
Natural Product and Natural Product-Derived Gamma Secretase Modulators from Actaea Racemosa Extracts

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Novel Gamma Secretase Modulators Based on Black Cohosh
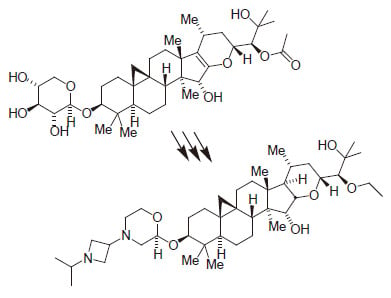
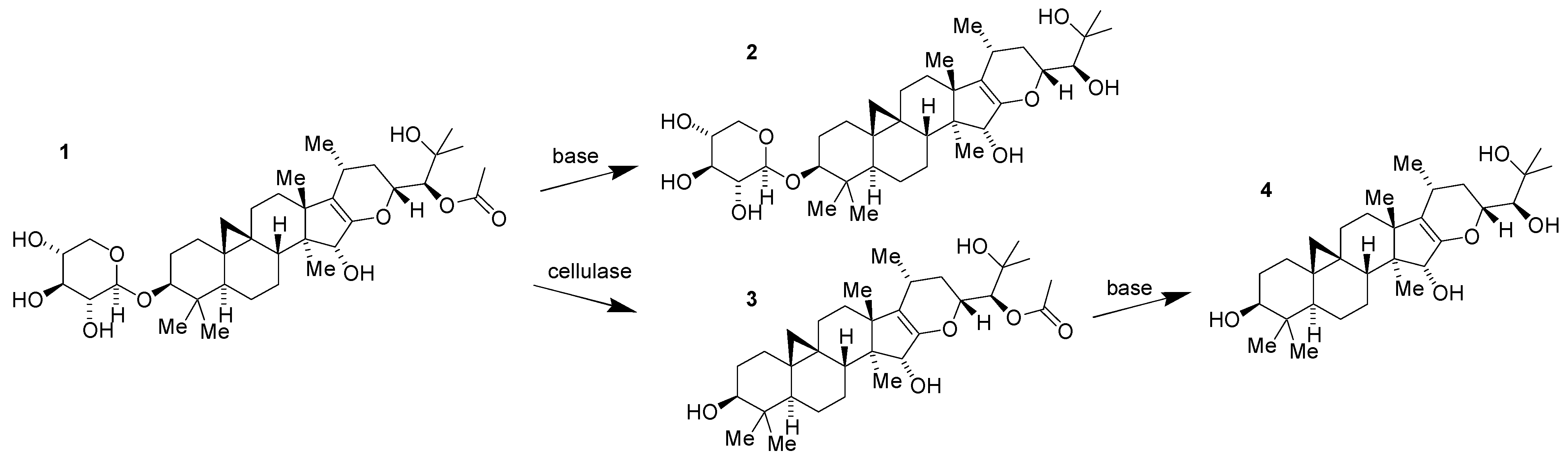
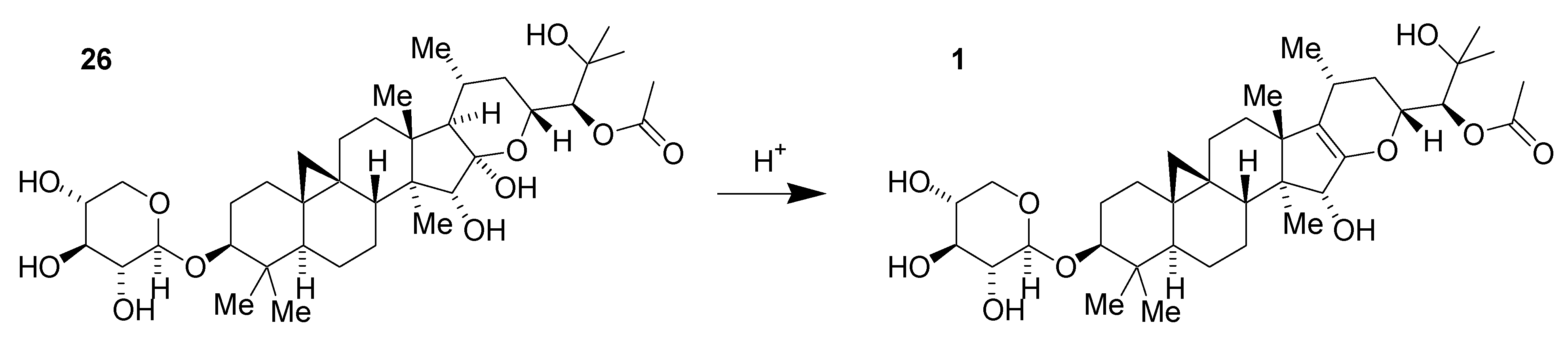
2.1. Isolation of 1 (SPI-014) from Black Cohosh

2.2. Preliminary Structure–Activity Relationships of 1 (SPI-014)





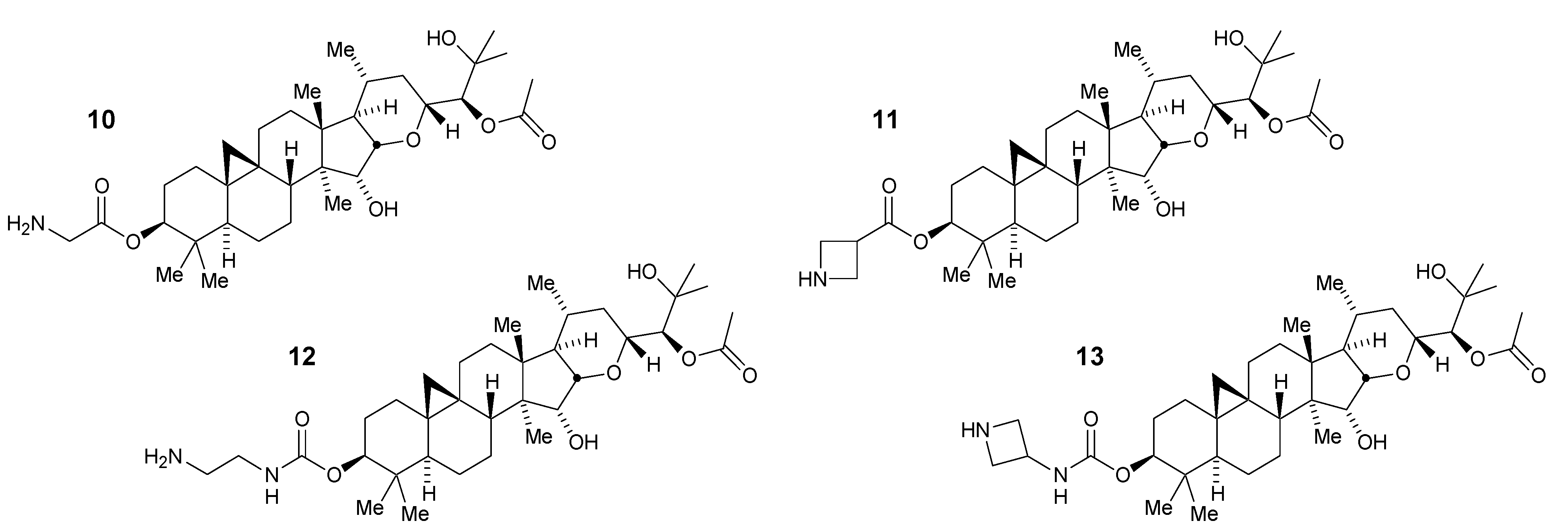
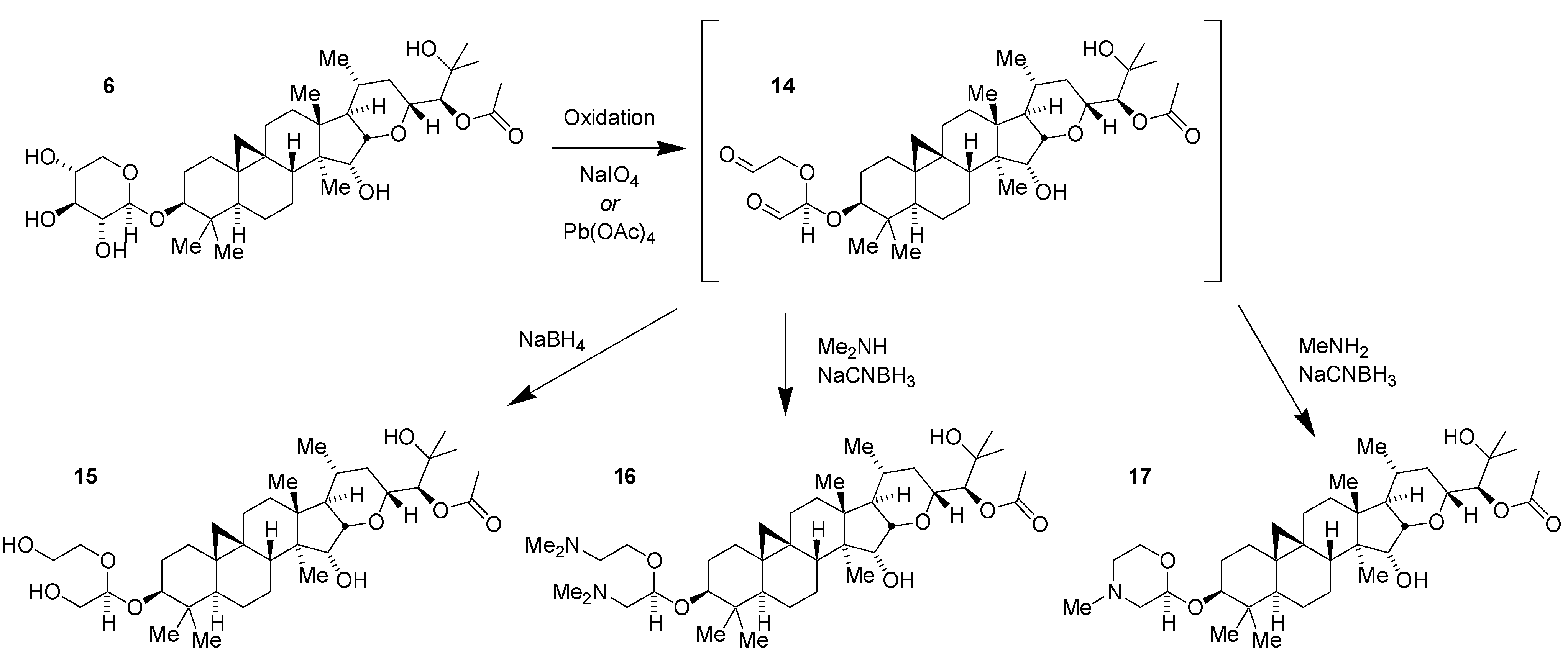
2.3. Lead Optimization


2.4. Gamma Secretase Inhibitors and Modulators—Comparative Pharmacology
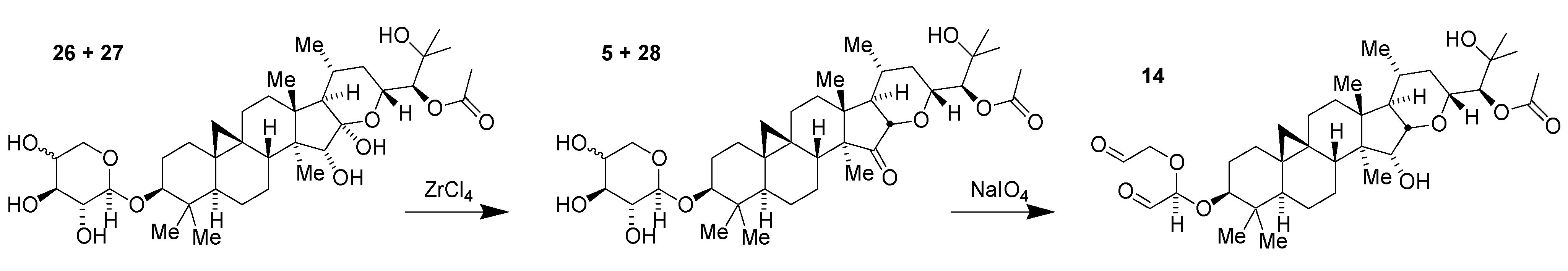
2.5. Scale-Up Chemistry


2.6. Development Status
3. Summary and Future Opportunities
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 2015, 11, 332–384. [Google Scholar]
- Alzheimer’s Disease International. Available online: http://www.alz.co.uk/research/statistics (accessed on 3 May 2015).
- Thomas, M.; Issac, M. Alois Alzheimer, a memoir. Trends Neurosci. 1987, 10, 306–307. [Google Scholar] [CrossRef]
- Boller, F.; Forbes, M.M. History of dementia and dementia in history: An overview. J. Neurol. Sci. 1998, 158, 125–133. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Selkoe, D.J. The Molecular Pathology of Alzheimer’s Disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Findeis, M.A. The role of amyloid β peptide 42 in Alzheimer’s Disease. Pharmacol. Ther. 2007, 116, 266–286. [Google Scholar] [CrossRef] [PubMed]
- Weggen, S.; Eriksen, J.L.; Das, P.; Sagi, S.A.; Wang, R.; Pietrzik, C.U.; Findlay, K.A.; Smith, T.E.; Murphy, M.P.; Bulter, T.; et al. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 2001, 414, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Haugabook, S.J.; Yager, D.M.; Eckman, E.A.; Golde, T.E.; Younkin, S.G.; Eckman, C.B. High throughput screens for the identification of compounds that alter the accumulation of the Alzheimer’s amyloid beta peptide (Abeta). J. Neurosci. Methods 2001, 108, 171–179. [Google Scholar] [CrossRef]
- Yager, D.; Watson, M.; Healy, B.; Eckman, E.A.; Eckman, C.B. Natural product extracts that reduce accumulation of the Alzheimer’s amyloid beta peptide: Selective reduction in A beta42. J. Mol. Neurosci. 2002, 19, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.M. Menopause: A standardized isopropanolic black cohosh extract (remifemin) is found to be safe and effective for menopausal symptoms. Holist. Nurs. Pract. 2012, 26, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Laakmann, E.; Grajecki, D.; Doege, K.; Eulenburg, C.Z.; Buhling, K.J. Efficacy of Cimicifuga racemosa, Hypericum perforatum and Agnus castus in the treatment of climacteric complaints: A systematic review. Gynecol. Endocrinol. 2012, 281, 703–709. [Google Scholar] [CrossRef] [PubMed]
- American Botanical Council. Taking a Closer Look at the US Black Cohosh Rhizome Trade. Available online: http://cms.herbalgram.org/heg/volume7/12December/BlackCohoshMono.html? ts=1431907669&signature=b40a8731dc3d427d2504227f6ffa2586 (accessed on 16 May 2015).
- Prendy, M.L.; de Angelis, P.; Chamberlain, J.L. Black Cohosh Actaea Racemosa: An Annotated Bibliography; General Technical Reports, SRS–97; Department of Agriculture Forest Service, Southern Research Station: Asheville, NC, USA, 2006; p. 99. Available online: http://www.srs.fs.usda.gov/ pubs/gtr/gtr_srs097.pdf (accessed on 16 May 2015).
- Findeis, M.A.; Schroeder, F.; McKee, T.D.; Yager, D.; Fraering, P.C.; Creaser, S.P.; Austin, W.F.; Clardy, J.; Wang, R.; Selkoe, D.; et al. Discovery of a novel pharmacological and structural class of gamma secretase modulators derived from the extract of Actaea racemosa. ACS Chem. Neurosci. 2012, 3, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Lupu, R.; Mehmi, I.; Atlas, E.; Tsai, M.S.; Pisha, E.; Oketch-Rabah, H.A.; Nuntanakorn, P.; Kennelly, E.J.; Kronenberg, F. Black cohosh, a menopausal remedy, does not have estrogenic activity and does not promote breast cancer cell growth. Int. J. Oncol. 2003, 23, 1407–1412. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Khan, I.A.; Willett, K.L.; Foran, C.M. In vivo effects of black cohosh and genistein on estrogenic activity and lipid peroxidation in Japanese Medaka (Oryzias latipes). J. Herb. Pharmacother. 2003, 3, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Amato, P.; Christophe, S.; Mellon, P.L. Estrogenic activity of herbs commonly used as remedies for menopausal symptoms. Menopause 2002, 9, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Liske, E. Physiological Investigation of a Unique Extract of Black Cohosh (Cimicifuga racemosa rhizome): A 6-month Clinical Study Demonstrates No Systemic Estrogenic Effect. J. Womens Health Gend. Based Med. 2002, 11, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Seidlova-Wuttke, D.; Hesse, O.; Jarry, H.; Christoffel, V.; Spengler, B.; Becker, T.; Wuttke, W. Evidence for selective estrogen receptor modulator activity in a black cohosh (Cimicifuga racemosa) extract: Comparison with estradiol-17beta. Eur. J. Endocrinol. 2003, 149, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Austin, W.F.; Hubbs, J.L.; Fuller, N.O.; Creaser, S.P.; McKee, T.D.; Loureiro, R.M.B.; Findeis, M.A.; Tate, B.; Ives, J.L.; Bronk, B.S. SAR investigations on a novel class of gamma-secretase modulators based on a unique scaffold. Med. Chem. Commun. 2013, 4, 569–574. [Google Scholar] [CrossRef]
- Fuller, N.O.; Hubbs, J.L.; Austin, W.F.; Creaser, S.P.; McKee, T.D.; Loureiro, R.M.B.; Tate, B.; Xia, W.; Ives, J.L.; Findeis, M.A.; et al. The Initial Optimization of a New Series of Gamma-Secretase Modulators Derived from a Triterpene Glycoside. ACS Med. Chem. Lett. 2012, 3, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Hubbs, J.L.; Fuller, N.O.; Austin, W.F.; Shen, R.; Creaser, S.P.; McKee, T.D.; Loureiro, R.M.B.; Tate, B.; Xia, W.; Ives, J.; et al. Optimization of a Natural Product-Based Class of γ-Secretase Modulators. J. Med. Chem. 2012, 55, 9270–9282. [Google Scholar] [CrossRef] [PubMed]
- Tate, B.; McKee, T.D.; Loureiro, R.M.; Dumin, J.A.; Xia, W.; Pojasek, K.; Austin, W.F.; Fuller, N.O.; Hubbs, J.L.; Shen, R.; et al. Modulation of gamma-secretase for the treatment of Alzheimer’s disease. Int. J. Alzheimers Dis. 2012. Available online: http://www.hindawi.com/journals/ijad/2012/210756/ (accessed on 16 May 2015). [Google Scholar] [CrossRef]
- Hubbs, J.L.; Fuller, N.O.; Austin, W.F.; Shen, R.; Ma, J.; Gong, Z.; Li, J.; McKee, T.D.; Loureiro, R.M.B.; Tate, B.; et al. Minimization of drug-drug interaction risk and candidate selection in a natural product-based class of gamma-secretase modulators. Bioorg. Med. Chem. Lett. 2015, 25, 1621–1626. [Google Scholar] [CrossRef] [PubMed]
- McKee, T.D.; Loureiro, R.M.B.; Dumin, J.A.; Zarayskiy, V.; Tate, B. An improved cell-based method for determining the γ-secretase enzyme activity against both Notch and APP substrates. J. Neurosci. Methods 2013, 213, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, R.M.; Dumin, J.A.; McKee, T.D.; Austin, W.F.; Fuller, N.O.; Hubbs, J.L.; Shen, R.; Jonker, J.; Ives, J.; Bronk, B.S.; et al. Efficacy of SPI-1865, a novel gamma-secretase modulator, in multiple rodent models. Alzheimers Res. Ther. 2013, 5. Available online: http://alzres.com/content/pdf/alzrt173.pdf (accessed on 16 May 2015). [Google Scholar] [CrossRef]
- Burton, C.R.; Meredith, J.E.; Barten, D.M.; Goldstein, M.E.; Krause, C.M.; Kieras, C.J.; Sisk, L.; Iben, L.G.; Polson, C.; Thompson, M.W.; et al. The amyloid-β rise and γ-secretase inhibitor potency depend on the level of substrate expression. J. Biol. Chem. 2008, 283, 22992–23003. [Google Scholar] [CrossRef] [PubMed]
- Ebke, A.; Luebbers, T.; Fukumori, A.; Shirotani, K.; Haass, C.; Baumann, K.; Steiner, H. Novel γ-secretase enzyme modulators directly target presenilin protein. J. Biol. Chem. 2011, 286. Available online: http://www.jbc.org/content/286/43/ 37181.full.pdf+html (accessed on 21 June 2015). [Google Scholar] [CrossRef]
- Wolfe, M.S. γ-Secretase inhibitors and modulators for Alzheimer’s disease. J. Neurochem. 2012, 120 (Suppl. 1), 89–98. [Google Scholar] [CrossRef] [PubMed]
- Ohki, Y.; Higo, T.; Uemura, K.; Shimada, N.; Osawa, S.; Berezovska, O.; Yokoshima, S.; Fukuyama, T.; Tomita, T.; Iwatsubo, T. Phenylpiperidine-type γ-secretase modulators target the transmembrane domain 1 of presenilin 1. EMBO J. 2011, 30. [Google Scholar] [CrossRef] [PubMed]
- Weggen, S.; Beher, D. Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer’s disease. Alzheimers Res. Ther. 2012, 4. Available online: http://www.alzres.com/content/pdf/ alzrt107.pdf (accessed on 21 June 2015). [Google Scholar] [CrossRef]
- Takeo, K.; Watanabe, N.; Tomita, T.; Iwatsubo, T. Contribution of the γ-secretase subunits to the formation of catalytic pore of presenilin 1 protein. J. Biol. Chem. 2012, 287. Available online: http://www.jbc.org/content/287/31/25834.full.pdf+html (accessed on 21 June 2015). [Google Scholar] [CrossRef]
- Shen, R.; Fuller, N.O.; Osswald, G.; Austin, W.F.; Hubbs, J.L.; Creaser, S.P.; Findeis, M.A.; Ives, J.L.; Bronk, B.S. Multikilogram-Scale Production of Cycloartenol Triterpenoid Glycosides as Synthetic Intermediates for a Gamma Secretase Modulator. Org. Process Res. Dev. 2014, 18, 676–682. [Google Scholar] [CrossRef]
- Fuller, N.O.; Hubbs, J.L.; Austin, W.F.; Shen, R.; Ives, J.L.; Osswald, G.; Bronk, B.S. Optimization of a Kilogram-Scale Synthesis of a Potent Cycloartenol Triterpenoid-Derived γ-Secretase Modulator. Org. Process Res. Dev. 2014, 18, 683–692. [Google Scholar] [CrossRef]
- Findeis, M.A.; Pal, K.; Schroeder, F. Compounds Useful for Treating Neurodegenerative Disorders. US 7,851,641, 4 December 2013. [Google Scholar]
- Findeis, M.A. Synthesis of Compounds Useful as Modulators of Amyloid-beta Production. US 8,263,755, 15 May 2013. [Google Scholar]
- Findeis, M.A.; Pal, K.; Schroeder, F. Compounds Useful for Treating Neurodegenerative Disorders. PCT Int. Appl. WO 06124956 A1 061123, 23 November 2006. [Google Scholar]
- Findeis, M.A. Compounds Useful for Treating Neurodegenerative Disorders. PCT Int. Appl WO 08063165 A1 080529, 29 May 2008. [Google Scholar]
- Findeis, M.A.; Creaser, S.P. Modulators of Amyloid Beta Production. PCT Int. Appl WO 08130449 A2 081030, 30 October 2008. [Google Scholar]
- Findeis, M.A. Synthesis of Compounds Useful as Modulators of Amyloid-beta Production. PCT Int. Appl. WO 08136863 A2 081113, 13 November 2008. [Google Scholar]
- Bronk, B.S.; Austin, W.F.; Creaser, S.P.; Findeis, M.A.; Fuller, N.O.; Hubbs, J.L.; Ives, J.L.; Shen, R. Compounds Useful for Treating Neurodegenerative Disorders. PCT Int. Appl. WO 2011/109657 A1 110909, 9 September 2011. [Google Scholar]
- Fierce Biotech. Available online: http://www.fiercebiotech.com/story/satori-pharmaceuticals shuts-down-after-failure-alzheimers-drug/2013-05-30 (accessed on 16 May 2015).
- CrunchBase. Available online: https://www.crunchbase.com/organization/satori-pharmaceuticals (accessed on 16 May 2015).
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Findeis, M.A.; Schroeder, F.C.; Creaser, S.P.; McKee, T.D.; Xia, W. Natural Product and Natural Product-Derived Gamma Secretase Modulators from Actaea Racemosa Extracts. Medicines 2015, 2, 127-140. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines2030127
Findeis MA, Schroeder FC, Creaser SP, McKee TD, Xia W. Natural Product and Natural Product-Derived Gamma Secretase Modulators from Actaea Racemosa Extracts. Medicines. 2015; 2(3):127-140. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines2030127
Chicago/Turabian StyleFindeis, Mark A., Frank C. Schroeder, Steffen P. Creaser, Timothy D. McKee, and Weiming Xia. 2015. "Natural Product and Natural Product-Derived Gamma Secretase Modulators from Actaea Racemosa Extracts" Medicines 2, no. 3: 127-140. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines2030127