
The Changing Landscape of Breast Cancer: How Biology Drives Therapy


Abstract
:
1. Introduction

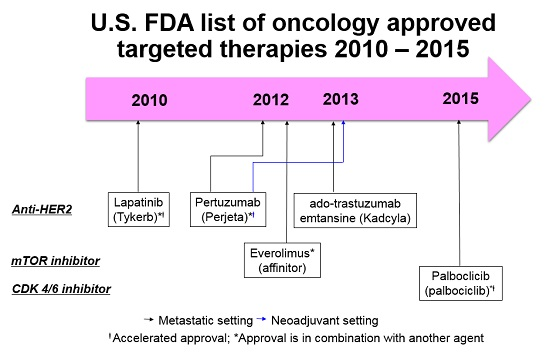
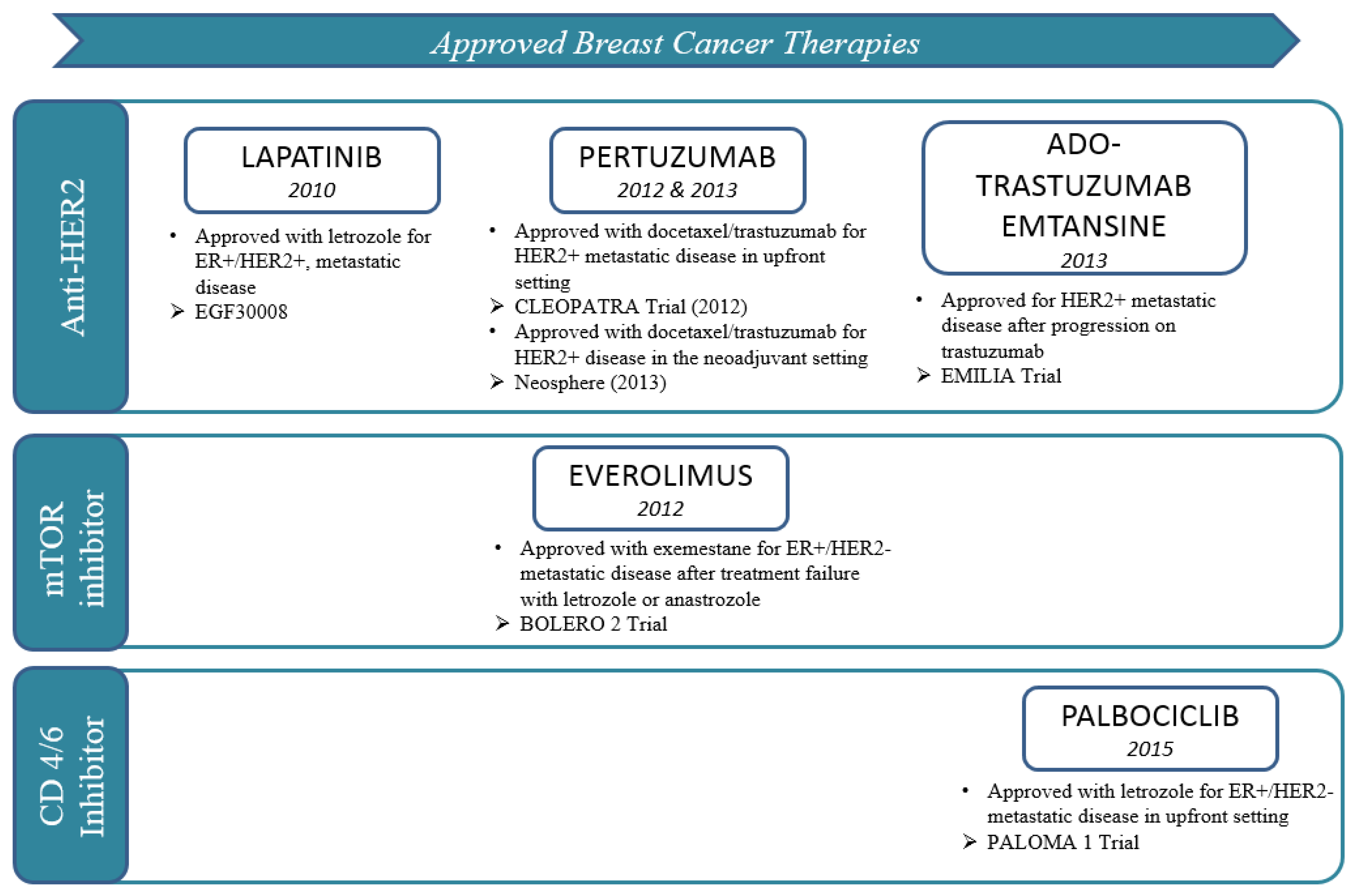
2. PI3K/AKT/mTOR Pathway: Everolimus, a Paradigm for Overcoming Endocrine Resistance
3. Cell Cycle Regulatory Machinery: Palbociclib, a Paradigm for Synergy with Endocrine Therapy

| Trials with CDK 4/5 Inhibitors | |||||
| Study Name/Identifier | Drug(s)/Novel Agent(s) | Study Phase | N | Primary Endpoint | Disease Setting |
| PALOMA-2 NCT01740427 | Letrozole +/− Palbociclib | II | 650 | PFS | Front-line, advanced/metastatic |
| PENELOPE-B NCT1864746 | Palbociclib + Endocrine | III | 800 | DFS | Residual disease after neoadjuvant |
| PEARL NCT02028507 | Palbociclib + exemestane vs. capecitabine | III | 348 | PFS | Metastatic after progression on AI |
| neoMONARCH NCT02441946 | Abemaciclib + Anastrozole vs. abemaciclib vs. anastrozole | II | 220 | Δ Ki-67 at 2 weeks | Neoadjuvant, Stage I-III |
| MONARCH NCT2107703 | Fulvestrant +/− Abemaciclib | III | 630 | PFS | Front-line, advanced/metastatic |
| MONALEESA-3 NCT02422615 | Fulvestrant +/− Ribociclib | III | 660 | PFS | 1st or 2nd line, advanced/metastatic |
| PALLAS NCT02513394 | Endocrine +/− Palbociclib | III | 4600 | DFS | Adjuvant, Stage II-III |
| Trials with CDK aurora kinase inhibitors | |||||
| Study Name/Identifier | Drug(s)/Novel Agent(s) | Study Phase | N | Primary Endpoint | Disease Setting |
| 2076-CL-005 NCT01639248 | ENMD-2076 | II | 37 | CBR | Metastatic TNBC |
| 13-033 NCT02187991 | Paclitaxel +/− Alisertib | II | 252 | TTP | ER+ or −/HER2−, locally recurrent or metastatic |
4. Antibodies beyond Trastuzumab: Pertuzumab and Ado-Trastruzumab-Emtansine (T-DM1)
| Immune Checkpoint Inhibitors | |||||
| Study Name/NCT Identifier | Drug(s)/Novel Agent(s) | Study Phase | N | Primary Endpoint | Disease Setting |
| TONIC NCT02499367 | Nivolumab | II | 84 | PFS | TNBC, ≥2nd-line Metastatic |
| 4147523 NCT02395627 | Pembroluzimab | II | 58 | ORR | Postmenopausal ER+, ≥2nd-line Metastatic |
| Vaccine, Small Molecule Inhibitors & Others | |||||
| Study Name/Identifier | Drug(s)/Novel Agent(s) | Study Phase | N | Primary Endpoint | Disease Setting |
| 11-202 NCT01570036 | NeuVax | II | 300 | DFS | HER2+, Adjuvant |
| OSU 13117 NCT01964924 | Trametinib + GSK2141795 | II | 41 | ORR | TNBC, ≥2nd-line Metastatic |
| NYU 11-00598 NCT01421017 | Imiquimod | I/II | 55 | ORR | ≥2nd-line; + skin lesion, advance/metastatic |
5. Challenges and Pitfalls Developing Targeted Agents for Breast Cancer: Lessons from Bevacizumab and Iniparib
5.1. An Almost Failed Attempt Targeting Angiogenesis with Bevacizumab
| TKI with Anti-Angiogenic Properties | |||||
| Study Name/NCT Identifier | Drug(s)/Novel Agent(s) | Study Phase | N | Primary Endpoint | Study Outcome |
| SCRI BRE 122 NCT00887575 | Neoadjuvant sunitinib + paclitaxel/carboplatin | I/II | 54 | pCR | Combo not recommended |
| ZACFAST NCT00752986 | Fulvestrant +/− vandetanib | II | 41 | EFS | Terminated |
| NSABP FB-6 NCT00849472 | Neoadjuvant AC ➔ +/− pazopanib | II | 101 | pCR | Increased toxicity; combo not recommended |
| A4061010 NCT00076024 | Docetaxel +/− Axitinib | I/II | 174 | TTP | Not significant |
| RESILIENCE NCT01234337 | Capecitabine +/− sorafenib | III | 519 | PFS | No advantage |
| Monoclonal Antibody | |||||
| Study Name/NCT Identifier | Drug(s)/Novel Agent(s) | Study Phase | N | Primary Endpoint | Study Outcome |
| Rose/TRIO-12 NCT00703326 | Docetaxel +/− ramucirumab | III | 1144 | PFS | No OS advantage |
5.2. Missing the Mark with the PARP Inhibitor, Iniparib
| Trials Focused Primarily in Breast Cancer with Brca Mutation | |||||
| Study Name/NCT Identifier | Drug(s)/Novel Agent(s) | Study Phase | N | Primary Endpoint | Disease Setting |
| EMBRACA Study NCT01945775 | Talazoparib vs. Physician’s Choice | III | 429 | PFS | Metastatic breast cancer patients with BRCA mutation |
| 2014-0045 NCT02282345 | Talazoparib | II | 20 | Toxicity, safety | Neoadjuvant, +BRCA mutation |
| Trials in Breast (with or without BRCA Mutation) and other Malignancies | |||||
| Study Name/NCT Identifier | Drug(s)/Novel Agent(s) | Study Phase | N | Primary Endpoint | Disease Setting |
| OlympiA NCT02032823 | Olaparib | III | 1320 | DFS | Adjuvant, TNBC in high risk BRCA 1/2 |
| ComPAKT NCT02338622 | Olaparib + AKT inhibitor (AZD5363) | I | 58 | Safety, tolerability | Advanced solid tumors, BRCA 1/2 mutation, TNBC or hyperactive PI3K-AKT pathway |
6. The Future of Novel Targeted Therapies: The Promise of Great Hope for Our Patients

{kind=link}
{kind=link}
{kind=link}

7. Conclusions
Author Contributions
Conflicts of Interest
References
- Bonotto, M.; Gerratana, L.; Iacono, D.; Minisini, A.M.; Rihawi, K.; Fasola, G.; Puglisi, F. Treatment of Metastatic Breast Cancer in a Real-World Scenario: Is Progression-Free Survival With First Line Predictive of Benefit From Second and Later Lines? Oncologist 2015, 20, 719–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonotto, M.; Gerratana, L.; Poletto, E.; Driol, P.; Giangreco, M.; Russo, S.; Minisini, A.M.; Andreetta, C.; Mansutti, M.; Pisa, F.E.; et al. Measures of outcome in metastatic breast cancer: Insights from a real-world scenario. Oncologist 2014, 19, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Xu, X.Y. PI3K/Akt/mTOR signaling pathway in cancer stem cells: From basic research to clinical application. Am. J. Cancer Res. 2015, 5, 1602–1609. [Google Scholar] [PubMed]
- Riggins, R.B.; Schrecengost, R.S.; Guerrero, M.S.; Bouton, A.H. Pathways to tamoxifen resistance. Cancer Lett. 2007, 256, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Yardley, D.A.; Noguchi, S.; Pritchard, K.I.; Burris, H.A., 3rd; Baselga, J.; Gnant, M.; Hortobagyi, G.N.; Campone, M.; Pistilli, B.; Piccart, M.; et al. Everolimus plus exemestane in postmenopausal patients with HR+ breast cancer: BOLERO-2 final progression-free survival analysis. Adv. Ther. 2013, 30, 870–884. [Google Scholar] [CrossRef] [PubMed]
- Piccart, M.; Hortobagyi, G.N.; Campone, M.; Pritchard, K.I.; Lebrun, F.; Ito, Y.; Noguchi, S.; Perez, A.; Rugo, H.S.; Deleu, I.; et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: Overall survival results from BOLERO-2. Ann. Oncol. 2014, 25, 2357–2362. [Google Scholar] [CrossRef] [PubMed]
- Bachelot, T.; Bourgier, C.; Cropet, C.; Ray-Coquard, I.; Ferrero, J.M.; Freyer, G.; Abadie-Lacourtoisie, S.; Eymard, J.C.; Debled, M.; Spaëth, D.; et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: A GINECO study. J. Clin. Oncol. 2012, 30, 2718–2724. [Google Scholar] [CrossRef] [PubMed]
- Treilleux, I.; Arnedos, M.; Cropet, C.; Wang, Q.; Ferrero, J.M.; Abadie-Lacourtoisie, S.; Levy, C.; Legouffe, E.; Lortholary, A.; Pujade-Lauraine, E.; et al. Translational studies within the TAMRAD randomized GINECO trial: Evidence for mTORC1 activation marker as a predictive factor for everolimus efficacy in advanced breast cancer. Ann. Oncol. 2015, 26, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Lazar, A.A.; Bondarenko, I.; Garin, A.M.; Brincat, S.; Chow, L.; Sun, Y.; Neskovic-Konstantinovic, Z.; Guimaraes, R.C.; Fumoleau, P.; et al. Randomized phase III placebo-controlled trial of letrozole plus oral temsirolimus as first-line endocrine therapy in postmenopausal women with locally advanced or metastatic breast cancer. J. Clin. Oncol. 2013, 31, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; O’Regan, R.; Ozguroglu, M.; Toi, M.; Xu, B.; Jerusalem, G.; Masuda, N.; Wilks, S.; Arena, F.; Isaacs, C.; et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014, 15, 580–591. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Andre, F.; Jiang, Z.; Shao, Z.; Mano, M.S.; Neciosup, S.P.; Tseng, L.M.; Zhang, Q.; Shen, K.; Liu, D.; et al. Combination of everolimus with trastuzumab plus paclitaxel as first-line treatment for patients with HER2-positive advanced breast cancer (BOLERO-1): A phase 3, randomised, double-blind, multicentre trial. Lancet Oncol. 2015, 16, 816–829. [Google Scholar] [CrossRef]
- Tamura, K. Development of cell-cycle checkpoint therapy for solid tumors. Jpn. J. Clin. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [PubMed]
- Clinicaltrials.gov. Available online: http://clinicaltrials.gov/ (accessed on 12 October 015).
- Baselga, J.; Swain, S.M. Novel anticancer targets: Revisiting ERBB2 and discovering ERBB3. Nat. Rev. Cancer 2009, 9, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.; Diéras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Baselga, J.; Kim, S.B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.; Schneeweiss, A.; Heeson, S.B.; et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Cameron, D.; Casey, M.; Oliva, C.; Newstat, B.; Imwalle, B.; Geyer, C.E. Lapatinib plus capecitabine in women with HER2-positive advanced breast cancer: Final survival analysis of a phase III randomized trial. Oncologist 2010, 15, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Scheuer, W.; Friess, T.; Burtscher, H.; Bossenmaier, B.; Endl, J.; Hasmann, M. Strongly enhanced antitumor activity of trastuzumab and pertuzumab combination treatment on HER2-positive human xenograft tumor models. Cancer Res. 2009, 69, 9330–9336. [Google Scholar] [CrossRef] [PubMed]
- Gianni, L.; Pienkowski, T.; Im, Y.H.; Roman, L.; Tseng, L.M.; Liu, M.C.; Lluch, A.; Staroslawska, E.; de la Haba-Rodriguez, J.; Im, S.A.; et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): A randomised multicentre, open-label, phase 2 trial. Lancet Oncol. 2012, 13, 25–32. [Google Scholar] [PubMed]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 362, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.; Wang, M.; Gralow, J.; Dickler, M.; Cobleigh, M.; Perez, E.A.; Shenkier, T.; Cella, D.; Davidson, N.E. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med. 2007, 357, 2666–2676. [Google Scholar] [CrossRef] [PubMed]
- Bear, H.D.; Tang, G.; Rastogi, P.; Geyer, C.E. Jr.; Liu, Q.; Robidoux, A.; Baez-Diaz, L.; Brufsky, A.M.; Mehta, R.S.; Fehrenbacher, L.; et al. Neoadjuvant plus adjuvant bevacizumab in early breast cancer (NSABP B-40 [NRG Oncology]): Secondary outcomes of a phase 3, randomised controlled trial. Lancet Oncol. 2015, 16, 1037–1048. [Google Scholar] [CrossRef]
- Cameron, D.; Brown, J.; Dent, R.; Jackisch, C.; Mackey, J.; Pivot, X.; Steger, G.G.; Suter, T.M.; Toi, M.; Parmar, M.; et al. Adjuvant bevacizumab-containing therapy in triple-negative breast cancer (BEATRICE): Primary results of a randomised, phase 3 trial. Lancet Oncol. 2013, 14, 933–942. [Google Scholar] [PubMed]
- Slamon, D.; Swain, S.M.; Buyse, M.; Martin, M.; Geyer, C.E.; Im, Y.-H.; Pienkowski, T.; Kim, S.-B.; Robert, N.J.; Steger, G.; et al. Abstract S1-03: Primary results from BETH, a phase 3 controlled study of adjuvant chemotherapy and trastuzumab ± bevacizumab in patients with HER2-positive, node-positive or high risk node-negative breast cancer. Cancer Res. 2013. [Google Scholar] [CrossRef]
- Hein, A.; Lambrechts, D.; von Minckwitz, G.; Haberle, L.; Eidtmann, H.; Tesch, H.; Untch, M.; Hilfrich, J.; Schem, C.; Rezai, M.; et al. Genetic variants in VEGF pathway genes in neoadjuvant breast cancer patients receiving bevacizumab: Results from the randomized phase 3 GeparQuinto study. Int. J. Cancer 2015. [Google Scholar] [CrossRef] [PubMed]
- Seimiya, H. Cancer therapy by PARP inhibitors. Nih. Rinsho Jpn. J. Clin. Med. 2015, 73, 1330–1335. [Google Scholar]
- Telli, M.L.; Jensen, K.C.; Vinayak, S.; Kurian, A.W.; Lipson, J.A.; Flaherty, P.J.; Timms, K.; Abkevich, V.; Schackmann, E.A.; Wapnir, I.L.; et al. Phase II Study of Gemcitabine, Carboplatin, and Iniparib As Neoadjuvant Therapy for Triple-Negative and BRCA 1/2 Mutation-Associated Breast Cancer With Assessment of a Tumor-Based Measure of Genomic Instability: PrECOG 0105. J. Clin. Oncol. 2015, 33, 1895–1901. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J.; Osborne, C.; Pippen, J.E.; Yoffe, M.; Patt, D.; Rocha, C.; Koo, I.C.; Sherman, B.M.; Bradley, C. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N. Engl. J. Med. 2011, 364, 205–214. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J.; Schwartzberg, L.; Danso, M.A.; Miller, K.D.; Rugo, H.S.; Neubauer, M.; Robert, N.; Hellerstedt, B.; Saleh, M.; Richards, P.; et al. Phase III study of iniparib plus gemcitabine and carboplatin versus gemcitabine and carboplatin in patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2014, 32, 3840–3847. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Cao, C.; Geng, X.; Tian, B.; Wu, Y.; Zhang, C.; Chen, Z.; Li, W.; Shen, H.; Ying, S. Effectiveness and safety of poly (ADP-ribose) polymerase inhibitors in cancer therapy: A systematic review and meta-analysis. Oncotarget 2015. [Google Scholar] [CrossRef]
- Zhou, Q.; Atadja, P.; Davidson, N.E. Histone deacetylase inhibitor LBH589 reactivates silenced estrogen receptor alpha (ER) gene expression without loss of DNA hypermethylation. Cancer Biol. Ther. 2007, 6, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Yardley, D.A.; Ismail-Khan, R.R.; Melichar, B.; Lichinitser, M.; Munster, P.N.; Klein, P.M.; Cruickshank, S.; Miller, K.D.; Lee, M.J.; Trepel, J.B. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J. Clin. Oncol. 2013, 31, 2128–2135. [Google Scholar] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friend, S.; Royce, M. The Changing Landscape of Breast Cancer: How Biology Drives Therapy. Medicines 2016, 3, 2. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines3010002
Friend S, Royce M. The Changing Landscape of Breast Cancer: How Biology Drives Therapy. Medicines. 2016; 3(1):2. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines3010002
Chicago/Turabian StyleFriend, Sarah, and Melanie Royce. 2016. "The Changing Landscape of Breast Cancer: How Biology Drives Therapy" Medicines 3, no. 1: 2. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines3010002