorodibenzo-p-dioxin increases reactive oxygen species production in human endothelial cells via induction of cytochrome P4501A1. Toxicol. Appl. Pharmacol. 2010, 245, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Costa, C.; Catania, S.; De Pasquale, R.; Stancanelli, R.; Scribano, G.; Melchini, A. Exposure of human skin to benzo[a]pyrene: Role of CYP1A1 and aryl hydrocarbon receptor in oxidative stress generation. Toxicology 2010, 271, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, B.; Kluza, J.; Antherieu, S.; Sotty, J.; Alleman, L.Y.; Perdrix, E.; Loyens, A.; Coddeville, P.; Guidice, J.-M.L.; Marchetti, P.; et al. Air pollution-derived PM2.5 impairs mitochondrial function in healthy and chronic obstructive pulmonary diseased human bronchial epithelial cells. Environ. Pollut. 2018, 243, 1434–1449. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.; Naidoo, R.N.; Robins, T.G.; Mentz, G.; Li, H.; London, S.J.; Batterman, S. GSTM1andGSTP1gene variants and the effect of air pollutants on lung function measures in South African children. Am. J. Ind. Med. 2012, 55, 1078–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gawda, A.; Majka, G.; Nowak, B.; Marcinkiewicz, J. Air pollution, oxidative stress, and exacerbation of autoimmune diseases. Cent. Eur. J. Immunol. 2016, 3, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.-N.; Xu, Z.; Wu, G.-C.; Mao, Y.-M.; Liu, L.-N.; Wu, Q.-; Dan, Y.-L.; Tao, S.-S.; Zhang, Q.; Sam, N.B.; et al. Emerging role of air pollution in autoimmune diseases. Autoimmun. Rev. 2019, 18, 607–614. [Google Scholar] [CrossRef]
- Lambert, A.L.; Trasti, F.S.; Mangum, J.B.; Everitt, J.I. Effect of Preexposure to Ultrafine Carbon Black on Respiratory Syncytial Virus Infection in Mice. Toxicol. Sci. 2003, 72, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Brandt, E.B.; Myers, J.M.B.; Acciani, T.H.; Ryan, P.H.; Sivaprasad, U.; Ruff, B.; Lemasters, G.K.; Bernstein, D.I.; Lockey, J.E.; LeCras, T.D.; et al. Exposure to allergen and diesel exhaust particles potentiates secondary allergen-specific memory responses, promoting asthma susceptibility. J. Allergy Clin. Immunol. 2015, 136, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Deo, S.S.; Mistry, K.J.; Kakade, A.M.; Niphadkar, P.V. Role played by Th2 type cytokines in IgE mediated allergy and asthma. Lung India 2010, 27, 66–71. [Google Scholar] [CrossRef]
- Paul, W.E.; Zhu, J. How are TH2-type immune responses initiated and amplified? Nat. Rev. Immunol. 2010, 10, 225–235. [Google Scholar] [CrossRef]
- Fu, H.; Liu, X.; Li, W.; Zu, Y.; Zhou, F.; Shou, Q.; Ding, Z. PM2.5 Exposure Induces Inflammatory Response in Macrophages via the TLR4/COX-2/NF-κB Pathway. Inflammation 2020, 43, 1948–1958. [Google Scholar] [CrossRef]
- Gawda, A.; Majka, G.; Nowak, B.; Śróttek, M.; Walczewska, M.; Marcinkiewicz, J. Air particulate matter SRM 1648a primes macrophages to hyperinflammatory response after LPS stimulation. Inflamm. Res. 2018, 67, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Orazzo, F.; Nespoli, L.; Ito, K.; Tassinari, D.; Giardina, D.; Funis, M.; Cecchi, A.; Trapani, C.; Forgeschi, G.; Vignini, M.; et al. Air pollution, aeroallergens, and emergency room visits for acute respiratory diseases and gastroenteric disorders among young children in six Italian cities. Environ. Health Perspect. 2009, 117, 1780–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leikauf, G.D.; Kim, S.; Jang, A.S. Mechanisms of ultrafine particle-induced respiratory health effects. Exp. Mol. Med. 2020, 52, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Perera, F.; Tang, W.-Y.; Herbstman, J.; Tang, D.; Levin, L.; Miller, R.; Ho, S.-M. Relation of DNA Methylation of 5′-CpG Island of ACSL3 to Transplacental Exposure to Airborne Polycyclic Aromatic Hydrocarbons and Childhood Asthma. PLoS ONE 2009, 4, e4488. [Google Scholar] [CrossRef]
- Ahn, K. The role of air pollutants in atopic dermatitis. J. Allergy Clin. Immunol. 2014, 134, 993–999. [Google Scholar] [CrossRef]
- Yoon, W.S.; Ryu, S.R.; Lee, S.S.; Chae, Y.S.; Kim, E.J.; Choi, J.H.; Oh, S.; Park, S.H.; Choung, J.T.; Yoo, Y.; et al. Suppression of Inflammation by Recombinant Salmonella typhimurium Harboring CCL22 MicroRNA. DNA Cell Biol. 2012, 31, 290–297. [Google Scholar] [CrossRef]
- Herberth, G.; Bauer, M.; Gasch, M.; Hinz, D.; Röder, S.; Olek, S.; Kohajda, T.; Rolle-Kampczyk, U.; Von Bergen, M.; Sack, U.; et al. Maternal and cord blood miR-223 expression associates with prenatal tobacco smoke exposure and low regulatory T-cell numbers. J. Allergy Clin. Immunol. 2014, 133, 543–550.e4. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A schematic diagram of PM exposure-induced innate immune responses. Lung mucosal exposure to PM can affect the innate immunity-associated human disease outcomes. PM exposure alters pattern recognition receptor (PRR)-linked innate immunity and subsequent adaptive immunity to infectious agents or allergens. During the pathologic process, various types of endogenous molecules, including reactive oxygen species (ROS) and cytokines, are involved in pulmonary tissue injuries or systemic immune dysfunctions via circulation. In particular, PM-insulted disruption of immunity may lead to infection, hypersensitivity, and chronic disorders. Excess oxidative stress can change the profile of Th cell-polarizing cytokines. Moreover, PM exposure can alter the expression of key transcription factors (GATA3 and T-bet) and disrupt the balance between Th1 and Th2 cells, influencing the disease severity.
Figure 1.
A schematic diagram of PM exposure-induced innate immune responses. Lung mucosal exposure to PM can affect the innate immunity-associated human disease outcomes. PM exposure alters pattern recognition receptor (PRR)-linked innate immunity and subsequent adaptive immunity to infectious agents or allergens. During the pathologic process, various types of endogenous molecules, including reactive oxygen species (ROS) and cytokines, are involved in pulmonary tissue injuries or systemic immune dysfunctions via circulation. In particular, PM-insulted disruption of immunity may lead to infection, hypersensitivity, and chronic disorders. Excess oxidative stress can change the profile of Th cell-polarizing cytokines. Moreover, PM exposure can alter the expression of key transcription factors (GATA3 and T-bet) and disrupt the balance between Th1 and Th2 cells, influencing the disease severity.

Figure 2.
Impacts of PM on macrophage polarization and disease outcomes. (A) There are two kinds of macrophages. Classically activated macrophages (M1) and alternatively activated macrophages (M2) are functionality corresponding to the Th1/Th2 paradigm. Macrophages differentiate into M1 type in response to INF-γ from Th1 cells and LPS, and M2 type in response to IL-4 and IL-13 from Th2 cells. Both M1 and M2 macrophages have a distinct phenotype, and they release pro-and anti-inflammatory cytokines, respectively. (B) Exposure to PM induced polarization towards M1 (exposure regime 1) or M2 (exposure regime 2) is associated with various respiratory and systemic immune disorders.
Figure 2.
Impacts of PM on macrophage polarization and disease outcomes. (A) There are two kinds of macrophages. Classically activated macrophages (M1) and alternatively activated macrophages (M2) are functionality corresponding to the Th1/Th2 paradigm. Macrophages differentiate into M1 type in response to INF-γ from Th1 cells and LPS, and M2 type in response to IL-4 and IL-13 from Th2 cells. Both M1 and M2 macrophages have a distinct phenotype, and they release pro-and anti-inflammatory cytokines, respectively. (B) Exposure to PM induced polarization towards M1 (exposure regime 1) or M2 (exposure regime 2) is associated with various respiratory and systemic immune disorders.

Figure 3.
Schematic diagram of mechanisms of PM-induced oxidative stress and inflammation in the lung. When PM is inhaled, oxidative stress is initially triggered in the airways. Redox-active chemicals are transition metals, quinones, and organic components like PAH, which are responsible for the generation of ROS and stimulate the production of endogenous through AhR-induced CYP and NADPH oxidase. Moreover, the AhR-XRE system can enhance the production of proinflammatory cytokines. In spite of the Nrf2-ARE-associated antioxidant system for antioxidant enzymes and phase II enzymes, it fails to activate protection against the oxidative and proinflammatory burst, leading to lung and extrapulmonary tissue damages during PM-induced hypersensitivity.
Figure 3.
Schematic diagram of mechanisms of PM-induced oxidative stress and inflammation in the lung. When PM is inhaled, oxidative stress is initially triggered in the airways. Redox-active chemicals are transition metals, quinones, and organic components like PAH, which are responsible for the generation of ROS and stimulate the production of endogenous through AhR-induced CYP and NADPH oxidase. Moreover, the AhR-XRE system can enhance the production of proinflammatory cytokines. In spite of the Nrf2-ARE-associated antioxidant system for antioxidant enzymes and phase II enzymes, it fails to activate protection against the oxidative and proinflammatory burst, leading to lung and extrapulmonary tissue damages during PM-induced hypersensitivity.


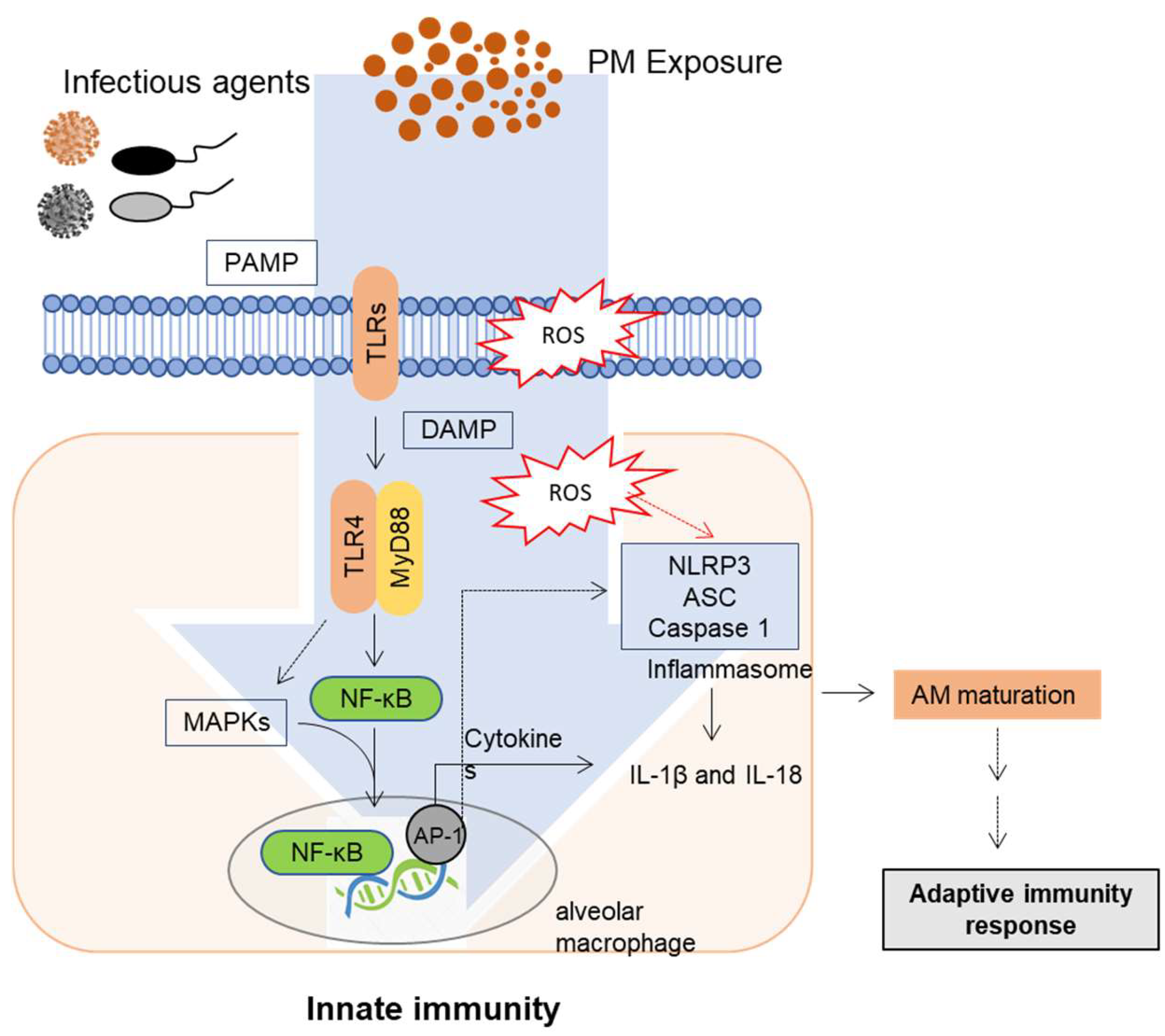{kind=link}
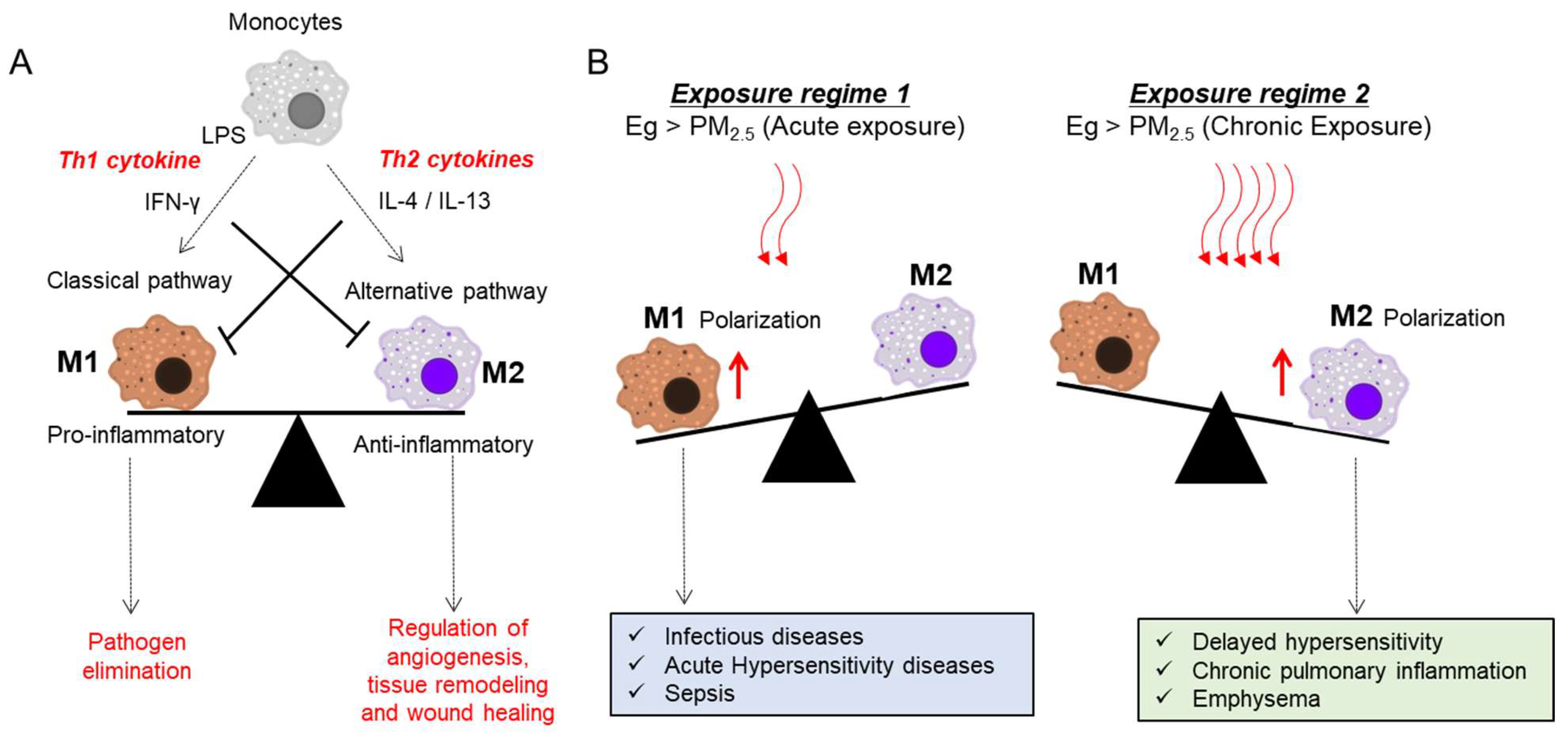{kind=link}
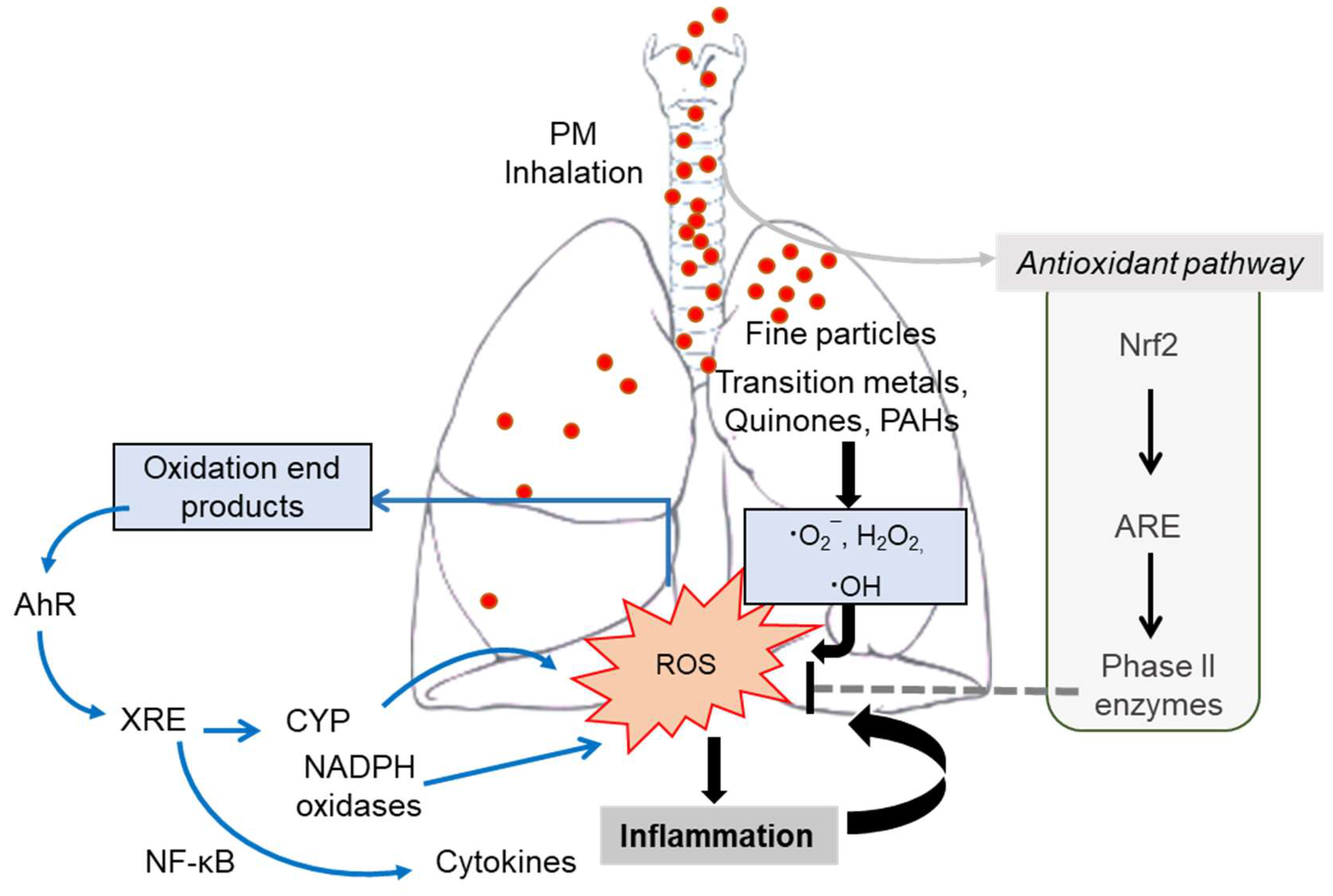{kind=link}
Table 1.
Main characteristics, primary sources, and properties of each particulate matter (PM) fraction.
Table 1.
Main characteristics, primary sources, and properties of each particulate matter (PM) fraction.
| Sl. No. | Characteristics | Ultrafine Particles (PM0.1) | Fine Particles (PM2.5) | Coarse Mode Particles (PM10) |
|---|---|---|---|---|
| 1. | Diameter | ≤ 0.1 μm | 0.1–2.5 μm | 2.5–10 μm |
| 2. | Sources | Diesel and automobile exhaust, emissions from the combustion of gas stove, vented gas dryer and candle, electric motors, residential burning | Emissions from the combustion of gasoline, oil, diesel fuel, wood burning, and coal burning | Abraded soil, dust from road and construction sites, landfills and agriculture, wildfires and brush/waste burning, industrial sources, fungi and bacteria, endotoxins, and pollen |
| 3. | Atmospheric half-life | Minutes to hours | Days to weeks | Minutes to days |
| 4. | Ability to travel (km) | 1 to 10 | 100 to 1000 | 1 to 100 |
| 5. | Redox activity | High | Medium | Low |
| 6. | Transition metal | Low | High | Medium |
| 7. | Polyaromatic hydrocarbon | High | Low | Low |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nagappan, A.; Park, S.B.; Lee, S.-J.; Moon, Y. Mechanistic Implications of Biomass-Derived Particulate Matter for Immunity and Immune Disorders. Toxics 2021, 9, 18. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics9020018
AMA Style
Nagappan A, Park SB, Lee S-J, Moon Y. Mechanistic Implications of Biomass-Derived Particulate Matter for Immunity and Immune Disorders. Toxics. 2021; 9(2):18. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics9020018
Chicago/Turabian StyleNagappan, Arulkumar, Su Bum Park, Su-Jun Lee, and Yuseok Moon. 2021. "Mechanistic Implications of Biomass-Derived Particulate Matter for Immunity and Immune Disorders" Toxics 9, no. 2: 18. https://0-doi-org.brum.beds.ac.uk/10.3390/toxics9020018
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.